

Abstract
Cell-autonomous neuronal functions of genes can be revealed by causing loss or gain of function of a gene in a small and sparse population of neurons. To do so requires generating a mosaic in which neurons with loss or gain of function of a gene are surrounded by genetically unperturbed tissue. Here, we combine the Cre-lox recombination system with in utero electroporation in order to generate mosaic brain tissue that can be used to study the cell-autonomous function of genes in neurons. DNA constructs (available through repositories), coding for a fluorescent label and Cre recombinase, are introduced into developing cortical neurons containing genes flanked with loxP sites in the brains of mouse embryos using in utero electroporation. Additionally, we describe various adaptations to the in utero electroporation method that increase survivability and reproducibility. This method also involves establishing a titer for Cre-mediated recombination in a sparse or dense population of neurons. Histological preparations of labeled brain tissue do not require (but can be adapted to) immunohistochemistry. The constructs used guarantee that fluorescently labeled neurons carry the gene for Cre recombinase. Histological preparations allow morphological analysis of neurons through confocal imaging of dendritic and axonal arbors and dendritic spines. Because loss or gain of function is achieved in sparse mosaic tissue, this method permits the study of cell-autonomous necessity and sufficiency of gene products in vivo.
Keywords: Neuroscience, Issue 129, Development, cerebral cortex, cell-autonomous, gene delivery, Cre recombinase, loxP, in utero electroporation, dendritic spines, mosaicism, gain of function, loss of function
Introduction
Generating a genetic mosaic is a classic experimental paradigm for understanding the function of a gene of interest. To determine if a gene is necessary for a cellular phenotype, the simplest approach is causing a loss of function of the gene throughout the organism (e.g. knockout). However, to determine if a gene is required specifically in a certain cell type, knockout of the gene throughout the organism is not a valid approach. Instead, a method is required that will cause the loss of function of a gene in a given cell while it is surrounded by wildtype (i.e. genetically unperturbed) tissue—in other words, creating mosaic tissue. If the mutant cell shows a mutant phenotype, but surrounding wildtype cells do not, the gene functions in a cell-autonomous manner. Analysis of mosaic tissue, in which mutant cells are surrounded by wildtype tissue, is ideal for understanding cell-autonomous functions of genes, especially in the brain where neurons and glia form a vast interconnected network of tissue.
Several forms of mosaic brain tissue have provided powerful models to investigate cell-autonomous functions of genes. Studies focused on neuronal transplantation1, female X-linked mosaicism2,3,4, and endogenous somatic mosaicism5,6 have drawn their conclusions based on mosaic brain tissue. Conditional deletion of a gene through the Cre-lox recombination system is a method that takes full advantage of the great availability of transgenic mouse lines. In this method, two loxP sites are introduced on either side of a required sequence of a gene (such as an exon), leaving it flanked by loxP sites that both face in the same direction ("floxed"). Cre recombinase excises the sequence between the loxP sites7. Cre-mediated recombination can be achieved by crossing floxed mice to another mouse line expressing Cre recombinase along with a fluorescent marker in a subset of cells ("Cre reporter line"). This has been demonstrated in a variety of ways to uncover the functions of a gene in subsets of cells, such as excitatory neurons or astrocytes8. Cre reporter lines can express CreERT2 to allow Cre-mediated recombination to be drug-inducible (single-neuron labeling with inducible Cre-mediated knockout, or SLICK)9. In another strategy called mosaic analysis with double markers (MADM)10,11, Cre-mediated interchromosomal recombination allows a homozygous mutant to be created alongside heterozygous tissue. In these approaches, a new line of mice needs to be produced each time for each candidate gene or cellular subtype that is tested. Alternatively, Cre recombinase can be introduced postnatally through iontophoresis12 or through viral vectors (e.g. adeno-associated viruses13 or lentiviruses14 carrying cellular subtype-specific promoters). This strategy creates strong and postnatal labeling. To target developing cerebral cortical neurons sparsely and prenatally, an ideal strategy is in utero electroporation of Cre recombinase with a fluorescent marker.
In addition to combining Cre-lox recombination through in utero electroporation to produce mosaic tissue in vivo, we introduce several adaptations to procedures from other published protocols15,16,17,18,19,20,21. We provide information to improve success in breeding timed-pregnant females. We also outline our two strategies to introduce sparse and bright labeling of neurons in cortical tissue: One strategy is to titrate the levels of a single construct coding for Cre recombinase and a fluorescent marker22. Another strategy is to use the "Supernova" system, designed specifically with these parameters in mind23,24. Additionally, we offer improvements on producing consistent microinjection pipettes and simplifications to the in utero electroporation surgery. Finally, we outline critical steps in a simplified histological preparation that permits the analysis of dendritic spines and dendritic and axonal arbors, without further staining or immunohistochemistry.
Protocol
Methods described here have been approved by the Animal Care and Use Committee (ACUC) of James Madison University, and are in accordance and compliance with all relevant regulatory and institutional guidelines.
1. Mouse Set-up
House a young (>P60) male and female homozygous floxed mouse together to set up a breeder pair25. NOTE: A good negative control is to set up an additional breeder pair of wildtype mice, in which Cre recombinase expression will not cause a mosaic26.
Allow the female to give birth and raise her first litter with the male. Keep the male together with the female to increase the litter's survival. After weaning the litter, e.g. at postnatal day (P) 21, separate the female and male.
- Murine breeding
- If a plug is found, separate, and weigh the female. Count the day as embryonic day (E) 0.5.
- Continue weighing the female every 3-5 days to determine if she is pregnant. Pregnant females will have a 10-20% increase in body weight 7-10 days after conception (E0). NOTE: This step confirms pregnancy earlier and more reliably than visual inspection25.
- If the female is not pregnant, repeat steps 1.3.1-1.3.3.
Once pregnancy is confirmed, choose the date of the in utero electroporation. To target layer II/III cortical pyramidal neuronal progenitors, perform electroporation at E15.5.
2. DNA set-up
Choose a single DNA construct that codes for Cre recombinase as well as a fluorescent marker, such as green fluorescent protein (GFP)28,29. Alternatively, use the "Supernova" system, described in detail in previous publications23,24. See list of materials for example constructs.
Amplify the DNA construct(s) from step 2.1 in E. coli using standard microbiological techniques30.
Purify the DNA construct with an endotoxin-free plasmid purification kit following the manufacturer's instructions. Elute the construct with endotoxin-free water. NOTE: Choosing an endotoxin-free purification kit over a standard purification kit is critical.
Concentrate the DNA eluate to 1-3 mg/mL depending on desired labeling density26. NOTE: A titer always needs to be established when working with a new DNA construct, given that they have different lengths and promoters. Consider injecting a construct at several different concentrations and/or amounts to assess expression. For example, injection of 1 µL or 1.5 µL of 2.0 mg/mL GFP.Cre is able to cause sparse (1 µL) or dense (1.5 µL) labeling of layer II/III visual cortical pyramidal neurons26.
Add 0.4% trypan blue in 1x phosphate buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4 in H2O, adjusted to pH 7.4 with HCl) 1:10 to the concentrated DNA solution.
Add 10x PBS 1:10 to the concentrated DNA solution.
Store the concentrated DNA solution at 4 °C. The solution may be stored indefinitely.
3. Pipette Set-up
- Using a glass capillary puller, pull a pipette with a very long (10-15 mm) taper and very fine tip, typical of pipettes for embryonic stem cell injection or nuclear transfer31. NOTE: To prevent debris from contaminating the pipettes, they should be made within 2 days of the surgery.
- Calibrate the glass capillary puller by performing a ramp test, following the manufacturer's instructions. Use the resulting heat value to program a pulling protocol using the following parameters: heat = (ramp test value + 15), pull = 30, vel = 120, time = 100, pressure = 200. Insert the glass capillary, fasten to clamps on puller, and activate the programmed pulling protocol, creating a taper on the glass capillary.
- Ensure that the taper length is between 10-15 mm using a compound light microscope (e.g. 10X objective)31.
- Break back the pipette to form a rough-edged tip to 20-25 µm diameter.
- Center a single-ply task wipe (see list of materials) over a 50 mL beaker and stretch wipe taut with one hand. With the other hand, hold and push a pipette (taper side down) perpendicularly and fully through the center of the wipe, causing a clean break in the taper31. NOTE: Pushing the pipette fully through the wipe will produce a 20-25 µm diameter tip about 70% of the time.
- Confirm under a compound light microscope (e.g. 20x objective). Caution: Use eye protection while breaking the glass.
Calibrate a pipette by aspirating 1 µL of 0.4% trypan blue in 1x PBS into a partially filled piptte, then graduating the pipette with 1 µL markings based on the height of 1 µL in the pipette, using a fine-tipped alcohol-resistant marker. Use the calibrated pipette to graduate the other pipettes before discarding. Keep pipettes in a pipette holder free from dust and other particles.
4. In Utero Electroporation
Place Graefe forceps, iris scissors, Hartman mosquito forceps, non-woven gauze sponges, ring-tipped forceps, and Petri dishes on a stainless steel tray, and wrap with aluminum foil. Place tray and 1 L 0.9% saline (0.9% wt/vol NaCl in water) in autoclave. Autoclave at 121 °C and 3.5 kPa for 40 min. Remove from autoclave and place in a disinfected surgical area. NOTE: It is critical to maintain sterile conditions during the surgery.
Warm the autoclaved 1 L saline bottle in a small water bath to 38 °C at least 2 h before the surgery.
Connect the tweezer-type electrodes and the foot pedal to the generator (see list of materials). Following the manufacturer's instructions, set the generator to deliver five 50 ms pulses of 50 V (with a 950 ms interval) when triggered by the foot pedal. Disinfect the tweezer-type electrodes and place on the disinfected surgical area.
Make an anesthesia nose cone as previously published32, but use the end of a nitrile or latex glove finger to form a diaphragm that fits securely over the nose cone opening. Cut a hole in the diaphragm large enough to fit over the mouse's snout.
Deeply anesthetize pregnant mice at E15.5 in an induction chamber filled with isoflurane (2-4%) in pure oxygen flowing at 1 L/min. After ~1 min, test the righting reflex (gently tilt the induction chamber and ensure that the mouse does not right itself). Weigh mouse to determine dosages of analgesia administered later during the surgery.
Transfer the mouse to the surgical area and administer inhaled isoflurane (1-2%) in pure oxygen flowing at 1 L/min through a nose cone to maintain mouse in surgical plane. Use forceps to test the pedal reflex (pinch the paw gently and ensure that the mouse does not return a reflex) to verify anesthetization. Monitor respiratory rate and effort, looking for deep, regular breathing (~70-100 breaths/min), and that mucous membranes are pink in color and moist.
Apply enough veterinary ophthalmic ointment over the eyes to cover them completely for protection against drying during the procedure.
Flex the metal activator disc of a sealed pouch filled with supersaturated salt solution (see list of materials), activating an exothermic crystallization of the salt. Lay 2 single-ply task wipes on top of the pouch, and place the mouse on the pouch to maintain body temperature.
Massage depilatory cream onto the abdominal area with cotton swabs until abdominal fur dissolves (approximately 20-40 s), then wipe off abdominal fur. Carefully clean off any remaining residue and depilatory cream with 70% ethanol and cotton swabs. Position a fenestrated sterile surgical drape over the abdominal area.
Using aseptic technique, pull the skin up and away from abdomen with Graefe forceps, and make a ~2 cm ventral midline incision on the skin using iris scissors, ensuring that underlying muscle is not cut. NOTE: An acceptable alternative to making an incision with Graefe forceps and iris scissors is to use a scalpel, as published previously17.
Find the linea alba to visualize the midline of the rectus abdominis. Pull muscle up and away from abdomen with Graefe forceps, and make another ~2 cm midline incision there with iris scissors, exposing the abdominal cavity (the uterus is translucent and embryos will be visible beneath). Ensure that underlying uterine and intestinal tissue is not cut. Make the incision only large enough (typically ~2 cm) to pull out and expose the uterus comfortably. NOTE: An acceptable alternative to making an incision with Graefe forceps and iris scissors is to use a scalpel, as published previously17.
Using a sterile 10 µL micropipette tip, dispense carprofen (dissolved in sterile 0.9% saline) at 4 mg/kg into the abdominal cavity for analgesia.
Clamp one Hartman mosquito forceps on the left edge of the rectus abdominis incision. Rest the forceps on overturned Petri dish to the left of the incision, keeping the left side of the incision open. Clamp another Hartman mosquito forceps on the right edge of the rectus abdominis incision and rest the forceps on an overturned Petri dish to the right of the incision, keeping the right side of the incision open. Place gauze sponge around the area of the incision.
Using ring forceps (without an attached limit screw), pull on the uterus between any two neighboring embryos without crushing or injuring any tissue. Begin pulling out all embryos through the incision, laying them on top of the gauze sponge. When pulling out the last embryos from the abdominal cavity, make sure not to pull on the ovaries or cervix. Once exposed, it is critical to keep the uterus moist with warm saline. The uterus typically contains between 5 and 8 embryos. NOTE: It is possible to add penicillin and streptomycin to the saline17.
Connect a calibrated pipette to the aspirator tube assembly (see list of materials), and inject exactly 1 µL of the DNA solution prepared in step 2 through the uterine wall into a lateral ventricle of each embryo. Lateral ventricles appear as two patches on the dorsal telencephalon of the embryo (patches are darker than surrounding tissue of telencephalon). Use the thumb and forefinger during microinjection and electroporation to manipulate the embryos, revealing the lateral ventricles and allowing the embryos to be pushed gently against the uterine wall during injection. Confirm successful injection by observing trypan blue filling the lateral ventricle.
Place the cathode of tweezer-type electrodes (see list of materials) on the uterus, directly over the medial and caudal cortex to target the visual cortex (targeting alternative areas of the brain will require different electrode placement )16,26. Place the anode on the uterus, just inferior and anterior to the embryo's head.
Confirm that the anode and cathode are touching the uterus surrounding the embryo at the appropriate locations. With a foot pedal, trigger the delivery of five 50 ms pulses of 50 V (with a 950 ms interval) across the electrodes. NOTE: The injected, negatively-charged DNA will move toward the cathode and will be incorporated into ventricular zone cells closest to the cathode.
Return the uterus to the abdominal cavity in the same orientation it was found. Use saline to lubricate the uterus while guiding it manually and extremely gently, taking care not to displace embryos from their position in the uterus.
Close the abdominal muscles with absorbable sutures (see list of materials) using a simple interrupted stitch, tying the ends with a surgeon's knot. Do not clip the ends too close to the knot or the knot will become unfastened. NOTE: It is critical to suture the abdominal muscles such that the edges of the wound are completely closed shut, but without causing blanching of the muscle. Knots should be tight so they cannot be unfastened.
Close the skin layer with absorbable sutures (simple interrupted stitch, surgeon's knot, as in step 4.19). Apply a small amount of tissue adhesive to seal the wound. Apply tissue adhesive on the knots to prevent unfastening. Use a micropipette to remove any tissue adhesive surrounding the wound that can be aspirated by the micropipette.
Lower isoflurane levels to 0.5-1.5%. Once tissue adhesive is dry (test by touching glove to adhesive), remove from isoflurane and allow female to recover alone in a warm cage. Administer buprenorphine intraperitoneally at 0.03 mg/kg using a 1 mL syringe with a 26G, ½" needle.
Continue to monitor the mouse until the mouse is fully recovered and behaving normally (i.e. grooming, exploring cage, eating, or drinking). Once recovered, place female back in cage with male. NOTE: If all steps are followed, the mouse will typically regain consciousness within 2 min and immediately begin moving about the cage, showing no signs of discomfort.
If the mouse exhibits signs of discomfort (e.g. hunched posture, porphyrin secretions)33, administer carprofen at 0.1 mg/mL in water supply. If several signs of discomfort are present, or if discomfort persists for more than 4 h despite carprofen administration, immediately euthanize the mouse by administering an intraperitoneal lethal injection of ketamine (240 mg/kg)-xylazine (48 mg/kg)-acepromazine (1.85 mg/kg) cocktail using a 1 mL syringe with a 26 G, ½" needle, and follow with an approved secondary form of euthanasia33.
To increase the survival of electroporated embryos after birth, keep the cage in a quiet holding room, free from noises and vibrations. Enrich the environment with plastic igloos, nesting materials, and dietary supplements such as sunflower seeds (see list of materials). Do not move or disturb the cage, particularly during the first 3 days after parturition.
5. Histological Preparation for Fluorescence Microscopy
NOTE: This histology protocol is optimized for preparing tissue from animals older than postnatal day (P) 13 that were electroporated in utero. To prepare tissue from younger postnatal animals (P0-P13), it is recommended to follow all steps (including transcardial perfusion), though the brain should be embedded in agar prior to preparing sections (step 5.9). Tissue may even be prepared from embryos within 1-2 days after electroporation, using methods described previously18,20.
- Prepare paraformaldehyde (PFA) solution. NOTE: It is imperative to prepare fresh PFA, within one day of the experiment. Caution: Paraformaldehyde is toxic and should be handled in a fume hood with appropriate personal protective equipment.
- To make 50 mL of 4% PFA, heat 40 mL water to 60 °C. Add 2 g PFA while stirring with glass rod or stirring bar. Add 10 µL of 10 M NaOH every 2 min until PFA dissolves.
- Add 5 mL of 10x PBS and bring solution to a final volume of 50 mL with water.
- After bringing solution to 50 mL, adjust pH to 7.4 as needed with 10 M HCl. Allow solution to cool and store at 4 °C.
Euthanize mouse with an intraperitoneal injection of ketamine (240 mg/kg)-xylazine (48 mg/kg)-acepromazine (1.85 mg/kg) cocktail using a 1 mL syringe with a 26G, ½" needle. Transcardially perfuse 10 or 20 mL ice-cold 1x PBS, followed by 25 or 40 mL freshly-made, ice-cold 4% PFA17. Use lower volumes for young postnatal (P0-P13) mice and higher volumes for older mice (P14 and above).
Immediately after perfusion, decapitate mouse carcass between occipital bone and C1 vertebra with large scissors and peel away and discard most of the skin surrounding skull using iris scissors, particularly skin surrounding frontal, parietal, and occipital bones. Keep the skull and brain intact.
Place the skull and brain in 10 mL 4% PFA in a 50 mL conical tube for 24 h at 4 °C to post-fix tissue. Then add 30 mL 1x PBS to conical tube to dilute the solution to 1% PFA for storage at 4 °C. NOTE: Use a separate conical tube for each skull and brain to be post-fixed. Do not incubate the brain in 1% PFA for longer than 2 weeks, or fluorescence will begin to fade.
Place the brain and skull on a flat surface with single-ply task wipes moistened with 1x PBS. Use a pair of tweezers (see list of materials) to remove any remaining skin or membrane surrounding the skull.
Using tweezers, first remove the occipital bone, then carefully remove the parietal bones, moving the tweezers out and away from the surface of the brain. Carefully remove any meninges to prevent damage to the cortex.
Wedge the back of the tweezers under the brain along the skull to sever any cranial nerves, and remove the brain.
- For young postnatal (P0-P13) brains
- Make 25 mL of 4% agar by heating 25 mL 1x PBS to 80 °C. Stir and dissolve 1 g agar with a glass rod or stirring bar).
- Cool solution to 35 °C while continuing to stir. Place brain in a small container (e.g. the cap of a 50 mL conical tube), and pour agar solution over brain. Allow agar to harden.
Make a coronal cut manually using a single-edge razor blade through the brain, approximately 0.5 mm rostral to bregma. Make another coronal cut through the brain approximately 0.5 mm caudal to visual cortex. Place a pool of cyanoacrylate glue with the same diameter as the rostral end of the brain on the center of the vibrating microtome specimen disc. Place the rostral end of the brain on top of the pool of glue, ensuring that the entire rostral surface of the tissue is bound to the specimen disc with glue.
Mount buffer tray onto vibrating microtome and secure with built-in clamping lever. Insert blade into knife holder, and mount knife holder onto vibrating microtome with built-in clamping screw. Secure specimen disc with brain onto buffer tray using built-in clamping device. Set speed setting to 5.70 (= 0.285 mm/s), and frequency setting to 5.33 (53.3 Hz).
Fill the chamber above the level of the brain with 1x PBS and lower the blade into the PBS. Begin taking 100 µm coronal sections through the tissue.
- If not requiring further processing, such as 4',6-diamidino-2-phenylindole (DAPI) nuclear staining or immunohistochemistry.
- Use a fine tipped paintbrush to transfer sections directly from the vibrating microtome onto a microscope slide, fitting 4-6 sections onto each slide.
- Allow the sections to dry by wicking away solution with a fine tipped paintbrush, such that the brain slices no longer drift on the slides.
- Then, place one drop of mountant (see list of materials) on each section on the slide.
- Gently lay a 24 x 50 mm coverslip on top, ensuring that 1) no air bubbles form on the sections, 2) the sections do not move, and 3) the mountant fully covers the area between the slide and coverslip. Allow the mountant to cure for at least 24-48 h.
- To stain nuclei with DAPI for histological examination of cortical laminae
- Use a fine tipped paintbrush to transfer each section into a solution containing 10 µg/mL DAPI (see list of materials) diluted in 1x PBS from stock solution (20 mg/mL DAPI in water).
- Incubate in DAPI solution for 5 min at room temperature, then use a fine tipped paintbrush to transfer section from DAPI solution to 1x PBS and incubate in 1x PBS for 5 min at room temperature. Transfer section two more times to fresh 1x PBS for 5 min each. Then, using a fine tipped paintbrush, transfer the section onto a microscope slide, follow steps 5.11.2-5.11.4, and proceed with step 5.13.
Heat a glass Petri dish containing Valap (equal parts Vaseline, lanolin, and paraffin) on a hot plate at a low heat setting until fully melted. Seal exposed edges of slides by brushing on melted Valap.
To inspect fluorescence in tissue, use a light microscope or confocal microscope (see list of materials) and a filter set adequate for viewing the fluorophore (e.g. DAPI: excitation 325-375 nm, emission 435-485 nm; GFP: excitation 450-490 nm, emission 500-550 nm)17. Confocal images may be taken with low- (4X, numerical aperture, NA = 0.20), medium- (20X, NA = 0.75), and high-power (60X, NA = 1.40) objectives.
Representative Results
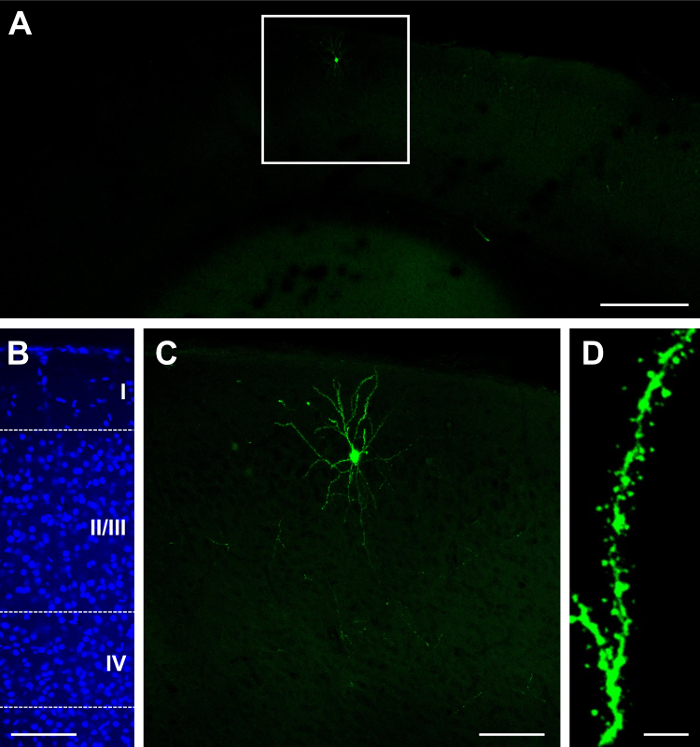
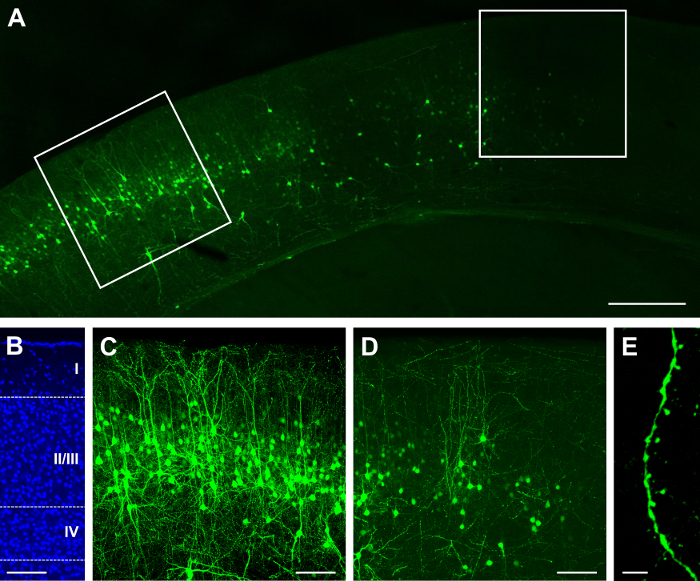
The single construct GFP.Cre (see list of materials) was electroporated at E15.5 and visualized at P14. Depending on the concentration of the construct and the volume of injection, a sparse or dense result can be obtained22,26. For example, injection of 1 µL of 2 mg/mL GFP.Cre results in a sparse distribution of labeled cells, some of which can be bright (Figure 1A), and localized in layer II/III (Figure 1B). Because the tissue is 100 µm thick, most of the dendritic arbors are preserved (Figure 1C). Dendritic spines can be observed at high magnifications (60X; Figure 1D). Injection of 1.5 µL at the same concentration results in very dense labeling (Figure 2A) in layer II/III (Figure 2B), which can be sub-optimal as it is difficult to track the source of neurites and dendritic spines (Figure 2C). However, it is still possible to image a neuron (Figure 2D) and its processes (Figure 2E) by selecting a bright cell in the periphery of the labeled area (Figure 2D).
Finally, it is possible to maximize the brightness of neurons while maintaining a sparse distribution of labeled cells. Here, Supernova-GFP (see list of materials) was electroporated at E15.5 and visualized at P23. Note that, based on observations of brain tissue taken at various postnatal ages, there does not appear to be an effect of age on the brightness of any fluorescent constructs used here23,24,26. Injection of 1 µL of a mixture containing 1 mg/mL Sn-GFP (CAG-loxP-stop-loxP-EGFP-ires-tTA-WPRE) and 10 µg/mL TRE-Cre results in a sparse distribution of mostly bright cells (Figure 3A) in layer II/III (Figure 3B). Dendritic and axonal processes (Figure 3B) and dendritic spines (Figure 3C) can be visualized. In our experience, targeting with either a single construct such as GFP.Cre or with "Supernova" constructs will reproducibly yield expression in at least 75% of electroporated embryos.
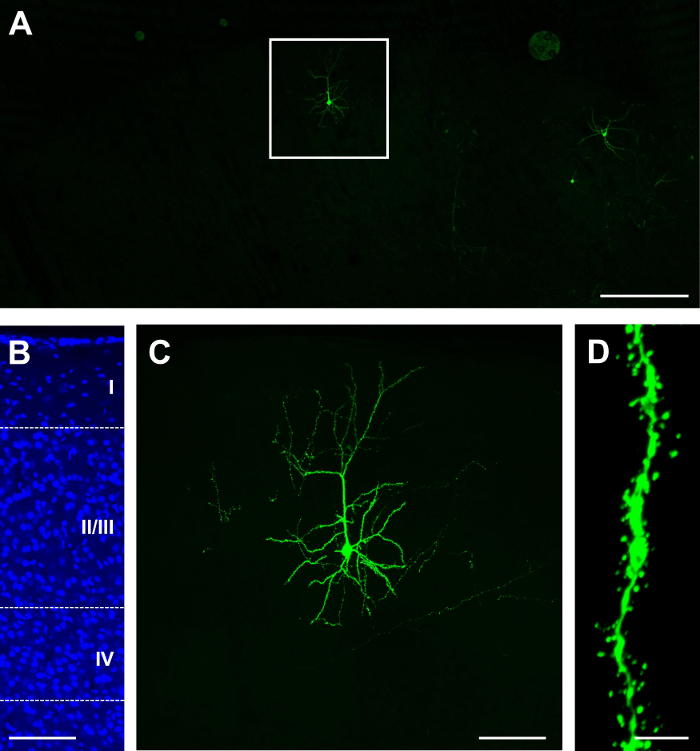
Figure 1:Sparse and bright expression after in utero electroporation with a single construct containing GFP and Cre recombinase. (A) Low-magnification (4X) fluorescence micrograph of cerebral cortex at P23, after electroporation at E15.5. (B) Medium-magnification (20X) fluorescence micrograph of DAPI nuclear stain, revealing cortical lamination (layers I, II/III, IV, and layers deep to IV). (C) Medium-magnification (20X) fluorescence micrograph of a single neuron. (D) High-magnification (60X) fluorescence micrograph of dendritic spines. Scale bars = 200 µm (A); 100 µm (B,C); and 5 µm (D). Please click here to view a larger version of this figure.
Figure 2:Dense and bright expression after in utero electroporation with a single construct containing GFP and Cre recombinase. (A) Low-magnification (4X) fluorescence micrograph of cerebral cortex at P14, after electroporation at E15.5. (B) Medium-magnification (20X) fluorescence micrograph of DAPI nuclear stain, revealing cortical lamination (layers I, II/III, IV, and layers deep to IV). (C) Medium-magnification (20X) fluorescence micrograph of neurons in the area of densest labeling. (D) Medium-magnification (20X) fluorescence micrograph of neurons taken at the periphery of the area of densest labeling. (E) High-magnification (60X) fluorescence micrograph of dendritic spines. Scale bars = 200 µm (A); 100 µm (B, C, D); and 5 µm (E). Please click here to view a larger version of this figure.
Figure 3:Sparse and bright expression after in utero electroporation with Supernova-GFP constructs containing GFP and Cre recombinase. (A) Low-magnification (4X) fluorescence micrograph of cerebral cortex at P14, after electroporation at E15.5. (B) Medium-magnification (20X) fluorescence micrograph of DAPI nuclear stain, revealing cortical lamination (layers I, II/III, IV, and layers deep to IV). (C) Medium-magnification (20X) fluorescence micrograph of a single neuron. (D) High-magnification (60X) fluorescence micrograph of dendritic spines. Scale bars = 200 µm (A); 100 µm (B,C); and 5 µm (D). Please click here to view a larger version of this figure.
Discussion
Here, we introduce the combination of in utero electroporation with Cre recombinase in floxed mice to generate mosaic brain tissue. An advantage of this approach is that a new mouse line does not need to be generated each time a different cellular subtype is to be targeted: in utero electroporation can be used to target excitatory neurons, inhibitory neurons, or glia depending on the time and location of electroporation15,16,17,18,19,20,21,34. In our approach, targeting the cerebral cortex is consistent and reproducible because we use 5 mm diameter electroporation electrodes that can be placed over a large area of the developing telencephalon. To target alternative sites in the brain (e.g. diencephalon, retina), or for more specific targeting of regions within the cerebral cortex, procedures and electrodes designed explicitly for this purpose may be used15,16. We also provide a single construct that provides bright labeling and guarantees that every fluorescently-labeled neuron contains Cre recombinase. A disadvantage of using a single construct to achieve bright labeling is that the labeled population can be too dense (Figure 2C), but this can be resolved by imaging cells at the periphery of the labeled area (Figure 2D). Alternatively, expression of a fluorescent construct can be designed to depend on Cre recombinase expression17, but low expression levels may require immunostaining for the fluorescent marker. This limitation is addressed by the "Supernova" system of Cre-dependent fluorescent marker expression (Figure 3A)23,24.
In addition to combining Cre-lox recombination with in utero electroporation, we offer several improvements for optimal breeding of timed-pregnant females. It is important to keep the mice in a holding room that is free from noises and vibrations caused by equipment such as laminar flow hoods. Enrich the environment with plastic igloos, nesting material, and dietary supplements like sunflower seeds (see list of materials). Apart from environmental considerations, we have noticed that the first litter of a female typically has the lowest survival rate. Thus, our protocol suggests waiting until the second pregnancy of the female to perform in utero electroporation. While this strategy increases the survival of the litter, it also tends to increase litter size, which can lengthen surgical time and thus lower the success of the surgery. Therefore, if encountering a particularly large number of embryos (e.g. more than 8), we suggest skipping the electroporation on some of the embryos, especially embryos closest to the ovaries and cervix, where the tissue is most prone to injury. Furthermore, using the same environmental enrichment strategy mentioned above (plastic igloos, nesting material, dietary supplements) tends to increase the survival of litters after birth. It is also important to note that estrus can occur in the female a few hours after parturition of the first litter. In this case, allow the female to give birth to her second litter, then separate after weaning in order to attempt timed mating. When looking for vaginal plugs, it is good practice to separate the female even if a plug is not found, particularly if the female was in estrus the night before. Plugs in certain strains are difficult to spot, and conception may have already occurred.
Another area of improvement is in producing adequate microinjection pipettes for in utero electroporation. Pulling pipettes for DNA injection into lateral ventricles is a critical step. Here, we provide a detailed protocol for setting up pipettes with a consistent tip size and rough tip edge. Other methods include breaking back the tip with a scalpel blade15 or pinching off the tip with forceps16,17. Note that if pipette tips are too small, they will correctly puncture the uterine wall but injection flow rates will be too low, lengthening the time in which the pipette is lodged in the embryonic brain. If pipette tips are too large, they may damage the uterine wall and embryonic brain. If pipette tips are too smooth, they will cause a large divot in the uterine wall and/or embryonic skull before breaking through, possibly damaging the embryo. Practice creating a rough break to produce a 20-25 µm tip and inspect tips under a simple light microscope (e.g. with a 10X objective) until a consistent result is achieved. Testing that liquid can be pipetted at a reasonable rate (e.g. ~0.5 µL/s) with an aspirator tube assembly is another method of ensuring that tip size is within an acceptable range. Because the injection pipettes are calibrated and graduated, the exact amount of DNA delivered to the lateral ventricle is known. This is the most critical step for achieving a desired sparseness reproducibly to an order of magnitude. In other words, a given concentration should yield labeling within a particular range, e.g. 10 to 100 labeled neurons.
The most important step in the protocol is the in utero electroporation procedure. Be extremely gentle while exposing the uterus. When pulling out the last embryos from the abdominal cavity, make sure not to pull on the ovaries or cervix. Using the thumb and forefinger during microinjection and electroporation allows gentle, dexterous manipulation of the embryos to reveal the lateral ventricle and allows the embryos to be pushed gently against the uterine wall. An extremely gentle touch is critical when handling the uterus and embryos. If it is difficult to achieve a gentle touch, use ring forceps with a built-in limit screw to prevent the forceps from clamping down on an embryo or the uterus, causing tissue damage. A further consideration is to administer both the carprofen and buprenorphine immediately before the surgery, a practice that appears to provide effective pain management during in utero surgery without affecting embryonic survival rates35.
In following our simplified histological procedure, several steps are critical. After perfusing experimental brains with fixative, keep the brain intact in the skull to protect the brain tissue from damage during post-fixation. While it is possible to store the brain in 1% PFA for days to weeks, note that fluorescence will decrease as the PFA solution polymerizes. Slicing 100 µm coronal sections is typically thin enough to permit confocal microscopy through the entire section while preserving much of the dendritic and axonal morphology. If fixation occurred properly, brains from animals older than P13 can be mounted onto the vibrating microtome simply with cyanoacrylate glue. However, if the brain is too pliable while being cut by the vibrating microtome, glue down a small agar or agarose block behind the brain to prevent it from bending while it is being sectioned. If the problem is not resolved, embed the brain in agar to form a block to cut with the vibrating microtome, as suggested for younger postnatal brains (step 5.8).
This method combines the Cre-lox recombination system with in utero electroporation in order to generate mosaics that can be used to study the cell-autonomous function of genes in neurons. This method could also be configured to support in utero electroporation of gene editing constructs (e.g. CRISPR/Cas9)6, or other site-specific recombinases (e.g. Flp/FRT or Dre/rox24), which could all be designed to produce mosaic brain tissue. One promising alternative technique that could be used to produce mosaic tissue is viral delivery via intraventricular injection to neonatal mice36. Combined with intraventricular injection at embryonic ages as demonstrated here and elsewhere15,16,17,18,19,20,21,22,37, viral vectors coding for Cre recombinase could be delivered before birth to infect floxed progenitor cells at the ventricular zone. One critical consideration would be to ensure that the viral vectors are diluted sufficiently to achieve sparse excision and labeling. However, this would also result in lower copy numbers of the construct being delivered, including fluorescent markers essential for neuroanatomical observations (e.g. dendritic spines or arborization)24. To counter this pitfall, new constructs using the same strategy as the "Supernova" constructs demonstrated here could be used to achieve sparse labeling without a corresponding decrease in the brightness of labeled cells24. Another critical consideration with viral delivery is to ensure that delivered constructs have promoters specific to the cell population being studied (e.g. excitatory pyramidal neurons). Intraventricular injection of viral vectors causes infection of all tissue surrounding the ventricles, including not only the ventricular zone of the dorsal telencephalon where cortical and hippocampal excitatory neurons are generated, but also the medial and lateral ganglionic eminences where many cortical interneurons are born34. Another feature of in utero electroporation compared to viral delivery of constructs is its relatively narrow time window of delivery. Viruses injected into the lateral ventricles can persist after the surgery, continuing to infect cells lining the ventricular zone. In contrast, electroporation occurs in seconds, targeting a far more specific set of cells. This may be an advantage or disadvantage to the investigator, depending on the subpopulation of cells to be studied.
Our approach supports Cre-lox recombination by introduction of either a single construct or "Supernova" constructs. In the case of the "Supernova" constructs, we urge investigators to become familiar with the advantages and limitations of using this strategy. For example, the timing of Cre deletion has been demonstrated to occur within 2 days in layer II/III excitatory neurons of the cerebral cortex24. Thus, it is plausible that Cre excision could occur over the 48 h following electroporation, rather than immediately following electroporation. Therefore, other forms of corroborating timing and location of Cre excision (e.g. using a Cre reporter mouse line) should be used to complement the use of this new technique, especially in studies where Cre excision within a small timeframe is a critical consideration. Another potential pitfall is that a small percentage of unlabeled cells in a "Supernova" experiment could express Cre recombinase. For example, in Figure 3, we co-electroporated two plasmids: a high concentration of CAG-loxP-stop-loxP-EGFP-ires-tTA-WPRE, and a low concentration of TRE-Cre. Because TRE is a leaky promoter, Cre recombinase could be expressed in cells that received the TRE-Cre plasmid but not the loxP-stop-loxP-EGFP-tTA plasmid, though this would be a rare occurrence. In an experiment where it is essential that all unlabeled cells have no Cre-mediated excision whatsoever, a "Supernova" experiment may need to be supplemented with a control experiment in which Cre recombinase expression outside of labeled neurons is assessed through other means (e.g. Cre recombinase immunohistochemistry).
In summary, our protocol is easily modified to accommodate these new constructs, making in utero electroporation an even more useful and adaptable method for mosaic analysis. Thus, in utero electroporation can be combined with the power of genetic recombination in multiple ways to study mosaic brain tissue in vivo.
Disclosures
The authors have no conflicts of interest to disclose.
Acknowledgments
The authors thank the generous support of the James Madison University Department of Biology and the James Madison University Light Microscopy and Imaging Facility. Dr. Mark L. Gabriele for helpful advice regarding young postnatal tissue preparation, and Drs. Justin W. Brown and Corey L. Cleland for generous coordination of surgical materials and space. This research was funded in part by a Collaborative Research Grant by 4-VA, a collaborative partnership for advancing the Commonwealth of Virginia (G.S.V.), and by a Virginia Academy of Science Small Project Research Grant (G.S.V.). Support has been generously provided by a Betty Jo Loving Butler '58 Endowment for Undergraduate Research Scholarship (to K.M.B.), a Farrell Summer Research Scholarship (to K.M.B.), a James Madison University Second Century Scholarship (to K.M.B.), a James Madison University Centennial Scholarship (to C.J.H.), a James Madison University Lucy Robinson Search '30 Memorial Scholarship (to Z.L.H.), and a James Madison University College of Science and Mathematics Faculty Assistance Grant (to G.S.V.).
References
- Southwell DG, Froemke RC, Alvarez-Buylla A, Stryker MP, Gandhi SP. Cortical plasticity induced by inhibitory neuron transplantation. Science. 2010;327(5969):1145–1148. doi: 10.1126/science.1183962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson JE, Madison DV. Presynaptic FMR1 genotype influences the degree of synaptic connectivity in a mosaic mouse model of fragile X syndrome. J. Neurosci. 2007;27(15):4014–4018. doi: 10.1523/JNEUROSCI.4717-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, Hays SA, Bureau I, Huber KM, Gibson JR. A Target Cell-Specific Role for Presynaptic Fmr1 in Regulating Glutamate Release onto Neocortical Fast-Spiking Inhibitory Neurons. J. Neurosci. 2013;33(6):2593–2604. doi: 10.1523/JNEUROSCI.2447-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel AB, Loerwald KW, Huber KM, Gibson JR. Postsynaptic FMRP promotes the pruning of cell-to-cell connections among pyramidal neurons in the L5A neocortical network. J. Neurosci. 2014;34(9):3413–3418. doi: 10.1523/JNEUROSCI.2921-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, et al. Mosaic Copy Number Variation in Human Neurons. Science. 2013;342(6158):632–637. doi: 10.1126/science.1243472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConnell MJ, et al. Intersection of diverse neuronal genomes and neuropsychiatric disease: The Brain Somatic Mosaicism Network. Science. 2017;356(6336):eaal1641. doi: 10.1126/science.aal1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Marth JD, Orban PC, Mossmann H, Rajewsky K. Deletion of a DNA polymerase beta gene segment in T cells using cell type-specific gene targeting. Science. 1994;265(5168):103–106. doi: 10.1126/science.8016642. [DOI] [PubMed] [Google Scholar]
- Hodges JL, et al. Astrocytic Contributions to Synaptic and Learning Abnormalities in a Mouse Model of Fragile X Syndrome. Biol. Psychiatry. 2016. pp. 1–11. [DOI] [PMC free article] [PubMed]
- Young P, Qiu L, Wang D, Zhao S, Gross J. Single-neuron labeling with inducible cre-mediated knockout in transgenic mice. Nat. Neurosci. 2011;11(6):721–728. doi: 10.1038/nn.2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong H, Espinosa JS, Su HH, Muzumdar MD, Luo L. Mosaic Analysis with Double Markers in Mice. Cell. 2005;121(3):479–492. doi: 10.1016/j.cell.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Hippenmeyer S, et al. Genetic mosaic dissection of Lis1 and Ndel1 in neuronal migration. Neuron. 2010;68(4):695–709. doi: 10.1016/j.neuron.2010.09.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De la Rossa A, Jabaudon D. In vivo rapid gene delivery into postmitotic neocortical neurons using iontoporation. Nat. Protoc. 2015;10(1):25–32. doi: 10.1038/nprot.2015.001. [DOI] [PubMed] [Google Scholar]
- Lu W, Bushong EA, Shih TP, Ellisman MH, Nicoll RA. The cell-autonomous role of excitatory synaptic transmission in the regulation of neuronal structure and function. Neuron. 2013;78(3):433–439. doi: 10.1016/j.neuron.2013.02.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan Y, et al. Semaphorin 5A inhibits synaptogenesis in early postnatal- and adult-born hippocampal dentate granule cells. eLife. 2014;3:1–24. doi: 10.7554/eLife.04390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, et al. Efficient Gene Delivery into Multiple CNS Territories Using In utero Electroporation. J. Vis. Exp. 2011. p. e2957. [DOI] [PMC free article] [PubMed]
- Matsui A, Yoshida AC, Kubota M, Ogawa M, Shimogori T. Mouse in utero Electroporation: Controlled Spatiotemporal Gene Transfection. J. Vis. Exp. 2011. p. e3024. [DOI] [PMC free article] [PubMed]
- Briz CG, Navarrete M, Esteban JA, Nieto M. In utero Electroporation Approaches to Study the Excitability of Neuronal Subpopulations and Single-cell Connectivity. J. Vis. Exp. 2017. [DOI] [PMC free article] [PubMed]
- Saito T, Nakatsuji N. Efficient gene transfer into the embryonic mouse brain using in vivo electroporation. Dev. Biol. 2001;240(1):237–246. doi: 10.1006/dbio.2001.0439. [DOI] [PubMed] [Google Scholar]
- Saito T. In vivo electroporation in the embryonic mouse central nervous system. Nature protocols. 2006;1(3):1552–1558. doi: 10.1038/nprot.2006.276. [DOI] [PubMed] [Google Scholar]
- Tabata H, Nakajima K. Efficient in utero gene transfer system to the developing mouse brain using electroporation: Visualization of neuronal migration in the developing cortex. Neuroscience. 2001;103(4):865–872. doi: 10.1016/s0306-4522(01)00016-1. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Cepko CL. Controlled expression of transgenes introduced by in vivo electroporation. Proc. Natl. Acad. Sci. U.S.A. 2007;10(3):1027–1032. doi: 10.1073/pnas.0610155104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacary E, et al. Visualization and Genetic Manipulation of Dendrites and Spines in the Mouse Cerebral Cortex and Hippocampus using In utero Electroporation. J. Vis. Exp. 2012. p. e4163. [DOI] [PMC free article] [PubMed]
- Mizuno H, et al. NMDAR-Regulated Dynamics of Layer 4 Neuronal Dendrites during Thalamocortical Reorganization in Neonates. Neuron. 2014;82(2):365–379. doi: 10.1016/j.neuron.2014.02.026. [DOI] [PubMed] [Google Scholar]
- Luo W, et al. Supernova: A Versatile Vector System for Single-Cell Labeling and Gene Function Studies in vivo. Sci. Rep. 2016;6:35747. doi: 10.1038/srep35747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mader SL, Libal NL, Pritchett-Corning K, Yang R, Murphy SJ. Refining timed pregnancies in two strains of genetically engineered mice. Lab animal. 2009;38(9):305–310. doi: 10.1038/laban0909-305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal GS, Djurisic M, Brown K, Sapp RW, Shatz CJ. Cell-Autonomous Regulation of Dendritic Spine Density by PirB. eNeuro. 2016;3(5):1–15. doi: 10.1523/ENEURO.0089-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Byers SL, Wiles MV, Dunn SL, Taft RA. Mouse estrous cycle identification tool and images. PLoS ONE. 2012;7(4):1–5. doi: 10.1371/journal.pone.0035538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreu-Agullo C, Maurin T, Thompson CB, Lai EC. Ars2 maintains neural stem-cell identity through direct transcriptional activation of Sox2. Nature. 2011;481:195–198. doi: 10.1038/nature10712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scotto-Lomassese S, et al. Fragile X mental retardation protein regulates new neuron differentiation in the adult olfactory bulb. J. Neurosci. 2011;31:2205–2215. doi: 10.1523/JNEUROSCI.5514-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plasmids 101: A Desktop Resource. 3rd ed. Cambridge, Massachusetts: Addgene; 2017. [Google Scholar]
- Pipette Cookbook. rev. ed. Novato, California: Sutter Instrument Company; 2015. [Google Scholar]
- Cuddington B, Verschoor M, Mossman K. Handling of the Cotton Rat in Studies for the Pre-clinical Evaluation of Oncolytic Viruses. J. Vis. Exp. 2014. p. e52232. [DOI] [PMC free article] [PubMed]
- Leary S, et al. AVMA Guidelines for the Euthanasia of Animals: 2013 Edition. Schaumburg, Illinois: American Veterinary Medical Association; 2013. [Google Scholar]
- Anderson SA, Eisenstat DD, Shi L, Rubenstein JLR. Interneuron Migration from Basal Forebrain to Neocortex: Dependence on Dlx Genes. Science. 1997;28(5337):474–476. doi: 10.1126/science.278.5337.474. [DOI] [PubMed] [Google Scholar]
- Parker JM, Austin J, Wilkerson J, Carbone L. Effects of Multimodal Analgesia on the Success of Mouse Embryo Transfer Surgery. J. Am. Assoc. Lab. Anim. Sci. 2011;50(4):466–470. [PMC free article] [PubMed] [Google Scholar]
- Kim J-Y, et al. Viral transduction of the neonatal brain delivers controllable genetic mosaicism for visualising and manipulating neuronal circuits in vivo. Eur. J. Neurosci. 2013;37(8):1203–1220. doi: 10.1111/ejn.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pierfelice TJ, Gaiano N. Ultrasound-Guided Microinjection into the Mouse Forebrain In Utero at E9.5. J. Vis. Exp. 2010. p. e2047. [DOI] [PMC free article] [PubMed]