

Abstract
This protocol describes a method developed to identify endogenous peptides in human cerebrospinal fluid (CSF). For this purpose, a previously developed method based on molecular weight cut-off (MWCO) filtration and mass spectrometric analysis was combined with an offline high-pH reverse phase HPLC pre-fractionation step.
Secretion into CSF is the main pathway for removal of molecules shed by cells of the central nervous system. Thus, many processes in the central nervous system are reflected in the CSF, rendering it a valuable diagnostic fluid. CSF has a complex composition, containing proteins that span a concentration range of 8 - 9 orders of magnitude. Besides proteins, previous studies have also demonstrated the presence of a large number of endogenous peptides. While less extensively studied than proteins, these may also hold potential interest as biomarkers.
Endogenous peptides were separated from the CSF protein content through MWCO filtration. By removing a majority of the protein content from the sample, it is possible to increase the sample volume studied and thereby also the total amount of the endogenous peptides. The complexity of the filtrated peptide mixture was addressed by including a reverse phase (RP) HPLC pre-fractionation step at alkaline pH prior to LC-MS analysis. The fractionation was combined with a simple concatenation scheme where 60 fractions were pooled into 12, analysis time consumption could thereby be reduced while still largely avoiding co-elution.
Automated peptide identification was performed by using three different peptide/protein identification software programs and subsequently combining the results. The different programs were complementary rather than comparable with less than 15% of the identifications overlapped between the three.
Keywords: Neuroscience, Issue 130, Cerebrospinal fluid, peptidomics, endogenous peptides, biomarkers, neurodegeneration, Alzheimer's disease, LC-MS/MS, pre-fractionation, multiplexed isobaric labelling
Introduction
Biomarkers in cerebrospinal fluid (CSF) are currently transforming research into neurodegenerative disorders. In Alzheimer's disease, the most common neurodegenerative disorder, affecting over 60 million people worldwide1,2, a biomarker triplet consisting of the peptide amyloid beta, microtubule-stabilizing protein tau, and a phosphorylated tau form, can detect the disease with high sensitivity and specificity, and has been included in the diagnostic research criteria3. In other neurodegenerative diseases, such as Parkinson's disease and Multiple Sclerosis, proteomic studies have identified numerous biomarker candidates, some of which are currently under evaluation in clinical studies4,5,6.
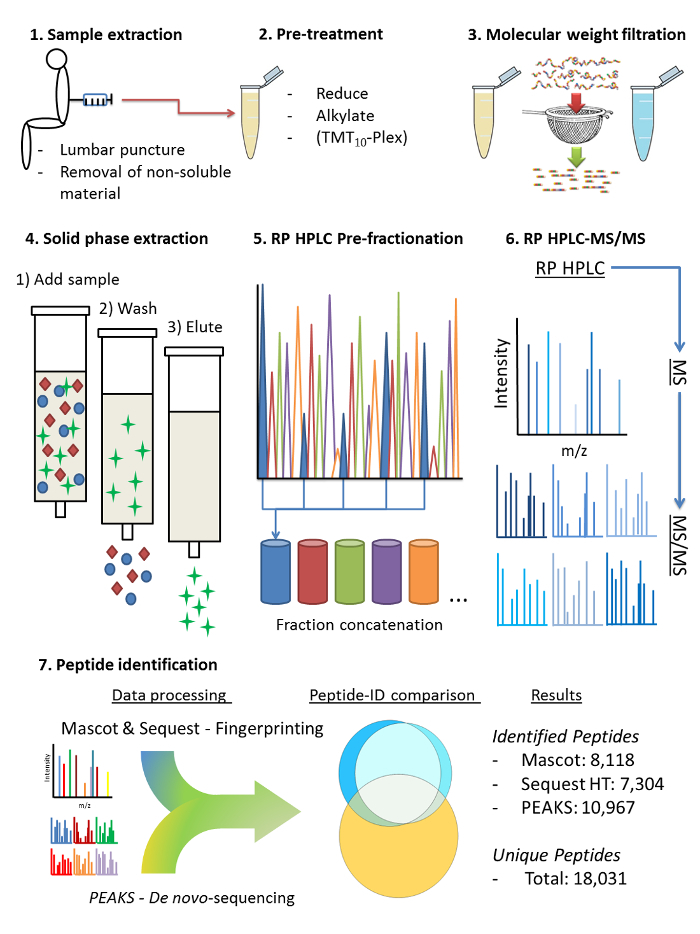
Alongside proteins, CSF also contains an abundance of endogenous peptides7,8,9,10,11,12. Constituting cleavage products of many brain-derived proteins, these peptides also represent a potentially important source of disease biomarkers. To increase the inventory of identified endogenous peptides in human CSF and enable CSF endopeptidomic analyses in clinical studies, a method was developed for sample preparation and LC-MS analysis (a brief protocol scheme has been included in Figure 1).The application of this method in a recent study resulted in the identification of nearly 16,400 endogenous CSF peptides in pooled CSF samples from several individuals of non-specific diagnosis, expanding the known CSF endopeptidome ten-fold13. The method can optionally be used in conjunction with isobaric labelling approach for quantification.
Sample Preparation
The main source of protein mass in CSF is plasma constituents (e.g. albumin and immunoglobulins) passing over the blood brain barrier14,15. Their high abundance hampers the detection of low-abundant, brain-derived sample components. Endogenous peptides can be readily separated from the high-abundant proteins, thereby allowing a significantly larger volume of CSF peptide extract to be used for LC-MS analysis, thereby enabling detection of lower-abundant peptides.
In the protocol presented here, molecular weight cut-off (MWCO) filtration was used to separate the CSF peptides from the protein fraction; a method that has been used in several previous studies8,9,10,11,12,16. The filtration step was followed by an offline RP HPLC pre-fractionation step performed over a high-pH mobile phase gradient. By performing two RP HPLC steps in tandem, with pH being the main distinction, the difference in selectivity between the two steps results mainly from altered peptide retention as a consequence of different peptide charge states. The application of high-pH peptide pre-fractionation prior to LC-MS under acidic conditions has proven efficient in increasing peptide identification17,18, and even to be superior for this purpose in complex biological samples compared to more orthogonal separation modes19, such as strong cat-ion exchange (SCX) and RP20. To shorten the analysis time, a concatenation scheme was used, pooling every 12th fraction (e.g., fractions 1, 13, 25, 37, and 49), which due to the high resolving power of RP HPLC still largely avoided co-elution of peptides from different fractions in the LC-MS step20,21.
Peptide identification
Peptide identification in peptidomic studies differs from that of proteomic studies in that no enzyme cleavage can be specified in the database search, and as a consequence, identification rates are usually lower11. A recent study13 showed that the identification rates for endogenous peptides obtained with Sequest and Mascot were substantially improved when the default scoring algorithm of the respective software program was modified using the adaptive scoring algorithm Percolator, indicating that optimal scoring algorithms for endogenous peptides differ from that of tryptic peptides13. In that study, identification based on automatic peptide de novo sequencing using the software PEAKS (BSI) was found to be complementary to the two fragment ion fingerprinting-based search engines, resulting in a significantly larger set of identified peptides.
Protocol
The protocol described below is a refined version of the one used in a previous study where a large amount of endogenous peptides were identified in human CSF15. Updates to the original protocol involve minor alterations to the chemical pre-treatment of CSF as well as optimisation of the gradient used for offline high-pH RP HPLC pre-fractionation.
Ethical considerations
All studies of Swedish patient and control materials have been approved by ethical committees: St. Göran (ref. 2005-554-31/3). CSF samples from the Amsterdam Dementia Cohort and samples collected at the National Hospital for Neurology and Neurosurgery, London were used for research with written consent from all participating patients and approval from regional ethics committees. The material here utilized mainly consisted of left-over CSF from samples taken for the purpose of diagnosis and it was de-identified before being included in our studies. There is no possibility of tracing back the sample to any individual donor or group of donors.
1. Extraction of Human Cerebrospinal Fluid (CSF):
Extract CSF through lumbar puncture (must be performed by a trained physician) using a standardized protocol22.
Remove cell debris and other non-soluble material by centrifugation at 2,500 x g for 20 min.
Visually inspect CSF samples for discoloration that may indicate blood contamination resulting from puncture bleeding. The significantly higher protein concentration in blood and the presence of proteases may greatly affect the analytical results.
2. Pre-treatment of CSF (1.5 mL Sample Volume, No Quantification):
Thaw 1.5 mL of CSF aliquots at room temperature (RT), transfer the contents to 10 mL polypropylene tubes and add 80 µL of 1 M Triethylammonium bicarbonate (TEAB) as a buffering agent.
Add 0.65 mL 8 M guanidinium hydrochloride (GdnHCl) (active concentration GdnHCl: 2.4 M) and vortex gently at RT for 10 min. NOTE: The chaotropic agent, GdnHCl, both affects solvent viscosity and interacts with the polypeptide chain, which results in protein unfolding being energetically favourable23. Thus, GdnHCl dissolves protein aggregates and increases recovery of endogenous peptides during subsequent filtration.
Add 60 µL of 200 mM of aqueous tris(2-carboxyethyl)phosphine hydrochloride (TCEP) (active concentration TCEP: 6 mM) and incubate at 55 °C for 1 h to reduce cysteine disulphides.
Add 35 µL of 400 mM iodoacetamide (IAA) (active concentration IAA: 4 mM) and incubate at RT in darkness for 30 min to alkylate cysteines. The addition of an alkyl group ensures that cysteine residues cannot spontaneously form new disulphide bridges at any point during subsequent sample preparation.
Add 3.25 mL of de-ionized water and vortex briefly to dilute the sample prior to MWCO filtration, resulting in a total sample volume of 5.5 mL. This step serves to dilute GdnHCl to below 1 M as a higher concentration has been observed to sometimes cause leaching of polymeric substances from the filter devices.
3. Pre-treatment of CSF (10 x 150 µL Sample Volume, Isobaric Labelling-quantification):
Thaw 150 µL of CSF aliquots from 10 individuals at RT and subsequently transfer the contents to individual 1.5 mL low-binding micro centrifuge tubes and add 8 µL of 1 M TEAB as a buffering agent.
Add 65 µL of 8 M GdnHCl (active concentration GdnHCl: 2.4 M) and vortex gently at RT for 10 min.
Add 6 µL of 200 mM of aqueous TCEP (active concentration TCEP: 6 mM) and incubation at 55 °C for 1 h to reduce cysteine disulphides.
Add 3.5 µL 400 mM aqueous IAA (active concentration IAA: 6 mM) and incubate at RT and darkness for 30 min to alkylate cysteines.
Prepare the isobaric labelling kit (e.g., Tandem Mass Tag 10plex isobaric labelling reagent). Allow the isobaric labelling-reagent vials to reach RT prior to opening them to avoid unnecessary reagent hydration, add 41 µL of HPLC-grade acetonitrile (AcN) and dissolve by gentle agitation for 5 min.
Transfer 30 µL of isobaric labelling-reagent solution to the corresponding sample and incubate for 1 h at RT under gentle agitation. The isobaric labelling reagent includes an NHS-ester group which reacts with the primary amines present at peptide N-termini as well as with Lysine residues.
Add 8 µL of 5% hydroxylamine (active concentration hydroxylamine: 0.16%) and shake gently at RT for 20 min to quench the labelling reaction. Since the separately labelled samples are to be combined, ensure that the labelling reaction is quenched. By addition of an abundance of amine groups in the form of hydroxylamine, the remaining isobaric labelling reagent is allowed to react and is thus rendered inert.
Combine the contents of each of the 10 individually labelled samples in a single 15 mL polypropylene tube.
Add 6.4 mL of de-ionized water to the combined sample and vortex briefly to reduce the AcN concentration from 12% to 3% and to the GdnHCl concentration GdnHCl to below 1 M as a higher concentration has been observed to sometimes cause leaching of polymeric substances from the filter devices.
4. Molecular Weight cut-off Filtration
In order to remove potential contaminants, condition the MWCO filters by loading 10 mL of aqueous 1 M GdnHCl, 25 mM TEAB and centrifuging for 15 min at 2,500 x g and RT, discard the flow-through (FT).
Load the whole sample volume on the filter (5 mL non-labelled CSF (step 2.5) or 10 mL isobarically-labelled CSF (step 3.9)) and centrifuge for 30 min at 2,500 x g and RT, leave the flow-through (FT) in the collection container. The result of the filtration is that peptides and small proteins are separated from larger proteins and residual cell debris. This step can be seen as the peptidomic equivalent to the use of proteolytic digestion of proteins to generate peptides in proteomic experiments.
Load 5 mL of 25 mM TEAB (aqueous) on the filters and centrifuge for 15 min at 2,500 x g and RT, in order to increase recovery of peptides.
Vortex the combined FTs from the two previous steps (a total 10 mL of non-labelled or 15 mL isobarically-labelled sample), and proceed to sample acidification.
5. De-salting and Sample Clean-up by Solid Phase Extraction
Acidify the filtered sample from step 4.4 prior to solid phase extraction (SPE). The acidification is performed in order to improve solute (peptide) interaction with the stationary phase of the SPE-cartridge.
For the non-labelled sample (volume 10 mL): add 550 µL of 1% Trifluoroacetic acid (TFA) - total volume of the sample: 10.55 mL with 0.05% TFA.
For the isobaric-labelled sample (volume 15 mL): add 20 mL 0.1% TFA to acidify and lower AcN concentration from 3% to 1% - total volume of the sample: 30 mL with 0.066% TFA. NOTE: TFA is also added for its function as an ion-pairing reagent, which improves retention of peptides not themselves capable of sufficiently strong hydrophobic interaction with the packing material of the SPE cartridge to be retained.
If pH >3, titrate the sample with 20% phosphoric acid until sample pH is <3.
Condition the SPE cartridges by addition of 1 mL of 84% AcN, 0.1% formic acid (FA), and discard the FT. Repeat once. Conditioning of the cartridge filter is required to remove unwanted substances which would otherwise elute along with the peptides in subsequent steps; further, conditioning increases permeability of the filter.
Equilibrate the SPE cartridge by addition of 1 mL of 0.1% TFA, discard FT. Repeat once. Ensure that the filter does not run dry after the last equilibration step - keep a small volume on top of the filter. Equilibration of the cartridge filter is performed in order to prepare the filter to retain peptides by removing the hydrophobic substance (acetonitrile) left from the conditioning step.
Load the whole sample volume (10.55 mL for non-labelled samples or 30 mL for isobarically-labelled samples), in several portions if necessary, and let the FT run into waste. Ensure that the cartridge does not run dry between sample loading rounds or after the last sample volume has been loaded - keep a small volume on top of the filter.
Pass 1 mL of 0.1% TFA over the filters to remove salts and reagents and discard the FT. Repeat once. Ensure that the cartridge does not run dry after each washing step - keep a small volume of liquid on top of the cartridge.
Place 1.5 of mL low binding micro centrifuge tubes under the cartridge and elute the sample by passing 1 mL of 84% AcN, 0.1% FA over the cartridge.
Remove the solvents from the de-salted sample by evaporation in a vacuum centrifuge run without active heating until dry, store at -80 °C or proceed immediately to high-pH fractionation.
6. Offline high-pH Reverse Phase HPLC Sample Fractionation
Prepare aqueous high-pH (HpH) mobile phases: HpH Buffer A: Pure water HpH Buffer B: 84% AcN HpH Buffer C: 25 mM NH4OH HpH Loading buffer: 2.5 mM NH4OH, 2% AcN Note: HpH Loading buffer is used as transport solution and sample buffer
Re-dissolve the sample in 16 µL HpH Loading buffer by gentle agitation for 20 min
Load 15 µL the sample on an HPLC system with an internal fraction collector for 96-deep-well plates configured according to Batth et al.20 with minor alterations. Fractionate at a flow of 100 µL/min over a pH-stable separation column (C18 3.5 µm, 2.1 mm x 250 mm) and collect one fraction per min over a linear 60 min gradient.
Use the following gradient time-points: t = 0 min, B = 1%, C = 10%; t = 4 min, B = 1%, C = 10%, start fraction collection; t = 76 min, B = 70%, C = 10%; end fraction collection; t = 76.5 min, B = 85%, C = 10%; t = 80 min, B = 85%, C = 10%; t = 80.5 min, B = 1%, C = 10% and t = 90 min, B = 1%, C = 10%.
Collect fractions repetitively in 12 wells in a circular pattern, thus concatenating fractions spaced by 12 min, resulting in 12 fractions, each containing 6 concatenated sub-fractions.
Remove the solvent from samples by vacuum centrifugation at RT and 3000 rpm until dry, and store the samples at -80 °C prior to LC-MS analysis.
7. LC-MS
Prepare aqueous mobile phases: Buffer A: 0.1% FA Buffer B: 0.1% FA, 84% AcN Loading buffer: 0.05% TFA, 2% AcN Note: Loading buffer is used as transport solution, sample buffer and mobile phase for the loading pump.
Re-dissolve each of the 12 fractions by addition of 6 µL loading buffer and shaking at RT for 20 min
Load 5 µL sample on a nano-flow HPLC, operating in trap column configuration (trap column: 75 µm x 2 cm, C18, 100 Å pore size, 3 µm particle size; separation column: C18, 75 µm x 500 mm, 100 Å pore size, 2 µm particle size, and perform peptide separation at a flow rate of 150 nL/min using the following gradient: t = 0 min, B = 2%; t = 10 min, B = 2%; t = 11 min, B = 7%; t = 100 min, B = 26%; t = 170 min, B = 45%; t = 175 min, B = 80%; t = 181 min, B = 2%, and t = 210 min, B = 2%.
- Perform MS on high-resolution hybrid mass spectrometer connected to the HPLC via a nano-ESI interface. Record full scan spectra in MS mode at a resolution setting of 120,000 (2.0e5 AGC target) over the m/z range 350 - 1,400.
- Operate the mass spectrometer in data-dependent acquisition mode, select MS/MS spectra from the top ten most intense peaks with m/z > 150 and within the intensity range 1.0e4-1.0e5 for fragment ion analysis. Isolate precursor ions using a quadrupole isolation window of 1 m/z, maximum injection time of 100 ms and the RF lens at 60%.
- Apply dynamic exclusion with an exclusion time of 15 s and an m/z tolerance of ±10 ppm. Perform fragmentation in the higher-collision energy dissociation (HCD) cell and record MS/MS acquisitions in the orbitrap at a resolution setting of 50,000 (5.0e4 AGC target value).
8. Peptide Identification
For peptide identification submit the resulting .raw-files from the mass spectrometric analysis to a proteomics search engine applying the following settings: NOTE: In our previous study15, three search engines; Mascot v2.4, Sequest HT, and PEAKS v7.5 were used in parallel and the settings here specified were employed for all three search engines, unless otherwise specified. The majority of the adjustable settings are universal for peptide/protein identification and should have corresponding settings in any given search engine. Spectrum selector: Min. precursor mass: 350 Da Max. precursor mass: 5,000 Da Scan type: Full Signal/Noise threshold: 1.5 Sequence database search Database: UniProt_SwissProt [version2015_11] Taxonomy: Homo sapiens Enzyme: none Max. missed cleavages: 0 Instrument (Mascot only): ESI-Trap Min. peptide length (SequestHT only): 6 Max. peptide length (SequestHT only): 144 Precursor mass tolerance: 15 ppm Fragment mass tolerance: 0.05 Da Static modifications: Carbamidomethyl (C); [If labelled] TMT10plex (N-Term) Dynamic modifications: Oxidation (M); [If labelled] TMT10plex (K) Peptide-spectrum match (PSM) validator Percolator (Mascot and Sequest HT only) or Decoy Fusion (PEAKS only) Target FDR: 0.01 Validation based on: q-value
Representative Results
The method described here has been applied and evaluated in three studies prior to the introduction of sample pre-fractionation (Table 1). The first study used offline LC for spotting CSF fractions on a MALDI target plate and resulted in 730 identified endogenous peptides11. In the two following studies, isobaric labelling was employed. Primarily in a case/control study for identification and characterisation of potential biomarkers in the CSF endopeptidome and proteome simultaneously24, and in the second study isobaric labelling was used to monitoring treatment effects in vivo of a γ-secretase inhibitor on the peptide expression in CSF over 36 h16. In the case/control study 437 endogenous peptides were identified, 64 of which significantly altered in concentration between individuals with AD and healthy controls. The third, treatment study, identified 1798 endogenous peptides, 11 of the monitored peptides could be shown to respond to the treatment.
In the fourth study, the aim was to increase the number of identified CSF peptides, particularly to identify lower-abundant peptides. Therefore, peptide pre-fractionation by HpH-RP chromatography was included and a 10-fold larger CSF sample volume was used, resulting in identification of 16,395 peptides. In this study, no isobaric labelling was performed. In addition to sample fractionation, the most recent study employed a combined peptide identification approach, whereas in the first three studies only a single database search was performed, which to some extent accounts for the larger number of peptides identified. Comparing the results obtained by the individual search engines (Mascot, Sequest HT or PEAKS) from the most recent study indicates that the used algorithms are to some extent complementary since a relatively small amount, less than 15% (2440), of peptides are identified by all three search engines (Figure 2). Further, the de novo-sequencing search engine PEAKS was the most efficient in identifying endogenous peptides, but more than 5,400 peptides would not have been identified if only PEAKS had been used (Figure 2). The application of several search engines on the same material has potential multiple testing issues and this was addressed with a test of identification correctness13. The acquired raw MS/MS data, as well as all results obtained in proteomic searches from the most recent trial have been made available in the PRIDE data repository via ProteomeXchange with identifier PXD004863.
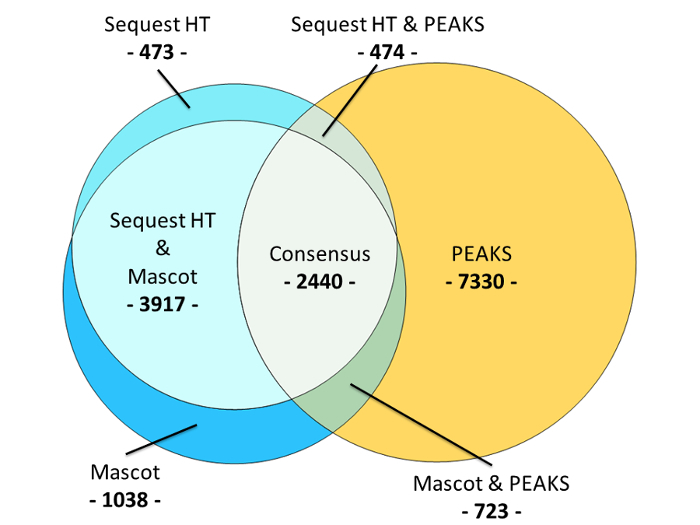
Figure 1: A protocol scheme visualizing the principal steps of the method. Extraction of cerebrospinal fluid by lumbar puncture followed by centrifugation to remove non-soluble material, 2) Addition of GdnHCl to the sample to dissociate peptide-protein aggregates, increasing the recovery of endogenous peptides; reduction and alkylation of cysteine disulphides; isobaric labelling for peptide quantification (optional) 3) molecular weight filtration to separate the endogenous peptides from the proteins, 4) solid phase extraction to remove salts and other polar contaminants, 5) RP HPLC pre-fractionation, alkaline mobile phase gradient and concatenation of every 12th fraction, 6) RP HPLC-MS/MS, acidic mobile phase gradient, each concatenated fraction run consecutively, 7) peptide identification performed by submitting MS/MS data from all 12 analysis runs as a single sample to the search engines, subsequently peptide IDs were compared and a summation of all unique peptide IDs was performed. Please click here to view a larger version of this figure.
| Study summary | TMT labelling (y/n) | HpH-RP fractionation (y/n) | Corresponding volume of CSF per MS-analysis (µl) | Number of identified peptides | Comment | Reference |
| Explorative CSF peptidome analysis | n | n | 500 | 730 | Offline LC MALDI target preparation, MALDI-MS; evaluation of MWCO filters | 4 |
| Quantitative comparison of CSF peptides; samples from 8 AD + 8 Ctrl | y | n | 200 | 437 | HPLC-ESI MS; combined peptidomic and proteomic protocol | 25 |
| AD gamma secretase inhibitor treatment study | y | n | 300 | 1798 | HPLC-ESI MS; CSF extracted at six time points after treatment | 17 |
| Expanding the CSF peptidome | n | y | 750-1000 | 18.031 | HPLC-ESI MS; combination of peptide identification softwares | 15 |
Table 1: A compilation of recent studies performed by this group which applies molecular weight filtration and mass spectrometric analysis for identification of endogenous peptides in human CSF.
Figure 2: A Venn diagram comparing the peptide identification results obtained from each of the three search engines Mascot, Sequest HT and PEAKS. A total of 16,395 endogenous peptides were identified. The de novo-sequencing search engine PEAKS identified 10,967 endogenous peptides; fragment-ion fingerprinting-based search engines Mascot and Sequest HT identified 8118 and 7304 endogenous peptides respectively. The identification consensus between all three search engines amounted to 2440 endogenous peptides, or 14.8%. There was a relatively large identification overlap between Mascot and Sequest HT, corresponding to 70% of their combined peptide identifications. PEAKS had a comparatively small identification overlap with both Mascot and Sequest HT; 20.5% and 18.9% respectively. Please click here to view a larger version of this figure.
Discussion
The introduction of an high-pH RP HPLC pre-fractionation step to a previously developed protocol for recovery of endogenous peptides by molecular weight ultrafiltration11 reduced relative sample complexity and thereby allowed for a 5-fold larger sample volume to be studied. This, in turn, increased the concentration of the subset of peptides present in each fraction and thereby improved the chances of detecting low abundant peptides.
By performing an identification strategy for endogenous peptides which employed three proteomic softwares in parallel, it was possible to expand the known CSF endopeptidome more than 10-fold. A total of 16,395 endogenous peptides were identified in a preliminary trial on a pooled CSF sample material. Among the identifications were a large number of endogenous peptides derived from proteins previously noted in the context of neurodegenerative disorders. Several peptides identified in the above studies are currently being evaluated as biomarkers in our laboratory. This process involves several steps, including verification of the peptides' identities by spiking CSF with synthetic analogues labelled with heavy isotopes, establishment of targeted mass spectrometric assays, assessing peptide stability during storage and freeze-thaw cycles, and analysing different clinical cohorts.
Modifications were made to the original protocol in order to avoid introduction of contaminants (steps 2.5 and 3.5) as a consequence of a high concentration of GdnHCl in the sample during MWCO-filtration. An update to the pre-fractionation gradient (step 6.3.1) was made, capacity prolonged, linear gradient is used.
The primary causes of analyte losses in the protocol here described can be attributed to the two RP chromatographic steps as well as the MWCO filtration and SPE sample clean up step.
The peptide losses due to interaction with the MWCO-filter, or proteins retained on it, during filtration are difficult to avoid and may be a source of inter sample variation.
Further selective losses likely arise in the RP-chromatographic steps. Since peptide hydrophobicity is pH-dependent, performing two consecutive RP chromatographic steps at high and low pH, respectively, may lead to losses of the subset of peptides that are too hydrophilic at pH ≥9 to be retained on the column and similarly a second subset too hydrophilic at pH ≤3 to be retained.
Compared to previously used methods the employment of pre-fractionation has led to a 10-fold increase in identification of endogenous peptides. It has allowed for successful detection of a large number of previously unidentified peptides and is therefore a valuable tool in the study and exploration of the CSF peptidome, and possibly other complex biological samples as well.
Combined with multiplexed isobaric labelling the protocol is intended to be further applied to identify biomarker candidates for various neurodegenerative disorders in CSF, blood and brain tissue.
Varying recovery in the sample preparation steps contributes to the analytical variation for the analysis of CSF proteins and peptides by LC-MS. Performing isobaric labelling of peptides at an early stage in the sample preparation decreases the influence of such variation greatly. Compared to previously reported sample preparation protocols, the implementation of high-pH reversed phase peptide pre-fractionation increased the number of identified peptides by a factor 5. Identification of endogenous peptides from MS/MS-data was improved significantly when combining different peptide identification software programs, employing different search algorithms.
Disclosures
No competing interests, financial or other, between authors have been reported.
Acknowledgments
Many thanks to Tanveer Batth and colleagues for advice in setting up the pre-fractionation method.
This work was supported by funding from the Swedish Research Council, the Wallström and Sjöblom Foundation, the Gun and Bertil Stohne Foundation Stiftelse, the Magnus Bergwall Foundation, the Åhlén Foundation, Alzheimerfonden, Demensförbundet, Stiftelsen för Gamla Tjänarinnor, the Knut and Alice Wallenberg Foundation, Frimurarestiftelsen, and FoU-Västra Götalandsregionen.
The main recipients of funding for this project were Kaj Blennow, Henrik Zetterberg and Johan Gobom.
References
- Wimo A, et al. The worldwide costs of dementia 2015 and comparisons with 2010. Alzheimers Dement. 2017;13(1):1–7. doi: 10.1016/j.jalz.2016.07.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scheltens P, et al. Alzheimer's disease. Lancet. 2016;388(10043):505–517. doi: 10.1016/S0140-6736(15)01124-1. [DOI] [PubMed] [Google Scholar]
- Dubois B, et al. Advancing research diagnostic criteria for Alzheimer's disease: the IWG-2 criteria. Lancet Neurol. 2014;13(6):614–629. doi: 10.1016/S1474-4422(14)70090-0. [DOI] [PubMed] [Google Scholar]
- Olsson B, et al. CSF and blood biomarkers for the diagnosis of Alzheimer's disease: a systematic review and meta-analysis. Lancet Neurol. 2016;15(7):673–684. doi: 10.1016/S1474-4422(16)00070-3. [DOI] [PubMed] [Google Scholar]
- Spellman DS, et al. Development and evaluation of a multiplexed mass spectrometry based assay for measuring candidate peptide biomarkers in Alzheimer's Disease Neuroimaging Initiative (ADNI) CSF. Proteomics Clin Appl. 2015;9(7-8):715–731. doi: 10.1002/prca.201400178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglund K, et al. Alzheimer's disease—Recent biomarker developments in relation to updated diagnostic criteria. Clin Chim Acta. 2015;449:3–8. doi: 10.1016/j.cca.2015.01.041. [DOI] [PubMed] [Google Scholar]
- Stark M, Danielsson O, Griffiths WJ, Jornvall H, Johansson J. Peptide repertoire of human cerebrospinal fluid: novel proteolytic fragments of neuroendocrine proteins. J Chromatogr B Biomed Sci Appl. 2001;754(2):357–367. doi: 10.1016/s0378-4347(00)00628-9. [DOI] [PubMed] [Google Scholar]
- Yuan X, Desiderio DM. Human cerebrospinal fluid peptidomics. J Mass Spectrom. 2005;40(2):176–181. doi: 10.1002/jms.737. [DOI] [PubMed] [Google Scholar]
- Berven FS, et al. Pre-analytical influence on the low molecular weight cerebrospinal fluid proteome. Proteomics Clin Appl. 2007;1(7):699–711. doi: 10.1002/prca.200700126. [DOI] [PubMed] [Google Scholar]
- Zougman A, et al. Integrated analysis of the cerebrospinal fluid peptidome and proteome. J Proteome Res. 2008;7(1):386–399. doi: 10.1021/pr070501k. [DOI] [PubMed] [Google Scholar]
- Holtta M, et al. Peptidome analysis of cerebrospinal fluid by LC-MALDI MS. PLoS One. 2012;7(8):e42555. doi: 10.1371/journal.pone.0042555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holtta M, et al. An integrated workflow for multiplex CSF proteomics and peptidomics-identification of candidate cerebrospinal fluid biomarkers of Alzheimer's disease. J Proteome Res. 2015;14(2):654–663. doi: 10.1021/pr501076j. [DOI] [PubMed] [Google Scholar]
- Hansson KT, et al. Expanding the cerebrospinal fluid endopeptidome. Proteomics. 2017;17(5) doi: 10.1002/pmic.201600384. [DOI] [PubMed] [Google Scholar]
- Guldbrandsen A, et al. In-depth characterization of the cerebrospinal fluid (CSF) proteome displayed through the CSF proteome resource (CSF-PR) Mol Cell Proteomics. 2014;13(11):3152–3163. doi: 10.1074/mcp.M114.038554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroksveen AC, Opsahl JA, Aye TT, Ulvik RJ, Berven FS. Proteomics of human cerebrospinal fluid: discovery and verification of biomarker candidates in neurodegenerative diseases using quantitative proteomics. J Proteomics. 2011;74(4):371–388. doi: 10.1016/j.jprot.2010.11.010. [DOI] [PubMed] [Google Scholar]
- Hölttä M, et al. A single dose of the γ-secretase inhibitor semagacestat alters the cerebrospinal fluid peptidome in humans. Alzheimers Res Ther. 2016;8(1):11. doi: 10.1186/s13195-016-0178-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Z, Tang HY, Wang H, Liu Q, Speicher DW. Systematic Comparison of Fractionation Methods for In-depth Analysis of Plasma Proteomes. J Proteome Res. 2012;11(6):3090–3100. doi: 10.1021/pr201068b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu CW, Chang CL, Chen SF. Evaluation of peptide fractionation strategies used in proteome analysis. J Sep Sci. 2012;35(23):3293–3301. doi: 10.1002/jssc.201200631. [DOI] [PubMed] [Google Scholar]
- Gilar M, Olivova P, Daly AE, Gebler JC. Orthogonality of separation in two-dimensional liquid chromatography. Anal Chem. 2005;77(19):6426–6434. doi: 10.1021/ac050923i. [DOI] [PubMed] [Google Scholar]
- Batth TS, Francavilla C, Olsen JV. Off-line high-pH reversed-phase fractionation for in-depth phosphoproteomics. J Proteome Res. 2014;13(12):6176–6186. doi: 10.1021/pr500893m. [DOI] [PubMed] [Google Scholar]
- Yang F, Shen Y, Camp DG, 2nd, Smith RD. High-pH reversed-phase chromatography with fraction concatenation for 2D proteomic analysis. Expert Rev Proteomics. 2012;9(2):129–134. doi: 10.1586/epr.12.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6(3):131–144. doi: 10.1038/nrneurol.2010.4. [DOI] [PubMed] [Google Scholar]
- Moglich A, Krieger F, Kiefhaber T. Molecular basis for the effect of urea and guanidinium chloride on the dynamics of unfolded polypeptide chains. J Mol Biol. 2005;345(1):153–162. doi: 10.1016/j.jmb.2004.10.036. [DOI] [PubMed] [Google Scholar]
- Hölttä M, et al. An Integrated Workflow for Multiplex CSF Proteomics and Peptidomics Identification of Candidate Cerebrospinal Fluid Biomarkers of Alzheimer’s Disease. J Proteome Res. 2014;14(2):654–663. doi: 10.1021/pr501076j. [DOI] [PubMed] [Google Scholar]