

Abstract
siRNA and shRNA-mediated knock down (KD) methods of regulating gene expression are invaluable tools for understanding gene and protein function. However, in the case that the KD of the protein of interest has a lethal effect on cells or the anticipated effect of the KD is time-dependent, unconditional KD methods are not appropriate. Conditional systems are more suitable in these cases and have been the subject of much interest. These include Ecdysone-inducible overexpression systems, Cytochrome P-450 induction system1, and the tetracycline regulated gene expression systems.
The tetracycline regulated gene expression system enables reversible control over protein expression by induction of shRNA expression in the presence of tetracycline. In this protocol, we present an experimental design using functional Tet-ON system in human cancer cell lines for conditional regulation of gene expression. We then demonstrate the use of this system in the study of tumor cell-monocyte interaction.
Keywords: Cancer Research, Issue 129, Conditional knock down, cell migration, doxycycline, shRNA, PAI-1, monocytes, cancer cell lines
Introduction
Tumor associated macrophages (TAMs) contribute to tumor development by promoting tumor growth, metastasis, and regulating the immune response2. Cancer cells recruit inflammatory monocytes, which infiltrate tumor and differentiate into pro-tumorigenic TAMs3. Infiltration of the tumor with TAMs correlates with poor clinical outcome and has been linked to the immunosuppressive role of macrophages4,5. However, the mechanisms of the recruitment of macrophages to the tumor are not well explored and a better understanding of the involved pathways is crucial for further advancement of the field and promising therapies. One of the challenges in studying interactions between tumor cells and normal cells in the tumor microenvironment (TME) is the complexity of the mechanisms and cells involved, requiring in vitro approaches that allow dissection of the crosstalk. Here we present a versatile methodology that can be applied to study the paracrine effect of a cancer cell-derived, secreted protein on migration of other cell types like macrophages, in vitro. Using a system where the expression of the shRNA against a cancer cell-derived protein involved in the recruitment of monocytes is under the control of the tetracycline inducible promoter, the paracrine effect of the secreted protein on monocytes is quantitated. In this protocol, cloning of shRNA sequences into the tetracycline regulated vector is presented followed by generation of stable cancer cell lines. Further, the purification of primary human monocytes and a Boyden chamber assay are used to analyze the paracrine effect of a cancer-cell derived protein on migration of monocytes.
Downregulation of protein coding genes is commonly applied with siRNA and shRNA techniques, though not without limitations in its use. The long-term knock-down (KD) of genes may elicit secondary adaptive responses of cells that interfere with experimental results. Lack of temporal control over gene expression makes it challenging to study the dynamic role of a protein over time or the role of a protein crucial for cell survival. This issue is especially important in in vivo settings, where the role of a protein in tumor development and progression might require downregulation of the expression of the protein of interest only after tumor is established. Conditional KD has the advantage of preventing a too early lethal effect of the KD on cells, and to enable analysis of the role of the protein within different stages of tumor growth, while unconditional KD could result in lack of tumor development.
A number of conditional KD systems have been developed to address the limitations of stable KD. The conditional gene expression systems include Ecdysone-inducible overexpression systems, the Cytochrome P-450 induction system1, and tetracycline regulated gene expression systems. The tetracycline-regulated gene expression systems allow control over expression of the shRNA upon addition of the antibiotic tetracycline (or its more stable analogue - doxycycline). In Tet-ON systems, the expression of shRNA is induced in the presence of tetracycline/doxycycline resulting in a gene expression KD, while in Tet-OFF systems, the expression of shRNA is suppressed in the presence of tetracycline resulting in gene expression. A drawback of tetracycline-inducible system is previously reported low levels of expression of shRNA in the absence of doxycycline - so-called leakiness6,7. In the Tet-ON system described here, upon administration of tetracycline, the binding of constitutively expressed tetracycline repressor (TetR) protein to the Tet-responsive element (TRE) sequence within the H1 promoter region of the shRNA of interest is suppressed. This results in expression of shRNA and inhibition of translation of the protein of interest in a tetracycline-dependent manner8,9.
Other available Tet-ON systems include simultaneous knock-in of the TetO sequence between TATA box and proximal sequence element (PSE) and between TATA box and transcription start site developed by Chan et al.10 This system requires less than toxic doses of tetracycline to regulate shRNA expression, however, under a non-induced state, low levels of shRNA are expressed. The Krüppel-associated box (KRAB) based Tet-ON system11 includes KRAB, a zinc finger protein, which subjects genes located within a 3 kb range of the KRAB binding site to transcriptional suppression. The chimeric protein tTRKRAB can bind to TetO, and due to the large-range of DNA controllable capacity, TetO do not need to be limited between the transcription start site and the promoter and have low impact on the activity of the promoter. Under the non-induced state, this controllable RNA interference system was reported to show a lower level of leaked expression of shRNA11,12; however, it requires a sequential, two-vector cloning approach. In comparison to previously developed conditional KD systems such as the Ecdysone-inducible overexpression system or Cytochrome P-450 induction system, the tetracycline-regulated system has the advantage of its robustness and reversibility, and therefore is the most routinely used system13. The system used in this protocol has the advantage over the dual TetO knock-in system and KRAB Tet-ON system as it requires straight forward, single vector cloning, allowing quick generation of multiple clones, and it exhibits very low levels of leakiness in the absence of doxycycline.
TME is critical for the development of cancer. To facilitate tumor growth, cancer cells recruit inflammatory monocytes by secreting chemotactic proteins. Recruited monocytes infiltrate the tumor and differentiate into pro-tumorigenic TAMs that contribute to tumor growth and metastasis. In vitro studies of the immune cell recruitment utilize migration assays, with Boyden chamber assay being widely used14,15,16,17. In this assay, the chemoattractant protein source, for example, cancer cells, or a purified protein, is placed in the bottom chamber. Immune cells are placed in the upper chamber separated with porous membrane from the bottom compartment. Cells migrating toward the increasing gradient of chemoattractant, and those found on the lower side of the membrane are stained and counted under the microscope. Here we test the chemoattractant function of Plasminogen Activator Inhibitor 1 (PAI-1) on monocytes by generating stable, inducible cancer cell lines where expression of PAI-1 is regulated by doxycycline addition. We use human primary monocytes in the Boyden chamber migration assay to assess the role of PAI-1 in monocyte migration. Differences between human primary monocytes and widely used monocytic cell lines like THP1 have been reported and include different cytokine expression patterns18; for example, 5 - 10 fold increase in TNF-α expression levels by THP1 cells compared to human monocytes19. THP1 cells are derived from human leukemia monocytic cells, are easy to maintain, and proliferate with an average doubling time of 19 - 50 h20. On the contrary, human monocytes are characterized by a short lifespan in the absence of growth factors. Since monocytes are purified from blood of donors, a fair amount of variability occurs among the individuals and depending on the purification method, contamination with other cell types may occur. Nevertheless, primary monocytes are relevant and it has been recommended to use or confirm the results obtained with the monocytic cell lines using primary monocytes in biological research19. Here we describe a protocol for purification of human primary monocytes from peripheral blood. Alternative methods of monocyte purification include density gradient and adhesion protocols and two step procedure with single gradients of Ficoll-Hypaque followed by a Percoll gradient21. The purity of the monocyte population obtained by those methods ranges between 70 - 90%. The method described here uses a density gradient followed by a negative immune selection22 and enables the purification of human monocytes without direct contact with antibodies, thereby avoiding their accidental activation and resulting in a >95% pure population of monocytes.
The protocol presented here is used for setting up a functional Tet-ON system for gene expression KD in human cancer cell lines to study the chemoattractant effect of the cancer-derived secreted protein PAI-1 on monocytes. PAI-1 is overexpressed by a variety of tumors and its expression paradoxically correlates with poor clinical outcome23,24. The pro-tumorigenic role of PAI-1 is a result of its pro-angiogenic and anti-apoptotic functions25,26. PAI-1 has been shown to contribute to inflammation by promoting the recruitment of macrophages to the site of inflammation27. PAI-1 was shown to promote smooth muscle cellmigration28,29 and to participate in the Mac-1 dependent macrophage migration30. PAI-1 overexpression has been also shown to significantly enhance the recruitment of Raw 264.7 macrophages into B16F10 melanoma tumors31. However, the role of PAI-1 in TAM migration has not been investigated in detail. We use the described protocol to answer the question of whether PAI-1 attracts monocytes to cancer cells. This methodology allows dissection of the crosstalk between tumor and TME by silencing the secreted protein in cancer cells and analyzing the components of the TME.
Protocol
The protocol section that uses human monocytes obtained from healthy volunteers follows the guidelines of the Children's Hospital Los Angeles Human Research Ethics Committee and has been approved by the Institutional Review Board under the Human Material Protocol number: CCI 08-00208.
1. Preparation of Cancer Cell Lines with Tetracycline-regulated shRNA Expression
- Cloning of shRNA PAI-1 into Tet-pLKO-puro vector 8 9
- Design shRNA oligomers that contain AgeI and EcoRI cleavage site at the 5' end, the sense sequence, a 6 nucleotide loop, and the antisense sequence. NOTE: Typical oligonucleotides are designed as follows: Forward oligo: 5' CCGG - 19 - 21 bp sense - CTCGAG - 19 - 21 bp antisense - TTTTT, Reverse oligo: 5' AATTAAAAA- 19-21 bp sense - CTCGAG - 19 - 21 bp antisense 3'. Detailed protocol for the oligomer design can be found in 8. In this study 3 shRNA sequences against PAI-1 were tested for the strongest silencing effect: shRNA 132, shRNA 2, shRNA 333, and scrambled33 are included in Table 1.
- Anneal the shRNA oligomers and scrambled shRNA oligomers.
- Reconstitute oligonucleotides to 100 µM in water and mix 1 µL Forward oligonucleotide, 1 µL Reverse oligonucleotide, and 8 µL H2O.
- To anneal the oligonucleotides, mix the following: 1 µL oligonucleotide mixture, 5 µL buffer 10 mM Tris(hydroxymethyl)aminomethane (Tris) pH 7.5, 50 mM Sodium Chloride (NaCl), 10 mM Magnesium Chloride (MgCl2), 1 mM dithioerythritol (DTE), and 44 µL H2O.
- Incubate the mixture in a polymerase chain reaction (PCR) thermal cycler at 95 °C for 5 min, switch off the instrument, and wait until room temperature (RT) is reached.
- Precipitate annealed oligonucleotides by adding 5 µL 3 M Sodium Acetate pH 5.2 to 50 µL annealed oligonucleotides.
- Add 100 µL cold 100% ethanol (EtOH) and incubate for 30 min at -80 °C. Centrifuge in a benchtop centrifuge at maximum speed for 30 min at 4 °C.
- Remove supernatant, add 500 µL cold 70% EtOH, centrifuge for 30 min, remove supernatant, and dissolve pellet in 20 µL H2O.
- Assess the concentration of annealed oligonucleotides by spectrophotometry, measuring the absorbance at 260 nm. Dilute oligonucleotides to the final concentration of 1 ng/µL.
- Prepare Luria Bertani (LB) medium and LB agar plates.
- For LB medium, dissolve 10 g tryptone, 5 g yeast extract, 10 g NaCl in 1 L of water. Adjust pH of the medium to pH 7.0 using 1 N NaOH and autoclave.
- For LB agar plates, add 15 g/L of agar into LB medium. Autoclave and cool down to approximately 50 °C. Add antibiotic and pour the plates.
- Streak bacteria transduced with Tet-pLKO-puro vector (see the Table of Materials) on LB agar plate supplemented with 100 µg/mL ampicillin and incubate overnight (O/N) at 37 °C.
- Inoculate 100 mL LB medium supplemented with 100 µg/mL ampicillin (LB/ampicillin) with 1 colony from the plate and grow O/N with shaking at 37 °C.
- IsolateTet-pLKO-puro from 100 mL bacterial culture using a plasmid isolation kit.
- Prepare the restriction reaction for the Tet-pLKO-puro vector with EcoRI and AgeI restriction enzymes as follows: 1 µL EcoRI, 1 µL AgeI, 5 µL 10x buffer, 1 µL plasmid DNA (1 µg), 42 µL H2O. Incubate 1 h at 37 °C. To obtain enough of cleaved vector, prepare up to 5 x 50 µL reactions.
- Use 1% agarose gel to purify cleaved vector from the agarose gel using a DNA purification kit. To increase the yield of DNA, minimize the agarose to DNA ratio in the gel by using an agarose gel comb for large wells and by carefully removing most of the agarose from the band using a scalpel.
- Prepare the ligation reaction mix as follows: 1 ng annealed oligonucleotides, 50 ng cleaved vector, 2 µL 10x ligase buffer, 1 µL ligase, and water up to 20 µL. Ligate the vector with annealed shRNA oligonucleotides at 16 °C O/N.
- Prepare in addition, a negative control reaction without oligonucleotides to account for uncleaved, partially cleaved, and self-ligated vector that will result in false positive colonies.
- Using standard transformation techniques, transform ligation mixtures into chemically competent bacterial cells designed to prevent homologous recombination of lentiviral vectors containing direct repeats.
- Grow bacteria on plates with ampicillin O/N at 37 °C. NOTE: If growth of satellite colonies occurs, repeat the transformation and grow bacteria at 30 °C to slow down the growth and thus prevent satellite colonies.
- To verify successful ligation, pick between 10 - 20 single colonies, inoculating 2 mL LB/ampicillin cultures and an ampicillin plate as a stock plate (to be used as a positive clone later). Grow liquid cultures with shaking O/N at 37 °C. Incubate the plate O/N at 37 °C.
- Seal the plate with parafilm and store at 6 °C.
- Isolate plasmid DNA from liquid cultures using a plasmid isolation kit.
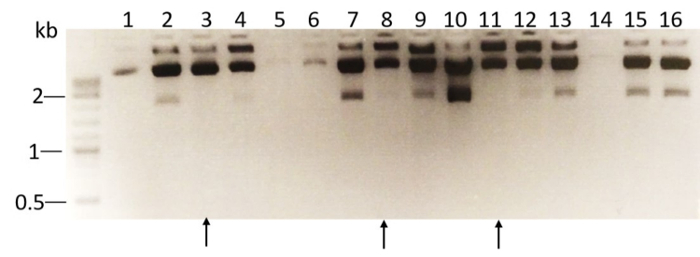
- Perform restriction analysis of plasmid DNA isolated from ampicillin-resistant colonies using XhoI enzyme. For each clone, prepare the following reaction mix: 0.5 µL XhoI, 2.5 µL 10x buffer, 1 µL plasmid DNA (0.5 µg), and 21 µL H2O. Incubate for 1 h at 37 °C. Analyze the result of the restriction reaction by running the reaction on a 1% agarose gel. NOTE: Restriction analysis of the WT vector cleaved by XhoI will generate 3 fragments: 8,447, 1,800, and 200 bp. The stuffer sequence in Tet-pLKO-puro vector of 1,800 bp is not present in positive clones containing shRNA-PAI-1 and the XhoI digest will result in DNA fragments of the size: 8,447, 190, and 130 bp. As shown in Figure 1, the 2,000 bp fragment is not found in DNA isolated from ampicillin resistant colonies number: 3, 8, and 11. Depending on the design of oligonucleotides, the length and number of obtained fragments in DNA from positive colonies may vary.
- Inoculate 100 mL LB/Ampicillin culture with the colony containing the correct clone and grow O/N at 37 °C.
- Isolate plasmid DNA using a plasmid isolation kit.
- Generation of lentivirus particles.
- Coat a 15 cm tissue culture dish with Poly-L-Lysine by covering the surface of the dish with sterile water solution of 0.01% Poly-L-Lysine and incubating at RT for 15 min. Remove the solution by aspiration and air-dry the dishes.
- Seed 5 x 106 Human Embryonic Kidney 293 cells (HEK293) cells in the Poly-L-Lysine-coated 15 cm dish, and culture in Dulbecco's Modified Eagle's Medium (DMEM) medium supplemented with 10% FBS and 1% Penicillin/Streptomycin mix (Pen/Strep) at 37 °C, 5% CO2 until they reach 70% confluency.
- To produce viral particles, transfect HEK293 cells with 25 µg pLKO-Tet-On-shRNA, 25 µg psPAX, (packaging plasmid), and 5 µg pMD2.G plasmids (envelope expressing plasmid) using transfection reagent. NOTE: psPAX and pMD2.G plasmids are a gift from the Didier Trono lab (Ecole Polytechnique Federale de Lausanne, Switzerland).
- Induce HEK293 cells the next day by changing the medium to DMEM supplemented with 10 mM sodium butyrate and 20 mM HEPES pH 7.2, and culturing for 8 h.
- Wash the cells with phosphate-buffered saline (PBS) and add 25 mL of fresh DMEM medium containing 20 mM 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES) pH 7.2. After incubation at 37 °C for 48 h, carefully collect virus-containing medium by pipetting; avoid spills.
- Filter the collected medium through a 0.45 µM syringe filter to remove HEK293 cells. The medium containing viral particles can be used immediately or stored at -80 °C for later use.
- Generation of stably transfected cell lines
- Seed HCT116 colon cancer cells and MDA-MB-231 breast cancer cells at 50,000 cells/well in DMEM medium supplemented with 10% fetal bovine serum (FBS) and 1% Pen/Strep in a 12-well plate. Incubate at 37 °C at 5% CO2 until cells are 60 - 70% confluent.
- Wash cells with PBS and add 1 mL of virus-containing medium to cancer cells and incubate O/N at 37 °C at 5% CO2. As a control, add 1 mL of fresh DMEM medium supplemented with 10% FBS and 1% Pen/Strep to 2 wells with cancer cells.
- Carefully remove virus-containing medium by aspiration. Wash the cells with PBS and change the medium to DMEM with 10% (v/v) tetracycline-free (Tet-free) FBS and 1% Pen/Strep.
- After 72 h, wash cells with PBS and change the medium to DMEM 10% Tet-free FBS 1% Pen/Strep, 1 µg/mL puromycin for selection of virus-transfected cells.
- Depending on the cell line used, adjust the puromycin concentration for optimal selection by testing the range of puromycin concentrations (0.1, 0.5, 1, 2, and 5 µg/mL) in non-transduced cells and choosing the lowest concentration where no control cells survive after 3 days of culture.
- Select virus-transduced cells by culturing cells in the presence of puromycin for 3 - 14 days. As controls, prepare two additional wells with non-transduced cells, one with medium containing puromycin and one without puromycin. Observe partial killing of the cells by puromycin in the well with virus-transduced cells compared to control wells. NOTE: Non-transduced cells will not grow in the presence of puromycin.
- Verify the efficiency of the conditional KD in the puromycin-resistant HCT116 colon cancer and MDA-MB-231 breast cancer cells. NOTE: The effective concentration of doxycycline will vary with the cell line used and must be titrated (typically from 100 ng/mL to 2 µg/mL).
- Determine the doxycycline concentration that is not toxic for the cells but effective in downregulating the expression of the protein of interest. In this protocol, 1 µg/mL was successfully used in vitro.
- Culture the cells for 72 h in the presence and absence of 0.1, 1, and 2 µg/mL doxycycline. Verify the level of expression of the downregulated protein (here, PAI-1) in the cell lysate by Western blot analysis. Compare HCT116 and MDA-MB-231 cells conditionally expressing shRNA to scrambled control cells.
- Preparation of conditioned medium
- Seed 1 x 106 of cells stably transfected with doxycycline regulated pLKO-shRNA containing lentiviruses in 10 cm dishes.
- Grow cell lines in Roswell Park Memorial Institute (RPMI) medium with 10% Tet-free FBS and 1% Pen/Strep in the absence and presence of 1 µg/mL doxycycline for 72 h at 37 °C at 5% CO2, adding fresh doxycycline daily. NOTE: RPMI medium is compatible with culture conditions of monocytes. Doxycycline is a light sensitive compound. Protect the cell cultures from light by covering with aluminum foil and avoid working under direct light exposure.
- Collect the medium by pipetting, filter through 0.45 µm filter, and freeze at -80 °C in aliquots.
2. Isolation of Monocytes from Human Blood
- Isolate monocytes from human peripheral blood. NOTE: Human primary monocytes are isolated from 7 mL of white blood cells concentrated in a leukocyte filter obtained from healthy platelet donors. Depending on the donor, one leukocyte filter produces between 1 x 109 and 2 x 109 of peripheral blood mononuclear cells (PBMCs), approximately 10% of which constitute monocytes.
- Prepare the workstation in the laminar flow cabinet.
- Sterilize scissors and the laminar flow cabinet with ultraviolet light (UV) for 30 min.
- Spray the leukocyte filter with 70% EtOH and air-dry inside the laminar flow cabinet.
- Cut both ends of the leukocyte filter with scissors and empty the content of the filter into a 50 mL tube.
- Dilute to 90 mL by adding PBS containing 1% (v/v) FBS (PBS/FBS).
- Prepare 3 x 50 mL tubes with 15 mL density gradient solution. Layer slowly 30 mL of PBS/FBS diluted blood over the density gradient solution using a pipette controller set on gravitational flow to avoid mixing of the layers.
- Centrifuge in a swing rotor at 400 x g at RT without brakes for 25 min.
- Dispose the top layer and transfer the middle layer containing PBMCs to a fresh 50 mL tube.
- Add PBS/FBS up to 50 mL and centrifuge at 120 x g at RT for 10 min. Remove the supernatant containing platelets.Repeat this step two more times.
- Resuspend the pellet in 50 mL of PBS/FBS and count the PBMCs using a hemocytometer. Centrifuge at 120 x g at RT and resuspend the pellet in PBS/FBS containing 1 mM ethylenediaminetetraacetic acid (EDTA) (PBS/FBS/EDTA) to a final concentration of 5 x 107 cells/mL.
- Use a negative selection kit to enrich monocytes by depletion of non-monocytic cells. NOTE: The negative selection kit uses a cocktail of antibodies (CD2, CD3, CD16, CD19, CD20, CD56, CD66b, CD123, and glycophorin A) against T cells, NK cells, neutrophils, B cells, granulocytes, and erythrocytes. The antibody complexes bind to the surface of these non-monocytic cells and to the dextran magnetic particles. When the tube is placed in a magnet, the beads adhere to the walls of the tube and remove the non-monocytic cells from the solution.
- Add 50 µL the antibody cocktail to 1 mL of 5 x 107 PBMCs/mL, mix with a pipette, and incubate for 10 min at RT. Add 50 µL magnetic beads to the PBMCs with the antibody cocktail, mix with a pipette, and incubate for another 10 min at RT.
- Add PBS/FBS/EDTA up to 25 mL and place the PBMC mixture in a magnet that can accommodate a 50 mL tube. Incubate for 10 min at RT. Magnetic beads will adhere to the walls of the tube and sequester the non-monocytic cells from the solution.
- Using a 25 mL pipette, remove the solution containing monocytes from the tube in the magnet. Be careful to not touch the sides of the tube with the pipette.
- Centrifuge the solution at 250 x g at RT, remove the supernatant, and resuspend the pellet containing monocytes in RPMI medium containing 10% Tet-free FBS and 1% Pen/Strep.
3. Boyden Chamber Migration Assay
Prepare 1% (w/v) bovine serum albumin (BSA) solution by dissolving 1 g of BSA in 100 mL of ultrapure water, and filter through a 0.2 µm pore size to sterilize.
- Incubate the porous membrane inserts in 1% sterile BSA solution O/N for improved adherence of macrophages to the membrane. For monocytes/macrophages, use 5 - 8 µm pore size inserts.
- Fill a sterile tissue culture dish with 1% BSA solution and place the inserts in the dish. Apply 200 µL 1% BSA solution inside the inserts.
Wash the inserts twice by placing them in a dish filled with sterile PBS and applying PBS inside the filter.
Seed 40,000 HCT116 or MDA-MB-231 cells in 0.5 mL RPMI medium containing 10% Tet-free FBS and 1% Pen/Strep per well, in a 24-well plate, in triplicate. Alternatively, place 0.5 mL of previously generated conditioned medium in the well as a source of chemoattractant.
On the next day, plate 8,000 monocytes in the prepared insert in 0.3 mL volume of the RPMI medium containing 10% Tet-free FBS and 1% Pen/Strep, in triplicate, and incubate at 37 °C at 5% CO2.
- Collect the inserts after 48 h. NOTE: The length of an experiment must be optimized depending on the specific conditions and cells used, by performing a time-course experiment where inserts are collected at different incubation times.
- Shake off the excess medium and precisely swipe the inner surface with a cotton-tipped applicator to remove cells that did not migrate.
- Stain the outer side of the inserts using the Wright-Giemsa method.
- Carefully mount 3 inserts/glass slide in a high viscosity microscope immersion oil, making sure that the side of the filter with migrated macrophages faces up.
Image the slides using a light microscope with 20X objective. Take pictures of 9 fields/filter. Count migrated macrophages in 9 fields/filter.
Representative Results
Three shRNA sequences were tested for the most efficient KD of PAI-1. For this, shRNA sequences against PAI-1 and scrambled (Table 1) were cloned into Tet-pLKO-puro expression vector following the protocol described above. The HT-1080 fibrosarcoma cell line was stably transfected with generated constructs and the cells were treated with doxycycline for 3 days. The expression of PAI-1 was verified by Western blotting (Figure 2A) and the intensity of the bands corresponding to PAI-1 and tubulin was analyzed by densitometry. Using the most effective KD sequence (shRNA 2), we additionally generated MDA-MB-231 epithelial breast adenocarcinoma cells (Figure 2B) and HCT116 colorectal carcinoma cells (Figure 2C) stably transfected with a tetracycline-regulated pLKO-shRNA2-PAI-1 expression vector (and shRNA scrambled as a control). We achieved a 74% decrease of PAI-1 expression in MDA-MB-231 cell line (Figure 2B) and 17.5% decrease in HCT116 cell line in the presence of doxycycline (1 µg/mL)(Figure 2C) as assessed by Western blotting and densitometric analysis of the bands. The Boyden chamber assay was used to quantify the migration of monocytes towards cancer cells. We hypothesized that cancer cell-derived PAI-1 promotes the migration of monocytes towards tumor cells, and therefore less migration will be observed when PAI-1 is downregulated by the addition of doxycycline. Cancer cells were treated with doxycycline for 3 days and were seeded in the bottom of a 24-well plate at 40,000 cells/well. Purified primary human monocytes were applied in the top chamber of the assay and after 24 h, the monocytes attached to the bottom of the membrane were stained and counted under the microscope. We demonstrate that in the absence of doxycycline and therefore the presence of PAI-1, in MDA-MB-231 and HCT116 cells (both PAI-1 shRNA and scrambled controls), significantly increased the migration of human monocytes (Figure 3). The migration of monocytes was then inhibited by 33% and 74% when doxycycline was added to the co-cultures and the production of PAI-1 by tumor cells was down-regulated (Figure 3A, C). No inhibition of monocyte migration was observed with tumor cells transduced with the scrambled shRNA controls, which also indicated that doxycycline had no inhibitory effect on monocyte migration. Similar results were obtained when the conditioned medium of doxycycline-treated cancer cells was placed in the bottom of the wells as a source of chemoattractants (Figure 3B, D).
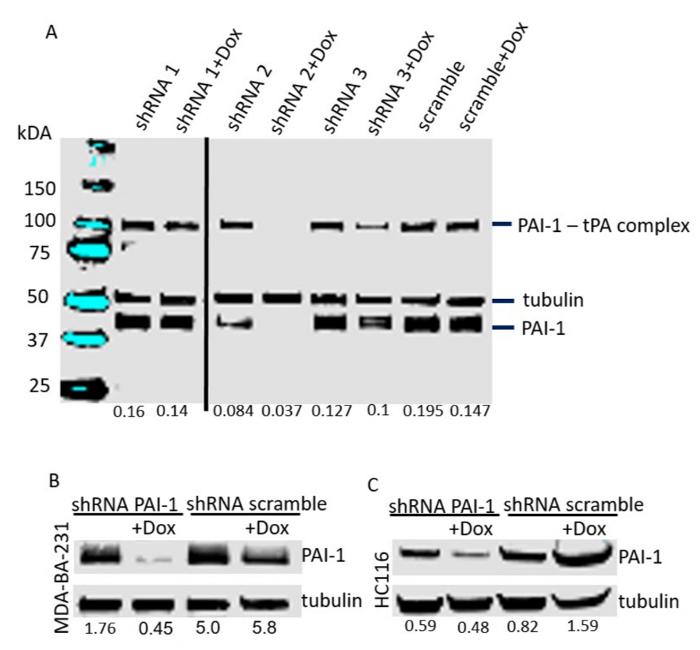
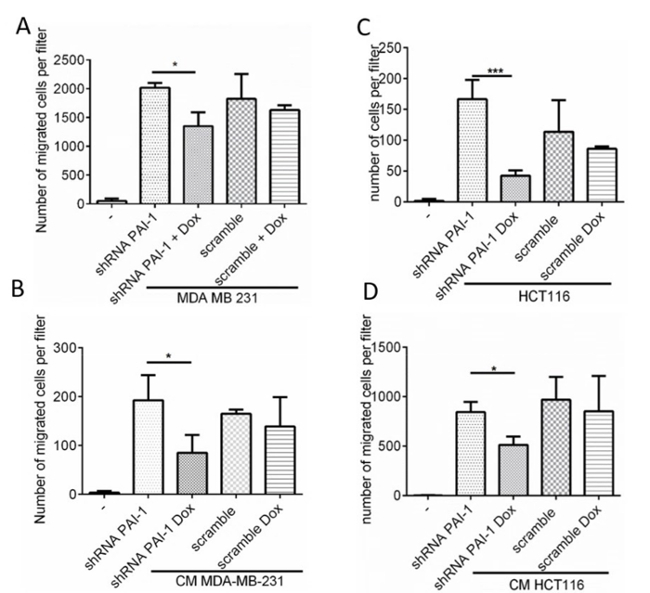
Figure 1. XhoI restriction analysis of DNA extracted from 16 bacterial colonies. DNA was extracted from 16 bacterial colonies and subjected to restriction reaction with XhoI enzyme. The resulting DNA fragments were analyzed by agarose gel electrophoresis. Positive clones (lanes 3, 8, and 11) that lack the 2 kb fragment after XhoI restriction digest are marked with arrows. Please click here to view a larger version of this figure.
Figure 2.Conditional KD of PAI-1 expression in cancer cell lines. Western blotting analysis of PAI-1 expression in cell lines stably transfected with tetracycline-regulated pLKO-shRNA-PAI-1 (shRNA 1-3) and scrambled expression vector after 3 days of doxycycline treatment. (A) HT1080. (B) MDA-MB-231. (C) HCT116. Densitometry analysis of the bands is indicated below the lanes of the Western blots as a ratio of the PAI-1 intensity over the tubulin band intensity. Please click here to view a larger version of this figure.
Figure 3.PAI-1 promotes macrophage migration. Migration of human monocytes towards (A) MDA-MB-231 and (C) HCT116 cell lines stably transfected with vector conditionally expressing shRNA 2 against PAI-1 and scrambled shRNA, measured using the Boyden chamber assay. Cancer cells were treated with doxycycline for 3 days and seeded in a 24-well plate in triplicate. Human primary monocytes were applied on the top of the insert. After 24 h, monocytes on the lower side of the filter were stained and counted under the microscope. Migration of human monocytes toward conditioned medium generated over 72 h by (B) MDA-MB-231 and (D) HCT116 cell lines conditionally expressing PAI-1 in the presence and absence of doxycycline. The data represent the mean of technical triplicates [+/- standard deviation (SD)]. For each replicate, migrated monocytes were counted in 9 fields at 20X objective magnification. Please click here to view a larger version of this figure.
| Name of the Oligo | Sequence | |
| shRNA 1 (anti PAI-1 sequence 1 - 8 ) | Forward: | shPAI-1A5' CCGGCTGACTTCACGAGTCTTTCCTCGAGGAAAGACTCGTGAAGTCAGTTTTT 3' |
| Reverse: | shPAI-1B 5' AATTAAAAACTGACTTCACGAGTCTTTCCTCGAGGAAAGACTCGTGAAGTCAG 3' | |
| shRNA 2: (anti PAI-1 sequence 2) | Forward: | shPAI-1C 5'-CCGGCAGACAGTTTCAGGCTGACTTCTCGAGAAGTCAGCCTGAAACTGTCTGTTTTT-3' |
| Reverse: | shPAI-1D 5'-AATTAAAAACAGACAGTTTCAGGCTGACTTCTCGAGAAGTCAGCCTGAAACTGTCTG-3' | |
| shRNA 3 (anti PAI-1 sequence - 3 9) | Forward: | shPAI-1E 5’-CCGGAAGGATGAGATCAGCACCACACTCGAGTGTGGTGCTGATCTCATCCTTTTTTT-3’ |
| Reverse: | shPAI-1F 5’-AATTAAAAAAAGGATGAGATCAGCACCACACTCGAGTGTGGTGCTGATCTCATCCTT-3’ | |
| Scramble (scramble sequence - 4 9) | Forward: | Scramble 1 5’- CCGGAATTCTCCGAACGTGTCACGTCTCGAGACGTGACACGTTCGGAGAATTTTTTT-3’ |
| Reverse: | Scramble 2 5’-AATTAAAAAAATTCTCCGAACGTGTCACGTCTCGAGACGTGACACGTTCGGAGAATT-3’ |
Table 1. shRNA sequences. Here 3 shRNA sequences against PAI-1 were tested for the strongest silencing effect: shRNA 1, shRNA 2, shRNA 3, and scrambled
Discussion
Composed of a variety of cell types, the TME is crucial for the development of cancer. To ascertain optimal growth conditions, cancer cells attract monocytes through secretion of chemotactic factors. Here we present a protocol for studying the mechanism of monocyte recruitment by tumor cells in vitro. For this purpose, a combination of inducible gene expression system, purification of primary human monocytes, and as an example of a migration assay, the Boyden chamber assay, is used.
The first critical step in the presented protocol is to confirm the correct cloning of the shRNA sequences into the Tet-ON vector, which should always be evaluated by sequencing. Further establishment of the functional stable Tet-ON cell lines needs to be confirmed by testing the KD in the presence of doxycycline by Western blotting. The amount of applied doxycycline may vary between cell lines, and therefore it is advisable to test the effective concentrations for KD and for toxicity on the cells. A known disadvantage of tetracycline-inducible systems is their leakiness6,7. The amount of protein detected in the presence and absence of doxycycline must be measured and compared to the scrambled controls to account for this effect. In the in vitro experiments, a low level of leakiness is observed with the shRNA2 and shRNA3 KD, as is shown in Figure 2. Additionally, it is important to always use tetracycline-free serum in vitro to avoid downregulation of protein levels in control conditions. The system used in this protocol has advantages over other Tet-ON systems as it requires a single vector cloning, allows quick generation of multiple clones, and exhibits very low levels of leakiness.
A critical step in obtaining primary human monocytes from blood is obtaining good gradient separation, which relies on slow layering of the diluted blood over the density gradient. Another crucial step of the protocol is to properly estimate the number of cells before the negative selection step because it will determine the amount of antibody cocktail and magnetic beads added to the blood suspension. To insure the reproducibility of experiments, more than one blood donor should be used in performing replicate experiments. This technique is superior to existing alternatives of monocyte purification including a density gradient and adhesion-based protocol34 and a two-step procedure with single gradients21, because it warrants low lymphocyte contamination level and high purity (>95%) of obtained monocytes. Since the non-monocytic cell types are removed from PBMC mixture by negative selection, monocytes do not come in direct contact with antibodies present in the antibody cocktail, and therefore the risk of monocytes becoming activated by interaction with antibodies is minimal. Alternative applications of obtained cells may include differentiation of monocytes into macrophages through in vitro and in vivo experiments where human monocytes are injected into immunodeficient mice. Modifications of the protocol commonly include use of the red blood cell lysis buffer during the washing steps of PBMCs. This modification aims at additional removal of red blood cells by applying osmotic pressure. Limitations of the technique include a limited number of macrophages that can be obtained using a specified amount of the reagents.
The Boyden chamber migration assay is a fast and straightforward way to analyze if a secreted protein or a cell line stimulates migration of other cell types. The pitfalls of this assay include low sensitivity and not accounting for cells undergoing cell death. The ways to address those concerns would be confirming the results with a different assay like a microfluidic based assay and a time-course experiment where migration is measured over shorter periods of time. Except for small populations, mature monocytes do not proliferate in vivo35,36; however if other cells are used, a timeline shorter than their division cycle should be chosen to avoid an increase in number of cells. Alternatively, reagents blocking cell division should be used to avoid changing the originally plated cell number.
The protocol presented here can be easily adapted to studies of other proteins secreted by cancer cells and their paracrine functions. One of the possible applications of conditional KD cancer cell lines is studying the role of proteins whose KD is toxic for cells not only in vitro but also in vivo, where Tet-regulated cell lines are xenotransplanted into mice, and the KD is induced after establishment of the tumor by the addition of doxycycline to the drinking water.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to acknowledge Jacqueline Rosenberg for proofreading the manuscript. This work was supported by the US Department of Health and Human Services/National Institutes of Health with a grant to YA DeClerck (grant 5R01 CA 129377) and the TJ Martell Foundation. MH Kubala is the recipient of a Research Career Development Fellowship award of The Saban Research Institute at Children's Hospital Los Angeles.
References
- Ryding AD, Sharp MG, Mullins JJ. Conditional transgenic technologies. J Endocrinol. 2001;171(1):1–14. doi: 10.1677/joe.0.1710001. [DOI] [PubMed] [Google Scholar]
- Mantovani A, Marchesi F, Malesci A, Laghi L, Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat Rev Clin Oncol. 2017. [DOI] [PMC free article] [PubMed]
- Franklin RA, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344(6186):921–925. doi: 10.1126/science.1252510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27(4):462–472. doi: 10.1016/j.ccell.2015.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P, Allison JP. The future of immune checkpoint therapy. Science. 2015;348(6230):56–61. doi: 10.1126/science.aaa8172. [DOI] [PubMed] [Google Scholar]
- Mizuguchi H, Hayakawa T. Characteristics of adenovirus-mediated tetracycline-controllable expression system. Biochim Biophys Acta. 2001;1568(1):21–29. doi: 10.1016/s0304-4165(01)00195-7. [DOI] [PubMed] [Google Scholar]
- Meyer-Ficca ML, et al. Comparative analysis of inducible expression systems in transient transfection studies. Anal Biochem. 2004;334(1):9–19. doi: 10.1016/j.ab.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Wiederschain D, et al. Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle. 2009;8(3):498–504. doi: 10.4161/cc.8.3.7701. [DOI] [PubMed] [Google Scholar]
- Wee S, et al. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A. 2008;105(35):13057–13062. doi: 10.1073/pnas.0802655105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Stamatoyannopoulos G, Song CZ. Down-regulation of CXCR4 by inducible small interfering RNA inhibits breast cancer cell invasion in vitro. Cancer Res. 2003;63(16):4801–4804. [PubMed] [Google Scholar]
- Wiznerowicz M, Trono D. Conditional suppression of cellular genes: lentivirus vector-mediated drug-inducible RNA interference. J Virol. 2003;77(16):8957–8961. doi: 10.1128/JVI.77.16.8957-8961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solari V, et al. MYCN-dependent expression of sulfatase-2 regulates neuroblastoma cell survival. Cancer Res. 2014;74(21):5999–6009. doi: 10.1158/0008-5472.CAN-13-2513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Tang L. Inducible RNAi system and its application in novel therapeutics. Crit Rev Biotechnol. 2016;36(4):630–638. doi: 10.3109/07388551.2014.1003030. [DOI] [PubMed] [Google Scholar]
- Chen HC. Boyden chamber assay. Methods Mol Biol. 2005;294:15–22. doi: 10.1385/1-59259-860-9:015. [DOI] [PubMed] [Google Scholar]
- Jungi TW. Assay of chemotaxis by a reversible Boyden chamber eliminating cell detachment. Int Arch Allergy Appl Immunol. 1975;48(3):341–352. doi: 10.1159/000231319. [DOI] [PubMed] [Google Scholar]
- Jung HS, et al. Monoclonal antibodies against autocrine motility factor suppress gastric cancer. Oncol Lett. 2017;13(6):4925–4932. doi: 10.3892/ol.2017.6037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H, et al. Effect of Sorbus commixta on the invasion and migration of human hepatocellular carcinoma Hep3B cells. Int J Mol Med. 2017. [DOI] [PubMed]
- Schildberger A, Rossmanith E, Eichhorn T, Strassl K, Weber V. Monocytes, peripheral blood mononuclear cells, and THP-1 cells exhibit different cytokine expression patterns following stimulation with lipopolysaccharide. Mediators Inflamm. 2013;2013:697972. doi: 10.1155/2013/697972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heil TL, Volkmann KR, Wataha JC, Lockwood PE. Human peripheral blood monocytes versus THP-1 monocytes for in vitro biocompatibility testing of dental material components. J Oral Rehabil. 2002;29(5):401–407. doi: 10.1046/j.1365-2842.2002.00893.x. [DOI] [PubMed] [Google Scholar]
- Tsuchiya S, et al. Establishment and characterization of a human acute monocytic leukemia cell line (THP-1) Int J Cancer. 1980;26(2):171–176. doi: 10.1002/ijc.2910260208. [DOI] [PubMed] [Google Scholar]
- de Almeida MC, Silva AC, Barral A, Barral Netto M. A simple method for human peripheral blood monocyte isolation. Mem Inst Oswaldo Cruz. 2000;95(2):221–223. doi: 10.1590/s0074-02762000000200014. [DOI] [PubMed] [Google Scholar]
- Flo RW, et al. Negative selection of human monocytes using magnetic particles covered by anti-lymphocyte antibodies. J Immunol Methods. 1991;137(1):89–94. doi: 10.1016/0022-1759(91)90397-x. [DOI] [PubMed] [Google Scholar]
- Grondahl-Hansen J, et al. Plasminogen activator inhibitor type 1 in cytosolic tumor extracts predicts prognosis in low-risk breast cancer patients. Clin Cancer Res. 1997;3(2):233–239. [PubMed] [Google Scholar]
- Borgfeldt C, Hansson SR, Gustavsson B, Masback A, Casslen B. Dedifferentiation of serous ovarian cancer from cystic to solid tumors is associated with increased expression of mRNA for urokinase plasminogen activator (uPA), its receptor (uPAR) and its inhibitor (PAI-1) Int J Cancer. 2001;92(4):497–502. doi: 10.1002/ijc.1215. [DOI] [PubMed] [Google Scholar]
- Devy L, et al. The pro- or antiangiogenic effect of plasminogen activator inhibitor 1 is dose dependent. FASEB J. 2002;16(2):147–154. doi: 10.1096/fj.01-0552com. [DOI] [PubMed] [Google Scholar]
- Bajou K, et al. Plasminogen activator inhibitor-1 protects endothelial cells from FasL-mediated apoptosis. Cancer Cell. 2008;14(4):324–334. doi: 10.1016/j.ccr.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Wang H, Wang Z, Xiao W. Plasminogen activator inhibitor-1 promotes inflammatory process induced by cigarette smoke extraction or lipopolysaccharides in alveolar epithelial cells. Exp Lung Res. 2009;35(9):795–805. doi: 10.3109/01902140902912519. [DOI] [PubMed] [Google Scholar]
- Ji Y, et al. Pharmacological Targeting of Plasminogen Activator Inhibitor-1 Decreases Vascular Smooth Muscle Cell Migration and Neointima Formation. Arterioscler Thromb Vasc Biol. 2016;36(11):2167–2175. doi: 10.1161/ATVBAHA.116.308344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmeliet P, et al. Inhibitory role of plasminogen activator inhibitor-1 in arterial wound healing and neointima formation: a gene targeting and gene transfer study in mice. Circulation. 1997;96(9):3180–3191. doi: 10.1161/01.cir.96.9.3180. [DOI] [PubMed] [Google Scholar]
- Cao C, et al. Endocytic receptor LRP together with tPA and PAI-1 coordinates Mac-1-dependent macrophage migration. EMBO J. 2006;25(9):1860–1870. doi: 10.1038/sj.emboj.7601082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thapa B, Koo BH, Kim YH, Kwon HJ, Kim DS. Plasminogen activator inhibitor-1 regulates infiltration of macrophages into melanoma via phosphorylation of FAK-Tyr(9)(2)(5) Biochem Biophys Res Commun. 2014;450(4):1696–1701. doi: 10.1016/j.bbrc.2014.07.070. [DOI] [PubMed] [Google Scholar]
- Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol. 2006;8(8):877–884. doi: 10.1038/ncb1448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang H, Placencio VR, DeClerck YA. Protumorigenic activity of plasminogen activator inhibitor-1 through an antiapoptotic function. J Natl Cancer Inst. 2012;104(19):1470–1484. doi: 10.1093/jnci/djs377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett S, Breit SN. Variables in the isolation and culture of human monocytes that are of particular relevance to studies of HIV. J Leukoc Biol. 1994;56(3):236–240. doi: 10.1002/jlb.56.3.236. [DOI] [PubMed] [Google Scholar]
- Geissmann F, et al. Development of monocytes, macrophages, and dendritic cells. Science. 2010;327(5966):656–661. doi: 10.1126/science.1178331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer M, Goldstein M, Heylmann D, Kaina B. Human monocytes undergo excessive apoptosis following temozolomide activating the ATM/ATR pathway while dendritic cells and macrophages are resistant. PLoS One. 2012;7(6):e39956. doi: 10.1371/journal.pone.0039956. [DOI] [PMC free article] [PubMed] [Google Scholar]