

Abstract
Reducing the complexity and cost of differentiation protocols is important for researchers. This interest fits with concerns about possible unintended effects that extrinsic patterning factors might introduce into human pluripotent stem cell (hPSC) models of brain development or pathophysiology, such as masking disease phenotype. Here, we present two cerebellar differentiation protocols for hPSCs, designed with simpler startup method, fewer patterning factors, and less material requirements than previous protocols. Recently, we developed culture procedures, which generate free-floating 3-dimensional (3D) products consistent with other brain "organoid" protocols, including morphologies relevant to modeling brain development such as sub/ventricular zone- and rhombic lip-like structures. The second uses an adherent, 2D monolayer procedure to complete differentiation, which is shown capable of generating functional cerebellar neurons, as products are positive for cerebellar-associated markers, and exhibit neuron-like calcium influxes. Together, these protocols offer scientists a choice of options suited to different research purposes, as well as a basic model for testing other types of streamlined neural differentiations.
Keywords: Developmental Biology, Issue 130, 3D, organoid, pluripotent stem cell, cerebellum, granule cell, cortex
Introduction
In vitro protocols for differentiating hPSCs toward cerebellar lineages initially operated on the principle of mimicking in vivo cerebellar development1,2,3,4. As such, they required a succession of factors introduced at specific times to drive pro-cerebellar patterning and maturation. Chief among these were WNT, bone morphogenetic proteins (BMPs), and fibroblast growth factors (FGFs) with known roles in mid-hindbrain development and formation of the isthmic organizer5,6,7. Of course, each additional step and factor means an increase in labor-intensive manipulations and greater expense for the researcher, and so developing simpler protocols capable of achieving equal results is of interest. This practical issue dovetails nicely with the hypothetical question, whether cells require such tight, external control over their development in vitro.
For cerebellar differentiation, a protocol published in 2015 addressed the necessity of using an extensive number of growth factors by using only FGF2, FGF19, and stromal cell-derived factor 1 (SDF1) for patterning purposes8. This study also differed from prior cerebellar protocols, by using a free-floating 3D culture system. In addition to producing cells positive for cerebellar markers, the brain "organoids" generated by their technique were shown to exhibit relevant morphology, unavailable in traditional 2D monolayer cultures, such as rhombic lip-like structures. Although less complex and costly with respect to growth factors, other features such as formation of uniform embryoid bodies (EBs) and culture in 96-well plates (96WPs), made it procedurally complex during initial steps. Another 3D protocol published the same year, reported successful differentiation to neural lineages using common and inexpensive cell culture techniques9. Although this group was investigating cortical rather than cerebellar differentiation, application of their concept for cerebellar differentiation could not be discounted.
We recently reported a 3D cerebellar differentiation protocol using a reduced number of patterning factors (namely, FGF2, 4, and 8), as well as a simplified setup by keeping cells in 6-well plates (6WPs) throughout to minimize medium requirements10. To assist production of granule cells, smoothened agonist (SAG) was used during the final maturation step. SAG is a less expensive chemical alternative to sonic hedgehog (SHH), which had been used in earlier cerebellar protocols, due to its role in promoting the growth of granule cell precursors (GCPs) in vivo1,2,11,12,13. Differentiation products were consistent with those from other 3D protocols, including the presence of cerebellar-associated markers in morphologically relevant structures8,9. Such results reinforce the earlier message that detailed mimicry of the in vivo environment may not be necessary for complex 3D in vitro differentiation protocols.
In addition to the 3D protocol, this report describes a 2D protocol, designed with the same quick setup, basic materials, and reduced number of growth factors. It is capable of producing cells from human embryonic stem cells (hESCs) or induced pluripotent stem cells (hiPSCs), positive for markers associated with early neural, cerebellar, and granule cell identities. In addition, calcium imaging indicates the presence of functional human neurons. The ability to choose between protocols, adds a level of flexibility for researchers, for those interested in either: (1) generating specific cell types, (2) modeling human brain development and associated structures, (3) analysis optimized in monolayer settings (e.g., patch-clamp recordings), or (4) cell-cell interactions in mixed neural cultures. Their simple and low-cost nature makes them accessible for researchers who are new to the hPSC field, or need base hPSC procedures from which to explore further differentiation options.
Protocol
1. Preparations
NOTE: For all steps see Table of Materials for specific items.
- Prepare 500 mL defined hPSC culture medium for hPSC culture NOTE: Use medium for steps 2.1-2.6.
- Thaw hPSC medium supplement overnight (o/n) at 4 °C. Remove 12.5 mL of hPSC base medium from medium bottle, then add 10 mL of supplement and 2.5 mL (making 100 U/L) Penicillin/Streptomycin (Pen/Strep) to the bottle.
- Store at 4 °C and use within 2 weeks. NOTE: The hPSC medium may be used to freeze down cells by adding 10 µM ROCK Inhibitor (RI) and 10% DMSO.
- Prepare 1 L neural maintenance medium (NMM) for differentiation culture NOTE: Use medium for steps 3.1-4.4.
- Mix glutamine fortified DMEM/F12 and neural basic medium (1:1 ratio) in a 1 L bottle, then supplement with (1x) N2 supplement, (1x) B27 supplement, 5 µg/mL insulin, 1.5 mM L-glutamine, 100 µM non-essential amino acids (NEAA), 100 U/L Pen/Strep, and 10 µM beta-mercaptoethanol.
- Store medium at 4 °C and use within 3 weeks. NOTE: Before adding supplements to mixed basal medium, remove the appropriate volume to adjust for the required volume of added components based on stock concentrations.
- Prepare 500 mL of 0.5 mM EDTA working solution for passaging hPSCs NOTE: Use medium for steps 2.4 and 2.5.
- Under a flow-hood, transfer 49 mL of Phosphate Buffered Saline (PBS) from a 500 mL sterile PBS bottle to a 50 mL tube. Add 0.5 mL of 0.5M EDTA and 0.9 g NaCl to the 50 mL tube. Mix gently to dissolve.
- Filter-sterilize solution using a 0.22 µm filter and transfer to the 500 mL sterile PBS bottle. Store at room temperature (RT).
- Prepare hPSC culture plates for hPSC culture NOTE: Use plates for steps 2.1-2.6.
- Make 50x working solution of hPSC-appropriate adherent coating (PAAC): Thaw a vial of PAAC o/n at 4 °C. Dilute PAAC in 1:1 ratio with DMEM/F12 and transfer as 400 µL aliquots into 1.5 mL tubes. Store 50x working solution PAAC at -80 °C. NOTE: (Important) PAAC solidifies quickly at RT, therefore it is necessary that all components (DMEM, tubes, etc.) are kept on ice (or at 4 °C).
- Thaw tube of 50x working solution PAAC at 4 °C, then dilute 50x in cold DMEM/F12. Add 750 µL/well of diluted PAAC to a 6WP. Incubate plate for at least 1 h at 37 °C. NOTE: PAAC plates may be stored for 1 week at 4 °C, by wrapping the plate after 1 h incubation period. Warm plate to 37 °C prior to use.
- Prepare anti-adhesive (AA) plates for differentiation culture NOTE: Use plates for steps 3.1-4.1.
- Make 5 mg/mL Poly (2-hydroxyethyl methacrylate) (poly-HEMA) solution in 95% Ethanol. Shake o/n at 37 °C until a clear solution is obtained. Store at RT. NOTE: Filtering through a 0.22 µm filter can remove undissolved poly-HEMA.
- Add poly-HEMA to the culture plate so that it covers the bottom of each well. Incubate the plate at 37 °C for 2 days, and inspect the plate to ensure complete evaporation of liquid/uniform coating of wells. AA plates may be wrapped and stored at RT.
- Prepare Poly-L-Ornithine/Laminin (PLO/LAM) plates for differentiation culture NOTE: Use plates for steps 4.2-4.4.
- Coat the surface area of wells, using 20 µg/mL PLO dissolved in sterile PBS. Incubate the plate o/n at 37 °C. Aspirate PLO and rinse 3 times with PBS. NOTE: (Optional) Incubated plate with PLO may be wrapped and stored at 4 °C until needed.
- Coat the surface areas of the PLO-coated wells, using 10 µg/mL LAM dissolved in sterile PBS. Incubate for at least 2 h at 37 °C or o/n at 4 °C. Remove LAM and wash wells 2-3x with PBS, then immediately add appropriate medium and/or cells. NOTE: (Optional) Removed LAM solution can be stored at 4 °C and re-used up to 2 times. (Important) Do not allow LAM-coated surfaces to dry out; to prevent this immediately add PBS or appropriate medium.
2. Protocol 1: Feeder-free hPSC Culture
NOTE: hESCs were obtained from a non-commercial organization (line H01, see Table of Materials). Three hiPSC control lines (hvs51, 60, and 88) were generated by reprogramming fibroblasts from three healthy human patients (fibroblasts were derived from anonymous, non-identifiable donors and therefore exempt from IRB approval)10,17.
- Maintain hPSCs in feeder-free culture
- After thawing and plating hPSCs onto PAAC plates in hPSC medium (see step 2.2), maintain hPSCs at 37 °C with 5% CO2. Refresh hPSC medium daily (see step 2.3), except the day after thawing or passaging, and examine cells under the microscope (objectives: 2.5x/0.06, 5x/0.12 Ph0, 10x/0.25 Ph1) to observe the growth rate and identify potential areas of differentiation (Figure 3, upper left panel shows example of differentiation).
- Passage hPSCs every 3-4 days, or when culture reaches >80% confluence. If less than 5% of cells exhibit differentiation, use normal hPSC passaging method (see step 2.4), otherwise use the gentle method (see step 2.5). When no longer needed in culture, hPSCs may be frozen for long term storage (see step 2.6).
- Thaw hPSCs in hPSC medium
- Transfer the required volume of hPSC medium to sterile tubes for the thawing process (9 mL/cryogenic tube) and to the prepared PAAC plate to receive thawed cells. Supplement the medium in both tubes with 10 µM RI.
- Retrieve the cryotube from LN2 storage and place directly into a water bath (37 °C). When only a small ice crystal remains, remove from the water bath and transfer the contents of the cryogenic tube to the tube for thawing process (total volume 10 mL). Centrifuge the tube at 290 x g for 5 min at RT.
- Use a serological pipette to remove the PAAC solution from the wells of the PAAC plate (see step 1.4) intended to receive cells, and add hPSC medium with 10 µM RI. NOTE: (Important) Do not aspirate PAAC solution with a suction needle, or it may solidify and clog lines to the vacuum pump.
- Remove the supernatant from the tube and resuspend cells in hPSC medium with 10 µM RI. Distribute the cells to the destination plate at ratio of 1 cryogenic tube/well of 6WP. Incubate at 37 °C with 5% CO2, and do not refresh medium for 1 day. NOTE: Starting up cells at 5% O2 may increase cell survival.
- Refresh hPSC medium
- Warm the necessary volume of hPSC medium in a sterile tube at RT or in a water bath; 2 mL/well of 6WP is suggested. NOTE: (Optional): By adding an extra amount of hPSC medium, hPSCs can remain an extra day without refreshing; however, do not allow this more than once a week.
- Aspirate the medium from wells containing hPSCs and add fresh hPSC medium.
- Culture the hPSCs in an incubator at 37 °C and 5% CO2.
- Passage hPSCs in hPSC medium
- Transfer the required volume of hPSC medium to sterile tubes, for passaging process and for preparation of PAAC plate to receive passaged cells. Supplement the medium for the destination plate with 10 µM RI. Warm the medium tubes at RT or in a water bath. NOTE: Preparation and handling will differ if using an alternative coating material than one listed in Table of Materials.
- Use a serological pipette to remove the PAAC solution from the wells of the PAAC plate (see step 1.4) intended to receive cells, and add hPSC medium with 10 µM RI. NOTE: (Important) Do not aspirate the PAAC solution with a suction needle, as it may solidify and clog lines to the vacuum pump.
- Aspirate the medium from wells with hPSCs to be passaged, wash cells twice with 0.5 mM EDTA, then add 0.5 mM EDTA and incubate for 2-5 min at 37 °C. NOTE: 1 mL/well of 6WP is a sufficient volume of EDTA for washing and incubation.
- Check the wells under the microscope (objectives: 2.5x/0.06, 5x/0.12 Ph0, 10x/0.25 Ph1). If cells are beginning to detach, aspirate EDTA solution and flush cells free using hPSC medium. NOTE: (Important) Take care not to remove whole hPSC colonies when aspirating EDTA (do not wait until whole colonies are detaching). Do not flush cells more than 5 times as this may harm hPSCs and affect pluripotency. Also, do not let cells stand in hPSC medium with RI before flushing cells from wells, as they may re-adhere to the plate.
- Based on empirical determination (usually related to confluence, size of colonies, and growth rate), transfer hPSCs to wells of the destination plate using a splitting ratio of 1:4-1:16 (i.e., 1 well from the original plate to 4 wells of the destination plate). Incubate at 37 °C with 5% CO2, and do not refresh medium for 1 day. NOTE: Splitting ratios as high as 1:16-1:20 are possible to prevent crowding and improve appearance of colonies.
- Passage hPSCs using the gentle-method (G-method)
- Transfer the required volume of hPSC medium to sterile tubes, for the passaging process and for preparation of PAAC plate to receive passaged cells. Supplement the medium for destination plate with 10 µM RI. Warm medium tubes at RT, or in water bath at 37 °C.
- Use serological pipette to remove PAAC solution from wells of PAAC plate (see step 1.4) intended to receive cells, and add hPSC medium with 10 µM RI. NOTE: (Important) Do not aspirate PAAC with a suction needle, or it may solidify and clog lines to the vacuum pump.
- Aspirate medium from the wells with hPSCs to be passaged, and wash the cells twice using 0.5 mM EDTA. On the second wash, wait 30 s before aspirating EDTA, then add 1 mL of PBS and incubate for 4-9 min at 37 °C. While waiting, prepare a sterile tube with 4 mL of PBS. NOTE: 1 mL/well of 6WP is sufficient volume of EDTA for washing.
- Check the wells under microscope (objectives: 2.5x/0.06, 5x/0.12 Ph0, 10x/0.25 Ph1). If cells are starting to detach, carefully tap the sides of the plate to help free colonies. When >50% of colonies are free floating, use a 5 mL serological pipette to transfer colonies in 1 mL PBS to the tube containing 4 mL PBS (do not triturate). NOTE: (Important) Since the purpose is to clean up hPSC culture, check to determine whether differentiated cells remain attached to the plate. Also, it is not necessary to passage all colonies in a well using this process, so colonies that remain attached may be left behind.
- Wait 5-10 min at RT for cells to settle in the tube (do not centrifuge). Aspirate PBS from the tube, taking care not to remove settled hPSCs. Carefully resuspend the cells in hPSC medium (do not triturate), and transfer the cells to the destination plate using a splitting ratio of 1:4-1:16. Incubate at 37 °C with 5% CO2, and do not refresh medium for 1 day.
- Freeze down hPSCs
- Depending on the confluence, use 1 well of hPSCs, at 2-3 days (maximum) in culture, to fill 1-2 cryopreservation vials for storage in LN2.
- At the end of passaging (step 2.5), use 500 µL or 1 mL of hPSC medium to transfer cells from 1 well of 6WP to 1 or 2 cryopreservation vials, respectively (500 µL/vial). To each tube, add 500 µL of 2x freezing medium containing hPSC medium, 20 µM RI, and 20% DMSO. NOTE: (Optional) Cells can be transferred directly in 1x freezing medium at 1 mL/vial.
- Place the cryogenic tubes in a cryogenic container (containing isopropanol) pre-cooled to 4 °C, and store immediately at -80 °C.
- On the next day, transfer the cryogenic tubes to an LN2 tank for long-term storage.

3. Protocol 2: 3D "Organoid" Differentiation
- Setup of differentiation with modified G-method of passaging of hPSCs
- Transfer the required volume of NMM for the number of destination wells to a sterile tube. Supplement with 4 ng/mL FGF2 and 10 µM RI. Warm the medium tube at RT, or in water bath at 37 °C. NOTE: Depending on confluence of the origination wells, hPSCs are concentrated during the distribution to the destination plate in a 2:1 or 3:1 ratio (i.e., 2 wells from the original plate to 1 well of the destination plate), with an end volume of 2.5 mL/well of 6WP.
- Aspirate the medium from wells containing hPSCs to be differentiated, and wash the cells twice using 0.5 mM EDTA. On the second wash, wait 30 s before aspirating EDTA, then add 1 mL of PBS and incubate for 4-9 min at 37 °C. While waiting, prepare a sterile tube with 4 mL of PBS. NOTE: 1 mL/well of 6WP is sufficient volume of EDTA for washing. (Important) It is preferable to use hPSCs that were no longer than 3 days in culture after the last passage, and at least 1-2 passages after thawing.
- Check wells under the microscope (objectives: 2.5x/0.06, 5x/0.12 Ph0, 10x/0.25 Ph1). If cells are starting to detach, free the cells by gentle tapping on the sides of the plate. Transfer the cells to a tube containing 4 mL PBS with a 5 mL pipette. NOTE: (Important) Light flushing and trituration is permitted to break up colonies and collect loose cells, but ignore cells that remain adhered to the plate.
- Allow the tube to sit for 10 min at RT, for gravity separation. Optionally, centrifuge the cells lightly (no greater than 200 x g at RT, for 5 min) if required.
- Aspirate the PBS from the tube, taking care not to remove settled hPSCs, resuspend the cells in NMM with 4 ng/mL FGF2 and 10 µM RI, and then distribute to the AA-coated plate in a 2:1 or 3:1 ratio. Incubate at 37 °C with 5% CO2, and do not refresh the medium for 3 days, unless required (see step 3.2.1). NOTE: (Optional) For convenience of transferring cells, add a portion of culture medium to wells of the destination plate prior to distribution, and resuspend the cells in a smaller volume. Also, hPSCs may be stained and counted to define the exact starting cell density. However, the final volume in the destination plate should be 2.5 mL/well of 6WP.
- Maintain free-floating differentiation culture at 37 °C (5% CO2)
- Check the plates every day for changes in medium color, accumulation of dead cells, clumping, and adherence to well bottoms.
- Optional: Regardless of the medium change schedule, refresh (including the first 3 days of no refreshing) the medium if it has turned yellow, and follow instructions for medium change/refresh (step 3.3). If the majority of cells appear dead, follow the instructions for medium change/refresh with gravity separation (step 3.4). NOTE: Formation of EBs and growth into large cell aggregates are expected, but cells and cell aggregates can clump together into large masses not due to individual growth/proliferation. If this is observed, light trituration to break up masses is permissible. If cells start to adhere to the AA-plate surface, the remaining floating contents of wells may be transferred directly to new wells, or transferred during the process of medium change/refresh. Do not attempt to transfer cells that have adhered to the plate.
- On day 3, change the medium to NMM with 4 ng/mL FGF2. Refresh the medium every other day.
- On day 7, change the medium to NMM with 1 µM Retinoic Acid (RA), 100 ng/mL FGF8B, and 4 ng/mL FGF2. Refresh the medium every other day. NOTE: (Important) RA is light-sensitive. Protect RA culture samples from light.
- On day 14, change the medium to NMM with 100 ng/mL FGF8B, 100 ng/mL FGF4, and 20 ng/mL FGF2. Refresh the medium every other day.
- On day 17, change the medium to NMM with 100 ng/mL FGF8B. Refresh the medium every other day.
- On day 21, change the medium to NMM with 100 ng/mL Brain Derived Neurotrophic Factor (BDNF) and 10 ng/mL Glial Derived Neurotrophic Factor (GDNF). Refresh the medium every other day.
- On day 28, change the medium to NMM with 100 ng/mL BDNF and 10 ng/mL GDNF, 3 ng/mL SAG, 100 ng/mL Neurotrophic Factor 3 (NT3), and 25 mM KCl. Refresh the medium every other day.
- On day 35, collect the 3D organoids for analysis.
- Change/refresh differentiation medium for 3D culture
- Transfer the required volume of NMM with the appropriate components (see steps 3.2.2-3.2.7 for component schedule) to a sterile tube. Warm in a water bath at 37 °C.
- Tip the plate and shake gently until the cells settle to the bottom edges of the wells. Carefully remove 2 mL of old medium using a serological pipette, avoiding removal of the cells, then add 2 mL of fresh medium. Incubate at 37 °C with 5% CO2. NOTE: (Important) The end volume should be 2.5 mL/well of 6WP. If evaporation occurs, do not remove 2 mL of old medium as this would further dry out the cell cultures; instead, add extra medium.
- Change/refresh differentiation medium with gravity separation
- Transfer the required volume of NMM with the appropriate components to a sterile tube. Warm in a water bath at 37 °C.
- Transfer the contents of the wells to a sterile tube, and allow the tube to sit for 10 min at RT, for gravity separation.
- Use a pipette to remove old medium from the tube, taking care not to remove settled cells, resuspend in NMM with appropriate components, and then distribute to new AA-coated wells. Incubate at 37 °C with 5% CO2. NOTE: (Optional) For convenience, a portion of culture medium may be added to the destination wells prior to distribution, with differentiating cells resuspended in a lower volume. The end volume should be 2.5 mL/well of 6WP.
4. Protocol 3: Alternative 2D Differentiation Culture
- Start and maintain culture using the steps as per Section 3 for 3D protocol through day 12
- Follow steps 3.1-3.2.3 and change/refresh medium as per steps 3.3 and 3.4.
- Switch to and maintain as 2D monolayer culture
- On day 13 of differentiation, follow the instructions for change/refresh medium with gravity separation (step 3.4), only distribute the cells/aggregates to PLO/LAM coated plate (see step 1.6) with an end volume of 2.5 mL/well of 6WP. NOTE: The medium may be supplemented with 10 µM RI during the initial plating to help adherence and survival of cells. (Important) It is desirable to evenly spread the cells in the wells, to avoid low density or crowding on the plates, and to passage (see step 4.4) as necessary. The preferred size of PLO/LAM coated plates (i.e., 6WP, 12WP, etc.) must be empirically determined, based on the proliferation rate for the cell line, and purpose for the product. Instructions will give volumes for 6WP, and can be converted by halving for each doubling of well number (i.e., 2 mL/well for 6WP, 1 mL/well for 12WP, etc.)
- On day 14, change the medium to NMM with 100 ng/mL FGF8B, 100 ng/mL FGF4, and 20 ng/mL FGF2. Refresh the medium every other day as described in step 4.3.
- On day 17, change the medium to NMM with 100 ng/mL FGF8B. Refresh the medium every other day as described in step 4.3.
- On day 21, change the medium to NMM with 100 ng/mL BDNF and 10 ng/mL GDNF. Refresh the medium every other day as described in step 4.3.
- On day 28, change the medium to NMM with 100 ng/mL BDNF and 10 ng/mL GDNF, 3 ng/mL SAG, 100 ng/mL NT3, and 25 mM KCl. Refresh the medium every other day as described in step 4.3.
- On day 35, collect cells for analysis, or maintain in the same medium as step 4.2.5 for extended culture (potential limit not tested).
- Change/refresh differentiation medium for 2D culture
- Transfer the required volume of NMM with appropriate components (see steps 4.2.2-4.2.5 for component schedule) to a sterile tube. Warm in a water bath at 37 °C.
- Aspirate the medium from wells, then add 2 mL new medium. Incubate at 37 °C with 5% CO2. NOTE: (Optional) The end volume can be kept at 2.5 mL/well of 6WP, using a pipette to remove 2 mL of old medium and adding 2 mL of fresh medium. Reserving a portion of old medium in the wells, and preventing cells from air contact, may lessen shock to the cells during change steps.
- Passage 2D differentiation culture
- Transfer the required volume of NMM with the appropriate components (see steps 4.2.2-4.2.5 for component schedule) to sterile tubes, for passaging process and, separately, to prepare PLO/LAM coated plate for receiving passaged cells. Supplement the medium of the destination plate with 10 µM RI. Warm the medium tubes at RT, or in a water bath at 37 °C. To save on components, it is possible to use NMM alone for washing cells during the passaging process. NOTE: (Important) If products will be used in calcium imaging experiments, ensure to passage cells between 2-6 days before the end of differentiation.
- Aspirate the medium from wells to be passaged. Add 300 µL/well of trypsin-based dissociation agent (see Table of Materials), swirl plate to cover wells, then immediately remove the dissociation agent.
- Allow the plate to sit for 2 min at RT, then loosen the cells by tapping on sides of the plate. Add 600 µL/well defined trypsin inhibitor (DTI) and transfer the cells in DTI to a sterile tube with 5 mL NMM.
- Centrifuge the tube at 290 x g for 15 min at RT. Aspirate the medium and add another 5 mL NMM to the tube.
- Centrifuge the tube at 290 x g for 15 min at RT. Aspirate the medium and resuspend the cells in the appropriate medium containing RI.
- Distribute the cells on the PLO/LAM plate using a splitting ratio of 1:1-1:12, depending on the initial confluence, proliferation rate, and size differential in origin to the destination plate, and maintain at 37 °C with 5% CO2.
Representative Results
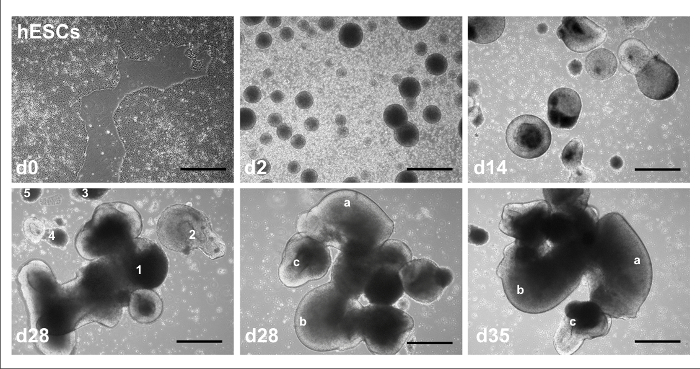
Visual Overview of Reduced Growth Factor 2D and 3D Cerebellar Differentiation ProtocolsFigure 1 shows the overall timeline for the 2D and 3D cerebellar differentiation protocols, identifying extrinsic factors and time of plating. The typical progress for hPSCs undergoing 3D cerebellar differentiation is depicted in Figure 2: with hESC line H01 starting as colonies in feeder-free culture at day 0 (upper left of figure); undergoing EB formation by day 2 (upper middle); growing into larger cell aggregates with apparent lumen following neural induction with RA and FGF8 at day 14 (upper right); forming aggregates of varying size and shape at day 28 (lower left); developing in complexity with different structures of a single aggregate indicated at day 28 (lower middle); and with continued morphological changes to the same structures at day 35 (lower right). The typical progress for hPSCs undergoing 2D cerebellar differentiation is depicted in Figure 3: with hESC line H01 as colonies in feeder-free culture at day 0 (upper left of figure, with circle indicating area of differentiated cells among hESC colonies); undergoing EB formation by day 2 (upper middle); growing into larger cell aggregates with apparent lumen following neural induction with RA and FGF8 at day 13 (upper right); proliferating as adherent cells after plating at day 14 (lower left); and then as a monolayer of cells with more complex/mature morphology at day 35 under low (lower middle) and high magnification (lower right).
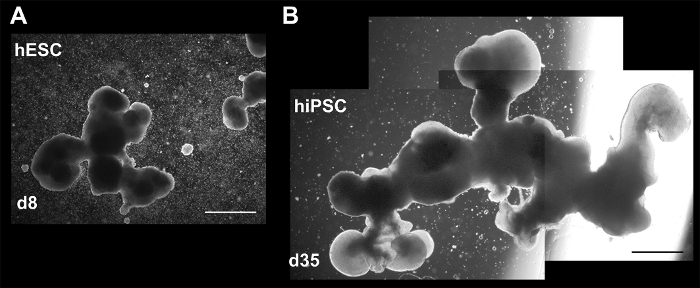
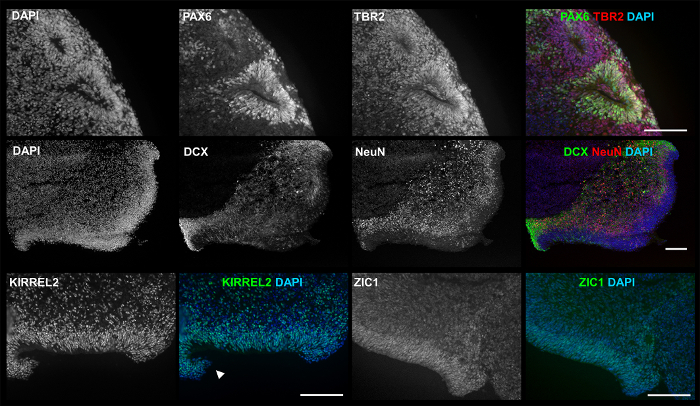
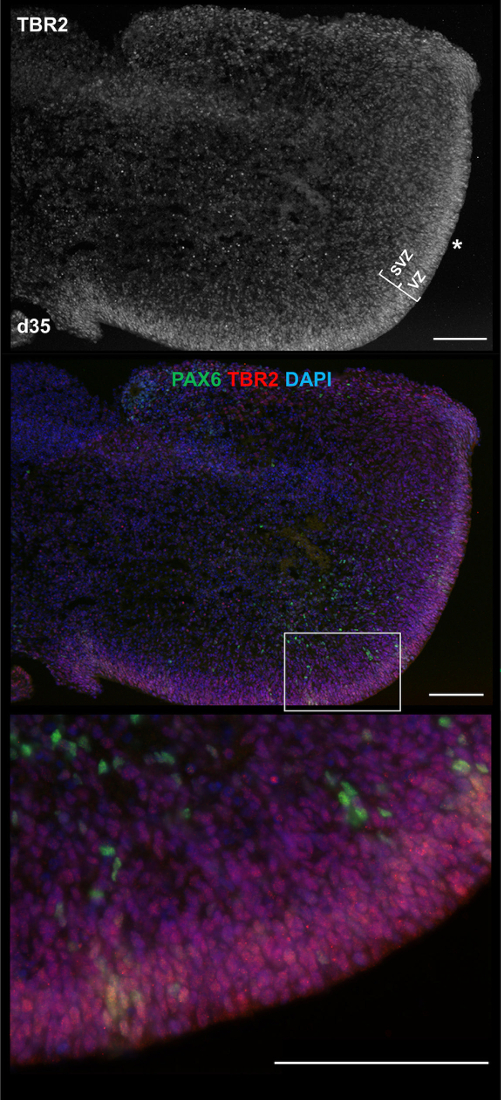
3D Products Exhibit Markers and Structures of Early NeuroepitheliumFigure 2 (lower left image) and Figure 4 show the heterogeneity of 3D aggregate morphology seen throughout culturing, due to varying growth and/or maturation rates, as well as the stochastic merging or breaking apart of aggregates. Despite heterogeneity, each differentiation produces aggregates exhibiting early neural and neuronal markers, including cerebellar granule marker ZIC1, as indicated by immunocytochemistry (ICC) staining in Figure 5. More importantly, Figure 5 and Figure 6 suggest that a simple 3D culture, with reduced growth factors, is capable of generating aggregates with complex structures related to brain development such as the early neuroepithelium and rhombic lip.
2D Products Exhibit Cerebellar Markers and Functional Neuronal Activity While 2D cultures cannot reproduce complex 3D structures, they are capable of generating cells exhibiting early neural and neuronal markers, including cerebellar granule cell marker ZIC1, as indicated by ICC staining in Figure 7. Gene expression analysis via RT-PCR, as seen in Figure 8, supports ICC staining results, though the presence of early granule cell marker ATOH1 is variable between experiments and lines. Calcium imaging is more easily handled in 2D culture. As seen in Figure 9, Supplemental Video 1, and Supplemental Video 2, electrically stimulated cells show calcium influxes that are typical of neuronal firing patterns, suggesting generation of functional neurons.
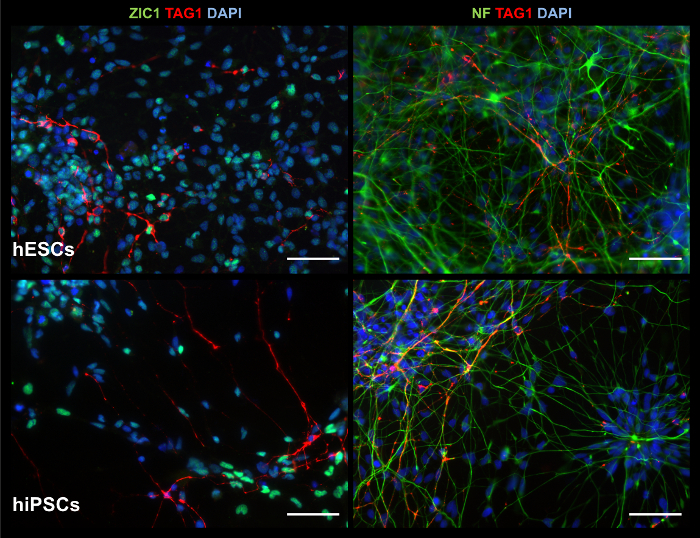
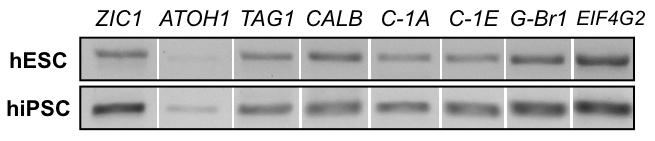
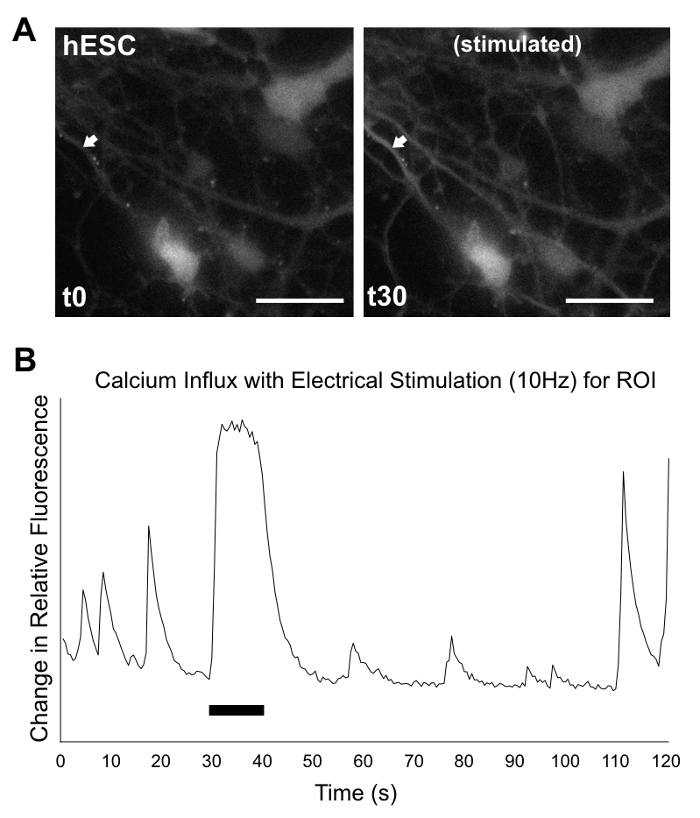
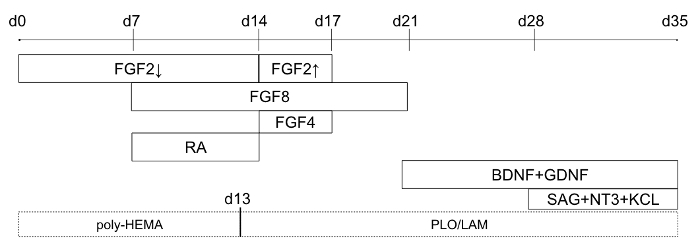
Figure 1: Timeline of differentiation protocol (starting at day 0 of differentiation). Solid line boxes indicate when specific factors are added to the culture medium, and dotted line boxes indicate plate-coating for optional 2D modification. For FGF2, the down arrow refers to lower concentrations (4 ng/mL), and the up arrow refers to higher concentrations (20 ng/mL). This figure has been modified from Holmes and Heine10. Please click here to view a larger version of this figure.
Figure 2: Representative brightfield images of 3D protocol. hESC colonies at d0, EBs at d2, aggregates following induction at d14, aggregates of different size and morphology (numbered 1-5) at d28, a single aggregate with unique, identifiable features (indicated by letters a-c) at d28, and visible changes in the same features at d35. Scale bar = 100 µm. This figure has been modified from Holmes and Heine10. Please click here to view a larger version of this figure.
Figure 3: Representative brightfield images of 2D protocol. hESC colonies a d0, EBs at d2, aggregates after induction at d13, after plating aggregates at d14, after maturation at d35 as seen at 5x magnification, and 20x magnification. The white circle in the upper left panel shows the area of differentiated cells among hESC colonies. Scale bar = 50 µm. Please click here to view a larger version of this figure.
Figure 4: Brightfield images showing varying size and complexity in 3D culture. Aggregates at (A) day 8 (hESCs), and (B) d35 (hiPSCs). The latter image is composed of three separate images to show the entire aggregate. Both aggregates may have been affected by merging of smaller aggregates or loss (breaking off) of structures. Scale bar = 200 µm. This figure is republished from Holmes and Heine10. Please click here to view a larger version of this figure.
Figure 5: ICC images of 3D products show relevant markers and structures. At culture d35, 3D products exhibit: PAX6 (green) and TBR2 (red) by lumen of neural rosette-like formation (first row); DCX (green) and NeuN (red) spreading from outer edge ventricular zone (VZ)-like structure (second row); KIRREL2, a marker associated with cerebellar neuroepithelium (third row, left); and ZIC1 a marker associated with cerebellar granule cells (third row, right). The experiment was conducted multiple times using four different hPSC lines: hESC line H01 (n = 5), and iPSC lines hvs88 (n = 4), hvs60 (n = 3), and hvs51 (n = 1). Arrows point to Rhombic lip (RL)-like structure. Scale bar = 100 µm. This figure is republished from Holmes and Heine10. Please click here to view a larger version of this figure.
Figure 6: Large scale ventricular zone-like structure in 3D product. At culture d35, an hESC-derived aggregate is positive for PAX6 (green) and TBR2 (red) commonly associated with early neurons found within ventricular zones (VZs) and subventricular zones (SVZs), in vivo. (Top) An asterisk (*) marks the apical side of a VZ-like region running along the edge of the aggregate, with brackets indicating depth/division of VZ/SVZs. (Middle) Merged signals show scattered sections of PAX6+/TBR2- cells increasing in size toward the upper right end of the VZ. (Bottom) Higher magnification image of the section indicated by a rectangle in the middle panel. Scale bar = 100 µm. This figure has been modified from Holmes and Heine10. Please click here to view a larger version of this figure.
Figure 7: ICC images of 2D products show relevant markers. At culture d35, 2D products exhibit, from hESCs (top row) and hiPSCs (bottom row), cells positive for: granule cell marker ZIC1 and migratory cerebellar neuron marker TAG1 (left column); and neuronal marker neurofilament (NF) and TAG1 (right column). Scale bar = 50 µm. Please click here to view a larger version of this figure.
Figure 8: RT-PCR of 2D products. mRNA expression analysis of hESC line H01 (top row) and hiPSC line hvs60 (bottom row) at the end of the 2D protocol shows products with gel electrophoresis for: granule cell marker ZIC1, granule cell marker ATOH1, migratory cerebellar neuron marker TAG1, Purkinje cell marker Calbindin (CALB), voltage-dependent calcium channel CACNA1A (C-1A), CACNA1E (C-1E), gamma-aminobutyric acid (GABA) B receptor 1 (G-Br1), and housekeeping gene EIF4G2).
Figure 9: Calcium imaging/analysis of 2D products. At culture d35, hPSC differentiation products were recorded for 2 min under the microscope, after incubation with fluor5 dye. At 30 s, cells were electrically stimulated for 10 s at 10 Hz. (A) Still images show hESCs at 0 s (left) and after the start of electrical stimulation at 30 s (right). Arrows indicate region of interest (ROI) for analysis of calcium influx. (B) Graphical analysis showing change in relative fluorescence versus time for ROI (change in fluorescence = (F-F0)/F0, where F0 = (∑F1-n)/n), with neuron-like spikes occurring before, during, and after stimulation. Black bar indicates length of electrical stimulation. Scale bar (white) = 50 µm. Full recording for hESC (as seen here) and an hiPSC line (not shown) are available in Supplementary Video 1 and Supplementary Video 2, respectively. Recordings are in AVI format, at 4x speed. Please click here to view a larger version of this figure.
Supplemental Video 1: Calcium imaging video of 2D product from hESC line H01. At culture d35, differentiation products from hESC line H01 were recorded for 2 min under the microscope, after incubation with fluor5 dye. At 30 s, cells were electrically stimulated for 10 s at 10 Hz. Recordings were made at 2 frames/s, and processed into AVI video at ~7 frames/s, producing a video lasting ~30 s, at ~4x speed. Please click here to download this file.
Supplemental Video 2: Calcium imaging video of 2D product from hiPSC line hvs51. At culture d35, differentiation products from hiPSC line hvs51 were recorded for 2 min under the microscope, after incubation with fluor5 dye. At 30 s, cells were electrically stimulated for 10 s at 10 Hz. Recordings were made at 2 frames/s, and processed into AVI video at ~7 frames/s, producing a video lasting ~30 s, at ~4x speed. Please click here to download this file.
Discussion
Complexity and costs are relevant factors for stem cell researchers when choosing or developing differentiation protocols. This is especially true as it is an open question of how much external control is required to generate desired cell types, or—to pose it differently—how competent hPSCs are at producing their own developmental environment, if left to themselves with sufficient nutrients. Introduction of extrinsic factors in vitro may very well produce desired cell products, but they could also interfere with the intrinsic developmental capacities cells would have exhibited in vivo. Such considerations are important, particularly if the goal is use of patient-derived iPSCs for disease modeling. Extensive use of patterning and/or growth factors could mask disease phenotypes. The protocols detailed in this report follow the trend of previous studies to reduce complexity, cost, and/or use of extrinsic patterning factors8,9.
Based on results reported by Muguruma et al., and our own recent study, it appears that it is possible to achieve differentiation towards cerebellar fates without concerted efforts to reproduce in vivo conditions, as earlier studies have done1,2,3,4,8,10. The intriguing part is that the two studies used different sets of growth factors, suggesting that neither set were necessary, though both used FGF2. We ran additional tests, where FGFs were selectively excluded from the protocol, and showed that cells were capable of generating the same products without extrinsic FGFs10. Differences between our studies were qualified by the fact that we used different hPSC lines and culture methods, induced neural differentiation with RA, and included components to support granule cell survival and maturation (BDNF, GDNF, SAG, and KCL)11-14. In addition, a less complex startup method, compared to Muguruma et al., was employed. Their protocol began by generating uniform EBs in 96WPs, which isolated them physically and chemically from each other. The protocol here had all PSCs relatively crowded together in 6WPs during EB formation, which allowed them to interact freely. How this may have differentially affected the physical and chemical environment of EBs and later organoids (including intrinsic production of signaling compounds) is unknown, and could be explored. Also, while we show expression of genes associated with—and so suggestive of—cerebellar origin, located within structures morphologically similar to those reported by Muguruma et al., we cannot exclude generation of neuronal-like structures that are of non-cerebellar identity. Future studies, using a large panel of antibodies like those reported by Muguruma et al. (i.e., ATOH1, CALB, etc.) would make such assignments, and comparison between products of both protocols, more conclusive.
Within the 3D protocol, it is important to start with and maintain a sufficient number of cells in culture to ensure sufficient numbers of end products for analysis. Given significant die-off early in the protocol, we recommend starting with more than 500 EBs/well during the first 3 days in culture (Figure 1). This should not be difficult to achieve given colony sizes for hPSCs in feeder-free culture, but might not be as easy for those still using feeder-dependent methods. Given the large number of cells, it is important to watch for color change in medium (indicating pH changes), and accumulation of dead cells. Both must be corrected to prevent collapse of the culture. There may also be clumping of cells and aggregates into massive structures. Although it may still result in aggregates that can be analyzed, product quantity will be greatly reduced, so breaking them up into smaller aggregates with gentle trituration can be useful. However, avoid disturbing normal aggregates, which themselves can grow to large sizes (Figure 4). If aggregates become too sparse, it is recommended to combine wells so that aggregates are not completely isolated. Product variability (in number, size, and morphology) is a well-known issue in 3D cell culture, including for those protocols starting with isolated, uniform EB formation steps, suggesting that a less complex startup procedure (such as the protocol described here) could be more practical8,15. While this heterogeneity is something researchers need to keep in mind, particularly during analysis, the reported protocol generated products consistent with those found in other 3D protocols8,9,15. Based on size and morphology, they fall within the range of neural rosette to cerebral organoid, as described in a recent review by Kelava and Lancaster15, with the most fitting the classification of spheroid. Particularly notable, are the presence of 3D structures suggestive of neural rosettes with lumen, (sub)ventricular zones, and rhombic-lip like features (Figure 5 andFigure 6) as identified by other groups8,15,16,17. Since every experiment produced at least one aggregate with putative VZ/SVZs and cerebellar-associated markers (ZIC1, KIRREL2), those are useful criteria for determining the success of a 3D differentiation using our protocol, with RL-like features providing additional support. Extending the length of culture past 35 days was not tested, but could be pursued to determine the maximum extent of growth, complexity, and maturity allowed by this technique.
The 2D protocol uses the same non-adherent EB formation and neural induction process as the 3D protocol and so the comments above also apply. Once plated, a different set of considerations should be considered. The EBs should adhere quickly for cells to proliferate outward onto the plate. If there are problems with adherence, addition of RI (if not already used), reduced volume of medium, or experimental changes in PLO/LAM concentration may be applied. It is important to keep cells from growing too dense or sparse (preferably grown between 20-80% confluency) in the wells; daily monitoring and timely passaging is important, to avoid over-confluency or floating cells. Unlike the 3D protocol, there should not be significant die-off during culture, though there may be areas of poor growth, or a slowing of proliferation rates. Passaging affects the maturation state of cells (for example, removing cell processes and developed networks between cells) and should be kept in mind when approaching points where cells will be collected or analyzed in some way. For example, for calcium imaging it is very important to passage cells between 2-6 days prior to analysis. Passaging too close to analysis might mean cells have not had time to connect and/or mature, and too far may result in cells overcrowding, making imaging difficult. Although variability between experiments may exist, results are consistent with those reported in initial 2D cerebellar protocols1,2. ICC staining and gene expression analysis corroborate the presence of cells positive for granule cell marker ZIC1, while also identifying markers associated with other neural and cerebellar identities (Figure 7 and Figure 8). Calcium imaging, which involves electrical stimulation of cells incubated with fluor5 dye, indicated functional neuronal activity (Figure 9, Supplemental Figure 1, and Supplemental Figure 2), though it is not confirmed if these were granule cells. It is arguable that by giving cells more time to mature by extending the length of culture past 35 days, the amount of functional neuronal activity should increase. This potential could be explored in the future.
In addition to the lines of research suggested above, it would be of interest to determine differences in product identities (quantity and quality) between the 2D and 3D protocols. The importance of extrinsic FGFs was not tested in the 2D protocol, and it would be useful to know if lack of 3D structure after plating, and so the associated signaling pathways, would make 2D cultures more or less dependent on those early patterning compounds. More stripped-down protocols (e.g., no RA, BDNF, SAG) are equally plausible lines for further investigation. Finally, future studies might benefit from new research tools to better characterize (and assess generation efficiency of) human-specific cerebellar neuronal subtypes.
With the given caveats in mind, both reported protocols may be used for cerebellar differentiations, with products suited to different purposes. They may serve as practical starting points for researchers conducting pilot studies, testing viability of cell lines for such differentiations, or as a basic model for other types of targeted neural differentiation.
Disclosures
The authors have nothing to disclose.
Acknowledgments
We are grateful to Gerbren Jacobs and Jurjen Broeke for their expert technical assistance, to Prisca Leferink for contributing to the generation and characterization of two control iPSC lines, and to Lisa Gasparotto for demonstrating our procedures.
References
- Erceg S, et al. Efficient differentiation of human embryonic stem cells into functional cerebellar-like cells. Stem Cells Dev. 2010;19:1745–1756. doi: 10.1089/scd.2009.0498. [DOI] [PubMed] [Google Scholar]
- Erceg S, Lukovic D, Moreno-Manzano V, Stojkovic M, Bhattacharya SS. Derivation of cerebellar neurons from human pluripotent stem cells. Curr Protoc Stem Cell Biol. 2012. Chapter 1, Unit 1H 5. [DOI] [PubMed]
- Su HL, Muguruma K, Matsuo-Takasaki M, Kengaku M, Watanabe K, Sasai Y. Generation of cerebellar neuron precursors from embryonic stem cells. Dev Biol. 2006;290:287–296. doi: 10.1016/j.ydbio.2005.11.010. [DOI] [PubMed] [Google Scholar]
- Salero E, Hatten ME. Differentiation of ES cells into cerebellar neurons. Proc Natl Acad Sci U S A. 2007;104:2997–3002. doi: 10.1073/pnas.0610879104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyner AL. Engrailed, Wnt and Pax genes regulate midbrain--hindbrain development. Trends Genet. 1996;12:15–20. doi: 10.1016/0168-9525(96)81383-7. [DOI] [PubMed] [Google Scholar]
- Joyner AL, Liu A, Millet S. Otx2, Gbx2 and Fgf8 interact to position and maintain a mid-hindbrain organizer. Curr Opin Cell Biol. 2000;12:736–741. doi: 10.1016/s0955-0674(00)00161-7. [DOI] [PubMed] [Google Scholar]
- Tam EWY, Benders MJNL, Heine VM. Cerebellar Development-The Impact of Preterm Birth and Comorbidities. Fetal and Neonatal Physiology. 5th ed 2017.
- Muguruma K, Nishiyama A, Kawakami H, Hashimoto K, Sasai Y. Self-organization of polarized cerebellar tissue in 3D culture of human pluripotent stem cells. Cell Rep. 2015;10:537–550. doi: 10.1016/j.celrep.2014.12.051. [DOI] [PubMed] [Google Scholar]
- Pasca AM, et al. Functional cortical neurons and astrocytes from human pluripotent stem cells in 3D culture. Nat Methods. 2015;12:671–678. doi: 10.1038/nmeth.3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes DB, Heine VM. Simplified 3D protocol capable of generating early cortical neuroepithelium. Biology Open. 2017;6:402–406. doi: 10.1242/bio.021725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Cooper MK, Beachy PA. Inhibition of Hedgehog signaling by direct binding of cyclopamine to Smoothened. Genes Dev. 2002;16:2743–2748. doi: 10.1101/gad.1025302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen JK, Taipale J, Young KE, Maiti T, Beachy PA. Small molecule modulation of Smoothened activity. Proc Natl Acad Sci U S A. 2002;99:14071–14076. doi: 10.1073/pnas.182542899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahmane N, Ruiz-i-Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development. 1999;126:3089–3100. doi: 10.1242/dev.126.14.3089. [DOI] [PubMed] [Google Scholar]
- Borghesani PR, et al. BDNF stimulates migration of cerebellar granule cells. Development. 2002;129:1435–1442. doi: 10.1242/dev.129.6.1435. [DOI] [PubMed] [Google Scholar]
- Kelava I, Lancaster MA. Stem Cell Models of Human Brain Development. Cell Stem Cell. 2016;18:736–748. doi: 10.1016/j.stem.2016.05.022. [DOI] [PubMed] [Google Scholar]
- Kadoshima T, et al. Self-organization of axial polarity, inside-out layer pattern, and species-specific progenitor dynamics in human ES cell-derived neocortex. Proc Natl Acad Sci U S A. 2013;110:20284–20289. doi: 10.1073/pnas.1315710110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Englund C, et al. Pax6, Tbr2, and Tbr1 are expressed sequentially by radial glia, intermediate progenitor cells, and postmitotic neurons in developing neocortex. J Neurosci. 2005;25:247–251. doi: 10.1523/JNEUROSCI.2899-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warlich E, et al. Lentiviral vector design and imaging approaches to visualize the early stages of cellular reprogramming. Mol Ther. 2011;19:782–789. doi: 10.1038/mt.2010.314. [DOI] [PMC free article] [PubMed] [Google Scholar]