

Abstract
Atherosclerosis is a major contributor to cardiovascular disease, the leading cause of death worldwide, which claims 17.3 million lives annually. Atherosclerosis is also the leading cause of sudden death and myocardial infarction, instigated by unstable plaques that rupture and occlude the blood vessel without warning. Current imaging modalities cannot differentiate between stable and unstable plaques that rupture. Peptide amphiphiles micelles (PAMs) can overcome this drawback as they can be modified with a variety of targeting moieties that bind specifically to diseased tissue. Monocytes have been shown to be early markers of atherosclerosis, while large accumulation of monocytes is associated with plaques prone to rupture. Hence, nanoparticles that can target monocytes can be used to discriminate different stages of atherosclerosis. To that end, here, we describe a protocol for the preparation of monocyte-targeting PAMs (monocyte chemoattractant protein-1 (MCP-1) PAMs). MCP-1 PAMs are self-assembled through synthesis under mild conditions to form nanoparticles of 15 nm in diameter with near neutral surface charge. In vitro,PAMs were found to be biocompatible and had a high binding affinity for monocytes. The methods described herein show promise for a wide range of applications in atherosclerosis as well as other inflammatory diseases.
Keywords: This Month in JoVE, Issue 129, Monocyte, peptide, micelles, atherosclerosis, nanoparticle, imaging, self-assembly, diagnostic
Introduction
Cardiovascular diseases remain to be the leading causes of death globally with approximately 17.3 million deaths worldwide1. Cardiovascular diseases are contributed by atherosclerosis, a condition in which plaques build up in the arteries, thereby inhibiting blood and oxygen flow to the cells of the body2,3. The progression of atherosclerosis involves the thickening and hardening of arteries by an inflammatory response, irregular lipid metabolism, and plaque build-up, leading to plaque rupture and myocardial infarction4,5. Endothelial cells express cytokines and adhesion molecules, which include MCP-1 that binds to the C-C chemokine receptor (CCR2) found on the surface of monocytes6,7,8. Oxidized cholesterol converts monocytes to macrophages during the early stage of plaque formation, which amplifies the inflammatory response in the region and leads to tissue injury and the formation of unstable or vulnerable plaques9,10.
Traditionally, atherosclerosis is evaluated by assessing luminal stenosis by anatomical imaging using angiography or ultrasound11,12. However, these methods can only determine severe narrowing of the arterial wall and not the early stage of atherosclerosis, as initial plaque growth causes arterial remodeling to maintain artery size and blood flow rate12,13,14. Therefore, angiograms underrepresent atherosclerosis prevalence. Additionally, noninvasive imaging techniques such as single photon emission computed tomography, positron emission tomography, and magnetic resonance imaging have recently been used to characterize plaque morphology as they can provide initial details and characterization of plaques. However, these modalities are often limited by the lack of sensitivity, spatial resolution, or require the use of ionizing radiation, making imaging plaque progression at different stages much more challenging15,16,17. An imaging delivery system that would specifically identify plaques at different stages of atherosclerosis remains to be developed.
Nanoparticles have shown to be an emerging platform for in vivo plaque targeting and diagnostics18,19,20,21. In particular, PAMs are advantageous due to their chemical diversity and ability to accommodate a variety of moieties, compositions, sizes, shapes, and surface functionalization22. Peptide amphiphiles (PAs) consist of a hydrophilic, peptide "headgroup" attached to a hydrophobic tail, which are typically lipids; this amphiphilic structure confers self-assembling capabilities and allows for a multivalent display of peptides on the surface of the particle22,23,24. The peptide headgroups can affect the particle shape through folding and hydrogen bonding between peptides25. Peptides that fold through β-sheet interaction have been shown to form elongated micelles, while α-helical confirmation can form both spherical and elongated micelles22,23,24,25,26,27. Polyethylene glycol (PEG) linkers that shield the surface charge of the peptide can be placed between the hydrophilic peptide and the hydrophobic tail of PAMs, enhancing the availability of the nanoparticle in systemic circulation28,29,30,31. PAMs are also advantageous because they are biocompatible and have been shown to have a broad range of applications32,33. The water solubility of micelles offers an advantage over other nanoparticle-based systems such as certain polymeric nanoparticles that are not soluble in water and have to be suspended in solubilizers for injections34. Additionally, the ability to create PAMs that disassemble in response to a specific stimuli makes PAMs an attractive candidate for controlled intracellular drug delivery35.
By binding to the CCR2 receptor and accumulating in the aortic arch, PAMs were previously developed for monocyte targeting to monitor different stages of atherosclerotic lesions in the aorta9. In ApoE-/- mice, monocyte accumulation increases proportionally to plaque progression36. Furthermore, it was found that patients with rupture-prone, late-stage plaques contain higher amounts of monocytes37. Therefore, the modification of PAMs to incorporate MCP-1 is useful because it allows for greater targeting specificity and differentiation between early- and late-stage atherosclerotic lesions. These proof-of-concept studies also verified that PAMs are safe enough to be used pre-clinically and are cleared renally38. Since monocytes and inflammation are characteristic to other diseases, MCP-1 PAMs have the potential to be used for therapeutic and diagnostic applications in other diseases beyond atherosclerosis8,39,40,41.
Herein, we report the fabrication of highly scalable and self-assembled MCP-1 PAMs that demonstrated the particle's optimal size, surface charge, and selective targeting to monocytes for enhanced imaging applications in atherosclerosis.
Protocol
NOTE: Read the MSDS for reagents and follow all chemical safety measures as required by local institution.
1. Preparation of MCP-1 PAMs
- Preparation of MCP-1 peptide
- Weigh out 0.25 mmol of Fmoc-L-Lys(Boc)-Wang in a reaction vessel (RV). Rinse the side of the RV with 5 mL dimethylforamide (DMF) in a chemical fume hood.
- Load the RV onto an automated benchtop peptide synthesizer. Load pre-packaged amino acid vials, N'-CYNFTINRKISVQRLASYRRITSS-C', from the C to N-terminus. Include an empty vial at the end for the final deprotection step. NOTE: A scrambled peptide with sequence, N'-CNNSIIRSKQVLRSTRYFRSATYK-C', can also be synthesized using the same protocol.
- Once synthesis is finished, transfer the peptides on resin from the RV to the scintillation vial with 2-3 mL methanol until all of the resins are transferred. After the resin has sunk to the bottom, remove the methanol supernatant and evaporate the remaining until dry. Vacuum dry for an additional 2 h or overnight at room temperature.
- Make 12 mL cleavage solution containing trifluoracetic acid (TFA):ethanedithiol:water:triisopropylsilane at 94:2.5:2.5:1 vol% in a chemical fume hood. Swirl the cleavage solution to make sure it is evenly mixed. CAUTION: Because triisopropylsilane and ethanedithiol are air-sensitive, argon purge stock vials of triisopropylsilane and ethanedithiol for 1 min prior to putting them back.
- Place the synthesis vessel (SV) onto an arm shaker by clamping near the bottom half of the SV. Transfer peptide-resin to the SV with 2 mL of cleavage solution until all of the peptide-resins are transferred.
- Wash the sides of the SV with 3 mL cleavage solution. Close the cap of the SV and shake at 300 oscillations/min for 4 h.
- Weigh out an empty 50 mL centrifuge tube. Record the mass.
- After 4 h, drain the cleaved peptide solution from the SV into the 50 mL centrifuge tube. Rinse the SV several times with the remaining cleavage solution.
- Evaporate cleaved peptide solution with nitrogen until there is less than 4 mL remaining in the centrifuge tube.
- Add 36 mL of ice-cold ether to the peptide-cleaved solution.
- Vortex the solution until it is completely white or yellow. Centrifuge at 3,000 x g for 5 min at 4 °C, and decant the supernatant.
- Add 40 mL ice-cold ether to the tube. Vortex and sonicate again. Repeat the centrifugation process. Decant the supernatant.
- Dry the crude PA pellet with nitrogen gas purging until the ether is visibly evaporated.
- Add 20 mL double-distilled water, vortex, and sonicate until the peptide is completely dissolved in solution.
- Freeze the solution and lyophilize until dry.
- Once the peptide is dried, weigh out the mass of the centrifuge tube containing the crude peptide. Dissolve the crude peptide in 5 mg/mL water.
- Dissolve 25 mg of crude MCP-1 peptide in 5 mL double-distilled water to make a total concentration of 5 mg/mL. Purify the crude MCP-1 peptide by high-performance liquid chromatography (HPLC) using 0.1% TFA in water (solvent A) and 0.1% TFA in acetonitrile (solvent B) as mobile phases on a C8 reverse-phase column. Inject the 5 mL of the peptide into the HPLC. NOTE: The initial mobile phase consisted of 100% solvent A at a flow rate of 9.0 mL/min. Decrease solvent A slowly to 30% in 27 min and hold at this composition for 3 min. Return the gradient to 100% solvent A over 3 min and hold at this composition for 1 min to give a total run-time of 34 min. Keep the column temperature at 55 °C. Use positive ion source mode for the analysis. Formic acid can be used in place of TFA, if TFA is too acidic for the HPLC column. It is recommended that the organic solvent composition ramp speed should not exceed 2%/min for better separation.
- Analyze each fraction using matrix-assisted laser desorption/ionization (MALDI) mass spectrometry for the expected product m/z at 2,890. NOTE: Dissolve α-cyano-4-hydroxycinnamic acid at 1 mg/mL in acetonitrile/water (50:50 vol%) as the matrix solution. Spot 0.75 µL of matrix solution onto the MALDI plate, followed by 0.75 µL of each fraction. Carry out the analysis using a positive reflective ion mode at a mass range of 500-5,000 Da.
- Combine product fractions, blow off acetonitrile, freeze, and lyophilize purified peptides until dry.
- Preparation of MCP-1 PA
- Prepare a 1.1 molar excess of 1,2-distearoyl-sn-glycero-3-phosphoethanolamine-N-[amino(polyethylene glycol)-2000] DSPE-PEG(2000)-maleimide to peptide in separate scintillation vials. Dissolve both compounds in double-distilled water to make ~ 15 mg/mL. Sonicate for 15 min or until both solutions are completely clear. NOTE: DSPE-PEG(2000)-Cy5 is synthesized using the same protocol with DSPE-PEG(2000)-amine and N-hydroxysuccinimide (NHS) ester-functionalized Cy5, except for step 1.2.3, where the pH of the solution is adjusted to 8.5.
- Add DSPE-PEG(2000)-maleimide solution to the peptide solution. Vortex and sonicate.
- Add 1 M NaOH dropwise (1 µL) at a time until the pH of the solution is 7.
- Nitrogen purge solution for 5 min. Stir the solution overnight.
- Freeze the solution and lyophilize until dry.
- Once the crude product is dried, dissolve the solid in 5 mg/mL water.
- Purify with HPLC as described above. NOTE: Unconjugated peptide and DSPE-PEG(2000) should elute between 15-30% organic concentration (vol%), while DSPE-PEG(2000)-MCP-1 conjugate should elute between 40-50% organic concentration.
- Analyze each fraction using MALDI mass spectrometry for the corresponding m/z. DSPE-PEG(2000)-MCP-1 should have a broad m/z peak at 5,760. NOTE: 2,5-dihydroxybenzoic acid is best used as the matrix for DSPE-PEG(2000)-MCP-1 with MALDI.
- Assembly of MCP-1 PAMs
- Dissolve 1.75 mg MCP-1 PAs with 3 mL methanol in a 1-dram vial. Sonicate until the MCP-1 PA is completely dissolved. NOTE: Cy5 amphiphiles can be incorporated at a molar ratio of 10:90 MCP-1 PA for imaging applications.
- Evaporate the methanol under nitrogen in a circular motion while rinsing the remaining methanol off the side of the vial until a uniform film is formed at the bottom of the vial. Vacuum-dry the film overnight.
- Hydrate the film with 3 mL of water or phosphate buffer saline (PBS) to make a 100 µM solution of MCP-1 PAMs. Gently vortex and sonicate the solution until clear. Incubate at 80 °C for 30 min. NOTE: The elevated temperature has been shown to accelerate the self-assembly process and allows the peptide component to form the most stable secondary structure, while lowering the critical micelle concentration (CMC)42,43.
- Cool the resulting dispersion to room temperature.
2. Characterization of MCP-1 PAMs
- Dynamic light scattering (DLS) and zeta potential
- Place 100 µM of MCP-1 PAM or scrambled PAM in a cuvette according to the instructions of the DLS or zeta potential instrument. NOTE: DLS and zeta potential cuvettes can be different based on the instrument's instructions.
- Equilibrate the instrument to room temperature. Measure the size and surface charge of the PAM.
- Transmission electron microscopy (TEM)
- Add 7 µL of 50 µM MCP-1 PAM or scrambled PAM onto a TEM grid suspended within a self-closing tweezer for 5 min. Wick away the droplet with a delicate task wipe.
- Add 7 µL of water to wash the grid. Wick away the droplet.
- Add 7 µL of 1 wt% phosphotungstic acid for 2 min. Wick away the droplet. Repeat step 2.2.2.
- Dry the TEM grid prior to TEM analysis.
- Circular dichroism (CD) spectroscopy
- Turn on the nitrogen for 5-10 min prior to use of the instrument.
- Place 300 µL of blank (water or PBS) in cuvette.
- Measure spectra at 190-265 nm, 0.2 mm path-length with a 1 s integration time, and a 1 nm bandwidth in room temperature. Collect 3 replicates.
- Measure 100 µM MCP-1 peptide, MCP-1 PAM, scrambled peptide, and scrambled PAM under the same condition described in step 2.3.3. Subtract from the blank.
- Fit the data using a linear interpolation of polylysine basis spectra.
- Turn off the instrument. Wait 10 min before turning off the nitrogen.
3. In Vitro Analysis of MCP-1 PAMs
- In vitro biocompatibility
- Culture and expand WEHI 274.1 murine monocytes in Dulbecco's Modified Eagle Medium supplemented with 10% fetal bovine serum (FBS), 1% penicillin-streptomycin, and 0.05 mM 2-mercaptoethanol in 37 °C under 5% CO2 to passage 5. NOTE: WEHI 274.1 are suspension cells. When culturing, the media should be replenished every 2-3 days.
- Pipet monocytes in media into a sterile 50 mL centrifuge tube. Centrifuge at 300 x g for 5 min at 4 °C.
- Aspirate the old media and resuspend in 10 mL fresh media into the centrifuge tube. Mix slowly until cells are evenly distributed in media. Count the cells using trypan blue staining and a hemocytometer.
- Dilute cells such that there are 4,000 monocytes per 90 µL of media per well. Add 90 µL of cells in wells of a 96-well plate. Incubate for 24 h. NOTE: Avoid using the wells on the exterior edge of the plate to avoid differences in evaporation and thermal changes in the plate. Fill the exterior wells with 100 µL PBS.
- Dissolve 0.15 µmol of MCP-1 peptide, MCP-1 PAM, scrambled peptide, or scrambled-PAM in 150 µL PBS in separate, sterilized microcentrifuge tubes. Perform serial dilution to make 65 µL of 1, 10, and 100 µM of each sample.
- Add 10 µL PBS of each concentration in wells containing WEHI 274.1 (total volume: 100 µL per well, 6 lines, 96 total wells). Incubate for 72 h.
- Add 10 µL (3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium) (MTS) in each well. Incubate for 1 h.
- Analyze the absorbance using a plate reader at 490 nm or based on the manufacturer's instructions.
- In vitro micelle binding
- Seed 500,000 monocytes in each well of a 6-well plate in 2 mL of media containing 100 µM Cy5-labeled MCP-1 PAM (10:90 molar ratio of DSPE-PEG(2000)-Cy5 and MCP-1 PA). Incubate for 1 h.
- Collect WEHI 274.1 by centrifugation at 300 x g for 5 min at 4 °C. Aspirate the media.
- Resuspend and wash the cells in 2 mL PBS. Centrifuge at 300 x g for 5 min at 4 °C. Aspirate the PBS. Repeat the washing process with 2 mL PBS. Aspirate the PBS.
- Add 2 mL of cold 4% paraformaldehyde to fix the cells. Incubate at room temperature for 10 min.
- Pipet the fixed cells onto a sterile glass coverslip within wells of a 6-well plate. Centrifuge at 400 x g for 5 min.
- Remove the paraformaldehyde. Wash and rinse with 2 mL PBS once. Resuspend with 1 mL PB NOTE: When rinsing, avoid disrupting the cells on the coverslip.
- Put 10 µL of 90% glycerol in PBS onto a slide. Use a needle and tweezer to pick up the coverslip. Dry the edge of the coverslip. Gently place the coverslip onto a slide facedown.
- Gently seal the edge of the coverslip with clear nail polish. Place in the refrigerator until ready for imaging.
- Use a confocal laser scanning microscope (CLSM) installed with lasers for Cy5 at λexcitation = 650 nm.
Representative Results
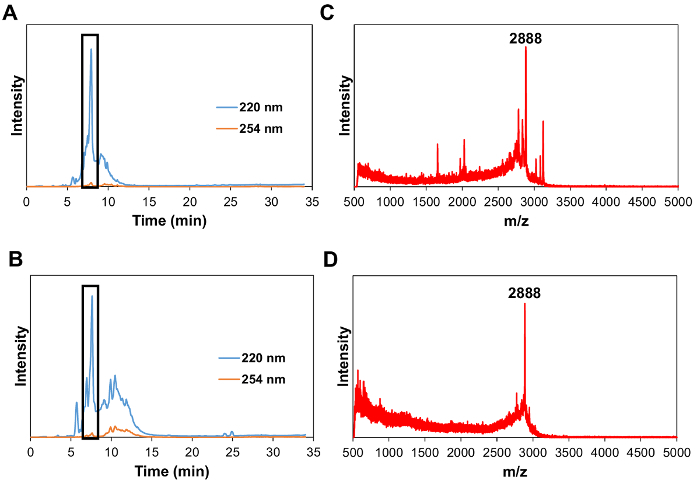
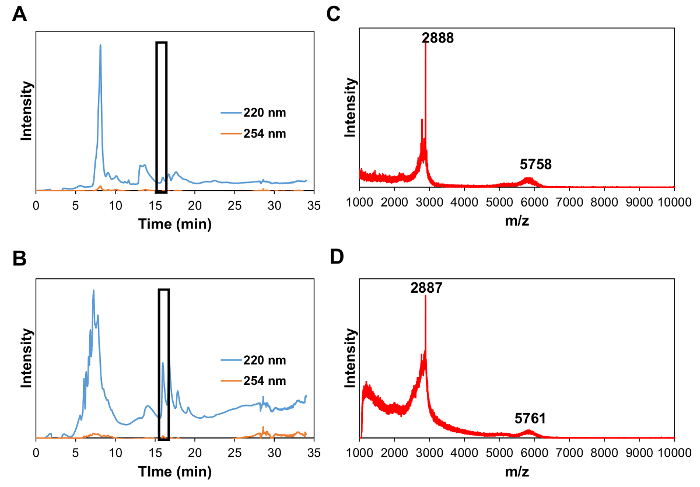
Preparation of MCP-1 PAM The CCR2-binding motif (residues 13-35) of the MCP-1 protein [YNFTNRKISVQRLASYRRITSSK] or scrambled peptide [YNSLVFRIRNSTQRKYRASIST] was modified by adding a cysteine residue on the N-terminus. The MCP-1 peptide was synthesized by a Fmoc-mediated solid-phase method using an automated peptide synthesizer. The crude peptide was purified by reverse-phase HPLC on a C8 column at 50 °C using 0.1% TFA in acetonitrile/water mixtures and characterized by MALDI mass spectrometry (Figure 1). The cysteine-containing peptide was conjugated onto DSPE-PEG2000-maleimide to yield DSPE-PEG(2000)-peptide conjugates via a thioether linkage. After 24 h at room temperature, the crude product was purified by HPLC (Figure 2). DSPE-PEG(2000)-Cy5 was synthesized using an NHS ester reaction. Monocyte-targeting PAMs co-assembled with DSPE-PEG(2000)-MCP-1 and DSPE-PEG(2000)-Cy5 were fabricated by dissolving in methanol that was then evaporated by N2, and the resulting film was dried under vacuum overnight and self-assembled in water or PBS through the hydrophobic interactions of the "tail groups" to form MCP-1 PAMs.
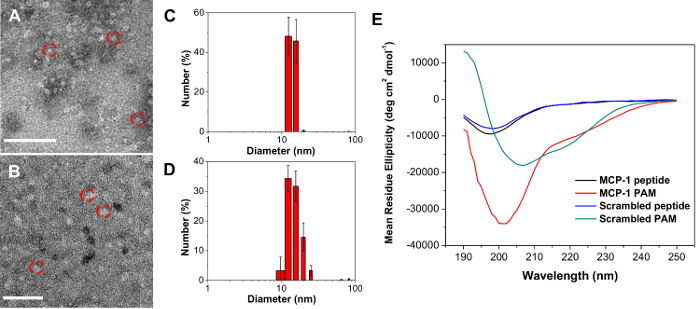
Characterization of MCP-1 PAM DLS data and TEM images of monocyte-targeting PAMs showed well-dispersed, spherical nanoparticle of 15.3 ± 2.0 nm in diameter (Figure 3 and Table 1). Both MCP-1 PAMs and scrambled PAMs showed a slight positive zeta potential of 12.1 ± 3.1 mV and 13.7 ± 1.9 mV, respectively (Table 1). CD showed that the incorporation of the MCP-1 peptide within the micelle enhanced the secondary structure (Table 2, MCP-1 PAMs: 46.2 ± 0.9% β-sheet, 53.1 ± 1.4% random coil, and free MCP1: 34.2 ± 3.5% β-sheet, 65.8 ± 3.5% random coil).
In Vitro Analysis of MCP-1 PAMIn vitro biocompatibility and monocyte binding of MCP-1 PAMs were observed by confocal microscopy. Mouse monocytes were incubated with increasing concentrations of MCP-1, MCP-1 PAM, scrambled peptide, and scrambled PAM for 72 h, and the cells were found to be viable and biocompatible up to 100 µM (Figure 4). Furthermore, confocal microscopy showed specific binding of monocytes to MCP-1 PAMs when compared to scrambled PAMs (Figure 5).
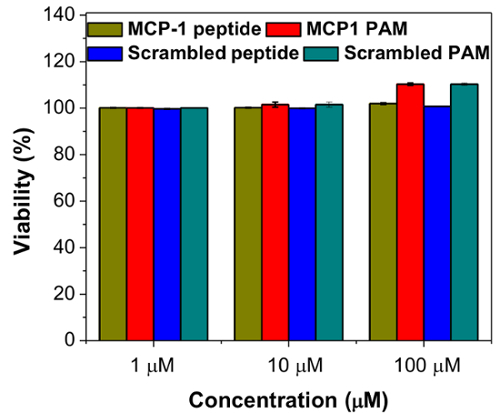
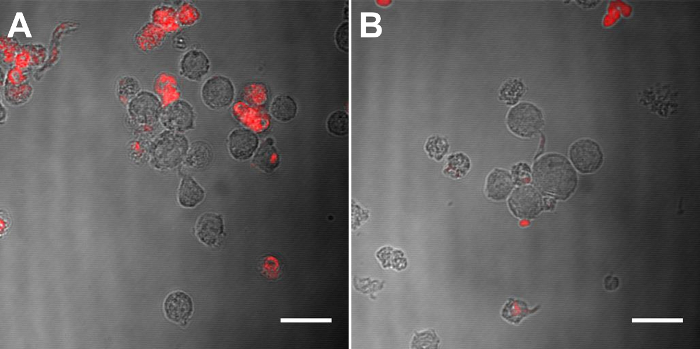
Figure 1: HPLC chromatogram at 220 nm (wavelength of peptide bonds) and 254 nm (tyrosine wavelength) of MCP-1 peptide (A) and scrambled peptide (B) (Boxed peak at 7.4 min). MALDI-TOF mass spectrum of MCP-1 peptide (C) and scrambled peptide (D) at a range of 500-5,000 Da showing the peak for [M+H]+ at m/z 2,888 (expected 2,890). Please click here to view a larger version of this figure.
Figure 2: HPLC chromatogram at 220 nm (wavelength of peptide bonds) and 254 nm (tyrosine wavelength) of DSPE-PEG(2000)-MCP-1 (A) and DSPE-PEG(2000)-scrambled (B). (Boxed peak at 16.9 min). (C) MALDI-TOF mass spectrum of DSPE-PEG(2000)-MCP-1 at a range of 1,000-10,000 Da showing the peaks for [M+H]+ at m/z 2,888 (expected 2,890) for MCP-1 and m/z 5,758 (expected 5,760) for DSPE-PEG(2000)-MCP-1. (D) MALDI-TOF mass spectrum of DSPE-PEG(2000)-scrambled at a range of 1,000-10,000 Da showing the peaks for [M+H]+ at m/z 2,887 (expected 2,890) for scrambled peptide and m/z 5,761 (expected 5,760) for DSPE-PEG-scrambled. Please click here to view a larger version of this figure.
Figure 3: TEM images of MCP-1 PAM (A) and scrambled PAM (B). Scale bar = 100 nm. Number-average size distribution of MCP-1 PAM (C) and scrambled PAM via DLS (D). Data are mean ± S.D. (n = 3). (E) CD analysis of MCP-1 and scrambled peptides and PAMs (n = 3). Please click here to view a larger version of this figure.
Figure 4:In vitro viability of WEHI 274.1 murine monocytes after 72 h exposure to MCP-1 peptide, MCP-1 PAM, scrambled peptide, and scrambled PAM. Data are mean ± S.D. (n = 6). Please click here to view a larger version of this figure.
Figure 5: CLSM images of MCP-1 PAM (A) and scrambled PAM (B) incorporated with 10 mol% Cy5 PA (red) incubated with WEHI 274.1 for 1 h. Scale Bar = 20 µm. Please click here to view a larger version of this figure.
| PAMs | Number-Ave diameter (nm) | PDI | Zeta Potential (mV) |
| MCP-1 | 15.3 ± 2.0 | 0.119 ± 0.006 | 12.1 ± 3.1 |
| Scrambled | 16.9 ± 1.4 | 0.232 ± 0.019 | 13.7 ± 1.9 |
| Measured in PBS buffer. Data are expressed as mean ± SD. |
Table 1: Size, polydispersity, and zeta potential of PAMs.
| a-Helix (%) | b-Sheet (%) | Random Coil (%) | |
| MCP-1 peptide | 0.0 ± 0.0 | 36.2 ± 2.2 | 63.8 ± 1.8 |
| MCP-1 PAM | 0.7 ± 0.6 | 46.2 ± 0.9 | 53.1 ± 1.4 |
| Scrambled peptide | 0.0 ± 0.0 | 43.7 ± 2.1 | 56.3 ± 3.5 |
| Scrambled PAM | 0.0 ± 0.0 | 60.7 ± 0.2 | 39.3 ± 0.2 |
Table 2: Secondary structure composition of peptides and PAMs.
Discussion
MCP-1 PAMs are a promising molecular imaging platform, consisting of a hydrophilic targeting peptide and hydrophobic tail that drives the self-assembled nature of the nanoparticle. This monocyte-targeting micelle can be prepared by simple synthesis and purification steps of the MCP-1 peptide and DSPE-PEG(2000)-MCP-1. PAMs have many beneficial characteristics for in vivo molecular imaging such as their self-assembly under mild conditions, intrinsic biodegradability, and structural and chemical diversity allowing for the incorporation of other imaging moieties or targeting peptides to selectively deliver to a specific site of interest. Their particle size, shape, and composition can be tuned by changing the peptide, hydrophobic tail, or micelle concentration, allowing optimal chemical and physical properties for a variety of applications22,33.
A few limitations of this protocol should be mentioned. First, since PAMs are driven by hydrophobic interactions, it is necessary to construct targeting peptides that are hydrophilic, thereby allowing the targeting moiety to be presented on the surface of the micelle. If the peptide sequence is hydrophobic, an addition of one or two hydrophilic amino acids at the C terminus can be accommodated, but the addition of amino acids could affect the peptide secondary structure within the micelle, thereby changing the properties of the peptide within its native state in the protein44,45. Second, at low concentrations, PAMs have poor stability in aqueous environments as they are highly dependent on the CMC, the minimum concentration needed for PAs to form a micellar structure46. Below the CMC, the micelle disassembles to individual PA monomers, and limits the micelle to act as a carrier and unload a drug at a specific site47. To alleviate this drawback, the CMC can be decreased by increasing the chain length of the hydrophobic tail48,49. Finally, the long-term storage of PAMs in solution is not recommended as PA secondary structure can change with time, thereby affecting the micellar structure and stability, as well as efficiency in ligand binding26.
In sum, this protocol demonstrates a robust, self-assembled peptide micelle-based nanomedicine strategy for imaging atherosclerosis. MCP-1 PAMs are biocompatible, biodegradable, and nontoxic as components of the micelle are inspired from elements endogenous to the body. More importantly, MCP-1 PAMs have preferential binding to monocytes, which increase proportionally to plaque progression, facilitating a non-invasive approach to differentiating stages of atherosclerotic plaques. Due to the modularity of this platform, PAMs can incorporate therapeutics, additional targeting peptides, imaging moieties, and nucleic acids. Hence, given such dynamic capabilities of these multifunctional micelles, we believe that these particles hold great promise for clinical translation in atherosclerosis and other diseases.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to acknowledge the financial support from the University of Southern California, the National Heart, Lung, and Blood Institute (NHLBI), R00HL124279, Eli and Edythe Broad Innovation Award, and the L.K. Whittier Foundation Non-Cancer Translational Research Award granted to EJC. The authors thank the Center for Electron Microscopy and Microanalysis, Center of Excellence in NanoBiophysics, Center of Excellence for Molecular Characterization, and Translational Imaging Center at the University of Southern California for assistance in instrumental setups.
References
- Mozaffarian D, et al. Heart Disease and Stroke Statistics-2016 Update: A Report From the American Heart Association. Circulation. 2016;133(4):e38–e60. doi: 10.1161/CIR.0000000000000350. [DOI] [PubMed] [Google Scholar]
- Falk E. Pathogenesis of atherosclerosis. J. Am. Coll. Cardiol. 2006;47(8, Suppl. C):C7–C12. doi: 10.1016/j.jacc.2005.09.068. [DOI] [PubMed] [Google Scholar]
- Rahmani M, Cruz RP, Granville DJ, McManus BM. Allograft Vasculopathy Versus Atherosclerosis. Circ. Res. 2006;99(8):801–815. doi: 10.1161/01.RES.0000246086.93555.f3. [DOI] [PubMed] [Google Scholar]
- Luis AJ. Atherosclerosis. Nature. 2000;407(6801):233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Ridker PM, Hansson GK. Progress and challenges in translating the biology of atherosclerosis. Nature (London, U. K) 2011;473(7347):317–325. doi: 10.1038/nature10146. [DOI] [PubMed] [Google Scholar]
- Boring L, Gosling J, Cleary M, Charo IF. Decreased lesion formation in CCR2-/- mice reveals a role for chemokines in the initiation of atherosclerosis. Nature (London) 1998;394(6696):894–897. doi: 10.1038/29788. [DOI] [PubMed] [Google Scholar]
- Szmitko PE, et al. New Markers of Inflammation and Endothelial Cell Activation. Circulation. 2003;108(16):1917–1923. doi: 10.1161/01.CIR.0000089190.95415.9F. [DOI] [PubMed] [Google Scholar]
- Deshmane SL, Kremlev S, Amini S, Sawaya BE. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009;29(6):313–326. doi: 10.1089/jir.2008.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung EJ, et al. Monocyte-Targeting Supramolecular Micellar Assemblies: A Molecular Diagnostic Tool for Atherosclerosis. Adv. Healthcare Mater. 2015;4(3):367–376. doi: 10.1002/adhm.201400336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung EJ. Targeting and therapeutic peptides in nanomedicine for atherosclerosis. Exp Biol Med (Maywood) 2016;241(9):891–898. doi: 10.1177/1535370216640940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz J, Fayad ZA. Imaging of atherosclerotic cardiovascular disease. Nature (London, U. K.) 2008;451(7181):953–957. doi: 10.1038/nature06803. [DOI] [PubMed] [Google Scholar]
- Tarkin JM, et al. Imaging Atherosclerosis. Circ. Res. 2016;118(4):750–769. doi: 10.1161/CIRCRESAHA.115.306247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennerici M, Baezner H, Daffertshofer M. Ultrasound and arterial wall disease. Cerebrovasc Dis. 2004;17 Suppl 1:19–33. doi: 10.1159/000074792. [DOI] [PubMed] [Google Scholar]
- Nissen SE. Application of intravascular ultrasound to characterize coronary artery disease and assess the progression or regression of atherosclerosis. Am J Cardiol. 2002;89(4A):24B–31B. doi: 10.1016/s0002-9149(02)02217-8. [DOI] [PubMed] [Google Scholar]
- Khalil MM, Tremoleda JL, Bayomy TB, Gsell W. Molecular SPECT Imaging: An Overview. Int J Mol Imaging. 2011;2011:796025. doi: 10.1155/2011/796025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerakis S, et al. Imaging of the vulnerable plaque: noninvasive and invasive techniques. Am J Med Sci. 2008;336(4):342–348. doi: 10.1097/MAJ.0b013e31816c7bdf. [DOI] [PubMed] [Google Scholar]
- Sun ZH, Rashmizal H, Xu L. Molecular imaging of plaques in coronary arteries with PET and SPECT. J Geriatr Cardiol. 2014;11(3):259–273. doi: 10.11909/j.issn.1671-5411.2014.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godin B, et al. Emerging applications of nanomedicine for the diagnosis and treatment of cardiovascular diseases. Trends Pharmacol. Sci. 2010;31(5):199–205. doi: 10.1016/j.tips.2010.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Branco de Barros AL, Tsourkas A, Saboury B, Cardoso VN, Alavi A. Emerging role of radiolabeled nanoparticles as an effective diagnostic technique. EJNMMI Res. 2012;2(1):31–39. doi: 10.1186/2191-219X-2-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayagopal A, Linton MF, Fazio S, Haselton FR. Insights into atherosclerosis using nanotechnology. Curr. Atheroscler. Rep. 2010;12(3):209–215. doi: 10.1007/s11883-010-0106-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodabandehlou K, Masehi-Lano JJ, Poon C, Wang J, Chung EJ. Targeting cell adhesion molecules with nanoparticles using in vivo and flow-based in vitro models of atherosclerosis. Exp Biol Med (Maywood) 2017;242(8):799–812. doi: 10.1177/1535370217693116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trent A, Marullo R, Lin B, Black M, Tirrell M. Structural properties of soluble peptide amphiphile micelles. Soft Matter. 2011;7(20):9572–9582. [Google Scholar]
- Hartgerink JD, Beniash E, Stupp SI. Peptide-amphiphile nanofibers: A versatile scaffold for the preparation of self-assembling materials. Proceedings of the National Academy of Sciences. 2002;99(8):5133–5138. doi: 10.1073/pnas.072699999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Missirlis D, et al. Effect of the Peptide Secondary Structure on the Peptide Amphiphile Supramolecular Structure and Interactions. Langmuir. 2011;27(10):6163–6170. doi: 10.1021/la200800e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong L, Johnson WC. Environment affects amino acid preference for secondary structure. Proc. Natl. Acad. Sci. U. S. A. 1992;89(10):4462–4465. doi: 10.1073/pnas.89.10.4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimada T, Lee S, Bates FS, Hotta A, Tirrell M. Wormlike Micelle Formation in Peptide-Lipid Conjugates Driven by Secondary Structure Transformation of the Headgroups. J. Phys. Chem. B. 2009;113(42):13711–13714. doi: 10.1021/jp901727q. [DOI] [PubMed] [Google Scholar]
- Missirlis D, et al. Linker Chemistry Determines Secondary Structure of p5314-29 in Peptide Amphiphile Micelles. Bioconjugate Chem. 2010;21(3):465–475. doi: 10.1021/bc900383m. [DOI] [PubMed] [Google Scholar]
- Elbert DL, Hubbell JA. Surface treatments of polymers for biocompatibility. Annu. Rev. Mater. Sci. 1996;26:365–394. [Google Scholar]
- Xue Y, O'Mara ML, Surawski PPT, Trau M, Mark AE. Effect of Poly(ethylene glycol) (PEG) Spacers on the Conformational Properties of Small Peptides: A Molecular Dynamics Study. Langmuir. 2011;27(1):296–303. doi: 10.1021/la103800h. [DOI] [PubMed] [Google Scholar]
- Canalle LA, Loewik DWPM, van Hest JCM. Polypeptide-polymer bioconjugates. Chem. Soc. Rev. 2010;39(1):329–353. doi: 10.1039/b807871h. [DOI] [PubMed] [Google Scholar]
- Hamley IW. PEG-Peptide Conjugates. Biomacromolecules. 2014;15(5):1543–1559. doi: 10.1021/bm500246w. [DOI] [PubMed] [Google Scholar]
- Acar H, et al. Self-assembling peptide-based building blocks in medical applications. Adv. Drug Delivery Rev. 2016. [DOI] [PMC free article] [PubMed]
- Busseron E, Ruff Y, Moulin E, Giuseppone N. Supramolecular self-assemblies as functional nanomaterials. Nanoscale. 2013;5(16):7098–7140. doi: 10.1039/c3nr02176a. [DOI] [PubMed] [Google Scholar]
- Barrett JC, et al. Modular Peptide Amphiphile Micelles Improving an Antibody-Mediated Immune Response to Group A Streptococcus. ACS Biomater. Sci. Eng. 2017;3(2):144–152. doi: 10.1021/acsbiomaterials.6b00422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toughrai S, et al. Reduction-Sensitive Amphiphilic Triblock Copolymers Self-Assemble Into Stimuli-Responsive Micelles for Drug Delivery. Macromol. Biosci. 2015;15(4):481–489. doi: 10.1002/mabi.201400400. [DOI] [PubMed] [Google Scholar]
- Swirski FK, et al. Monocyte accumulation in mouse atherogenesis is progressive and proportional to extent of disease. Proc. Natl. Acad. Sci. U. S. A. 2006;103(27):10340–10345. doi: 10.1073/pnas.0604260103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi M, et al. Association of monocyte subsets with vulnerability characteristics of coronary plaques as assessed by 64-slice multidetector computed tomography in patients with stable angina pectoris. Atherosclerosis (Amsterdam, Neth) 2010;212(1):171–176. doi: 10.1016/j.atherosclerosis.2010.05.004. [DOI] [PubMed] [Google Scholar]
- Chung EJ, Tirrell M. Recent Advances in Targeted, Self-Assembling Nanoparticles to Address Vascular Damage Due to Atherosclerosis. Adv. Healthcare Mater. 2015;4(16):2408–2422. doi: 10.1002/adhm.201500126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L, Pollard JW. Cancer immunosurveillance: role of patrolling monocytes. Cell Res. 2016;26(1):3–4. doi: 10.1038/cr.2015.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams CB, Yeh ES, Soloff AC. Tumor-associated macrophages: unwitting accomplices in breast cancer malignancy. NPJ Breast Cancer. 2016;2 doi: 10.1038/npjbcancer.2015.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richards DM, Hettinger J, Feuerer M. Monocytes and Macrophages in Cancer: Development and Functions. Cancer Microenviron. 2013;6(2):179–191. doi: 10.1007/s12307-012-0123-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schick MJ. Effect of temperature on the critical micelle concentration of nonionic detergents. Thermodynamics of micelle formation. J. Phys. Chem. 1963;67(9):1796–1799. [Google Scholar]
- Marullo R, Kastantin M, Drews LB, Tirrell M. Peptide contour length determines equilibrium secondary structure in protein-analogous micelles. Biopolymers. 2013;99(9):573–581. doi: 10.1002/bip.22217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilbich C, Kisters-Woike B, Reed J, Masters CL, Beyreuther K. Substitutions of hydrophobic amino acids reduce the amyloidogenicity of Alzheimer's disease βA4 peptides. J. Mol. Biol. 1992;228(2):460–473. doi: 10.1016/0022-2836(92)90835-8. [DOI] [PubMed] [Google Scholar]
- Trevino SR, Scholtz JM, Pace CN. Amino Acid Contribution to Protein Solubility: Asp, Glu, and Ser Contribute more Favorably than the other Hydrophilic Amino Acids in RNase Sa. J. Mol. Biol. 2007;366(2):449–460. doi: 10.1016/j.jmb.2006.10.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SW, Shi YZ, Kim JY, Park KN, Cheng JX. Overcoming the barriers in micellar drug delivery: Loading efficiency, in vivo stability, and micelle-cell interaction. Expert Opin Drug Delivery. 2010;7(1):49–62. doi: 10.1517/17425240903380446. [DOI] [PubMed] [Google Scholar]
- Rangel-Yagui CO, Pessoa A, Jr, Tavares LC. Micellar solubilization of drugs. J. Pharm. Pharm. Sci. 2005;8(2):147–163. [PubMed] [Google Scholar]
- Batrakova E, et al. Fundamental relationships between the composition of pluronic block copolymers and their hypersensitization effect in MDR cancer cells. Pharm. Res. 1999;16(9):1373–1379. doi: 10.1023/a:1018942823676. [DOI] [PubMed] [Google Scholar]
- Chen LJ, Lin SY, Huang CC. Effect of Hydrophobic Chain Length of Surfactants on Enthalpy-Entropy Compensation of Micellization. J. Phys. Chem. B. 1998;102(22):4350–4356. [Google Scholar]