

Abstract
Mammalian meiosis is a dynamic developmental process that occurs in germ cells and can be studied and characterized. Using a method to spread nuclei on the surface of slides (rather than dropping them from a height), we demonstrate an optimized technique on mouse spermatocytes that was first described in 1997. This method is widely used in laboratories to study mammalian meiosis because it yields a plethora of high quality nuclei undergoing substages of prophase I. Seminiferous tubules are first placed in a hypotonic solution to swell spermatocytes. Then spermatocytes are released into a sucrose solution to create a cell suspension, and nuclei are spread onto fixative-soaked glass slides. Following immunostaining, a diversity of proteins germane to meiotic processes can be examined. For example, proteins of the synaptonemal complex, a tripartite structure that connects the chromosome axes/cores of homologs together can be easily visualized. Meiotic recombination proteins, which are involved in repair of DNA double-strand breaks by homologous recombination, can also be immunostained to evaluate progression of prophase I. Here we describe and demonstrate in detail a technique widely used to study mammalian meiosis in spermatocytes from juvenile or adult male mice.
Keywords: Cellular Biology, Issue 129, Meiosis, mouse, spermatocytes, meiotic chromosome spreads, immunostaining
Introduction
Mice are used extensively as a model organism to study meiosis in mammals. Both the normal developmental processes and defects that occur during meiosis can be evaluated. The timeline and progression of salient features that occur during prophase I and substages, as well as a multitude of specific proteins involved in processes crucial to regulation of meiosis can be characterized in both wild type and mutant mice. Several specialized processes that occur during meiosis I can be studied in detail (reviewed in Handel and Schimenti1). These include DNA double-strand break (DSB) formation and repair, recombination, synaptonemal complex formation, and chromosome segregation.
An important aspect of studying meiotic processes is the ability to examine nuclei containing homologous chromosomes that are visually and optically resolved to better distinguish and identify the substages of prophase I. Prophase I is comprised of 5 substages that are characterized by specific features including formation of the synaptonemal complex (SC). The SC is a tripartite protein scaffold that enables pairing and DNA double-strand break repair of homologs. During leptonema, homologs align as axial elements of the SC are laid down. Then attachment of central elements to the synaptonemal complex during zygonema facilitates pairing and physical connection (synapsis) between pairs of homologs. During pachynema, synapsis of homologs becomes complete and DNA crossovers are formed from repair of a select population of DNA double-strand breaks via homologous recombination. The SC disassembles during diplonema, allowing homologs to desynapse but remain attached at their centromeres and at sites of DNA crossovers. Finally during diakinesis, homologs recondense and the transition to metaphase occurs1.
Historically, surface-spreading of meiotic chromatin yielded few nuclei, especially those from early stages of prophase I2. As a result, these methods were modified to improve the separation of cells from tissues (i.e. testis or ovary), the technique of spreading meiotic chromatin, and the yield of high quality meiotic nuclei for evaluation2,3,4. In addition, this method proved to be useful in preserving nuclear and chromatin bound proteins in meiotic nuclei as demonstrated by published immunolocalization techniques5,6,7. Therefore, the method initially described by Peters2 and demonstrated here yields many separate burst spermatocyte nuclei containing homologs undergoing the leptotene, zygotene, pachytene, diplotene, and diakinesis substages of prophase I.
The technique of preparing surface-spread nuclei (also called chromatin spread analysis) is widely used to study meiosis in the fields of reproductive and cell biology. Slides containing surface-spread nuclei can be subsequently immunostained with antibodies to proteins of interest or subjected to silver staining, and then analyzed with microscopy. The method is similar for both male and female mice, although modifications have been described for female mice2. It is crucial not to overmince seminiferous tubules. Nuclei must also be well spread so that following immunostaining homologs and associated proteins are clearly seen with microscopy. Surface-spread nuclei can be prepared in just a few hours and either immunostained immediately or stored at -80 °C for a maximum of 3 weeks before thawing and immunostaining. However, optimal results are best obtained for some proteins if immunostaining is performed immediately or within 2 weeks of preparing surface-spread nuclei. Slides can be immunostained with a standard protocol using antibodies to proteins of interest, followed by incubation with secondary antibodies conjugated to fluorescent proteins or dyes, then imaged with fluorescence microscopy8. At postnatal day 15 - 21 there is sufficient tissue per pair of wild type testes to yield 6 - 10 slides, and older mice (including adults) will yield more slides. However, wild type mice 6 weeks of age and older will also yield more post-meiotic cells (i.e. spermatids) on the slides following immunostaining. In addition, all substages of prophase I can be observed in juvenile and adult mice. Testes from males at postnatal days 10 through 15 will yield many more leptotene and zygotene cells. Mice older than postnatal day 14 to 15 will yield more pachytene and diplotene nuclei.
Here we describe and demonstrate a widely-used method of preparing surface-spread nuclei from mouse spermatocytes.
Protocol
All methods involving mice utilized high ethical and welfare standards, and were approved by the Institutional Animal Care and Use Committee at Drexel University College of Medicine.
1. Preparation of solutions and instruments needed
Prepare Hypotonic Extraction Buffer (HEB) in 50 mL of ultrapure laboratory grade water, pH 8.2 - 8.4 (Table 1). Make the solution fresh each time, keep on ice, and use within 2 h of preparing spreads; this is necessary to optimize the reducing and protease inhibiting activities of DTT and PMSF, respectively. Use 10 mL of HEB for each pair of mouse testes.
Prepare a 100 mM sucrose solution (fresh each time) by adding 0.342 g of sucrose to 10 mL of ultrapure laboratory grade water. Adjust the pH to 8.2
- Prepare fixative solution (1% paraformaldehyde (PFA), 0.15% Triton X-100, pH adjusted to 9.2 with 1 N HCl or 1 N NaOH) fresh monthly and store it at room temperature protected from light. Dilute a 16% commercially available PFA starting stock to 4% in 1X PBS for use as a working stock, and then store 4% aliquots at -20 °C.
- To prepare the fixative solution, add 10 mL of 4% PFA solution and 600 µL of 10% Triton X-100 to 30 mL of 1X PBS, mix well, and adjust the pH to 9.2 with 1 N HCl or 1 N NaOH. Store the fixative solution in a 50-mL conical tube wrapped in aluminum foil at room temperature (for no more than 4 weeks).
Prepare wash solutions (while slides are drying the last 30 min) with Photo-Flo 200: 0.4% Photo-Flo 200/PBS (320 µL of Photo-Flo 200 in 80 mL of 1X PBS) and 0.4% Photo-Flo 200/dH2O (160 µL of Photo-Flo 200 in 40 mL of ultrapure laboratory grade water). Use a 50 mL Coplin jar filled with 40 mL of solutions to wash 10 or fewer slides at a time.
- Prepare the following instruments: 1 pair of surgical scissors (to incise the skin), 1 pair of fine tip surgical scissors (to incise the peritoneum), 2 pairs of straight fine tip forceps (to dissect out each testis), 1 straight edge razor blade (to incise each testis), 2 pairs of curved tip forceps (to immobilize each testis and squeeze out the seminiferous tubules), 50 mL Coplin jars, Teflon printed 3 ring slides, 1 scalpel with size 11 blade, humidified chamber, 100 x 15 mm Petri dishes, fine tip permanent marker or pencil, and a platform rotating shaker.
- Place premium uncharged precleaned frosted end glass slides upright in a 50 mL Coplin jar filled with fixative solution until use. NOTE: Pencil is preferred if slides will be used subsequently in procedures requiring ethanol, such as fluorescence in situ hybridization.
2. Spermatocyte nuclei spreading
Euthanize a male mouse according to institutional guidelines (e.g. cervical dislocation with or without CO2 asphyxiation).
Use P10 - P20 males to evaluate meiotic cells in the first wave of spermatogenesis; we routinely use P15 - P17 males to obtain cells from all substages of prophase I. Adult testes will provide cells from prophase I too, but they will also yield more postmeiotic cells, such as spermatids.
Harvest and weigh the testes (record the paired weights); then place them into a Petri dish containing a few drops of cold HEB to keep them moist.
- Hold the testis against the bottom of the dish with curved tip forceps and use a straight edge razor blade to make a small vertical midline incision through the capsule of the testis. While still holding the testis against the bottom of the dish with one forceps, use a second set of curved tip forceps or straight edge razor blade to gently squeeze the seminiferous tubules from the testis.
- Place the tubules into a 15 mL conical tube containing 10 mL of cold HEB, taking care to avoid putting pieces of capsule into the tube. Repeat these steps with the second testis.
Incubate the decapsulated seminiferous tubules in the conical tube containing HEB on ice for 30 - 60 min, depending on the size of the testes. Incubate seminiferous tubules from a pair of testes weighing 30 mg or less for 30 min, tubules from a pair of testes weighing 31 to 50 mg for 30 - 45 minutes, and larger testes for 60 minutes. Do not alter the times for a single testis. NOTE: The optimal incubation time may vary depending on the experience of the experimenter, the reagents used, and the mice being analyzed.
While seminiferous tubules are incubating in HEB, use a marker or pencil to label the frosted ends of precleaned slides with mouse number, slide number, and date. Then place the pre-cleaned slides in a Coplin jar containing the fixative solution.
Following incubation, pour tubules and HEB into a clean Petri dish, and separate tubules into approximately 3 mm diameter clumps. Each clump yields 2 slides and the number of slides yielded depends on the size of testes. NOTE: We obtain 8 slides from wild type and 6 slides from subfertile Chtf18-null P16 testes (which are smaller due to decreased numbers of meiotic and mitotic germ cells8). One adult testis weighing 100 mg or more would be expected to yield 16 slides.
Place 23 µL of 100 mM sucrose solution into one ring of a Teflon printed, 3 ring slide and add one clump of seminiferous tubules to the ring. Then using curved tip forceps to hold the clump, gently mince the tubules with a size 11 razor blade until the solution becomes cloudy. NOTE: Do not overmince as this will result in fragmented chromosomes. Note that fragmented chromosomes could also be indicative of a mutant phenotype.
Remove debris, add another 23 µL of sucrose solution, and use a micropipette to gently pipette the mixture up and down a few times to help separate the cells in the suspension.
- Remove a slide soaking in the Coplin jar containing fixative solution and tilt the slide over the jar so that one generous droplet collects at the bottom corner of the slide.
- Pipet 20 µL of sucrose cell suspension from the ring of the Teflon printed 3 ring slide into the fixative droplet on a premium frosted glass slide, and pipet the suspension up and down 2 - 3 times. NOTE: The fixative droplet should be large enough in volume so that addition of 20 μL of the sucrose cell suspension to it allows coverage across the entire slide when the mixture is spread (see 2.11 below).
Slowly tilt the slide to the side opposite the droplet to spread the cell suspension along the length of the slide. Then slowly tilt the slide back to spread the cell suspension along the width of the slide to cover it completely. Avoid spreading the suspension over the same area of the slide more than once otherwise the cells will overlap with each other.
Immediately place slide flat in a humidified chamber and repeat steps 2.8 to 2.11 until all clumps of seminiferous tubules have been used to make slides. It is important not to move the humidified chamber during the drying process to avoid tearing and overlapping cells.
Allow the slides to incubate in a covered humidified chamber at room temperature for 2.5 h so that they dry slowly and not completely. High humidity and slow drying in this step are important to ensure adequate spreading of the chromatin. Then remove the cover of the chamber and allow slides to air-dry completely an additional 30 min.
Place slides (back to back or in a zigzag pattern) in a Coplin jar containing 0.4% Photo-Flo 200/1X PBS solution and wash the slides twice, 5 min each, by gently agitating the Coplin jar on a platform rotating shaker. Use fresh solution for each wash and keep slides from different mice in separate jars for each of the washes.
Wash the slides in a Coplin jar containing 0.4% Photo-Flo 200/dH2O solution for 5 min once while agitating.
Remove slides from the Coplin jar (last wash), carefully wipe the edges and bottoms of the slides to remove excess liquid, and place slides at an angled, almost-upright position to dry. An experimenter who is proficient in this technique can process and prepare surface-spread nuclei from 4 or 5 mice in one day.
- Once dry (15 - 20 min), process slides immediately for immunofluorescence staining or store at -80 °C in a tightly closed slide box protected from light for a maximum of 3 weeks (depending on the target proteins being evaluated; optimal results may be obtained if stained the same day or within 2 weeks).
- If stored at -80 °C, then allow slides to come to room temperature and wash in a Coplin jar containing 1X PBS or Block for 5 min prior to staining. NOTE: Many different immunofluorescence staining protocols will provide good results; refer to Berkowitz et al. for an immunostaining protocol8.
Representative Results
The ability of this method to provide large numbers of surface spread nuclei containing intact homologs depends on three main factors: 1) appropriate incubation time of seminiferous tubules in hypotonic buffer (i.e. HEB) to obtain adequately swelled spermatocyte nuclei that burst but do not disintegrate from prolonged incubation in HEB, 2) micropipetting the sucrose droplets of cells to obtain a separated suspension of nuclei that are not clumped together, and 3) tilting each fixative coated slide in one direction at a time and not back and forth. Once prepared, surface spread nuclei can be immunostained with fluorescently labeled antibodies to proteins of interest and then imaged with microscopy (representative examples are shown in Figure 1 and Figure 2. Figure 1 shows examples of wild type pachytene nuclei that are well spread (A), nuclei containing unevenly immunostained, tattered appearing homologs due to incubation in HEB too long (B), poorly spread overlapping nuclei (C), and nuclei containing fragmented homologs due to "overmincing" of seminiferous tubules (D). When the technique is performed well, a plethora of nuclei representing the main 5 substages of prophase I can be obtained. Figure 2 demonstrates the leptotene, zygotene, pachytene, diplotene, and diakinesis stages of prophase I in nuclei of wild type spermatocytes from juvenile males.
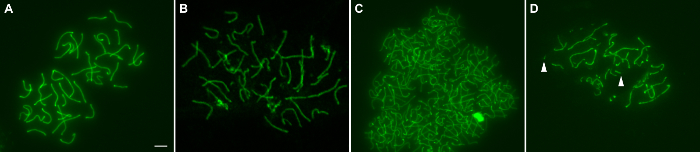
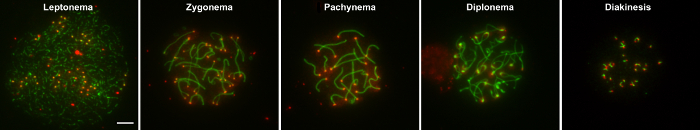
Figure 1: Surface spread nuclei of wild type spermatocytes from juvenile mice. Pachytene spermatocyte nuclei were stained with anti-SYCP3 (green), which stains the axial/lateral elements of the synaptonemal complex (A) Well spread nuclei. (B) Nuclei from seminiferous tubules incubated in HEB too long. (C) Poorly spread overlapping nuclei. (D) Fragmented homologs due to "overmincing" of seminiferous tubules (arrowheads indicate two small homolog fragments). Scale bar is 5 μm. Please click here to view a larger version of this figure.
Figure 2: Examples of nuclei from substages of prophase I. Wild type spermatocytes from juvenile males were stained with CREST serum (red), which stains centromeres, and anti-SYCP3 (green), which stains the axial/lateral elements of the synaptonemal complex. The leptotene, zygotene, pachytene, diplotene, and diakinesis stages of prophase I, respectively, are shown. Scale bar is 5 μm. Please click here to view a larger version of this figure.
| Reagent | Quantity | Final concentration |
| 1M Tris-Cl (pH 8.2) | 1.5 mL | 30 mM |
| Sucrose | 0.85575 g | 50 mM |
| Trisodium citrate dihydrate | 0.25 g | 17 mM |
| 0.5 M EDTA | 500 μL | 5 mM |
| Dithiothreitol (DTT) | 0.00385 g | 0.5 mM |
| 0.2 M Phenymethylsulfonyl fluoride (PMSF) | 25 μL | 0.1 mM |
Table 1: Recipe for Hypotonic Extraction Buffer (HEB). Add reagents to 50 mL ultrapure laboratory grade water and adjust to pH 8.2 - 8.4 (if necessary use 1 N NaOH solution or 1 N HCl). Prepare fresh HEB each time, keep on ice, and use within 2 h of preparation.
Discussion
To obtain high numbers of well spread, good quality spermatocyte nuclei, the most critical steps of this protocol include appropriate incubation time of seminiferous tubules in HEB, appropriate mincing of seminiferous tubules, and the technique employed to spread the sucrose cell suspension across the fixative coated slide(s). We incubate seminiferous tubules from testes of wild type males ranging from postnatal day 15 to adulthood in HEB for 45 min, while testes from comparably aged Chtf18-null mice are incubated for only 30 min because testes are half the size and contain significantly fewer germ cells8. In our experience, incubation of Chtf18-null testes for longer than 30 min compromises the integrity of the cells, resulting in poorly resolved homologs. Seminiferous tubules are minced only to obtain a cloudy sucrose cell suspension, and each slide must contain a generous droplet of fixative prior to spreading (see 2.10). Slides are tilted in one direction at a time and not back and forth. The method is ideal to study meiosis I, but it is largely limited to substages of prophase I. However, nuclei in premeiotic S phase, metaphase I and II can be observed and have been reported7,9.
This is a straightforward method that necessitates some practice to gain competence and best results. Suboptimal results could occur for several different reasons. Nuclei may not be well spread, resulting in clustered or overlapping cells. This can occur if the sucrose cell suspension is not adequately pipetted or there is not enough fixative on the slide to spread the nuclei well. If slides are not coated with enough residual fixative or the cell suspension is not spread across the slide in one smooth direction or repeatedly spread back and forth, then cells will overlap and clump together. If seminiferous tubules are "overminced" then homologs may be fragmented or appear somewhat shredded. Because mutant testes may be smaller, contain fewer spermatocytes, and be more susceptible to "overmincing" or fragmentation of seminiferous tubules, we recommend practicing a few times on control (e.g. wild type) testes first to optimize the technique in the hands of a novice.
Preparing surface spread nuclei is a fundamental technique used to examine key regulatory steps and progression of meiosis, as well as to characterize meiotic phenotypes in mice. Therefore, we use this method extensively and teach all new lab members how to perform it. Once this technique is mastered, a myriad of other techniques can be used subsequently and in conjunction with it. For example, several types of immunostaining and/or microscopy including widefield or confocal with epifluorescence, or electron microscopy can be performed. In addition to studying meiosis-specific proteins such as components of synaptonemal complex, those interacting with the SC, such as cohesins, multiprotein complexes that mediate cohesion, can be evaluated (reviewed in 10,11,12). Regulation of DNA crossover formation, processing, and maturation can be and have also been studied with this technique13. In addition, this technique has been performed following the use of methods to obtain specific populations of mouse spermatocytes: 1) following culture of spermatocytes in okadaic acid to induce progression to the G2/MI transition, yielding increased numbers of diplotene, diakinesis, and metaphase I spreads14,15 and 2) after methods used to isolate an enriched population of pachytene spermatocytes14.
The technique of preparing surface-spread nuclei from mouse spermatocytes described and demonstrated here is an extremely useful and straightforward technique. It is quintessential to every lab that studies meiosis using mice as a model organism, but requires some practice and finesse to achieve the best results.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors acknowledge the technical assistance of Abigail Harris. They also acknowledge Paula Cohen and Kim Holloway for helping to optimize the protocol of preparing surface-spread nuclei from mouse spermatocytes. The authors also appreciate critical reading of the manuscript by Karen Schindler.
This work was supported by NIH R01 GM106262 to K.M.B.
References
- Handel MA, Schimenti JC. Genetics of mammalian meiosis: regulation, dynamics and impact on fertility. Nat Rev Genet. 2010;11:124–136. doi: 10.1038/nrg2723. [DOI] [PubMed] [Google Scholar]
- Peters AH, Plug AW, van Vugt MJ, de Boer P. A drying-down technique for the spreading of mammalian meiocytes from the male and female germline. Chromosome Res. 1997;5:66–68. doi: 10.1023/a:1018445520117. [DOI] [PubMed] [Google Scholar]
- Counce SJ, Meyer GF. Differentiation of the synaptonemal complex and the kinetochore in Locusta spermatocytes studied by whole mount electron microscopy. Chromosoma. 1973;44:231–253. doi: 10.1007/BF00329119. [DOI] [PubMed] [Google Scholar]
- Speed RM. Meiosis in the foetal mouse ovary. I. An analysis at the light microscope level using surface-spreading. Chromosoma. 1982;85:427–437. doi: 10.1007/BF00330366. [DOI] [PubMed] [Google Scholar]
- Ashley T, et al. Dynamic changes in Rad51 distribution on chromatin during meiosis in male and female vertebrates. Chromosoma. 1995;104:19–28. doi: 10.1007/BF00352222. [DOI] [PubMed] [Google Scholar]
- Baker SM, et al. Involvement of mouse mLh1 in DNA mismatch repair and meiotic crossing over. Nat Genet. 1996;13:336–342. doi: 10.1038/ng0796-336. [DOI] [PubMed] [Google Scholar]
- Plug AW, Xu J, Reddy G, Golub EI, Ashley T. Presynaptic association of Rad51 protein with selected sites in meiotic chromatin. Proc Natl Acad Sci U S A. 1996;93:5920–5924. doi: 10.1073/pnas.93.12.5920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkowitz KM, et al. Disruption of CHTF18 causes defective meiotic recombination in male mice. PLoS Genet. 2012;8:e1002996. doi: 10.1371/journal.pgen.1002996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez R, et al. Sororin loads to the synaptonemal complex central region independently of meiotic cohesin complexes. EMBO Rep. 2016;17:695–707. doi: 10.15252/embr.201541060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brooker AS, Berkowitz KM. The roles of cohesins in mitosis, meiosis, and human health and disease. Methods Mol Biol. 2014;1170:229–266. doi: 10.1007/978-1-4939-0888-2_11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNicoll F, Stevense M, Jessberger R. Cohesin in gametogenesis. Curr Top Dev Biol. 2013;102:1–34. doi: 10.1016/B978-0-12-416024-8.00001-5. [DOI] [PubMed] [Google Scholar]
- Rankin S. Complex elaboration: making sense of meiotic cohesin dynamics. FEBS J. 2015;282:2426–2443. doi: 10.1111/febs.13301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holloway JK, Sun X, Yokoo R, Villeneuve AM, Cohen PE. Mammalian CNTD1 is critical for meiotic crossover maturation and deselection of excess precrossover sites. J Cell Biol. 2014;205:633–641. doi: 10.1083/jcb.201401122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- La Salle S, Sun F, Handel MA. Isolation and short-term culture of mouse spermatocytes for analysis of meiosis. Methods Mol Biol. 2009;558:279–297. doi: 10.1007/978-1-60761-103-5_17. [DOI] [PubMed] [Google Scholar]
- Wiltshire T, Park C, Caldwell KA, Handel MA. Induced premature G2/M-phase transition in pachytene spermatocytes includes events unique to meiosis. Dev Biol. 1995;169:557–567. doi: 10.1006/dbio.1995.1169. [DOI] [PubMed] [Google Scholar]