

Abstract
Human norovirus exacts considerable public health and economic losses worldwide. Emerging in vitro cultivation advances are not yet applicable for routine detection of the virus. The current detection and quantification techniques, which rely primarily on nucleic acid amplification, do not discriminate infectious from non-infectious viral particles. The purpose of this article is to present specific details on recent advances in techniques used together in order to acquire further information on the infectivity status of viral particles. One technique involves assessing binding of a norovirus ssDNA aptamer to capsids. Aptamers have the advantage of being easily synthesized and modified, and are inexpensive and stable. Another technique, dynamic light scattering (DLS), has the advantage of observing capsid behavior in solution. Electron microscopy allows for visualization of the structural integrity of the viral capsids. Although promising, there are some drawbacks to each technique, such as non-specific aptamer binding to positively-charged molecules from sample matrices, requirement of purified capsid for DLS, and poor sensitivity for electron microscopy. Nonetheless, when these techniques are used in combination, the body of data produced provides more comprehensive information on norovirus capsid integrity that can be used to infer infectivity, information which is essential for accurate evaluation of inactivation methods or interpretation of virus detection. This article provides protocols for using these methods to discriminate infectious human norovirus particles.
Keywords: Immunology, Issue 129, Virus capsid, norovirus, aptamer, infectivity, dynamic light scattering, transmission electron microscopy
Introduction
Human norovirus is responsible for a considerable public health burden globally, causing about 685 million illnesses and 212,000 deaths annually1, at a cost in the billions of dollars2,3. Today, norovirus detection and quantification involves genome amplification and/or ligand-based platforms. The former is generally preferred as it is more sensitive, provides quantitative information, and has generally lower risk of false positive results4. The most common norovirus detection and quantification genome amplification technique is reverse transcriptase quantitative polymerase chain reaction (RT-qPCR). Because it targets and amplifies a small segment of the human norovirus genome, RT-qPCR alone is not capable of discriminating between infectious and non-infectious particles, as free genomic RNA, damaged capsids containing genomes, and capsids with fatally mutated/truncated genomes can still be amplified by RT-qPCR. While two new in vitro cultivation techniques for human norovirus5,6 have been reported recently, both still rely on RT-qPCR for virus quantification and are not yet feasible methods for routine clinical/environmental testing for human noroviruses. Thus, RT-qPCR remains the gold standard for clinical/environmental testing.
Other in vitro methods have been developed in an effort to better estimate the infectivity status of norovirus particles. These methods can be generally categorized as those that address the integrity of the norovirus capsid and those evaluating genome integrity. The former approach is more popular. A commonly used method is RNase pretreatment prior to extraction of viral genomic RNA, however this does not account for viral particles that remain intact but lack the ability to bind host receptors/co-factors, as well as viral particles that have fatally mutated or have truncated or marginally damaged genomes7. Capsid integrity can also be evaluated based on the ability of the virus to bind to human norovirus co-receptor/co-factors, which are carbohydrates known as histo-blood group antigens (HBGAs). HBGAs are present in host intestinal epithelial cells as part of glycoproteins or glycolipids (among multiple other tissues) and may be secreted in bodily fluids like saliva. Enteric bacteria containing HBGA-like substances on their surfaces have also been shown to bind human norovirus8,9,10, and such interactions may promote norovirus infection5. The basic concept of utilizing HBGA binding is that only viral particles capable of binding the putative receptor/co-factor would be capable of infecting cells. Multiple studies suggest that bead-based HBGA binding preceding nucleic acid amplification is a promising method for infectivity discrimination11,12,13,14,15,16. A major challenge regarding HBGAs is that no single HBGA type can bind all human norovirus genotypes. To address this, porcine gastric mucin, which contains HBGAs, has been used in place of purified HBGA carbohydrates in the interest of ease of synthesis, consistency, and reduced cost.
A recent publication by Moore et al.17 introduced a set of new techniques for estimating human norovirus capsid integrity. In place of HBGAs, Moore et al.17 investigated binding of a broadly reactive nucleic acid aptamer (M6-2)18 and broadly reactive monoclonal antibody (NS14)19 to heat-treated GII.4 Sydney norovirus capsids (virus-like particles or VLPs) and compared this to the binding to synthetic HBGAs. Nucleic acid aptamers are short (~20 - 80 nt) single stranded nucleic acids (ssDNA or RNA) that fold into unique three-dimensional structures as a consequence of their sequence and bind a target. Because they are nucleic acids, they are less costly; easily chemically synthesized, purified, and modified; and stable to heat. Several reports of aptamers generated against noroviruses exist, with some of them showing broad reactivity to a variety of strains18,20,21,22. By way of example, Moore et al.17 demonstrated that aptamer M6-2 bound to purified, assembled human norovirus capsids (VLPs) and behaved similarly to HBGA in the reliance on the viral capsid to maintain higher order (e.g., secondary and tertiary) protein structure for binding to occur. On the other hand, a significant proportion of norovirus VLP binding to antibody NS14 remained after capsids were completely denatured. Evaluation of norovirus binding in the study by Moore et al.17 was done using a simple, plate-based method similar to ELISA, with the exception that aptamers are used in place of antibodies (hence the method was called ELASA). This high throughput experimental method was used to evaluate the effects of different treatments on the norovirus capsid, work that is valuable for understanding the mechanism of viral inactivation upon exposure to physical or chemical stressors. However, one drawback to this method is the lower sensitivity and lack of tolerance for matrix-associated contaminants at higher levels that cause non-specific binding by aptamers.
Moore et al.17 used another method, dynamic light scattering (DLS), to monitor aggregation of viral particles in response to heat treatment. DLS is commonly used to evaluate the size of nanoparticle suspensions and has been extended to proteins23,24 and viruses25,26,27. The intensity of light scattered by particles in solution fluctuates as a function of particle size, allowing for the calculation of diffusion coefficient and then particle diameter using well-established formulae. The DLS technique can distinguish the hydrodynamic diameter of dispersed particles down to the nanoscale, which allows detection of dispersed viruses, individual capsid proteins or dimers, and virus aggregates based on size27. Virus aggregation, represented by an increase in particle size, is indicative of loss of capsid integrity. As the capsid is denatured and its structure disrupted, hydrophobic residues become exposed and cause the particles to stick together and form aggregates. DLS can be used to measure particle size after a specified treatment or the kinetics of aggregation in real time17,28. This method has the advantage of allowing observation of capsid behavior in solution but requires high concentrations of purified capsid, which may not be entirely representative of the virus in its natural state.
The final method utilized by Moore et al.17 was transmission electron microscopy (TEM). Although this method lacks sensitivity and does not produce quantitative data, it allows for visualization of the effects of different treatments on the viral capsid structure. Although not yet ideal for clinical/environmental settings, the use of these methods in combination is valuable in understanding human norovirus inactivation. The purpose of this article is to provide in-depth protocols for the plate-based binding assay (ELASA), DLS, and TEM preparation methods used to investigate the effects of heat treatments on the norovirus capsid in the context of the treatments' effects on capsid integrity as presented in Moore et al.17.
Protocol
1. Plate-based Binding Assay for Evaluating Loss of Higher Order Norovirus Capsid Structure
NOTE: A very similar ELASA protocol to the one presented here has been published29, and similar ELASA assays have previously been reported as part of research articles elsewhere17,18,22.
- Heat treatment of human norovirus GII.4 Sydney capsids
- Obtain purified human norovirus major capsid protein (VP1) of GII.4 Sydney (Accession: JX459908) assembled in capsids. NOTE: Capsids in the study were obtained courtesy of R. Atmar (Baylor College of Medicine, Houston, TX).
- Dilute capsids to 50 µg/mL in 1x phosphate-buffered saline (PBS, pH 7.2) to a maximum volume of 15 µL in individual capped PCR tubes. Ensure that the tube contains at least 600 ng of capsid. Ensure that there is 600 ng of capsid per treatment-ligand combination.
- Preheat thermal cycler to desired heat treatment. NOTE: For the purposes of this protocol, treatment at 68 °C for different times (0 - 25 min) was chosen.
- Preheat an additional thermal cycler to 4 °C for immediate cooling. Add tubes with capsid solution to the thermal cycler for the desired time. Immediately transfer to pre-cooled 4 °C cycler for 5 min after heating.
- Include 95 °C for 5 min treatment as a denatured capsid control. Include untreated (23 °C) capsid solution as a positive control. Include PBS with no capsid as an additional negative control.
- Briefly spin down in a centrifuge to get all droplets to the bottom of the tube and dilute the treated capsid solutions in PBS to 3 µg/mL. Ensure that there is at least 200 µL of diluted, treated capsid (100 µL/well).
Apply 100 µL of capsid solution to each well, ensuring there are at least two wells per treatment. Additionally, include 2 control wells with 100 µL PBS for each ligand; this provides the background absorbance of the assay. Cover the plate with a lid, seal the edges with paraffin film to reduce evaporation, and leave overnight on a shaking incubator at 4 °C or for 2 h at room temperature.
Prepare the blocking buffer by making 5% skim milk solids (w/v) in PBS with 0.05% Tween 20 (PBST).
Remove the treated capsid solutions from the plate. Add 200 µL/well of blocking buffer. Incubate for 2 h at room temperature or overnight, sealed at 4 °C.
- Prepare the ligand binding solutions for the biotinylated aptamer M6-218,29 and biotinylated synthetic HBGA blood type A or biotinylated type H.
- Dilute the biotinylated aptamer to 1 µM in nuclease-free water. Ensure that there is enough solution for 200 µL total for each treatment. Dilute the chosen biotinylated HBGA to 30 µg/mL in the blocking buffer. Ensure that there is enough solution for 200 µL total volume for each treatment. NOTE: To use less HBGA, 10 µg/mL of HBGA in 0.25% skim milk-PBST can be substituted. Concentrations for both biotinylated M6-2 and HBGA have been previously optimized and reported.
Remove blocking buffer and wash the plates 3 times with 200 µL/well PBST. Pat the plate upside down on sterile paper towels to dry the residual moisture.
Add 100 µL/well of the ligand binding solutions, ensuring that there are 2 wells per heat treatment-ligand combination. Incubate the plate covered with a lid on an orbital shaker at about 60 rpm for 1 h at room temperature. NOTE: Be sure to include 2 wells each of the untreated capsids and completely denatured capsids for each ligand as positive and negative controls, respectively. Additionally, include 2 "no capsid" wells for each ligand as the plate background control.
Remove the ligand solutions and wash the wells 3 times with 200 µL/well PBST. Pat the plate upside down on sterile paper towels to dry residual moisture.
Add 100 µL/well of solution of 0.2 µg/mL streptavidin-horseradish peroxidase in PBS. Incubate for 15 min at room temperature with gentle shaking on an orbital shaker. NOTE: Different streptavidin-horseradish peroxidase conjugates will have different concentrations. Consult the user's manual for the ideal range of concentrations for ELISA applications of enzyme and be sure it is optimal.
Remove the conjugate solution. Wash 3 times with 200 µL/well PBST. Pat the plate upside down on sterile paper towels to dry residual moisture.
Add 100 µL/well 3,3',5,5'-tetramethylbenzidine (TMB) substrate, and allow the color to develop for 5 - 15 min. Observe negative control wells for any color and be sure to consistently develop for the same time between replicate plates. Be aware of the absorbance range of the plate reader used, as well. NOTE: The developing times may differ based on the quality of the ligands/reagents used.
Stop the development of the reaction with 100 µL/well 1M phosphoric acid. Immediately read plate at 450 nm. Save the data. NOTE: Introduction of the acid generally slows the reaction drastically but does not completely stop it. Do not leave the stopped plate for an extended amount of time before reading the absorbance.
2. Analysis of Plate-based Assay Results
Save the raw absorbance data in a spreadsheet program. Produce the average absorbances for each well pair with the specific treatment and ligand. Use these averages for all subsequent analyses. NOTE: Compare both well absorbances to be sure that no pairs of wells have significantly disparate values.
To analyze the absolute capsid integrity (all binding), subtract the raw absorbance of the "no capsid" control from every other sample absorbance value. NOTE: Alternatively, loss of binding due to loss of higher order (secondary, tertiary, quaternary) structure can directly be analyzed. Instead of subtracting the absorbance of the negative (no capsid) control, subtract the value of the completely denatured capsid control. This removes all binding signal due to nonspecific binding to the apparatus as well as binding due to the capsid sequence. HBGA and aptamer M6-2 display little binding to completely denatured capsid, so either method produces similar results. False positive signal attributed to nonspecific aptamer binding must be considered when choosing which analysis to use.
With the negative or denatured control-adjusted absorbances, divide the treatment absorbances by their respective positive (no treatment) control absorbances and multiply by 100. This provides the percentage of signal of the treated capsid relative to the untreated control.
To estimate the degree of binding signal for each ligand attributable to the capsid sequence, subtract the "no capsid" negative control from the initial absorbances, and take the adjusted absorbance of the denatured capsid. Divide this by the adjusted absorbance of the positive control signal, and multiply it by 100. This gives the apparent percentage of signal due to ligand binding to denatured capsid/capsid sequence.
3. DLS for Detecting Virus Aggregation After Heat Treatment
NOTE: The steps below may be specific to the software and instrument used, but can be adapted and applied to similar devices.
Create a new size SOP in the DLS instrument software using: Material = Protein, Dispersant = PBS, Temperature = 25 °C, Equilibration time = 0 sec, Cell type = disposable cuvette (small volume, ZEN0040), Measurement angle = 173° Backscatter, Measurement duration = automatic, Number of measurements = 3, Delay between measurements = 0 sec, Data processing: Analysis model = General purpose (normal resolution). Use the default settings for all other parameters.
Select the green run arrow within the software, and name the sample.
Follow steps 1.1.1 through 1.1.4 for the heat treatment of VLPs.
Dilute the heat treated VLPs 1:10 in 1x PBS for a final concentration of 5 µg/mL. (Optional) Filter the sample with a 1 µm pore size to remove dust particles; DLS is very sensitive to dust, as large particles scatter much more light than small particles.
Transfer 50 µL of diluted VLPs into a small volume disposable cuvette. Insert the cuvette into the sample chamber of the instrument.
- Start the run, and use the 'Correlogram', 'Cumulants Fit', and 'Expert Advice' tabs to evaluate data quality.
- During the run, check that the polydispersity index value is less than 0.3, representing a quality measurement. In the ‘Multi-view’ tab, make sure the correlation coefficient is constant and close to, but not more than 1, at a short time, and drops off sharply to 0 at a later time, characteristic of particle size. Use the ‘Expert Advice’ tab for feedback on the data quality during the measurements.
- After the measurement is complete, check again that the polydispersity index value is less than 0.3. Make sure that the cumulants fit line follows the data points closely in the ‘Cumulants fit’ tab.
Take the average of the Z-average particle diameter reported for each measurement as the particle size of each sample. Plot the diameter in nm vs. temperature in °C.
4. DLS for Detecting Virus Aggregation in Real Time
NOTE: The steps below may be specific to the software and instrument used, but can be adapted and applied to similar devices.
Create a new size SOP in the DLS instrument software using: Material = Protein, Dispersant = PBS, Temperature = as desired, Equilibration time = 0 sec, Cell type = quartz cuvette, Measurement angle = 173° Backscatter, Measurement duration = automatic, Number of measurements = 50 (may be increased or decreased depending on aggregation rate), Delay between measurements = 0 sec, Data processing: Analysis model = Multiple narrow modes (high resolution). Use the default settings for all other parameters.
Select the run arrow within the software to open and name a new sample, and allow the instrument to reach the temperature equilibrium.
Suspend the VLPs in 1X PBS in a microcentrifuge tube at a concentration of 5 µg/mL and a volume between 200 - 500 µL. Vortex the suspension.
Spin down the VLP suspension for 5 - 10 s in a tabletop centrifuge to de-gas. This helps to prevent bubble formation during the heat treatment that interferes with size measurements.
Transfer the VLP suspension to a small volume quartz cuvette-avoid bubbles and pipette slowly against the wall of the cuvette. After the instrument has reached temperature equilibrium, insert the cuvette into the sample chamber.
Wait 10 s for the sample temperature to equilibrate, then start the run. Start a timer at the start of the run. Stop the timer at the end of the first measurement. Take this time as the first time point. Obtain subsequent time points from the measurement times recorded by the software.
- Start the run, and monitor the correlogram, distribution fit, and expert advice to evaluate the data quality. NOTE: The PDI will not necessarily be less than 0.3 for these measurements as the sample is expected to be polydisperse during aggregation.
- Make sure the correlation coefficient is constant and close to, but not more than 1 at short time, and drops off sharply at one or more later times, characteristic of particle size.
- Make sure the distribution fit line follows the data points closely.
- Analyze the size data.
- Determine each time point from the difference in time between each measurement and the initial time point. Measurement durations are not all exactly the same.
- Using an intensity distribution, record the size of the peaks with the largest areas for each measurement/time point.
- Plot the peak diameters in nm vs. time in min.
5. TEM Sample Preparation
Obtain carbon support film (nickel) grids designed for TEM. NOTE: Consult with the core facility on recommended reagents and their handling for use with the facility microscope. Be sure to store grids in a box containing desiccant or a vacuum chamber to minimize exposure to moisture.
Tear approximately 5 cm x 5 cm pieces of filter paper. Obtain glass Petri dishes and proper self-closing reverse tweezers designed for use in microscopy.
Cut approximately 2.5 cm x 5 cm piece of paraffin film. Place on the benchtop.
- Heat treatment of norovirus GII.4 Sydney capsids
- Follow the heat treatment procedure exactly as described above in steps 1.1.1 - 1.1.5 except: use 10 mM HEPES (pH 7.4) for treatment instead of PBS and do not further dilute capsids after treatment (keep solution at 50 µg/mL). Pipette the entire ~15 µL drop of treated capsid solution onto paraffin film.
- Grab the grid edge with tweezers, allowing them to close on the edge to hold the grid, and place the grid carbon-side-down on top of the drop of treated solution. Let incubate for 10 min to allow the capsid to attach to the grid.
- Depending on the purity of the capsid preparation, add washes of the 10 mM HEPES solution if needed in the form of 10-15 µL droplets placed in line after the treated capsid droplet.
- After 10 min, pick up the grid with the droplet. Place the grid nearly perpendicular to a piece of filter paper to wick away the droplet. Pick the droplet of 10 - 15 µL HEPES buffer up with the grid, hold for 30 s, then wick away with another piece of filter paper to wash.
- Place a 15 µL droplet of 2% uranyl acetate solution on the paraffin film in an empty area.
- Pick up the droplet of uranyl acetate with the grid and hold for 45 s. Wick away the uranyl acetate, being sure to remove most of the solution. Place the grid carbon-side up on a piece of filter paper, being careful that the grid does not "stick" to the moisture on the tweezers. Place the filter paper with the grid on it in an open glass Petri dish. NOTE: Do not use plastic Petri dishes, as the grids will be electrostatically attracted to them and become stuck to dish.
Repeat for all treatments. Place the Petri dish halves with the grids in a desiccator overnight to dry prior to observing with TEM.
Consult the microscopy facility at the respective institution regarding observing the prepared grids. Some facilities permit the user to be trained on the operation of their facility's instrument. Specific details regarding the setup and observation of a specific microscope is beyond the scope of this article.
Representative Results
The structural integrity of human norovirus GII.4 Sydney capsids (VLPs) was assessed using several novel methods as presented by Moore et al.17. First, an experiment was done in which the integrity of the capsids as a function of their ability to bind a putative receptor/co-factor (HBGA) or ssDNA aptamer (M6-2) were compared (Figure 1). The results displayed have been previously presented by Moore et al.17, and demonstrate that the aptamer M6-2-capsid binding behavior was similar to that of virus-HBGA binding upon exposure to 68 °C heat treatment for various exposure times. While the HBGA binding signal was lost slightly earlier than that of the aptamer M6-2, M6-2 displayed a similar rate of signal loss calculated after VLPs were exposed to 65 °C and 68 °C for various time points (Table 1)17. This suggests that both HBGA and aptamer M6-2 binding was abolished in a similar manner.
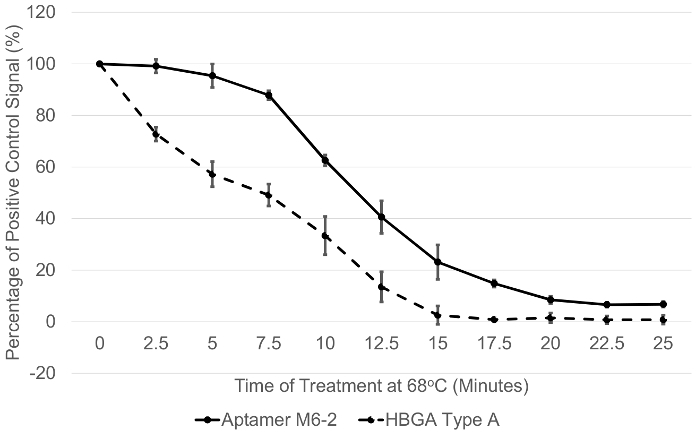
DLS measurements demonstrated that aggregation due to protein denaturation likely corresponded with loss of ligand binding signal (Figure 2), as a hydrodynamic diameter greater than 100 nm was observed after about 18 min of treatment at 68 °C. These were the conditions at which complete loss of HBGA binding and nearly all loss of binding signal for aptamer M6-2 (>80%) occurred (Figure 1)17. TEM results supported these observations as morphological changes and clumping of the capsids became increasingly apparent as the temperature was increased incrementally between 60 °C and 80 °C for 1 min (Figure 3a). A similar phenomenon was observed after heating the capsids at 68 °C for up to 25 min, but the resolution of the TEM was less pronounced (Figure 3b)17.
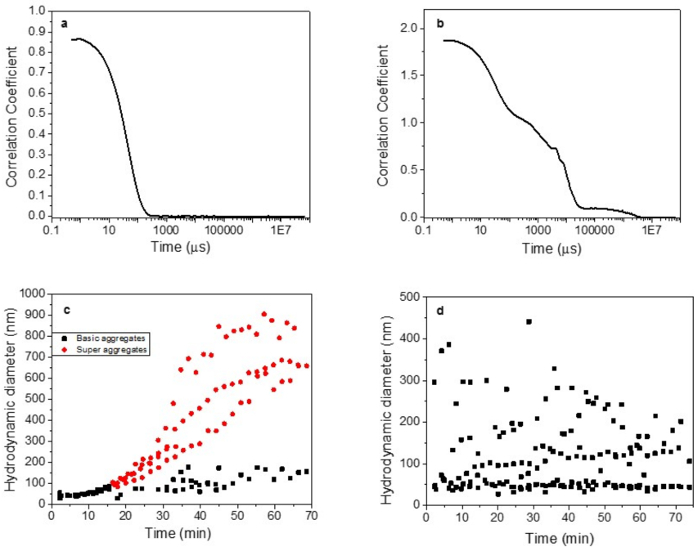
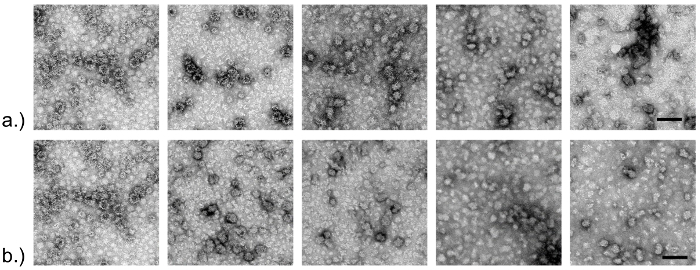
Figure 1: Conformation-dependent binding of two different ligands to norovirus GII.4 Sydney capsids subjected to heat treatment. Purified GII.4 Sydney capsids (VLPs) were subjected to heat treatment at 68 °C for different amounts of time, and the binding to blood type A HBGA and aptamer M6-2 was evaluated using a plate-based ELASA binding assay. The Y-axis displays the absorbance signal observed for a treatment (exposure time) as the percentage of an untreated capsid control. This figure has been previously presented and this figure has been modified from Moore et al.17 Error bars represent ± 1 standard deviation of % signal. Data are for at least three replicate plates. Please click here to view a larger version of this figure.
Figure 2: Example DLS data for heat-treated norovirus GII.4 Sydney capsids showing ideal and ambiguous/low quality results. Capsids (VLPs) were subjected to heat treatment at 65 °C and 68 °C, and their size was monitored in real time. Panel (a) corresponds to a high quality, clear correlation coefficient measurement; and (b) to a low quality, ambiguous correlation coefficient measurement. Panel (c) displays particle size data, showing a clear aggregation profile for capsids treated at 68 °C; and (d) shows ambiguous particle size data for heat-treated capsids. Some of these data are presented, and thus this image is modified from Moore et al.17 Please click here to view a larger version of this figure.
Figure 3: TEM of norovirus GII.4 Sydney capsids subjected to different heat treatments. Purified capsids (VLPs) were subjected to various heat treatments and observed using transmission electron microscopy. Panel (a) shows data for capsids heated at different temperatures (60, 65, 70, 75, and 80 °C) for 1 min. Panel (b) shows capsids subjected to 68 °C treatment for 0 - 25 min. Scale (bar) on right-hand photos represents 100 nm. These images are presented in Moore et al.17 Please click here to view a larger version of this figure.
| Exponential Decay Rate (% signal/min) | ||
| Temperature | 65 °C | 68 °C |
| Ligand | ||
| HBGA Type A | 2.50 ± 0.24 | 13.30 ± 1.15 |
| Aptamer M6-2 | 2.47 ± 0.66 | 13.18 ± 0.47 |
Table 1: Exponential decay rates for norovirus GII.4 Sydney capsids treated at 65 °C and 68 °C. Capsids were subjected to heat treatments for various times at 65 °C and 68 °C , cooled at 4 °C , and their ability to bind synthetic HBGA or aptamer evaluated. The rate of signal loss over time (decay rate) was then calculated with each ligand for each temperature. These data have previously been presented in Moore et al.17 Data are presented as % signal/min ± 1 standard deviation of % signal.
Discussion
The protocol and techniques described here provide a means of assessing human norovirus capsid integrity/functionality. These methods can be used for obtaining mechanistic insight into the effects of inactivation treatments against the virus, and can potentially supplement more traditional inactivation evaluation methods such as RT-qPCR or plaque assay. For instance, the evaluation of chemical or physical inactivation by plaque assay alone provides a measure of degree of virus inactivation but not the underlying mechanism of that inactivation. Also, virus aggregation can result in underestimation of titer and therefore overestimate treatment efficacy30. These methods can further inform such a study by providing information about the effects of a treatment on the integrity of the norovirus capsid. Recently, two in vitro (cell culture) cultivation methods for human norovirus have been reported5,6; these still relied on PCR for virus enumeration, though theoretically the enteroid model presented could be utilized for TCID50 using antibody staining for relative reduction determination. The methods presented here could have utility for understanding the complete mechanism of viral inactivation for multiple different types of treatments beyond just heat treatment. For example, plate-based ligand binding and TEM were used as tools to supplement RT-qPCR and plaque assay data in determining the mechanism of norovirus inactivation on copper surfaces31,32, upon exposure to silver dihydrogen citrate33, and high hydrostatic pressure treatment14.
There are numerous steps in these protocols in which attention to methodological detail is crucial. Specifically, for the plate assay, great attention must be paid to the buffer used to dilute the ligand for binding. Aptamer M6-2, and ssDNA aptamers in general, are sensitive to salt concentrations and buffer conditions. Using a non-optimal buffer with the aptamer may ablate binding. Although aptamer M6-2 has successfully been used in diluted human stool, too much stool/matrix interference can interfere with binding, which is consistent with observations for other human norovirus aptamers18,22. One major factor causing this is that aptamers can exhibit a degree of nonspecific binding to positively charged molecules. Thus, this could lead to some degree of false positive signal in the assay described here. This can be accounted for by selecting a negative control that is either a complex matrix without virus or an unrelated protein. Likewise, the HBGA concentration and the amount of skim milk solids used in the binding buffer can significantly affect positive and background signals. Note that different HBGAs have varying degrees of affinity to different strains of human norovirus, so the binding buffers may need to be optimized depending on HBGA type14,31,33. Similar consideration should be given to the buffer used for the streptavidin-horseradish peroxidase conjugate, as concentrations from different manufacturers can differ; however, much less variability would be expected given the high affinity of the streptavidin-biotin interaction34. Because of the high degree of adaptability of this plate-based method, troubleshooting suboptimal results is relatively straightforward. For example, in the case of high background signal, optimization by increasing the skim milk concentration and decreasing ligand concentration can be investigated, and vice versa for plates with low positive signals. For TEM, one of the most critical considerations is the use of non-static materials (like glass) when storing/handling grids, as the grids are extremely difficult to handle around materials such as disposable Petri dishes. If such materials are used, grids will tend to "jump" and stick to the material. Additionally, much care should be taken to ensure that there is no residual moisture on the grids. Ideally, a vacuum desiccator should be used if available.
When performing experiments using DLS, the purity of the sample is a very important consideration. The functions used to correlate changes in light scattering intensity to diameter rely on having monomodal or narrow multimodal particle distributions. Impurities down to the nanoscale, including proteins, introduce polydispersity and alter size calculations. Also, the intensity of light scattered is proportional to the diameter raised to a power of 6, so large particles such as dust substantially interfere with measurements. Bubble formation during real-time heat treatments results in nonsensical results due to light scattered by the fluctuating bubbles. Capsid (VLP) concentration must also be considered when using DLS. Sample concentrations must be high enough to produce sufficient signal and low enough to avoid multiple scattering. It is also of vital importance to input the correct material and dispersant properties, as these values are used by the software for size calculations. In particular, dispersant viscosity is a function of temperature and must be adjusted accordingly during heat treatments (the software used has temperature-dependent viscosities of water built in).
Although the methods described here offer multiple opportunities for improved ways to assess capsid integrity, they are not perfect. Because these methods require very high concentrations of virus, they must be used with purified, assembled capsids instead of native infectious viruses. The VLPs used here consist of the major capsid protein of human norovirus (VP1), which assembles into the norovirus capsid without viral nucleic acid. Although these VLPs exhibit antigenically similar behavior to infectious virus capsids, they may be more susceptible to inactivation. Further, VLPs are difficult to produce and purify and are not readily available to many investigators. Use of purified human norovirus (e.g., using ultracentrifugation) is possible but availability of norovirus-positive human fecal specimens is limited and even with purification, virus titers may not be high enough to accommodate the methods described here. Future work at optimizing assays for use of semi-purified infectious virus in stool is currently underway. In fact, this has already been demonstrated for the plate assay18,22, but would undoubtedly be more complicated for DLS. It should also be noted that these methods were only applied to evaluate capsid integrity after application of a physical (heat) inactivation approach, which is known to directly affect the capsid. Results may be more ambiguous were these techniques used to evaluate the capsid integrity using other inactivation approaches, such as chemical agents or methods that mostly target destruction of the viral genome. However, some reports have favorably demonstrated the performance of HBGA binding for evaluating chemical inactivation31,33,35,36,37. At the current time, the protocols reported here are best used in concert with traditional technique(s) like RT-qPCR and plaque assays with surrogate viruses.
These protocols provide a simple alternative means by which to understand the effects of a physical virus inactivation method (heat) on human norovirus. These methods could likely be expanded for use with other non-enveloped viruses (e.g., hepatitis A and E viruses), as the overall principle of detecting loss of higher order protein structure is the same. Additionally, the behavior and potential use of other ligands for indicating loss of capsid integrity should be evaluated. Adapting the protocols to more complex sample matrices, and improving their analytical sensitivity (detection limits) is also a logical future direction. Overall, the methods described here provide a more accessible means of determining human norovirus capsid integrity and gaining mechanistic insight into the effects of inactivation treatments against this important pathogen.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by the Agriculture and Food Research Initiative Competitive Grant no. 2011-68003-30395 from the United States Department of Agriculture, National Institute of Food and Agriculture through the NoroCORE project. Funding for open access charge was provided by the United States Department of Agriculture. We would like to thank Robert Atmar (Baylor College of Medicine, Houston, TX) for kindly providing us the purified capsids and Valerie Lapham for her assistance with the TEM images. We would also like to thank Frank N. Barry for helping us start the experiments presented.
References
- Kirk MD, et al. World Health Organization estimates of the global and regional disease burden of 11 foodborne bacterial, protozoal, and viral diseases, 2010: A data synthesis. PLOS Med. 2015;12(12):e1001920. doi: 10.1371/journal.pmed.1001921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharff RL. Economic burden from health losses due to foodborne illness in the United States. J. Food Prot. 2012;75(1):123–131. doi: 10.4315/0362-028X.JFP-11-058. [DOI] [PubMed] [Google Scholar]
- Lee BY, Mcglone SM, Bailey RR, Zachary S, Umscheid CA, Muder RR. Economic impact of outbreaks of norovirus infection in hospitals. Infect. Control Hosp. Epidemiol. 2011;32(2):191–193. doi: 10.1086/657910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law JW-F, Ab Mutalib N-S, Chan K-G, Lee L-H. Rapid methods for the detection of foodborne bacterial pathogens: principles, applications, advantages and limitations. Front. Microbiol. 2015;5:770. doi: 10.3389/fmicb.2014.00770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones MK, et al. Enteric bacteria promote human and mouse norovirus infection of B cells. Science. 2014;346(6210):755–759. doi: 10.1126/science.1257147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ettayebi K, et al. Replication of human noroviruses in stem cell - derived human enteroids. Science. 2016 Aug;5211 doi: 10.1126/science.aaf5211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MD, Goulter RM, Jaykus L-A. Human norovirus as a foodborne pathogen: challenges and developments. Annu. Rev. Food Sci. Technol. 2015;6(1):411–433. doi: 10.1146/annurev-food-022814-015643. [DOI] [PubMed] [Google Scholar]
- Miura T, et al. Histo-blood group antigen-like substances of human enteric bacteria as specific adsorbents for human noroviruses. J. Virol. 2013;87(17):9441–9451. doi: 10.1128/JVI.01060-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almand EA, et al. Human norovirus binding to select bacteria representative of the human gut microbiota. Plos One. 2017;12(3):e0173124. doi: 10.1371/journal.pone.0173124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubio-del-Campo A, et al. Noroviral P-Particles as an in vitro model to assess the interactions of noroviruses with probiotics. PLoS ONE. 2014;9(2):e89586. doi: 10.1371/journal.pone.0089586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirneisen KA, Kniel KE. Comparison of ELISA attachment and infectivity assays for murine norovirus. J. Virol. Methods. 2012;186(1-2):14–20. doi: 10.1016/j.jviromet.2012.07.018. [DOI] [PubMed] [Google Scholar]
- Dancho BA, Chen H, Kingsley DH. Discrimination between infectious and non-infectious human norovirus using porcine gastric mucin. Int. J. Food Microbiol. 2012;155(3):222–226. doi: 10.1016/j.ijfoodmicro.2012.02.010. [DOI] [PubMed] [Google Scholar]
- Li X, Chen H. Evaluation of the porcine gastric mucin binding assay for high-pressure-inactivation studies using murine norovirus and Tulane virus. Appl. Environ. Microbiol. 2015;81(2):515–521. doi: 10.1128/AEM.02971-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou F, et al. High-pressure inactivation of human norovirus virus-like particles provides evidence that the capsid of human norovirus is highly pressure resistant. Appl. Environ. Microbiol. 2012;78(15):5320–5327. doi: 10.1128/AEM.00532-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Tian P. Inactivation conditions for human norovirus measured by an in situ capture-qRT-PCR method. Int. J. Food Microbiol. 2014;172:76–82. doi: 10.1016/j.ijfoodmicro.2013.11.027. [DOI] [PubMed] [Google Scholar]
- Wang D, Xu S, Yang D, Young GM, Tian P. New in situ capture quantitative (real-time) reverse transcription-PCR method as an alternative approach for determining inactivation of Tulane virus. Appl. Environ. Microbiol. 2014;80(7):2120–2124. doi: 10.1128/AEM.04036-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MD, Bobay BG, Mertens B, Jaykus L. Human norovirus aptamer exhibits high degree of target conformation-dependent binding similar to that of receptors and discriminates particle functionality. mSphere. 2016;1(6):e00298–e00216. doi: 10.1128/mSphere.00298-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore MD, Escudero-Abarca BI, Suh SH, Jaykus L-A. Generation and characterization of nucleic acid aptamers targeting the capsid P domain of a human norovirus GII.4 strain. J. Biotechnol. 2015;209:41–49. doi: 10.1016/j.jbiotec.2015.06.389. [DOI] [PubMed] [Google Scholar]
- Kitamoto N, et al. Cross-Reactivity among Several Recombinant Calicivirus Virus-Like Particles (VLPs) with Monoclonal Antibodies Obtained from Mice Immunized Orally with One Type of VLP. J. Clin. Microbiol. 2002;40(7):2459–2465. doi: 10.1128/JCM.40.7.2459-2465.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giamberardino A, et al. Ultrasensitive norovirus detection using DNA aptasensor technology. PloS one. 2013;8(11):e79087. doi: 10.1371/journal.pone.0079087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beier R, et al. Selection of a DNA aptamer against norovirus capsid protein VP1. FEMS Microbiol. Lett. 2014;351(2):162–169. doi: 10.1111/1574-6968.12366. [DOI] [PubMed] [Google Scholar]
- Escudero-Abarca BI, Suh SH, Moore MD, Dwivedi HP, Jaykus L-A. Selection, characterization and application of nucleic acid aptamers for the capture and detection of human norovirus strains. PloS one. 2014;9(9):e106805. doi: 10.1371/journal.pone.0106805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharjee S. DLS and zeta potential - What they are and what they are not? Journal Control. Release. 2016;235:337–351. doi: 10.1016/j.jconrel.2016.06.017. [DOI] [PubMed] [Google Scholar]
- Khodabandehloo A, Chen DDY. Particle sizing methods for the detection of protein aggregates in biopharmaceuticals. Bioanalysis. 2017;9(3):313–326. doi: 10.4155/bio-2016-0269. [DOI] [PubMed] [Google Scholar]
- Mattle MJ, et al. Impact of virus aggregation on inactivation by peracetic acid and implications for other disinfectants. Environ. Sci. Technol. 2011;45:7710–7717. doi: 10.1021/es201633s. [DOI] [PubMed] [Google Scholar]
- Samandoulgou I, Fliss I, Jean J. Zeta potential and aggregation of virus-like particle of human norovirus and feline calicivirus under different physicochemical conditions. Food Environ. Virol. 2015;7(3):249–260. doi: 10.1007/s12560-015-9198-0. [DOI] [PubMed] [Google Scholar]
- Mertens BS, Velev OD. Soft matter norovirus interactions. Soft Matter. 2015;11:8621–8631. doi: 10.1039/c5sm01778e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panyukov Y, Yudin I, Drachev V, Dobrov E, Kurganov B. The study of amorphous aggregation of tobacco mosaic virus coat protein by dynamic light scattering. Biophys. Chem. 2007;127(1-2):9–18. doi: 10.1016/j.bpc.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Moore MD, Escudero-Abarca BI, Jaykus L-A. An Enzyme-Linked Aptamer Sorbent Assay to Evaluate Aptamer Binding. Methods in Molecular Biology. 2017;1575:291–302. doi: 10.1007/978-1-4939-6857-2_18. [DOI] [PubMed] [Google Scholar]
- Langlet J, Gaboriaud F, Gantzer C. Effects of pH on plaque forming unit counts and aggregation of MS2 bacteriophage. J. Appl. Microbiol. 2007;103:1632–1638. doi: 10.1111/j.1365-2672.2007.03396.x. [DOI] [PubMed] [Google Scholar]
- Manuel CS, Moore MD, Jaykus LA. Destruction of the Capsid and Genome of GII.4 Human Norovirus Occurs during Exposure to Metal Alloys Containing Copper. Appl. Environ. Microbiol. 2015;81(15):4940–4946. doi: 10.1128/AEM.00388-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warnes SL, Summersgill EN, Keevil CW. Inactivation of murine norovirus on a range of copper alloy surfaces is accompanied by loss of capsid integrity. Appl. Environ. Microbiol. 2014. Dec, [DOI] [PMC free article] [PubMed]
- Manuel C, Moore MD, Jaykus L-A. Efficacy of a disinfectant containing silver dihydrogen citrate against GI.6 and GII.4 human norovirus. J. Appl. Microbiol. 2017;122(1):78–86. doi: 10.1111/jam.13331. [DOI] [PubMed] [Google Scholar]
- Green NM. Avidin and streptavidin. Methods Enzymol. 1990;184:51–67. doi: 10.1016/0076-6879(90)84259-j. [DOI] [PubMed] [Google Scholar]
- Kingsley DH, Vincent EM, Meade GK, Watson CL, Fan X. Inactivation of human norovirus using chemical sanitizers. Int. J. Food Microbiol. 2014;171:94–99. doi: 10.1016/j.ijfoodmicro.2013.11.018. [DOI] [PubMed] [Google Scholar]
- Tian P, Yang D, Quigley C, Chou M, Jiang X. Inactivation of the Tulane virus, a novel surrogate for the human norovirus. J. Food Prot. 2013;76(4):712–718. doi: 10.4315/0362-028X.JFP-12-361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Baert L, Van Coillie E, Uyttendaele M. Critical studies on binding-based RT-PCR detection of infectious noroviruses. J. Virol. Methods. 2011;177(2):153–159. doi: 10.1016/j.jviromet.2011.07.013. [DOI] [PubMed] [Google Scholar]