

Abstract
The popularity of the CRISPR/Cas9 system for both genome and epigenome engineering stems from its simplicity and adaptability. An effector (the Cas9 nuclease or a nuclease-dead dCas9 fusion protein) is targeted to a specific site in the genome by a small synthetic RNA known as the guide RNA, or gRNA. The bipartite nature of the CRISPR system enables its use in screening approaches since plasmid libraries containing expression cassettes of thousands of individual gRNAs can be used to interrogate many different sites in a single experiment.
To date, gRNA sequences for the construction of libraries have been almost exclusively generated by oligonucleotide synthesis, which limits the achievable complexity of sequences in the library and is relatively cost-intensive. Here, a detailed protocol for CORALINA (comprehensive gRNA library generation through controlled nuclease activity), a simple and cost-effective method for the generation of highly complex gRNA libraries based on enzymatic digestion of input DNA, is described. Since CORALINA libraries can be generated from any source of DNA, plenty of options for customization exist, enabling a large variety of CRISPR-based screens.
Keywords: Genetics, Issue 130, gRNA library, CRISPR, Cas9, nuclease, library generation, genome-wide
Introduction
The adaptation of the bacterial CRISPR/Cas9 system as a molecular targeting tool caused the most recent revolution in molecular biology. Never before has it been so easy to manipulate chromatin at defined genomic locations. Common applications of CRISPR include targeted gene mutations1, genome engineering2, epigenome editing3, transcriptional activation and gene silencing4. One particular advantage of the CRISPR system is that its applications are not limited to well-studied candidate sites, as gRNA libraries make less biased screens possible. These facilitate the discovery of functional loci in the genome without any prior experimental knowledge. However, gRNA library construction is currently mostly based on oligo-nucleotide synthesis, and there are limited options to purchase gRNA libraries that are not of human or mouse origin or target regions outside open reading frames. Thus, although CRISPR screens have already proven incredibly potent5,6,7,8, their full potential has not yet been exploited.
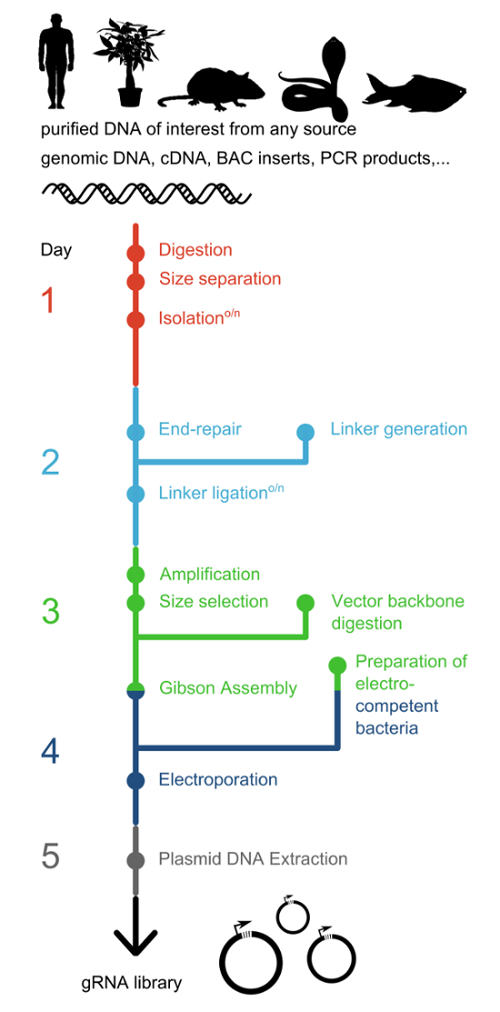
To overcome the limitation of classical gRNA generation methods two strategies have recently been developed. Both are based on controlled enzymatic digestion of target DNA rather than relying on custom oligonucleotide synthesis. While CORALINA9 employs micrococcal nuclease, the only currently available alternative method, CRISPR-EATING10, makes use of restriction enzymes (HpaII, ScrFI, BfaI and MmeI). Importantly, both techniques can be applied to any input DNA, which serves as the source of gRNA protospacer sequences. While the CRISPR-EATING method employs a strategy to decrease the number of cloned gRNAs whose targeting sites are not followed by the required S.pyogenes PAM (protospacer adjacent motive), it generates only a small fraction of all possible functional gRNAs for a given region. CORALINA, on the other hand, is able to generate all potential gRNAs for the source sequence, but also incorporates a higher fraction of non-functional guides. gRNA library generation through controlled nuclease activity enables the production of comprehensive gRNA libraries for any species, any Cas9-protein or -effector system in a simple and cost-effective manner. Moreover, CORALINA is adaptable to customization, as appropriate input and vector choices define the library type, size and content. Here, a detailed protocol is presented that can be used for the generation of comprehensive gRNA libraries from diverse sources of DNA (Figure 1), including bacterial artificial chromosomes (BACs) or genomic DNA9. The representative results accompanying this protocol were derived by applying the CORALINA protocol to BAC DNA.
Protocol
1. Digestion of DNA with Micrococcal Nuclease
Perform an optimization reaction for each new batch of micrococcal nuclease enzyme (MNase). NOTE: The number of units of MNase used should be tested (using a serial dilution, Figure 2A). Usually, 5-10 U of MNase digest 1 µg of purified genomic or BAC DNA down to a range of 5-100 bp with the conditions described below.
Per reaction, set up 1 µL of 10x MNase Buffer, 1x bovine serum albumin (BSA), 1 µg of target DNA, 1 µL of MNase (0.1-50 units) in a 10 µL reaction volume.
Incubate at 37 °C for 15 min.
Immediately inactivate the enzyme by adding 1 µL of 500 mM ethylene glycol-bis(β-aminoethyl ether)-N,N,N',N'-tetraacetic acid (EGTA).

2. Separation of DNA Fragments Using Polyacrylamide Gel Electrophoresis (PAGE)
Add sample buffer/gel loading dye to DNA samples and load onto a 20% PAGE gel. Load an appropriate DNA ladder for sizing (5 bp DNA Ladder). Do not overload the gel (1 µg DNA per well).
Run gels in appropriate 1x TRIS-Borate-EDTA (TBE) running buffer at 150 V (constant) for around 1.5 h or until the lower dye front (bromophenol blue, dark blue color) that travels at around 15 bp, reaches the lower end of the gel.
Stain the gel using an ultra-sensitive nucleic acid stain and visualize under UV light.
From the gel image, determine the optimum concentration of MNase for digesting DNA down to 20-30 bp in size.
Use the optimized concentration of MNase to digest the target DNA by repeating steps 1.2-2.2. Usually, setting up 10-12 reactions (i.e. 10-12 µg of total starting material) will yield enough digested DNA following PAGE gel extraction to proceed to subsequent steps.
Using a sterile scalpel, cut the gel next to the marker lane and stain only the part of the gel containing the ladder with fresh 1x TBE running buffer containing 1X of an ultra-sensitive nucleic acid stain. Visualize the DNA ladder and excise MNase-digested DNA fragments in the size range between approximately 18-30 bp using a razor blade. NOTE: Avoid exposing the MNase digested fragments to UV light. It is possible to use a blue light source (instead of UV) or take an image of the ladder under UV, print it to scale and use this to guide excision of MNase-digested DNA fragments). This step avoids staining and exposure of DNA fragments to UV light. Always use an unused, sterile disposable scalpel or razor blade for this step to avoid contamination.
Transfer the gel slice to a microcentrifuge tube.
Stain the remainder of the gel with nucleic acid stain as above, expose to UV light and record an image to keep a record of the gel excision step.
3. Isolation of DNA Fragments from PAGE-gels Using the Crush and Soak Method
NOTE: This step has been adopted from Sambrook et al.11
Prepare PAGE gel solubilization buffer (1.88 mL of 4 M ammonium acetate, 150 µL of 1 M magnesium acetate, 30 µL of 0.5 M ethylenediaminetetraacetic acid (EDTA) (pH 8) in ultrapure H2O to a total volume of 15 mL).
Crush the excised gel slice against the wall of the micro-centrifuge tube using a sterile pipette tip.
Add 2 gel volumes of PAGE solubilization buffer and incubate at 37 °C for 16 h on a rotating platform.
Centrifuge the samples for 1 min at maximum speed in a microcentrifuge. Transfer the supernatant to a new microcentrifuge tube, taking care not to transfer any crushed gel pieces.
Add 0.5 volumes of PAGE solubilization buffer to the gel pellet, vortex, and repeat the centrifugation (step 3.4). Combine the supernatants.
- Extract the DNA fragments using standard phenol-chloroform extraction. CAUTION: Phenol is toxic if it comes into contact with skin or if swallowed. Safety precautions such as gloves, protective eyewear, a lab coat, and working in a fume hood are critical. Dispose of all phenol-containing waste according to the institute's regulations.
- Add one volume of phenol:chloroform:isoamyl alcohol (25:24:1) to the sample and vortex thoroughly.
- Centrifuge for 10 min at maximum speed (16,000 x g) in a tabletop centrifuge at room temperature. Carefully transfer the upper aqueous phase to a fresh microcentrifuge tube. Take care not to carry over any phenol during pipetting.
- Repeat steps 3.6.1 and 3.6.2 once.
- Add a small amount (0.2 µL) of glycogen, 0.1 volumes of 3 M sodium acetate (pH 5.2) and 2 volumes of 100% ethanol (EtOH).
- Vortex and incubate at -20 °C for several hours or overnight.
- Centrifuge for 30 min at maximum speed at 4 °C using a tabletop centrifuge.
- Carefully remove the supernatant and wash the DNA pellet with 70% EtOH.
- Centrifuge for 30 min at maximum speed at 4 °C using a tabletop centrifuge.
- Remove the supernatant and air-dry the DNA pellet. Make sure all the EtOH has evaporated but be careful not to over-dry the pellet.
- Dissolve DNA pellet in 12 µL of H2O. NOTE: PAGE gel extraction is very inefficient. For every 10 µg of starting DNA digested with MNase, expect to recover 1-20 ng of purified fragments after gel extraction. Control amount and integrity of fragments by loading 1/6th (2 µL) on a PAGE gel (Figure 2C).
4. End Repair of MNase-digested, Gel-purified Fragments
Set up the following reaction using a DNA blunting kit: 10 µL of purified DNA from step 3.6.10, 1.5 µL of 10X blunting buffer, 1.5 µL of 1 mM dNTP mix, 0.6 µL of blunting enzyme, 1.4 µL of H2O.
Incubate at 22 °C for 30 min, and then heat-inactivate the enzyme by incubation at 70 °C for 10 min.
Add 85 µL of H2O and perform a reaction clean-up using standard phenol/chloroform- extraction and EtOH-precipitation (as described in section 3.6). Proceed immediately to linker ligation.
5. Linker Generation
NOTE: Linkers need to be amplified in parallel with section 3 to be able to proceed immediately with linker ligation. Primer sequences used below must be appropriate for the chosen gRNA expression vector. Those presented here have been designed for the vector pgRNA-pLKO.1.9 For amplification of the 5' linker from pgRNA-pLKO.1, use the primer sequences 5'-linker-F (TTGGAATCACACGACCTGGA) and 5'-linker-R (CGGTGTTTCGTCCTTTCCAC), yielding a 689 bp amplicon. For amplification of the 3' linker from pgRNA-pLKO.1, use the primers 3'-linker-F: (GTTTTAGAGCTAGAAATAGCAAGTTAAAATA) and 3'-linker-R: (ACTCGGTCATGGTAAGCTCC), which yield an 848 bp amplicon.
PCR-amplify the adapter sequences from the gRNA expression vector (using reagents of choice and custom primer sequences, if necessary). For pgRNA-pLKO.1 set up the following 50 µL PCR reaction: 25 µL of PCR master mix, 2.5 µL of primer F (10 µM), 2.5 µL of primer R (10 µM), 0.1 ng of gRNA expression vector (pgRNA-pLKO.1) in H2O.
Incubate reactions on a thermocycler using the following conditions: 1 cycle 98 °C for 30 s, 32 cycles 98 °C for 10 s, 59 °C for 10 s, 72 °C for 30 s, 1 cycle 72 °C for 10 min.
Purify PCR reactions using solid phase reversible immobilization beads according to the manufacturer's instructions. Elute in 30 µL of H2O.
- To enforce directional ligation of the linker to the end-repaired, MNase-digested DNA fragments, digest linkers with appropriate restriction enzymes (here HindIII and SacII). Set up the following reactions:
- For digestion of 5' linker with HindIII, use 30 µL of purified 5' linker amplicon from step 5.3., 5 µL of Buffer, 3 µL of HindIII (20U/µL), and 12 µL of H2O.
- For digestion of 3' linker with SacII, use 30 µL of purified 3' linker amplicon from step 5.3., 5 µL of Buffer, 3 µL of SacII (20 U/µL), and 12 µL of H2O.
Incubate digests at 37 °C for 3 h.
Add DNA gel loading dye and run the restriction enzyme digests on a 1% agarose gel. Excise the bands at 637 bp (5' linker digest) and 295 bp (3' linker digest).
Purify the DNA from the excised gel pieces using a gel extraction kit.
6. Linker Ligation and Amplification of Inserts
- Set up a 14 µL ligation reaction using equimolar amounts of MNase-digested, end-repaired fragments and linker sequences. NOTE: Linker to fragment ratios may be optimized. It is recommended to include a no-fragment control (NFC) reaction.
- Use 5 ng of MNase-digested fragments (end-repaired and purified), 120 ng of 5'linker (HindIII digest, purified), 55 ng of 3' linker (SacII digest, purified), 1.4 µL of T4 ligase Buffer, and 1.4 µL of concentrated T4 DNA ligase in H2O.
Incubate the ligation reactions at 16 °C for 16 h. Do not heat-inactivate the enzyme. Proceed immediately to nick-translation. NOTE: The end-repaired MNase-digested fragments provide the 5' phosphates necessary for ligation of linkers, as the linker themselves are un-phosphorylated.
To the ligation reaction (and the NFC reaction) add the following: 25 µL of Taq 2X master mix (capable of nick translation), 2.5 µL of primer Linker-Minus450-F (10 µM, GGGCAAGTTTGTGGAATTGG), 2.5 µL of primer Linker-Plus275-R (10 µM, AAGTGGATCTCTGCTGTCCC) and 6 µL of H2O.
Include a no-template control (NTC). Incubate reactions on a thermocycler using the following conditions: 1 cycle (nick translation): 72 °C for 20 min, 1 cycle: 95 °C for 5 min, 3-4 cycles: 95 °C for 15 s, 58 °C for 15 s, 72 °C for 30 s, 1 cycle: 72 °C for 5 min.
Perform a reaction clean-up using solid phase reversible immobilization beads with a sample to bead ratio of 1:1. Elute in 40 µL of H2O.
- Further amplify the desired fragment (5' linker + MNase fragment +3' linker) using appropriate PCR reagents and the following primers:
- Use 12.5 µL of PCR master mix, 1.25 µL of primer Linker-Minus450-F (10 µM, GGGCAAGTTTGTGGAATTGG), 1.25 µL of primer Linker-Plus275-R (10 µM, AAGTGGATCTCTGCTGTCCC), 2.5 µL of purified nick translation product from step 6.4. and 7.5 µL of H2O. Use the following conditions: 1 cycle: 98 °C for 30 s, 10-16 cycles: 98 °C for 10 s, 63° C for 10 s, 72° C for 15 s, 1 cycle: 72 °C for 10 min. NOTE: If necessary, several reactions can be set up in parallel to ensure there is enough PCR product for subsequent steps. To visualize small amounts of the PCR product on an agarose gel, set up additional reactions as in step 6.5 and increase the cycle number from 15 to 32. This reaction can be used as quality control, if amplicons are not visible after 15 cycles. Include also a no template control (NTC).
7. Size Selection
NOTE: This step separates MNase-fragments with the correctly attached 5' and 3' linker from fragments with two 5' or two 3' linkers based on size.
Combine all 15-cycle PCR reactions from step 6.5., add DNA loading dye and run on a 0.8% agarose gel. Excise the prominent band at 869 bp and purify DNA using a gel extraction kit.
Quantify the amount of DNA (e.g. using a spectrophotometer).
8. Cloning of PCR-amplified Fragments into the gRNA Expression Vector by Gibson Assembly
- Prepare assembly master mix as follows: NOTE: The following steps are adapted from Gibson et al.12.
- Create 6 mL isothermal reaction buffer by combining 3 mL of 1 M Tris(hydroxymethyl)aminomethane (Tris)-HCl pH 7.5, 300 µL of 1 M MgCl, 60 µL of 100 mM deoxyguanosine triphosphate (dGTP), 60 µL of 100 mM deoxyadenosine triphosphate (dATP), 60 µL of 100 mM deoxythymidine triphosphate (dTTP), 60 µL of 100 mM deoxycytidine triphosphate (dCTP), 300 µL of 1 M dithiothreitol (DTT), 1.5 g of polyethylene glycol (PEG)-8000, 300 µL of 100 mM nicotinamide adenine dinucleotide (NAD) in ultrapure H2O. NOTE: This buffer can be aliquoted and stored at -20 °C.
- Create 1.2 mL assembly master mix by combining 320 µL of 5X isothermal reaction buffer, 3 µL of 10 U/µL T5 exonuclease, 20 µL of 2 U/µL DNA polymerase, 160 µL of 40 U/µL DNA ligase in ultrapure H2O. Use 15 µL of assembly master mix with 5 µL of insert. NOTE: The assembly master mix can be aliquoted and stored at -20 °C, where it is stable for more than a year and can tolerate multiple freeze-thaw cycles. The chosen amount of T5 exonuclease is ideal for use with long overhangs.
- Vector backbone digestion NOTE: Make sure to digest a sufficient amount of vector as input for the desired number of assembly reactions in step 8.3.
- Per reaction, add the following: 1.5 µg of gRNA expression vector (pgRNA-pLKO.1), 5 µL of buffer, 1.5 µL of AgeI (5U/µL), and 38.5 µL of H2O. Incubate digest at 37 °C for 2 h.
- Dephosphorylation of the linearized vector. NOTE: This step is advised, but not strictly necessary. T5 exonuclease in the master mix will mostly remove the AgeI overhangs before the Taq DNA ligase has had a chance to act. Therefore, excessive re-ligation of the vector is not expected.
- Add 2.5 µL of shrimp alkaline phosphatase enzyme (rSAP, 1 U/µL).
- Incubate at 37 °C for 30 min, and then inactivate the enzyme by incubating at 65 °C for 5 min.
Perform DNA purification NOTE: It is recommended to perform an agarose gel extraction step. Alternatively, column- or bead purification can be used. It is important to check that the vector digestion is complete, e.g. by agarose gel electrophoresis. For comparison, undigested vector should be run in parallel.
Quantify the amount of purified digested vector in the sample.
Set up the assembly with 2-fold molar excess of inserts to vector. Per 20 µL reaction use 100 ng of vector (AgeI digest from step 8.5), 12.2 ng of insert (from step 7.1.), 15 µL of assembly master mix from step 8.1.2. in H2O.
Incubate at 50 °C for 1 h.
Purify the reactions using column purification. Resuspend the DNA in 75 µL of H2O (or appropriate volume depending on scale of electroporation). NOTE: The transformation efficiency is greatly dependent on DNA purity. Additional purification steps (e.g. phenol/chloroform extraction) might improve efficiency.
9. Preparation of Electro-competent TG1 E. coli Cells
Ensure that all centrifuge bottles and flasks are free from detergents by rinsing and subsequent filling with distilled water before autoclaving. NOTE: This step helps to remove any impurities which may affect transformation efficiency. Water should be discarded immediately before use. Alternatively, use of disposable centrifugation bottles might be advisable.
Ensure that centrifuge bottles, tubes and solutions used for the preparation of electrocompetent cells are chilled on ice prior to use. It is best to conduct the following steps in a cold room to minimize temperature fluctuations which may affect the transformation efficiency.
Prepare 2TY medium. To 16 g of bacto tryptone, 10 g of yeast extract, and 5 g of NaCl, add distilled water to 1 L, mix and autoclave. Store medium at room temperature.
Prepare 2TY-agar coated bioassay dishes and 10 cm Petri dishes containing the appropriate antibiotic. Store plates at 4 °C. NOTE: The choice of antibiotic depends on the nature of the gRNA expression vector, use 100 µg/mL ampicillin for pgRNA-pLKO.1.
Scrape from a glycerol stock of TG1 cells to inoculate 10 mL of 2TY medium (without antibiotics).
Incubate culture at 37 °C overnight (~16 h) with shaking at 225 rpm.
Inoculate 1 L of 2TY medium (without antibiotics) with the 10 mL of overnight culture (1/100 dilution) and divide it equally between two 2 L flasks (containing baffles).
Incubate culture at 37 °C, 225 rpm until an OD600 nm of 0.55 is reached (approximately after 1.5 - 2 h). Use a spectrophotometer to check OD600 nm regularly.
Chill cultures on ice for 30 min.
Split the culture equally between four 500 mL centrifuge bottles (pre-chilled on ice).
Centrifuge for 15 min at 4,000 x g at 4 °C in a pre-chilled centrifuge.
Decant supernatants and add 1 volume (i.e. 250 mL) of pre-chilled ice-cold sterile distilled H2O to each of the centrifuge bottles. Resuspend the bacterial pellet by swirling or inverting the bottle (or by gentle pipetting, if necessary). NOTE: It is easier to resuspend the pellet by first adding a small volume of water. Ensure the pellet is completely resuspended eventually.
Centrifuge for 15 min at 4,000 x g at 4 °C.
Repeat the wash two times (steps 9.12. and 9.13). Remove the supernatant. Be careful when decanting as the bacterial pellet becomes increasingly loose after washing.
Resuspend the pellet in 50 mL of sterile, ice-cold 10% glycerol and transfer it to a pre- chilled 50 mL centrifuge tube.
Centrifuge cells for 15 min at ~4,000 x g at 4 °C. Carefully remove the supernatant.
Gently resuspend the bacteria in 2 mL of ice-cold sterile 10% glycerol.
Keep on ice if the cells are to be used immediately for electroporation. NOTE: The cells can be frozen in aliquots of 50 µL in 0.5 mL tubes in a dry-ice ethanol bath and stored at -80 °C, but this is not recommended.
10. Electroporation of TG1 Electrocompetent E. coli Cells
NOTE: Electroporation is one of the bottlenecks in comprehensive library generation. To preserve the library representation, it is recommended to conduct as many individual electroporation reactions as necessary/practicable and to perform the quality control steps described below (10.6. and 10.8.).
Aliquot the purified reactions (from step 8.8) into sterile and pre-chilled PCR tubes and keep them on ice (1 µL per tube). Chill 1 mm gap electroporation cuvettes on ice.
Add 25 µL of freshly prepared TG1 cells directly to one aliquot of DNA and immediately transfer the mixture into an electroporation cuvette. Flick or tap the cuvette to ensure the cells/DNA mix is distributed along the length of the cuvette chamber (without any trapped air or bubbles).
Place the cuvette in the slide chamber and start the appropriate electroporation program (e.g. 1 pulse of 1.8 kV (EC1)). NOTE: The time constant should lie between 5.7 and 6.0 ms. In case the electroporator arcs, flick the cuvette, ensure there are no air bubbles in the chamber and try again.
Immediately add 975 µL of room temperature 2TY medium to the cuvette.
Using a transfer pipette, move the electroporated bacteria to a 50 mL tube. Repeat from step 10.2. and collect all cells transformed with one library in one 50 mL tube.
Document the total volume.
- Quality control step
- To quantify the competence of the freshly generated electro-competent cells, perform a separate electroporation reaction using a defined quantity of uncut plasmid DNA (e.g. 10 pg pUC19 control plasmid). Transfer this to a 1.5 mL microfuge tube. NOTE: The competency should be at least 1010 colony-forming units (cfu) per µg DNA. Freshly prepared cells usually perform better than this.
Incubate transformed bacteria at 37 °C for 60 min shaking at 225 rpm.
- Quality control step:
- Plate a defined small amount of bacteria transformed with the library (e.g. 10 µL of a 10-100 fold dilution) on a 10 cm agar plate with the appropriate antibiotic selection. NOTE: Knowing the total culture volume (from step 10.6.), the obtained colony number can be used to estimate the total number of colonies for the entire library. 20-30 fold representation of the library should ideally be maintained.
Disperse the remainder of the bacteria onto 2TY agar coated bioassay dishes containing the appropriate antibiotic selection. NOTE: The volume of the culture can be reduced by centrifugation at ~4,000 x g for 10 min (or until a visible pellet has formed and the supernatant appears clear). This reduces the time plates need to dry after spreading of the culture. Use of plates rather than liquid culture minimizes disproportionate growth of individual colonies.13
Spread an appropriate volume of the pUC19 control electroporation reaction on an additional 10 cm agar dish with appropriate antibiotic selection.
Incubate agar plates overnight (16 h) at 37 °C.
Count obtained colonies on the control plates (pUC19 and control library plate) to estimate CFU/µg DNA for the bacteria as well as library complexity.
11. Extraction of Plasmid DNA
Add 10 mL of 2-TY media to the overnight plates, scrape the bacterial layer off the plate using a disposable spreader and collect it in a 50 mL tube. Repeat a few times until all of the plate appears clean.
Extract DNA using a Plasmid Maxi kit (2-3 columns needed per Bio-assay plate).
Representative Results
Using the protocol at hand, CORALINA gRNA libraries have been generated from human and mouse genomic DNA9 and BAC DNA (Figure 1). To produce fragments of input DNA suitable for cloning into gRNA expression vectors, optimal conditions for controlled nuclease digestion have to be determined. A typical result for the optimization of micrococcal nuclease digestion is depicted in Figure 2A. Insufficient amount of nuclease (0.1, 2, 3, 4, 4.5 or 5 units) produces no noticeable products in the required size range (10-100 bp) and 5.5-7.5 units still produced fragments that are on average too long. Larger amounts of enzyme (50 units) lead to excessive degradation of input DNA after 10 min. Consequently, an intermediate amount was chosen (10 units). The digest was scaled up to produce enough digested fragments for subsequent purification and cloning (Figure 2B). While it is recommended to blindly select DNA fragments by size and only rely on the DNA ladder for orientation to minimize exposure of DNA fragments to UV light, gels can be stained afterwards for quality control of digestion and cutting. Figure 2B shows a representative example of a PAGE gel from which DNA fragments between 20 and 30 bp have been excised. Gel purified MNase fragments were loaded onto a 20% PAGE gel to check successful size selection and purification of MNase-digested fragments (Figure 2C). The protocol at hand is compatible with the use of customized linker sequences, allowing to clone the MNase-digested fragments into gRNA expression vectors of choice. Here, gRNA-PLKO9 was used as backbone. The linkers are amplified from the gRNA expression vector using standard PCR. Figure 2D depicts a representative example of amplified linker sequences devoid of additional, incorrect or no template amplicons. Next, linker amplicons are digested with restriction enzymes to ensure linkers are ligated onto the MNase-digested fragments in the correct orientation. Figure 2D shows agarose gels of 5' and 3' linkers before and after digestion with HindIII and SacII respectively, indicating complete digestion of the linkers to the predicted 637 and 295 bp. The right-hand portion of the gel image documents the excision of the digested linker fragments. Following gel extraction of digested linkers, the next step in the protocol is the ligation of linkers to the end-repaired MNase-digested fragments. Because linker sequences are generated by PCR using unphosphorylated primers, self-ligation of linkers should not occur. Only the end-repaired MNase-digested DNA fragments provide the phosphate groups necessary for ligation. Following nick translation, the ligation product is amplified by PCR. In order to avoid excessive PCR amplification bias that could skew the representation of gRNA sequences in the library, amplification is limited to less than 20 cycles in total. Following PCR, the amplification products are difficult to visualize on agarose gels. Separate control PCRs with 32 cycles are therefore performed to detect the products (but are not used for library preparation). Results from this control PCR are shown in Figure 2E. This allows to optimize the ligation reactions and to ensure reactions are devoid of PCR artefacts, which sometimes occur in "no fragments controls" (NFC). Figure 2E shows the desired amplicon (5' linker + DNA fragment + 3' linker, length: 869 bp) following amplification of ligation reactions using equimolar (1:1) ratios between fragments and linker sequences.
Figure 1: Suggested timeline for preparing a gRNA library. CORALINA offers a simple and cost-efficient strategy for the generation of comprehensive gRNA libraries from a plethora of different DNA sources from any organism. The protocol at hand can be brought to completion during one working week. Linker generation can be performed in parallel with DNA end-repair. Preparation of electrocompetent bacteria takes two days and includes an overnight growth step and should therefore be started before assembly reactions are set up. Please click here to view a larger version of this figure.
Figure 2: Critical steps during the protocol.(A) Controlled digestion of BAC DNA enables generation of fragments of different sizes. Shown here is the optimization of MNase digestion. Purified BAC DNA was treated with different amounts of MNase for 10 min. 10 U of MNase generate DNA fragments of the desired length (20-30 bp). (B) Size selection of fragments between 20 and 30 bp using excision from polyacrylamide gels. Purified BAC DNA was treated with 10 U of MNase for 10 min. The image was recorded following excision. (C) Quality control of gel-purified fragments. After gel-purification, 1/6th of the purified MNase fragments was loaded onto a 20% PAGE gel to check successful size selection and purification. (D) Amplification of linker sequences for assembly and restriction enzyme digestion of linkers to ensure directional cloning. 5' and 3' linkers were amplified and cut with HindIII and SacII, respectively. No-template controls (NTC) were included to control for PCR artefacts and DNA contamination. Left: analytical sample application; right: preparative sample application. Image was recorded after gel excision. (E) Successful ligation of linkers to DNA fragments can be analyzed using PCR with an increased number of PCR cycles (32) and controlled by performing no template controls with H2O (NTC) or using the NTC from the previous nick translation step as input (NTC NT)). It is important to include a no fragment control (NFC), which is an amplification from a ligation and nick translation reaction from which the MNase fragments were omitted. Only samples in which MNase fragments have been combined with linker DNA produce the expected amplicon (869 bp). Please click here to view a larger version of this figure.
Discussion
CORALINA can be used to generate large scale gRNA libraries by controlled nuclease digestion of target DNA and bulk cloning of resulting double stranded fragments. Statistical inference indicates that many more than 107 individual gRNA sequences have already been successfully cloned using the protocol at hand9. CORALINA can be customized in multiple ways. The choice of template DNA defines the target region and the maximal complexity of the generated library. Using this protocol, CORALINA libraries have previously been generated from human and mouse genomic DNA9. Representative results presented here depict the generation of a CORALINA library from purified BAC DNA. Further customization can be achieved by the choice of gRNA expression vector and linker sequences. We have previously tested three different pairs of linker lengths for Gibson assembly with little variations in efficiency9.
Due to their origin from bulk digested DNA, protospacer of CORALINA gRNAs are usually not exactly 20 bp in length, but show a length distribution with a mean that depends both on the parameters of the MNase digestion as well as the size of the excision made from the PAGE gels. The representative example shown in Figure 2B and C, depicts fragments with a median length between 19 and 27 bp. In our experience, the length of the fragments is faithfully preserved by the generated gRNA protospacer9. While fragments shorter than 20 bp should be avoided due to higher off-target rate of resulting gRNAs, longer fragments are likely much less of a problem for downstream applications, since it has been demonstrated that gRNAs with protospacers as long as 45 bp are still functional9.
The two most critical steps in the CORALINA protocol are the size selection of MNase-digested fragments and cloning steps. Generation of fragments that are too short (e.g. average below 18 bp) or incorporation of too many empty gRNA expression vectors will render the library useless. Thus, it is important to optimize the MNase digestion step (Figure 2A), to monitor excision (Figure 2B, C), check for complete digestion of the gRNA vector backbone and including no fragment controls throughout the protocol. Special care has also to be taken to preserve the representation of the gRNA library. One common bottleneck of library generation in general is the efficient transfer of plasmids into bacteria for amplification. Thus, large quantities of bacteria with excellent competency and a large number of individual electroporation events are necessary to achieve a high number of gRNA clones.
New strategies for gRNA library production will be necessary to harvest the full potential of CRISPR-based screening approaches over the next decades. There is a significant demand for cost-effective, simple and customizable methods to generate large-scale libraries, a pre-requisite to make screening amenable to a larger number of model systems and different CRISPR-based engineering approaches. CORALINA is providing a first step toward this. The potential uses are manifold, especially to produce comprehensive libraries of genomes, cDNA derived libraries of less common model systems, highly focused libraries and experimental set-ups in which different CRISPR proteins (with differing PAM requirements) are used in combination.
Unlike other methods, CORALINA generates all possible gRNAs from the input DNA. However, one drawback of the method is that gRNAs lacking the required PAM sequence are also included in the library, a feature that it shares with a second enzymatic method for gRNA library generation, CRISPR-EATING (Table 1). The choice of the ideal method for gRNA library generation depends on the specifications of the planned screening experiment, especially the nature (genic, regulatory, intergenic) and size of the target region (single locus, multiple regions, genome-wide). We see a special upside in using CORALINA when a large number of non-coding or regulatory regions are to be analyzed, if there is incomplete or unreliable sequence information (exotic model systems, mixtures of species (e.g. microbiomes) or experimentally obtained input), if different CRISPR endonucleases are combined or if saturating analysis is performed on a short and defined locus (e.g. represented by BACs).
Disclosures
The authors have nothing to disclose.
Acknowledgments
The authors would like to thank Prof. Dr. Stephan Beck and Prof. Dr. Magdalena Goetz for their input, help and support in developing the CORALINA method, Maximilian Wiessbeck and Valentin Baumann for helpful comments. The work has been supported by DFG (STR 1385/1-1).
References
- Mali P, Esvelt KM, Church GM. Cas9 as a versatile tool for engineering biology. Nat Methods. 2013;10(10):957–963. doi: 10.1038/nmeth.2649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang H, et al. One-step generation of mice carrying reporter and conditional alleles by CRISPR/Cas-mediated genome engineering. Cell. 2013;154(6):1370–1379. doi: 10.1016/j.cell.2013.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stricker SH, Koferle A, Beck S. From profiles to function in epigenomics. Nat Rev Genet. 2017;18(1):51–66. doi: 10.1038/nrg.2016.138. [DOI] [PubMed] [Google Scholar]
- Dominguez AA, Lim WA, Qi LS. Beyond editing: repurposing CRISPR-Cas9 for precision genome regulation and interrogation. Nat Rev Mol Cell Biol. 2016;17(1):5–15. doi: 10.1038/nrm.2015.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Wei JJ, Sabatini DM, Lander ES. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343(6166):80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koike-Yusa H, Li Y, Tan EP, Velasco-Herrera Mdel C, Yusa K. Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nat Biotechnol. 2014;32(3):267–273. doi: 10.1038/nbt.2800. [DOI] [PubMed] [Google Scholar]
- Hart T, et al. High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell. 2015;163(6):1515–1526. doi: 10.1016/j.cell.2015.11.015. [DOI] [PubMed] [Google Scholar]
- Koferle A, et al. CORALINA: a universal method for the generation of gRNA libraries for CRISPR-based screening. BMC Genomics. 2016;17(1):917–940. doi: 10.1186/s12864-016-3268-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane AB, et al. Enzymatically Generated CRISPR Libraries for Genome Labeling and Screening. Dev Cell. 2015;34(3):373–378. doi: 10.1016/j.devcel.2015.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambrook J, Russell DW. Isolation of DNA fragments from polyacrylamide gels by the crush and soak method. CSH Protoc. 2006. [DOI] [PubMed]
- Gibson DG. Enzymatic assembly of overlapping DNA fragments. Methods Enzymol. 2011;498:349–361. doi: 10.1016/B978-0-12-385120-8.00015-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsaesser R, Paysan J. Liquid gel amplification of complex plasmid libraries. Biotechniques. 2004;37(2):200–202. doi: 10.2144/04372BM04. [DOI] [PubMed] [Google Scholar]