

Abstract
Core cellular processes such as DNA replication and segregation, protein synthesis, cell wall biosynthesis, and cell division rely on the function of proteins which are essential for bacterial survival. A series of target-specific dyes can be used as probes to better understand these processes. Staining with lipophilic dyes enables the observation of membrane structure, visualization of lipid microdomains, and detection of membrane blebs. Use of fluorescent-d-amino acids (FDAAs) to probe the sites of peptidoglycan biosynthesis can indicate potential defects in cell wall biogenesis or cell growth patterning. Finally, nucleic acid stains can indicate possible defects in DNA replication or chromosome segregation. Cyanine DNA stains label living cells and are suitable for time-lapse microscopy enabling real-time observations of nucleoid morphology during cell growth. Protocols for cell labeling can be applied to protein depletion mutants to identify defects in membrane structure, cell wall biogenesis, or chromosome segregation. Furthermore, time-lapse microscopy can be used to monitor morphological changes as an essential protein is removed and can provide additional insights into protein function. For example, the depletion of essential cell division proteins results in filamentation or branching, whereas the depletion of cell growth proteins may cause cells to become shorter or rounder. Here, protocols for cell growth, target-specific labeling, and time-lapse microscopy are provided for the bacterial plant pathogen Agrobacterium tumefaciens. Together, target-specific dyes and time-lapse microscopy enable characterization of essential processes in A. tumefaciens. Finally, the protocols provided can be readily modified to probe essential processes in other bacteria.
Keywords: Microbiology, Issue 129, Agrobacterium, essentiality, time-lapse microscopy, agarose pads, fluorescent d-amino acids, DNA staining, membrane staining, epifluorescence microscopy
Introduction
Progression through the bacterial cell cycle requires the coordination of many processes including membrane and cell wall biosynthesis, DNA replication and segregation, and cell division. To fully understand the complexity of bacterial cell biology, it is necessary to study these essential events; however, this is a non-trivial task since cell viability is compromised when key components of these pathways are mutagenized. Epifluorescence microscopy coupled with target-specific dyes is a powerful approach to probe these essential processes in wildtype and mutant bacterial strains.
Peptidoglycan-specific dyes include fluorescent antibiotics (vancomycin-FL, bocillin-FL) and fluorescent-d-amino acids (for example, 7-hydroxycoumarin-3-carboxylic acid-3-amino-d-alanine, HADA; 4-chloro-7-nitrobenzofurazan-3-amino- d-alanine, NADA; tetramethylrhodamine-3-amino-d-alanine; TADA). In Gram-positive bacteria, the use of sublethal concentrations of fluorescent antibiotic analogs to probe sites of peptidoglycan biosynthesis has been an effective strategy to reveal peptidoglycan insertion patterns1,2,3,4. While fluorescent vancomycin labeling has been used to gain insights into the peptidoglycan insertion patterns in fixed Gram-negative bacteria5, the outer membrane generally provides a permeability barrier that prevents the use of fluorescent antibiotics as a probe for peptidoglycan biosynthesis in live cells. In contrast, short pulses of fluorescent-d-amino acids or d-amino acids with biorthogonal functional groups covalently label regions of recent peptidoglycan insertion in a wide range of living bacterial cells6,7. Patterns of peptidoglycan insertion that have been observed with synthetic d-amino acids include punctate and septal (Escherichia coli and Bacillus subtilis), polar and septal (Agrobacterium tumefaciens and Listeria monocytogenes), septal only (Staphylococcus aureus), and apical (Streptomyces venezuelae)6,7. These observations indicate that bacteria exhibit diverse patterns of cell wall biogenesis and that the use of synthetic d-amino acids as probes for examining growth patterning is a valuable strategy in many bacteria.
Dyes that label bacterial chromosomes include the deoxyribonucleic acid (DNA) specific minor groove binder (4,6-diamidino-2-phenylindole; DAPI) and high affinity cyanine dyes (Green and Orange; see materials list). DAPI staining of fixed cells assists in enumeration of bacteria from environmental samples8, whereas DAPI staining of live cells is used to indicate bacterial viability9. In contrast, cyanine dyes such as Orange and Green are frequently described as membrane impermeant "dead" cell stains to enumerate non-viable cells9. Remarkably, when these reagents are used to probe the morphology of the bacterial nucleoid during cell growth, DAPI, Orange, and Green were all shown to be membrane permeant and capable of labeling live cells10. In live E. coli cells, DAPI staining of DNA appears diffuse due to auto-fluorescence from the cytoplasm and repeated exposures of DAPI-stained cells to ultraviolet (UV) light perturbs the nucleoid structure10. Staining E. coli or B. subtilis with Orange reveals that this dye is membrane permeant and provides long-lasting fluorescence upon binding to DNA in live cells without impacting cell growth, DNA replication, or chromosome segregation10. These observations suggest that cyanine DNA dyes can be used to monitor the morphology of nucleoids during cell growth in many bacteria.
Phospholipid-specific stryl dyes such as N-(3-triethylammoniumpropyl) -4-(6-(4-(diethylamino) phenyl)hexatrienyl)pyridinium dibromide (4-64; see materials list) are cationic compounds and associate preferentially with negatively charged phospholipids such as cardiolipin and phosphatidylglycerol11. Distinct patterns are observed when 4-64 is used to label the membrane of different bacteria. In Escherichia coli, 4-64 is enriched in the poles, in bands along the lateral wall, and in the division sites of late pre-divisional cells12. In Bacillus subtilis, 4-64 labeling enables the visualization of lipid spirals13. In Agrobacterium tumefaciens, 4-64 labels the outer membrane and is observed in a characteristic "horseshoe" pattern in which the growth pole is devoid of labeling14,15. These observations indicate that these bacteria exhibit heterogeneous lipid distributions due to the presence of lipid domains which contribute to cellular asymmetry. Changes in 4-64 labeling patterns such as the presence of diffuse labeling, blebs or vesicles, invaginations, or membrane shrinkage can be informative in characterizing mutants that impact the distribution or biosynthesis of lipids.
Beyond staining cells, determining the function of proteins participating in essential processes is necessary. The characterization of essential proteins is technically challenging because it is not possible to delete essential genes and study the phenotypic consequences. Thus, alternative approaches that deplete the protein have emerged. For example, an essential gene can be put under the control of an inducible promoter rather than its native promoter. Inducible promoters are responsive to small molecules such as; zinc16, isopropyl β-d-1-thiogalactopyranoside (IPTG)17,18,19,20,21, arabinose22, vanillate17,23, and xylose23, thus the transcription of the target gene ceases and the protein of interest is depleted when the inducer is removed. Alternative approaches for depleting essential proteins of interest include synthetic riboswitches24 which use small molecule-RNA interactions to hinder transcription of target genes, CRISPR interference25,26 to block transcription of target genes, and inducible protein degradation27,28 which uses peptide tags to target proteins for degradation by the ClpXP protease. Depletion strains provide only a short time for characterization before the cells lose viability, therefore, microscopic imaging of cells over time during protein depletion is a powerful approach for characterization. Indeed, microscopy of living bacterial cells has enabled researchers to gain insights into fundamental biological processes, including the mechanisms of cell shape maintenance, secretion, and compartmentalization29.
A. tumefaciens is a bacterial plant pathogen30 and natural genetic engineer31,32. Thus, mechanisms related to pathogenicity, including host-pathogen interactions33,34,35, secretion36, and host transformation30,31,37 have been extensively investigated. To design strategies that prevent A. tumefaciens mediated disease or enhance plant transformation, the processes essential for A. tumefaciens survival need to be better understood. The use of target-specific dyes and the recent development of a protein depletion strategy for A. tumefaciens18 provides a means to investigate essential processes.
Here, detailed protocols for microscopic analysis of wildtype, mutant, and protein depletion strains of A. tumefaciens are provided. The first two protocols describe how to prepare cells and label them with target-specific dyes. The third protocol provides step-by-step directions for preparing agarose pads (Figure 1) and imaging the bacterial cells (Figure 2, Figure 3, Figure 4). These protocols may also be suitable for other bacteria with additional adaptations to account for different media conditions, growth rates, oxygen requirements, and cell structures.
Protocol
1. Growth of A. tumefaciens Strains
- Culturing A. tumefaciens strains
- Use a sterile wooden stick or pipet tip to inoculate 1 mL of ATGN growth media (see materials list for the recipe) with a single colony of the desired strain. NOTE: For A. tumefaciens depletion strains, the ATGN should contain 1 mM IPTG as an inducer to maintain biosynthesis of the essential protein.
- Grow the A. tumefaciens strains overnight in ATGN at 28 °C with shaking at 225 rpm.
- Measure the optical density of the cells at 600 nm (OD600) using a spectrophotometer. Dilute the cell culture to an OD600 = ~0.2 and continue to grow until OD600 = ~0.6 is reached. For depletion strains, continue to grow with inducer until OD600 = ~0.6 is reached.
- Use wildtype and mutant strains of A. tumefaciens at OD600 = ~0.6 for target-specific staining and/or time-lapse microscopy (see sections 2 and 3.1). For depletion strains, wash out the inducer (see section 1.2.)
- Removal of inducer for characterization of A. tumefaciens protein depletion strains
- Pellet 1 mL of exponential culture (OD600 = ~0.4-0.6) by centrifugation at 7,000 x g for 5 min in a desktop centrifuge at room temperature.
- To wash, remove supernatant and resuspend the pellet in fresh media without inducer and pellet the cells as described above (1.2.1). Wash the cells a total of 3 times in fresh media.
- Resuspend the final pellet in fresh media and concentrate the cells to an OD600 = ~0.8 for immediate staining with target specific dyes (section 2 and Figure 4B-D) or time-lapse microscopy (section 3.2 and Figure 4A). Alternatively, pre-deplete the cells for the desired amount of time by growing the cells in media without inducer prior to staining and imaging of the cells.
2. Target-specific Staining of A. tumefaciens Cells
- Fluorescent d-amino acid cell wall labeling NOTE: FDAAs are non-toxic fluorescent d-amino acids which are readily incorporated into the A. tumefaciens peptidoglycan. This simple labeling procedure probes growth patterning in live cells6. Protocols for synthesis of four FDAAs are available38.
- Pellet 500 µL of exponential culture (OD600 = ~0.4-0.6) by centrifugation at 7,000 x g for 5 min in a desktop centrifuge. Resuspend the cell pellet in 100 µL of fresh media.
- Add 5 µL of 5 mM FDAA to washed and concentrated cells and incubate for 2 min in the dark. NOTE: Limit FDAA exposure to light. The time of incubation will vary depending on the growth rate of the strain. Typically, incubation times should be 5-10% of the doubling time6.
- Pellet the cells by centrifugation and wash cell pellets with phosphate buffered saline (PBS) three times.
- Resuspend pellet in ~50 µL PBS. NOTE: The volume of PBS used for resuspension may vary based on pellet size.
- Apply 0.8-1 µL of cells to an agarose pad and image using epifluorescence microscopy immediately (See sections 3.1 and 3.2).
- Alternatively, stop further label incorporation by fixing the cells with ice-cold 70% ethanol. Resuspend the cell pellet in 1 mL of ice-cold 70% ethanol and incubate on ice for 15 minutes.
- Collect cells by centrifugation and resuspend in a small volume of PBS for imaging at a later time. Store cell suspensions at 4 °C and image within 48 hours.
- DNA staining with DAPI or Orange stain NOTE: To observe DNA structure, cells are labeled with either DAPI or Orange (dead cell stain). Although classically described as a "dead-cell stain", Orange is permeant, photostable, and does not affect the growth of bacterial cells, enabling live-cell DNA imaging10. In A. tumefaciens, Orange labeling works well in living cells and is suitable for time-lapse microscopy (Figure 3). In contrast, the ultraviolet (UV) light exposure necessary for imaging DAPI-stained cells is phototoxic (Figure 3) and thus DAPI staining is best suited for staining live cells for immediate imaging or staining fixed cells.
- DAPI staining of cells
- Pellet 1 mL of exponential culture (OD600 = ~0.4-0.6) by centrifugation at 7,000 x g for 5 min in a desktop centrifuge.
- Optionally, to fix cells prior to staining, resuspend the cell pellet in 1 mL of 70% ice-cold ethanol. Incubate on ice for 10-15 min. Collect the cells by centrifugation as described in 2.2.1.1.
- Resuspend the cell pellet in 1 mL of PBS containing 1 µL of DAPI stock solution (1 mg/mL; see Materials Table). Mix gently by pipetting and incubate for 5 minutes in the dark.
- Pellet cells by centrifugation at 7,000 x g for 5 min and resuspend in 1 mL of PBS to remove excess DAPI. Wash the cells two more times and resuspend the pellet in 50 µL PBS or media.
- Apply 0.8-1 µL of cells to an agarose pad and image using epifluorescence microscopy immediately. See sections 3.1 and 3.2. NOTE: This protocol is not optimal for time-lapse microscopy to observe chromosome dynamics (Figure 3). Consider using Orange staining as an alternative (see section 2.2.2).
- Orange staining of live cells
- Pellet 1 mL of exponential culture (OD600 = ~0.4-0.6) by centrifugation at 7,000 x g for 5 min in a desktop centrifuge. Resuspend cell pellet in 1 mL of PBS containing 1 µL of Orange stock solution (see materials list). Mix gently by pipetting and incubate for 5 min in the dark.
- Pellet cells by centrifugation and resuspend in 1 mL of PBS to remove excess Orange. Wash the cells two more times and resuspend the pellet in 50 µL PBS.
- Apply 0.8-1 µL of cells to an agarose pad and image using epifluorescence microscopy immediately. See sections 3.1 and 3.2. NOTE: This protocol works well with live cells and is suitable for time-lapse microscopy to observe chromosome dynamics (Figure 3). If it is not convenient to image immediately, cells may be fixed in ice-cold 70% ethanol (see section 2.2.1.2).
- Membrane labeling NOTE: The lipophilic styryl fluorescent dye 4-64 has been used extensively to observe the membrane of bacterial cells. In contrast to many bacteria, 4-64 labeled A. tumefaciens cells do not label uniformly, but rather frequently exhibit a "horseshoe" pattern14,15. Remarkably, the growth pole is nearly devoid of stain whereas the old pole is intensely labeled. Thus, 4-64 enables visualization of both cell growth patterns and membrane structure in A. tumefaciens.
- Add FM 4-64 to a final concentration of 8 µg/mL in 1 mL of exponential phase cell culture and incubate at room temperature for 5 minutes in the dark.
- Pellet labeled cells by centrifugation at 7,000 x g for 5 minutes in a desktop centrifuge and resuspend cell pellet in 1 mL of PBS. Wash the cells a total of three times to remove excess dye.
- Resuspend cells in a small volume of PBS and spot on an agarose pad to image immediately. (See sections 3.1 and 3.2) CAUTION: Cells must be imaged immediately to observe characteristic labeling patterns.
3. Imaging of A. tumefaciens Cells
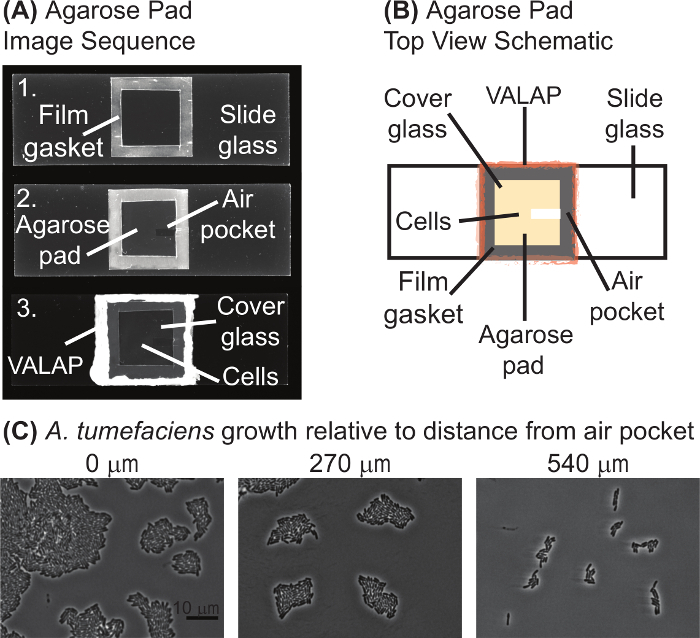
- Agarose pad preparation NOTE: Figure 1 contains an image sequence (panel A) and schematic (panel B) of a typical agarose pad prepared for time-lapse microscopy. Agarose pads are typically prepared as needed.
- 3.1.1. Use a coverslip as a guide and cut a 22 mm x 22 mm square of laboratory film (see materials list) by running a scalpel around the edges.
- Cut a square out of the center of the laboratory film leaving a ~2-5 mm border to serve as a gasket for the agarose pad. Discard the center cutout.
- Place the film gasket onto a glass slide (75 mm x 25 mm) cleaned with an ammonia and alcohol-free cleaner (see materials list) and heat until the film is slightly melted onto glass (top image in Figure 1A). To melt laboratory film, use the edge of a heat block set to 70 °C or melt lightly over a flame.
- Prepare agarose solution by mixing ~0.075 g agarose (see materials list) in 5 mL of media in a small flask. Heat the solution in a microwave with periodic swirling to mix until the agarose is dissolved and the solution is clear. Keep the agarose solution at 55-70 °C and use for construction of multiple agarose pads within 48 hours.
- Pipette media containing 1.2-1.5% agarose into the center of the gasket. NOTE: The volume of media is typically 50 - 60 µL but will vary based on gasket size. Adding media to a cold slide can cause the agarose to solidify too quickly, thus the slide should be kept on a warm surface. The edge of a heat block set to 70 °C works well. Water agarose or buffered agarose solutions such as phosphate buffered saline (PBS) can be used instead of media containing agarose for applications in which continued cell growth is not required. For imaging depletion strains, inducer can be present or absent in the media. Presence of inducer can be used as a control whereas the absence of inducer will reveal the depletion phenotype.
- Place a coverslip over the gasket to evenly distribute the agarose.
- Place slide on a cool, level surface to solidify for ~2 min. Avoid overdrying of the agarose pad as this will result in a wrinkled surface on the agarose pad and pooling of the cell suspension when applied to the surface.
- Carefully slide the coverslip off the agarose pad. Caution: Do not rush this step. The agarose pad can easily tear and an uneven agarose pad can make imaging difficult.
- Allow the agarose pad to air dry for 1-2 min at room temperature until the surface of the pad appears dry. Using a scalpel, remove a small strip of agarose to create an air pocket (middle image, Figure 1A); the air pocket is typically ~2 mm x 7-10 mm and A. tumefaciens cells tend to grow best near the air pocket (Figure 1C). NOTE: For imaging of fixed cells or under conditions where growth is not monitored, an air pocket is not necessary.
- Spot 0.8-1 µL of cells on the agarose pad and cover with a new coverslip. Gently place the coverslip over the top of the agarose pad to distribute the cells across the surface of the agarose pad.
- Seal the edges of the coverslip using melted VALAP (see Materials Table for recipe; Figure 1A, bottom image). Failure to seal along all the edges and corners of the coverslip can cause drying of the agarose pad, which will lead to the drifting of cells during imaging. NOTE: Sealing of the coverslip is only needed for long-term time-lapse imaging.
Figure 1: Agarose pad preparation. (A) Image sequence of the agarose pad preparation protocol. Image 1 is a slide with the laboratory film gasket. In image 2, the agarose pad and air pocket are visualized. Finally, image 3 shows the complete agarose pad with the cells under a coverglass and sealed with VALAP. (B) A schematic of an agarose pad for time-lapse microscopy is provided. Key features of the agarose pad are labeled on the schematic. (C) The air pocket promoted growth of A. tumefaciens on agarose pads. Images of wildtype A. tumefaciens cells grown on an agarose pad for 20 hours are shown. Images were taken at positions increasingly distant from the air pocket. The distance from the closest edge of the image to the air pocket is shown above each image. Scale bar = 10 µm. Please click here to view a larger version of this figure.
- Imaging NOTE: Differential interference contrast, phase contrast, and epifluorescence imaging is performed with an inverted microscope equipped with hardware-based autofocusing, an automated stage, standard filters, an LED light source, 60X oil immersion objectives (1.4 NA) for phase contrast or differential interference contrast (DIC), and a 1K electron-multiplying charge-coupled-device (EMCCD) camera. The microscope should be placed in a temperature controlled room. Alternatively, a stage warmer or chamber can be used to maintain consistent temperatures during imaging.
- Epifluorescence microscopy NOTE: For fluorescence imaging of live cells, it is necessary to limit the number of image acquisitions and optimize the exposure time to minimize photobleaching and phototoxicity. For imaging of each dye, it is recommended to find an optimal exposure time which provides sufficient fluorescence detection but does not lead to photobleaching or phototoxicity. In Figures 2-4, exposure times of 200 ms were used for all fluorescence images. For the time-lapse sequences shown in Figure 3, images were acquired every 10 min for 3 h.
- Place immersion oil on the desired objective and place the inverted slide into the slide holder on the stage. Use the focusing knobs to bring the cells into focus.
- Time-lapse microscopy NOTE: During time-lapse microscopy the following features are computer controlled: x, y, and z position, shutters, and fluorescence filters. A hardware-based auto-focus system is optimal to maintain focus during time-lapse imaging. Alternatively, a software-based auto-focusing loop can be used.
- Place immersion oil on the desired objective and place the inverted slide into the slide holder on the stage. Use the focusing knobs to bring the cells into focus.
- Optionally: Acquire multiple (x,y) positions. Randomly select 10 fields of cells in close proximity to the air pocket of the agarose pads.
- Set-up the time sequence acquisition to image in phase or DIC at the desired time interval. NOTE: For the time-lapse sequence shown in Figure 4, DIC images were acquired with 30 ms exposure every 10 min for 14 h. When using fluorescence imaging during time-lapse microscopy, adjust the acquisition interval and exposure time to minimize photobleaching and phototoxicity.
Representative Results
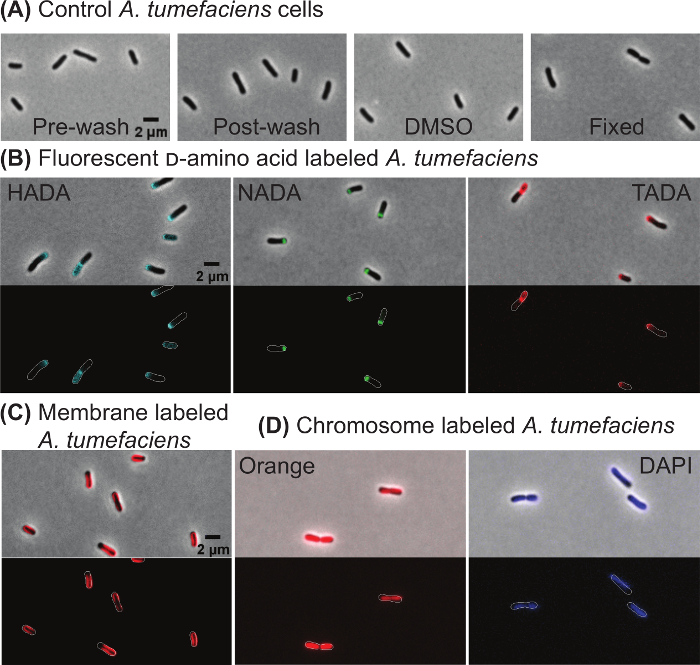
Target-specific labeling of wildtype A. tumefaciens cells In order to illustrate that cell morphology is not impacted by the wash steps or treatment with 1% DMSO (which is used to dilute the fluorescent dyes), cells were imaged directly from culture (Figure 2A, far left panel), after washing the cells by centrifugation as described in 1.2 (Figure 2A, left panel), or after incubating the cells with 1% DMSO for 10 min and washing the cells (Figure 2A, right panel). These images illustrate that morphology is not impacted by washing or the presence of DMSO. In addition, cell growth is not impacted by washing or DMSO treatment based on growth curve analysis (data not shown). In addition, fixing A. tumefaciens with ice-cold ethanol does not cause gross changes in morphology (Figure 2A, far right panel).
Next, target specific dyes were used to observe cell wall biogenesis, membrane domains, and DNA within wildtype A. tumefaciens cells. The patterns of new peptidoglycan insertion were observed following labeling with three fluorescent-d-amino acids: HADA (Figure 2B, left panel), NADA (Figure 2B, center panel), and TADA (Figure 2C, right panel). In all three labeling experiments, new peptidoglycan labeling was enriched at the growth pole or septum of the cells. These growth patterns were consistently observed with all three FDAAs, indicating that the FDAA selection can be modified to enable dual labeling with other stains or cells expressing fluorescently labeled proteins. The lipophilic dye 4-64 preferentially labels the old pole region of A. tumefaciens cells resulting in a characteristic horseshoe pattern (Figure 2C). Both Orange (Figure 2D, left panel) and DAPI (Figure 2D, right panel) label DNA within live A. tumefaciens cells. In late pre-divisional cells, two distinct nucleoids are observed with Orange staining, whereas DNA labeling appears more diffuse with DAPI staining (Figure 2D) consistent with experiments results observed in E. coli10.
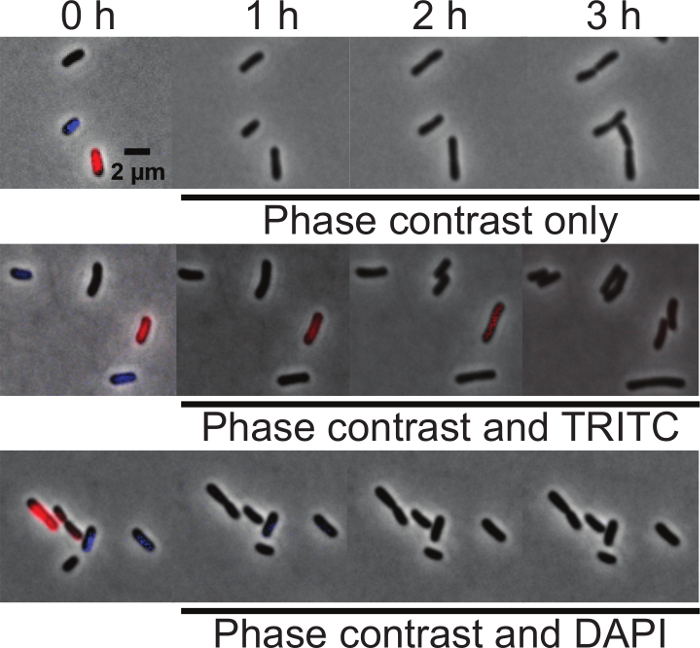
Suitability of DNA dyes for time-lapse microscopy In order to determine if either DAPI or Orange are suitable for time-lapse imaging of nucleoid morphology during cell growth, an equal proportion of unlabeled, DAPI-labeled, and Orange-labeled wildtype A. tumefaciens cells were mixed and spotted on agarose pads. At time 0, initial images were taken using phase contrast, DAPI, and TRITC filters to determine if each cell was labeled. The cell mixtures were then imaged under three different conditions: (1) phase contrast microscopy only, (2) phase contrast and epifluorescence microscopy using the TRITC filter and (3) phase contrast and epifluorescence microscopy using the DAPI filter. Images were acquired every 10 minutes for 3 hours. All cells grew well irrespective of the fluorescent stain used when imaged with phase contrast microscopy indicating that neither Orange or DAPI staining impair cell growth (Figure 3, top panel). All cells also grew well when imaged by phase microscopy and epifluorescence microscopy using the TRITC filter (Figure 3, center panel). The Orange label is subject to photobleaching but can be observed for at least 2 h (13 total exposures of 200 ms), indicating the suitability of this dye for short-term microscopy of live cells. Irrespective of labeling, all cells stop growing within an hour when imaged by phase microscopy and epifluorescence microscopy using the DAPI filter (Figure 3, bottom panel). This observation demonstrates that A. tumefaciens is sensitive to UV exposure and indicates that dyes requiring a UV filter for imaging should be avoided for time-lapse microscopy experiments.
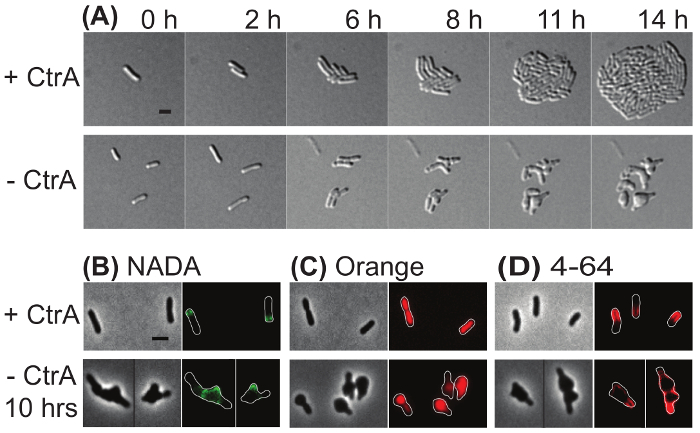
Time-lapse imaging and target-specific labeling of an A. tumefaciens depletion strain Both saturating transposon mutagenesis39 and failing to construct a deletion strain18,40 indicate that the master regulator protein, CtrA, is essential in A. tumefaciens. To demonstrate the value of microscopic analysis of depletion strains, a previously described ctrA depletion strain18 was subjected to characterization by microscopy. In Figure 4A, time-lapse microscopy was used to compare the growth of ctrA depletion cells in which ctrA expressionwas induced (top) and uninduced (bottom). In the presence of CtrA, a single cell gave rise to a microcolony within 14 h. In contrast, when CtrA was depleted, cells either lysed or failed to divide. Cells that failed to divide exhibited gross morphological changes including rounding from mid-cell and at the cell poles. These observations suggest that CtrA has an important function in the regulation of cell division.
In Figure 4B-D, fluorescent dyes were used to characterize the ctrA depletion strain after induction or depletion of ctrA for 10 h. Cells were pulse labeled with NADA for 2 min (Figure 4B). When CtrA was present (top panel), polar peptidoglycan biosynthesis was observed. In contrast, extensive NADA labelling occurred in the poles and large mid-cell swellings occurred when CtrA was depleted. This observation is consistent with continued peptidoglycan synthesis despite a failure of the cells to divide. The cyanine DNA dye Orange was used to characterize DNA distribution in the ctrA depletion strain (Figure 4C). When CtrA was present, growing cells had an even distribution of DNA and segregation of nucleoids was apparent in a late pre-divisional cell; in contrast, when CtrA was depleted, DNA was unevenly distributed throughout the cells. These observations suggest that CtrA contributes to proper DNA replication or segregation. Finally, ctrA depletion cells were labeled with 4-64 (Figure 4D). In the presence of CtrA, the cell membrane was labeled with a characteristic horseshoe pattern in which the growth pole was devoid of stain. In cells depleted of CtrA, 4-64 appeared to label the entire membrane, although one pole was more intensely stained. This observation suggests that membranes remain intact although the lipid microdomain organization may be disrupted when CtrA is depleted.
Figure 2: Representative images of wildtype Agrobacterium tumefaciens cells labeled with target specific dyes. (A) Phase contrast images of A. tumefaciens cells before and after the washing protocol, when treated with 1% DMSO, or after fixation with ice-cold ethanol. (B) Polar growth of A. tumefaciens cells is shown by labeling with the fluorescent d-amino acids HADA, NADA, and TADA. (C) Staining of A. tumefaciens with the lipophilic 4-64 stain. (D) Staining of A. tumefaciens cells with the DNA-specific dyes Orange and DAPI. For panels B-D, phase contrast (top) and epifluorescence (bottom) images are shown. Cell outlines are provided in fluorescence images for reference. Scale bar = 2 µm. Please click here to view a larger version of this figure.
Figure 3: Comparison of DAPI and Orange labeling of DNA in live A. tumefaciens cells. Equal proportions of unlabeled, DAPI-labeled, and Orange-labeled wildtype A. tumefaciens cells were mixed and spotted on agarose pads. At time 0, initial images were taken using phase contrast, TRITC, and DAPI filters to determine if the cells were labeled. (A) Time-lapse of unlabeled, DAPI-labeled, and Orange-labeled cells imaged by phase contrast microscopy. (B) Time-lapse of unlabeled, DAPI-labeled, and Orange-labeled cells imaged by phase microscopy and epifluorescence microscopy using the TRITC filter. (C) Time-lapse of unlabeled, DAPI-labeled, and Orange-labeled cells imaged by phase microscopy and epifluorescence microscopy using the DAPI filter. Scale bar = 2 µm. Please click here to view a larger version of this figure.
Figure 4: Representative images of the ctrA depletion strain in induced and uninduced conditions. (A) Time-lapse microscopy of the ctrA depletion strain under conditions where ctrA was induced (top) or depleted (bottom). The numbers above each panel indicate the time in hours. (B) Phase contrast (left) and epifluorescence (right) images of cells labeled with NADA. (C) Phase contrast (left) and fluorescence (right) images of cells labeled with Orange. (D) Phase contrast (left) and fluorescence (right) images of cells labeled with 4-64. Cell outlines were provided in fluorescence images for reference. Scale bar = 2 µm. Please click here to view a larger version of this figure.
Discussion
This protocol contains a series of procedures for the investigation of A. tumefaciens wildtype, mutant, and depletion strains. It is worth noting that all of the procedures listed in the protocol section can be readily adapted for other bacterial strains with additional modifications to account for growth media, temperatures, and growth rates.
The use of target-specific dyes is a valuable tool for providing a detailed characterization of cell-cycle events in bacterial cells. Here, peptidoglycan insertion patterns were observed using fluorescent-d-amino acids, lipid domains were visualized with 4-64, and nucleoids were labeled with either DAPI or Orange. Each of the protocols can be adapted for use in other bacteria after optimizing the wash protocol and ensuring that the DMSO treatment does not impair growth or morphology. Key considerations include the selection of a buffer for wash steps, incubation times for label incorporation, and the selection of an appropriate fluorophore to avoid auto-fluorescence or to enable multiplex labeling studies. For example, an alternative to the lipophilic red-fluorescent membrane stain (4-64) is the green-fluorescent membrane stain (1-43). Similarly, blue-, green-, and red-fluorescent-d-amino acids are available6,38 and d-amino acids with biorthogonal handles can be conjugated to common fluorophores through click chemistry6,7. Finally, in addition to the orange-fluorescent cyanine DNA dye, a green fluorescent cyanine DNA dye is available and performs well with live bacterial cells10. Visualization of key cell structures labeled with target-specific dyes can be combined with the observation of fluorescent proteins to gain mechanistic insights into essential processes in bacteria.
These protocols include the conditions necessary for microscopic observation of A. tumefaciens depletion stains and it should be possible to extend these approaches to protein depletion strains in other bacteria. The characterization of depletion strains can be particularly challenging as there is a limited timeframe to complete investigations before the cells stop growing or lyse. It is crucial to adequately wash the cells to remove all traces of the excess dye during labeling; however, some cells depleted of essential proteins have defects in their cell surfaces which can cause them to be sensitive to wash steps. Increasing the starting volume of cell cultures can help compensate for loss of cells during the wash steps. Depletion strains which have compromised cell membranes or cell walls may be prone to cell lysis when grown in liquid culture without inducer. Inclusion of an osmoprotectant (5% sucrose works well for A. tumefaciens) can help to maintain cell morphology and limit cell lysis during depletion. It is necessary to empirically determine the appropriate amount of time to pre-deplete the cells prior to staining with fluorescent dye. Cells with permeablized membranes or cell walls may be prone to cell lysis or non-selective labeling with fluorescent reagents making imaging of late depletion time points particularly challenging.
A critical step for time-lapse microscopy of A. tumefaciens (or other bacterial cells) is the preparation of the agarose pad. The agarose pad needs to be level to ensure good focus throughout time-lapse microscopy experiments. In addition, the coverslip must be completely sealed to prevent the agarose pad from drying and changing the focal plane. For A. tumefaciens, oxygen is required for cell growth. Imaging near an air pocket is necessary to get robust growth of A. tumefaciens during long time-lapse microscopy experiments (Figure 1C). If cells fail to grow or stop growing after just a few hours, it is advisable to prepare an agarose pad with a larger air pocket and image near the air pockets. Even a well-constructed agarose pad with a proper air pocket can only support A. tumefaciens growth for a maximum of ~24 h. If longer time-lapse sequences are necessary, alternative approaches such as microfluidics or flow cells should be considered. If the cells drift out of focus during imaging, ensure that the agarose pad is level and properly sealed under the coverslip. Note that even with a near perfect agarose pad, it is possible that some cells will drift out of focus, thus imaging multiple fields and frequent refocusing are highly recommended. For other bacteria, alternative approaches of preparing agarose pads should be considered41,42,43 to best meet the growth requirements for the bacterium. Agarose pads are also useful for immobilizing cells during imaging of labeled or fixed cells when time-lapse is not necessary. In this case, sealing the coverslip is not necessary and alternative approaches for constructing agarose pads which can be stored until later may be appropriate43.
Overall, the use of target-specific dyes and time-lapse microscopy can provide insights about fundamental processes in bacteria. Here, we illustrate the usual horseshoe pattern of labeling with the lipophilic 4-64 dye and the characteristic polar and septal labeling with fluorescent-d-amino acids in A. tumefaciens. Observations of these patterns in A. tumefaciens are consistent with the polar growth of this bacterium44 and it is likely that similar studies may reveal information about growth patterning in other bacteria. The finding that cyanine dyes such as Orange are suitable for time-lapse microscopy studies10 (Figure 3) will enable careful studies of nucleoid morphology during cell growth in wildtype and mutant strains. Imaging fluorescent target-specific dyes during the depletion of an essential protein can provide key observations to determine the function of the protein. Finally, since many of these dyes are available with different spectral properties, studies using multiple labels simultaneously should enable insights into the coordination of essential processes during cell cycle progression.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We thank Michael VanNieuwenhze (Indiana University) for the gift of the FDAAs used in Figure 2 and Figure 4. We thank members of the Brown lab for feedback during the preparation of this manuscript. Research in the Brown lab on A. tumefaciens cell growth and division is supported by the National Science Foundation (IOS1557806).
References
- Daniel RA, Errington J. Control of cell morphogenesis in bacteria: two distinct ways to make a rod-shaped cell. Cell. 2003;113(6):767–776. doi: 10.1016/s0092-8674(03)00421-5. [DOI] [PubMed] [Google Scholar]
- Tiyanont K, et al. Imaging peptidoglycan biosynthesis in Bacillus subtilis with fluorescent antibiotics. Proc Natl Acad Sci U S A. 2006;103(29):11033–11038. doi: 10.1073/pnas.0600829103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner RD, et al. Peptidoglycan architecture can specify division planes in Staphylococcus aureus. Nat Commun. 2010;1:26. doi: 10.1038/ncomms1025. [DOI] [PubMed] [Google Scholar]
- Wheeler R, Mesnage S, Boneca IG, Hobbs JK, Foster SJ. Super-resolution microscopy reveals cell wall dynamics and peptidoglycan architecture in ovococcal bacteria. Mol Microbiol. 2011;82(5):1096–1109. doi: 10.1111/j.1365-2958.2011.07871.x. [DOI] [PubMed] [Google Scholar]
- Turner RD, Hurd AF, Cadby A, Hobbs JK, Foster SJ. Cell wall elongation mode in Gram-negative bacteria is determined by peptidoglycan architecture. Nat Commun. 2013;4:1496. doi: 10.1038/ncomms2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuru E, et al. In Situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem Int Ed Engl. 2012;51(50):12519–12523. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegrist MS, et al. (D)-Amino acid chemical reporters reveal peptidoglycan dynamics of an intracellular pathogen. ACS Chem Biol. 2013;8(3):500–505. doi: 10.1021/cb3004995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kepner RL, Pratt JR. Use of fluorochromes for direct enumeration of total bacteria in environmental samples: past and present. Microbiol Rev. 1994;58(4):603–615. doi: 10.1128/mr.58.4.603-615.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson MB, Criss AK. Fluorescence microscopy methods for determining the viability of bacteria in association with mammalian cells. J Vis Exp. 2013. [DOI] [PMC free article] [PubMed]
- Bakshi S, et al. Nonperturbative imaging of nucleoid morphology in live bacterial cells during an antimicrobial peptide attack. Appl Environ Microbiol. 2014;80(16):4977–4986. doi: 10.1128/AEM.00989-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barak I, Muchova K. The role of lipid domains in bacterial cell processes. Int J Mol Sci. 2013;14(2):4050–4065. doi: 10.3390/ijms14024050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fishov I, Woldringh CL. Visualization of membrane domains in Escherichia coli. Mol Microbiol. 1999;32(6):1166–1172. doi: 10.1046/j.1365-2958.1999.01425.x. [DOI] [PubMed] [Google Scholar]
- Barak I, Muchova K, Wilkinson AJ, O'Toole PJ, Pavlendova N. Lipid spirals in Bacillus subtilis and their role in cell division. Mol Microbiol. 2008;68(5):1315–1327. doi: 10.1111/j.1365-2958.2008.06236.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cameron TA, Anderson-Furgeson J, Zupan JR, Zik JJ, Zambryski PC. Peptidoglycan synthesis machinery in Agrobacterium tumefaciens during unipolar growth and cell division. MBio. 2014;5(3):e01219. doi: 10.1128/mBio.01219-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zupan JR, Cameron TA, Anderson-Furgeson J, Zambryski PC. Dynamic FtsA and FtsZ localization and outer membrane alterations during polar growth and cell division in Agrobacterium tumefaciens. Proc Natl Acad Sci U S A. 2013;110(22):9060–9065. doi: 10.1073/pnas.1307241110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eberhardt A, Wu LJ, Errington J, Vollmer W, Veening JW. Cellular localization of choline-utilization proteins in Streptococcus pneumoniae using novel fluorescent reporter systems. Mol Microbiol. 2009;74(2):395–408. doi: 10.1111/j.1365-2958.2009.06872.x. [DOI] [PubMed] [Google Scholar]
- Iniesta AA, Garcia-Heras F, Abellon-Ruiz J, Gallego-Garcia A, Elias-Arnanz M. Two systems for conditional gene expression in Myxococcus xanthus inducible by isopropyl-beta-D-thiogalactopyranoside or vanillate. J Bacteriol. 2012;194(21):5875–5885. doi: 10.1128/JB.01110-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Figueroa-Cuilan W, Daniel JJ, Howell M, Sulaiman A, Brown PJ. Mini-Tn7 Insertion in an Artificial attTn7 Site Enables Depletion of the Essential Master Regulator CtrA in the Phytopathogen Agrobacterium tumefaciens. Appl Environ Microbiol. 2016;82(16):5015–5025. doi: 10.1128/AEM.01392-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacob F, Monod J. Genetic regulatory mechanisms in the synthesis of proteins. J Mol Biol. 1961;3:318–356. doi: 10.1016/s0022-2836(61)80072-7. [DOI] [PubMed] [Google Scholar]
- Khan SR, Gaines J, Roop RM, 2nd, Farrand SK. Broad-host-range expression vectors with tightly regulated promoters and their use to examine the influence of TraR and TraM expression on Ti plasmid quorum sensing. Appl Environ Microbiol. 2008;74(16):5053–5062. doi: 10.1128/AEM.01098-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yansura DG, Henner DJ. Use of the Escherichia coli lac repressor and operator to control gene expression in Bacillus subtilis. Proc Natl Acad Sci U S A. 1984;81(2):439–443. doi: 10.1073/pnas.81.2.439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177(14):4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thanbichler M, Iniesta AA, Shapiro L. A comprehensive set of plasmids for vanillate- and xylose-inducible gene expression in Caulobacter crescentus. Nucleic Acids Res. 2007;35(20):e137. doi: 10.1093/nar/gkm818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Topp S, et al. Synthetic riboswitches that induce gene expression in diverse bacterial species. Appl Environ Microbiol. 2010;76(23):7881–7884. doi: 10.1128/AEM.01537-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JM, et al. A Comprehensive, CRISPR-based Functional Analysis of Essential Genes in Bacteria. Cell. 2016;165(6):1493–1506. doi: 10.1016/j.cell.2016.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qi LS, et al. Repurposing CRISPR as an RNA-guided platform for sequence-specific control of gene expression. Cell. 2013;152(5):1173–1183. doi: 10.1016/j.cell.2013.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith KL, Grossman AD. Inducible protein degradation in Bacillus subtilis using heterologous peptide tags and adaptor proteins to target substrates to the protease ClpXP. Mol Microbiol. 2008;70(4):1012–1025. doi: 10.1111/j.1365-2958.2008.06467.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGinness KE, Baker TA, Sauer RT. Engineering controllable protein degradation. Mol Cell. 2006;22(5):701–707. doi: 10.1016/j.molcel.2006.04.027. [DOI] [PubMed] [Google Scholar]
- Schneider JP, Basler M. Shedding light on biology of bacterial cells. Philos Trans R Soc Lond B Biol Sci. 2016;371(1707) doi: 10.1098/rstb.2015.0499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar MA, Dandekar AM. Agrobacterium tumefaciens as an agent of disease. Trends Plant Sci. 2003;8(8):380–386. doi: 10.1016/S1360-1385(03)00162-6. [DOI] [PubMed] [Google Scholar]
- Gelvin SB. Agrobacterium-mediated plant transformation: the biology behind the "gene-jockeying" tool. Microbiol Mol Biol Rev. 2003;67(1):16–37. doi: 10.1128/MMBR.67.1.16-37.2003. table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nester EW. Agrobacterium: nature's genetic engineer. Front Plant Sci. 2014;5:730. doi: 10.3389/fpls.2014.00730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imam J, Singh PK, Shukla P. Plant Microbe Interactions in Post Genomic Era: Perspectives and Applications. Front Microbiol. 2016;7:1488. doi: 10.3389/fmicb.2016.01488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramoni S, Nathoo N, Klimov E, Yuan ZC. Agrobacterium tumefaciens responses to plant-derived signaling molecules. Front Plant Sci. 2014;5:322. doi: 10.3389/fpls.2014.00322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan ZC, Williams M. A really useful pathogen, Agrobacterium tumefaciens. Plant Cell. 2012;24(10) doi: 10.1105/tpc.112.tt1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Martinez CE, Christie PJ. Biological diversity of prokaryotic type IV secretion systems. Microbiol Mol Biol Rev. 2009;73(4):775–808. doi: 10.1128/MMBR.00023-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pitzschke A. Agrobacterium infection and plant defense-transformation success hangs by a thread. Front Plant Sci. 2013;4:519. doi: 10.3389/fpls.2013.00519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuru E, Tekkam S, Hall E, Brun YV, Van Nieuwenhze MS. Synthesis of fluorescent D-amino acids and their use for probing peptidoglycan synthesis and bacterial growth in situ. Nat Protoc. 2015;10(1):33–52. doi: 10.1038/nprot.2014.197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curtis PD, Brun YV. Identification of essential alphaproteobacterial genes reveals operational variability in conserved developmental and cell cycle systems. Mol Microbiol. 2014;93(4):713–735. doi: 10.1111/mmi.12686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Heindl JE, Fuqua C. Coordination of division and development influences complex multicellular behavior in Agrobacterium tumefaciens. PLoS One. 2013;8(2):e56682. doi: 10.1371/journal.pone.0056682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jong IG, Beilharz K, Kuipers OP, Veening JW. Live Cell Imaging of Bacillus subtilis and Streptococcus pneumoniae using Automated Time-lapse Microscopy. J Vis Exp. 2011. [DOI] [PMC free article] [PubMed]
- Turnbull L, et al. Super-resolution imaging of the cytokinetic Z ring in live bacteria using fast 3D-structured illumination microscopy (f3D-SIM) J Vis Exp. 2014. p. e51469. [DOI] [PMC free article] [PubMed]
- Zeng L, Golding I. Following cell-fate in E. coli after infection by phage lambda. J Vis Exp. 2011. p. e3363. [DOI] [PMC free article] [PubMed]
- Brown PJ, et al. Polar growth in the Alphaproteobacterial order Rhizobiales. Proc Natl Acad Sci U S A. 2012;109(5):1697–1701. doi: 10.1073/pnas.1114476109. [DOI] [PMC free article] [PubMed] [Google Scholar]