

Abstract
We present a modified native chromatin immunoprecipitation sequencing (ChIP-seq) experimental protocol compatible with a Gaussian mixture distribution based analysis methodology (nucleosome density ChIP-seq; ndChIP-seq) that enables the generation of combined measurements of micrococcal nuclease (MNase) accessibility with histone modification genome-wide. Nucleosome position and local density, and the posttranslational modification of their histone subunits, act in concert to regulate local transcription states. Combinatorial measurements of nucleosome accessibility with histone modification generated by ndChIP-seq allows for the simultaneous interrogation of these features. The ndChIP-seq methodology is applicable to small numbers of primary cells inaccessible to cross-linking based ChIP-seq protocols. Taken together, ndChIP-seq enables the measurement of histone modification in combination with local nucleosome density to obtain new insights into shared mechanisms that regulate RNA transcription within rare primary cell populations.
Keywords: Genetics, Issue 130, Epigenomics, chromatin immunoprecipitation, histone modification, nucleosome density, cellular heterogeneity, micrococcal nuclease
Introduction
The eukaryotic genome is packaged into chromatin via repeating nucleosome structures that consist of two copies of four histone proteins (e.g., H2A, H2B, H3, and H4) circumscribed by 146 base pairs of DNA1,2. Chromatin remodeling complexes control nucleosome position within gene promoter boundaries and participate in the regulation of gene expression by altering accessibility of the DNA to transcription factors and to the RNA polymerase machinery3,4.
Amino terminal tails of histones within the nucleosome are subjected to various covalent modifications, including acetylation, methylation, phosphorylation, ubiquitylation, sumoylation, formylation, and hydroxylation of specific amino acids5,6,7,8. Positions and degrees of these modifications dictate a chromatin state that influence chromatin structure and control access of the molecular complexes that allow activation of transcription7. Given that both nucleosome density and histone modification play a role in the local control of gene transcription, we developed a native ChIP approach that enables the simultaneous measurement of nucleosome density and histone modification9,10.
Native ChIP-seq takes advantage of the endonuclease micrococci nuclease (MNase) to digest intact chromatin in its native state within the nucleus11,12, a property that has been leveraged to map nucleosome positioning13,14,15. Nucleosome density ChIP-seq (ndChIP-seq) takes advantage of the property of preferential access of MNase to open regions of chromatin to generate measurements that combine MNase accessibility with histone modification10. ndChIP-seq is suitable for the profiling of histone modifications in rare primary cells, tissues, and cultured cells. Here, we present a detailed protocol that enables the generation of sequence datasets suitable for a previously described analytical frame work10 that integrates fragment size post immunoprecipitation, determined by paired-end read boundaries, to simultaneously investigate MNase accessibility with histone modification measurements. Previously, application of this protocol to 10,000 primary human cord blood derived CD34+ cells and human embryonic stem cells revealed unique relationships between chromatin structure and histone modifications within these cell types10. Given its ability to simultaneously measure nucleosome accessibility and histone modification, ndChIP-seq is capable of revealing epigenomic features in a cell population at a single nucleosome level, and resolving heterogeneous signatures into their constituent elements. An example of the exploration of heterogeneous cellular populations by ndChIP-seq is investigation of bivalent promoters, where both H3K4me3, an active mark, and H3K27me3, a repressive mark, are present10.
Protocol
NOTE: The minimum input for this protocol is 10,000 cells per single immuno-precipitation (IP) reaction. Print out the supplied experimental worksheet and utilize as a guideline to plan out the experiment. Incubations at room temperature are assumed to be at ~22 °C. All of the buffer recipes are provided in Table 1. All of the buffers should be stored at 4 °C and kept on ice during the procedure, unless stated otherwise.
1. Cell Preparation
- Cultured cells
- Wash the cells with 1 mL of phosphate buffer saline (PBS) and accurately determine the cell concentration using a hemocytometer. If there are more than one million cells, increase the volume of PBS.
- Based on the cell count, aliquot an equivalent of 70,000-100,000 cells into a sterile 1.5 mL tube, and spin down at 500 x g for 6 min at 4 °C. Using a pipette, slowly remove and discard the supernatant (without disturbing the cell pellet) and resuspend the pellet in ice-cold lysis buffer + 1x protease inhibitor cocktail (PIC) to a concentration of 1,000 cells/µL. Mix well by pipetting up and down 20-30 times. It is critical that cell clumps are disrupted. Try not to create bubbles.
- Proceed directly to Day 1 (Section 2) of the ndChIP-seq protocol or flash freeze the cell pellet in liquid nitrogen and store at -80 °C.
- Sorted cells
- Collect 70,000-100,000 sorted16 cells into a 1.5 mL tube with 350 µL of Hank's buffered salt solution (HBSS), or PBS + 2% fetal bovine serum (FBS).
- Spin down each cell aliquot at 500 x g for 6 min at 4 °C. Using a pipette, slowly remove the supernatant (without disturbing the cell pellet) and resuspend in ice-cold lysis buffer + 1x PIC to a final concentration of 1,000 cells/µL. Mix well in lysis buffer by pipetting up and down 20-30 times. Ensure that the cell clumps are disrupted. Try not to create bubbles.
- Proceed directly to Day 1 (Section 2) of the ndChIP-seq protocol, or flash freeze the cell pellet in liquid nitrogen and store at -80 °C.
2. DAY 1: ndChIP-seq
- Preparation of the antibody-bead complex
- Prepare a 37 °C water bath and an ice bucket. Working on ice, prepare 1x IP buffer/1x PIC and 1x antibody (Ab) dilution buffer, and keep them on ice. Retrieve Protein A (or G) magnetic beads (see Discussion for selection criteria) from the 4 °C storage, and mix very well by gentle pulse-vortexing. Keep it on ice. NOTE: Pulse-vortexing means vortexing is stopped every time a full vortex is formed in the tube.
- Transfer 24 µL of Protein A (or G) magnetic beads per IP reaction into a new 2 mL tube. Record the volume of beads and keep the tube on ice. For example, for 7 IPs, use 24 µL x 7 = 168 µL.
- Place the tube on the tube magnet and wait for the solution to become clear indicating bead separation. Without disturbing the beads, carefully remove the supernatant using a pipette and discard. Take the tube off the tube magnet and place it on ice.
- Add an equal volume (i.e., initial volume of beads) of ice-cold IP buffer + 1x PIC mix. Mix by pipetting up and down. Do not vortex.
- Place the IP buffer+ 1x PIC + beads back onto the tube magnet and wait for the solution to become clear indicating bead separation. Without disturbing the beads, carefully remove the supernatant using a pipette and discard. Repeat cold IP buffer + 1X PIC wash two more times for a total of 3 washes.
- After the final wash, resuspend the beads in an equal volume (i.e., initial volume of beads) of ice-cold IP buffer + 1x PIC mix. Mix by pipetting up and down. Do not vortex. Keep the tube on ice.
- On ice, pour 10 mL of IP buffer + PIC into a 25 mL V-shaped reservoir. Using a multichannel pipette, add 130 µL of ice-cold IP buffer + 1x PIC mix into individual wells of a clean V-bottom 96-well plate. Fill one well per IP. Label the plate as "Ab-bead complex".
- Add 12 µL of the washed Protein A (or G) magnetic beads into each well containing IP buffer + PIC, and mix by pipetting up and down. Keep the remaining washed beads on ice.
- Obtain validated antibodies from their cold storage and thaw on ice, if required. Working on ice, dilute the antibodies with 1x Ab dilution buffer to the concentrations shown in Table 2.
- To each well of the Ab-bead complex plate, add 1 µL of the appropriate, diluted antibody (Table 1). Record the well to the antibody key. Using multichannel pipette, mix each row by pipetting up and down 20 times. Change tips between rows. Seal the Ab-bead complex plate very well with an aluminum plate cover and incubate at 4 °C on a rotating platform for at least 2 h. NOTE: This incubation can proceed until ready to start step 2.4.
- Cell lysis and chromatin digestion
- Working on ice, prepare 1x lysis buffer + 1x PIC and 1 mL of MNase I dilution buffer (Table 3) and keep them on ice .
- Retrieve the cell pellets from their -80 °C storage (or from ice, if freshly prepared). Thaw each cell pellet in a 37 °C water bath for a 10 s, then transfer to ice.
- To each cell pellet immediately add ice-cold 1x lysis buffer + 1x PIC to a final concentration of 10,000 cells/20 µL, and mix 10 times by pipetting up and down, without creating bubbles. For example, for 70,000 cells the final volume is 140 µL.
- Working on ice, aliquot 20 µL/well of the resulting lysates into a 96-well plate. Cover the plate with a plastic seal and incubate on ice for 20 min. Label the plate "MNase digestion" and record the wells to a template key. In order to assure an exact timing of the chromatin digestion reaction, do not proceed to the digestion with more than 2 rows of samples at a time.
- Just before the 20 min lysis is complete, dilute the MNase I enzyme with MNase I dilution buffer, to a final concentration of 20 U/µL, and keep it on ice.
- Working on ice, prepare the MNase I digestion master mix according to Table 4 and aliquot 20 µL of the mix per each row of samples plus 5 µL dead volume into one row of a 96-well reservoir plate and keep it on ice. For two rows, the volume should be: (20 µL x 2) + 5 µL = 45 µL.
- After the lysates finish incubating, remove the MNase digestion plate from ice. Using a multichannel pipette, add 20 µL of MNase I digestion master mix into each row of samples, and mix 10 times by pipetting up and down. Change tips between rows. Incubate at room temperature for exactly 5 min.
- After 5 min, to stop the reaction, use a multichannel pipette to add 6 µL of 250 µM ethylenediaminetetraacetic acid (EDTA) into each row of samples and mix up and down a few times. Change tips between rows. After EDTA addition, switch the setting of the pipette to 20 µL and mix 10 times by pipetting up and down to assure a complete stop of the digestion reaction. Change tips between rows.
- Using a multichannel pipette, to each row of the MNase digested samples, add 6 µL of 10x lysis buffer and mix well by pipetting up and down 10 times. Cover with a plastic seal and incubate on ice for 15 min.
- Input separation and pre-clearing
- Following the 15 min incubation, working on ice, pool all wells for the same cell pellet/template into one sterile, pre-chilled on ice, 1.5 mL tube (pre-labeled with the template ID) and mix thoroughly but slowly using a pipette to avoid detergent foaming.
- For each cell pellet/template, transfer 8 µL of the digested chromatin into a new sterile, 0.5 mL tube (pre-labeled with the template ID), and store at 4 °C overnight. This will serve as the input control.
- Distribute the remaining volume of the digested chromatin in 48 µL aliquots, into a new 96-well plate. Cell pellet/template 1 goes into wells A01-A06, cell pellet/template 2 goes into wells A07-A12, etc. Record the wells to a template key. Label the plate "Pre-clearing".
- Using a multichannel pipette, add 120 µL of 1x IP Buffer + 1x PIC and 12 µL of washed Protein A (or G) magnetic beads into each well of the Pre-clearing plate from step 2.3.3. Mix each row by pipetting up and down 10 times. Change tips between rows. Seal the plate very well with an aluminum plate cover and incubate on a rotating platform at 4 °C for a minimum of 2 h.
- Immunoprecipitation reaction
- Before starting this step make sure that the Ab-bead complex plate (step 2.1.10) and the Pre-clearing plate (step 2.3.4) have incubated for at least 2 h. Quick spin both plates by centrifuging for 10 s at 200 x g.
- Place the Ab-bead complex plate (from step 2.1.10) on a plate magnet and wait 15 s for the solution to become clear. Carefully remove and discard the supernatant using a pipette without disturbing the beads. Remove the plate from the plate magnet and keep it on ice.
- Place the Pre-clearing reaction plate (from step 2.3.4) on a plate magnet and wait 15 s for the beads to separate and for the solution to become clear. Without disturbing the beads, carefully transfer the supernatant using a pipette to the corresponding wells of the Ab-bead complex plate kept on ice and mix gently 15 times by pipetting up and down. Seal the plate well with an aluminum plate cover and incubate overnight (12-18 h) at 4 °C on a rotating platform. Re-label the plate "IP reaction".
3. DAY 2: ndCHIP-seq
- Washes and elution
- Set the heating mixer to 65 °C. On ice, prepare a low-salt wash buffer and high-salt wash buffer. Quick spin the IP reaction plate from step 2.4.3 for 10 s at 200 x g.
- Place the IP reaction plate on a plate magnet and wait 15 s for the solution to become clear. Using a multichannel pipette, without disturbing the beads, carefully remove and discard the supernatant. Take the plate off the plate magnet and place it on ice.
- To each row of samples in the IP reaction plate, add 150 µL of ice-cold low-salt wash buffer and mix slowly 10 times up and down to fully resuspend the beads.
- Place the IP reaction plate back onto the plate magnet, wait for the beads to separate, and using a multichannel pipette, without disturbing the beads remove and discard the supernatant. Place the plate back on ice and repeat steps 3.1.3 and 3.1.4 for a total of 2 washes.
- On ice, to each row of samples in the IP reaction plate, add 150 µL of the high salt wash buffer and mix slowly 10 times by pipetting up and down to fully resuspend the beads.
- Place the IP reaction plate back onto the plate magnet, wait for the beads to separate, and using a multichannel pipette, without disturbing the beads remove and discard the supernatant. Place the IP reaction plate on ice and pre-chill a new 96-well plate beside it.
- To each row of samples in the IP reaction plate, add 150 µL of the high salt wash buffer and mix slowly 10 times by pipetting up and down to fully resuspend the beads. After resuspension, transfer each row of samples to the corresponding row of the new, pre-chilled, 96-well plate. Label the plate "IP reaction". Discard the old plate.
- Place the new IP reaction plate on the plate magnet and wait for the beads to separate. Using a multichannel pipette, without disturbing the beads, carefully remove and discard the supernatant. Keep the plate at room temperature.
- To each row of samples in the IP reaction plate, add 30 µL of the ChIP elution buffer (EB) and mix slowly 10 times by pipetting up and down. Make sure to prevent bubble formation.
- Seal the plate well with a PCR cover and incubate in a heating mixer at 65 °C for 1.5 h with a mixing speed of 1,350 rpm.
- After 1.5 h incubation, spin down the IP reaction plate at 200 x g for 1 min at 4 °C. Change the setting of the heating mixer to 50 °C.
- After the spin, place the IP reaction plate on a plate magnet and wait for the solution to clear. Using a multichannel pipette, without disturbing the beads, carefully transfer 30 µL of the supernatant into a new 96-well plate. Use fresh tips for each row. Label the plate "IP reaction" and keep it at room temperature.
- Protein digestion
- Retrieve 30% PEG/1 M NaCl magnetic bead17 (buffer composition table) solution from the 4 °C refrigerator and keep it at room temperature for at least 30 min. Critical: Ensure that the bead solution fully reaches room temperature before proceeding.
- Retrieve input control samples from 4 °C storage (step 2.3.2) and quick spin. Measure the volume using a pipette and add ultrapure water to each input control for a final volume of 30 µL. Transfer the input control samples to the pre-selected input wells (empty wells) in the IP reaction plate (step 3.1.12). Record the well to the sample key.
- On ice, prepare the Protein Digestion Master Mix as shown in Table 5 and aliquot equal volume into one row of a 96-well reservoir plate.
- Using a multichannel pipette, to each row of samples in the IP reaction plate, add 40 µL of the Protein Digestion Master Mix and mix slowly 10 times up and down. Seal the plate well with a PCR cover, spin down at 200 x g for 1 min, and incubate in a heating mixer at 50 °C for 30 min set at 650 rpm. While the plate is incubating, prepare fresh 70% ethanol (EtOH) and keep it at room temperature.
- After the 30 min incubation is complete, spin the IP reaction plate at 200 x g for 1 min, 4 °C. Keep the plate at room temperature. NOTE: The total volume should be now ~70 µL/well.
- Bead clean-up using 30% PEG/1 M NaCl magnetic bead solution
- Aliquot the room temperature 30% of PEG/1 M NaCl magnetic bead solution into one row of a clean 96-well reservoir plate (75 µL per sample x # of rows).
- Working at room temperature, using a multichannel pipette, add 70 µL (1:1 ratio) of the 30% PEG/1 M NaCl magnetic bead solution to each row of samples in the IP reaction plate and mix 10 times by pipetting up and down. Change tips between rows. Incubate the plate for 15 min at room temperature.
- Place the plate on the plate magnet and incubate for 5 min to allow the beads to separate.
- Using a pipette, without disturbing the beads, carefully remove and discard the supernatant. Keep the beads.
- While the IP reaction plate is still on the plate magnet, using a multichannel pipette, to each row of samples, add 150 µL of room temperature 70% EtOH and incubate for 30 s. After 30 s, using a multichannel pipette, remove and discard the supernatant. Repeat this step one more time for a total of 2 washes.
- After the second EtOH wash, incubate the 'dry' plate on the plate magnet for 3 min. Visually inspect the plate to make sure that all the EtOH is evaporated. If not, incubate the plate for 1 min. Over-drying the beads results in a lower yield.
- Take the plate off the plate magnet and using a multichannel pipette, add 35 µL EB (Table of Materials). Mix well by pipetting 10 times up and down. Change tips between rows. Incubate at room temperature for 3 min to elute.
- After incubation, place the IP reaction plate back onto the plate magnet and incubate for 2 min. The beads should separate and the solution will become clear.
- Using a multichannel pipette, without disturbing the beads, carefully transfer the supernatant into the corresponding wells of a new 96-well plate.
- Seal the plate with an aluminum plate cover, label "IPed + Input DNA", and store at 4 °C overnight, or at -20 °C for long term (>48 h) storage.
4. DAY 3: Library Construction
- End repair and phosphorylation
- The input for the End Repair/Phosphorylation reaction is the IPed + Input plate from step 3.3.10. After thawing, spin the plate down at 200 x g for 1 min at 4 °C and keep it on ice.
- Retrieve from -20 °C freezer all of the reagents (except enzymes) required for the End Repair/Phosphorylation reaction (Table 6) and thaw them at room temperature, then immediately transfer to ice.
- Working on ice, follow Table 6 to set up the End Repair/Phosphorylation Master Mix in a sterile, 1.5 mL microcentrifuge tube. After the addition of all of the non-enzyme components, mix well by pulse-vortexing and place the tube back on ice.
- Retrieve the relevant enzymes from their cold storage and transport to the bench in a chilled tube cool rack. Mix each enzyme by flicking the tube, quick spin, and place it back in the chilled tube cool rack. When pipetting the enzyme, aspirate slowly to assure an accurate volume transfer. After the addition, wash the tip in the master mix by pipetting up and down.
- Once the enzymes are added, gently pulse-vortex the master mix 5 times to assure an even distribution of all the components, then quick spin and immediately place the tube back on ice.
- On ice, aliquot an appropriate volume of the End Repair/Phosphorylation Master Mix into one row of a new 96-well reservoir plate; volume to be aliquoted: (15 µL x # of rows) + 5 µL dead volume. An example calculation for two rows: (15 µL x 2) + 5 µL = 35 µL/well.
- Using a multichannel pipette, add 15 µL of the End Repair/Phosphorylation Master Mix to each row of samples and mix 10 times by pipetting up and down. Change tips between rows. Seal the plate with a plastic cover, spin down at 200 x g for 1 min at 4 °C, and incubate at room temperature for 30 min.
- Bead clean-up after end repair and phosphorylation
- From the 4 °C refrigerator, retrieve the 20% PEG/1 M NaCl magnetic bead solution and 30% PEG/1 M NaCl solution and incubate at room temperature for at least 30 min. NOTE: The 30% PEG/1 M NaCl solution DOES NOT contain magnetic beads.
- After both solutions reach room temperature, for each sample prepare 80 µL of 1:2 mix of 30% PEG/1 M NaCl and 20% PEG/1 M NaCl magnetic bead solutions. An example for 24 samples: 80 µL x 24 = 1,920 µL (640 µL of 30% PEG/1 M NaCl and 1,288 µL of 20% PEG/1 M NaCl magnetic bead solutions).
- Aliquot the bead solution mix, equal volume, into one row of a 96-well reservoir plate and keep it at room temperature. Aliquot EB buffer (40 µL per sample x # of rows) into one row of a clean 96-well reservoir plate. An example for two rows: 40 µL x 2 = 80 µL/well.
- Using a multichannel pipette, add 75 µL of the bead mix prepared in step 4.2.2 into each row of samples from step 4.1.7 and mix up and down 10 times. Change tips between rows. Incubate at room temperature for 15 min. Continue with the bead clean-up procedure as outlined in steps 3.3.3-3.3.10. Cover the plate with a plastic seal, quick spin and place it on ice. Proceed to step 4.3.
- A-Tailing reaction
- Retrieve from the -20 °C freezer all of the reagents (except enzymes) required for the A-Tailing reaction (Table 7), thaw them at room temperature then immediately transfer to ice.
- Working on ice, follow Table 7 to set up the A-Tailing Master Mix in a sterile, 1.5 mL microcentrifuge tube. Follow the general enzymatic brew setup instructions as outlined in steps 4.1.4-4.1.6.
- Using a multichannel pipette, add 15 µL of the A-Tailing Master Mix to each row of samples and mix 10 times by pipetting up and down. Change tips between rows. Seal the plate with a PCR cover, spin down at 200 x g for 1 min at 4 °C, and incubate in a thermocycler at 37 °C for 30 min. After the incubation, spin the plate down at 200 x g for 1 min and keep it at room temperature.
- Bead clean-up after A-Tailing
- Perform the steps described in step 4.2. Cover the plate with a plastic seal, label "A-tail + BC", quick spin, and place it on ice. Proceed to step 4.5.
- Adaptor ligation
- Retrieve from the -20 °C freezer all of the reagents (except enzymes) required for the adapter ligation reaction (Table 8), thaw them at room temperature then immediately transfer to ice.
- Retrieve 10 µM PE adapter (Supplemental Table 1) stock solution and dilute to 0.5 µM using EB. Mix well by pulse vortexing. The volume required is 3 µL x # of samples. Working on ice, aliquot the 0.5 µM PE adapter, equal volume, into 12 wells of a clean 96-well reservoir plate. Keep it on ice.
- Working on ice, follow Table 8 to set up the Adapter Ligation Master Mix in a sterile, 1.5 mL microcentrifuge tube. Follow the general enzymatic brew setup instructions as outlined in steps 4.1.4-4.1.6. Make sure that the 5X Quick Ligation Buffer is fully thawed and very well mixed before use.
- On ice, aliquot an appropriate volume of the Adapter Ligation Master Mix into one row of a new 96-well reservoir plate. For example, calculation for two rows: (23 µL x 2) + 5 µL = 51 µL/well. Keep the plate on ice.
- Using a multichannel pipette, add 2 µL of the 0.5 µM Paired End (PE) Adapter into each row of samples from step 4.4.1 and mix. Change tips between rows.
- Using a multichannel pipette, add 23 µL of the Adapter Ligation Master Mix to each row of samples and mix 15 times by pipetting up and down. Change tips between rows. Seal the plate with a metal cover, label "Ligation", spin down at 200 x g for 1 min at 4 °C, and incubate at room temperature overnight.
- Day 4: Bead clean-up #1 after adapter ligation
- From the 4 °C refrigerator, retrieve the 20% PEG/1 M NaCl magnetic bead solution and incubate at room temperature for at least 30 min. While the solution is equilibrating prepare fresh 70% EtOH.
- Aliquot the 20% PEG/1 M NaCl magnetic bead solution, 55 µL per sample, into one row of a 96-well reservoir plate and keep it at room temperature. An example for two rows: 55 µL x 2 = 110 µL/well.
- Aliquot EB buffer, 50 µL per sample, into one row of a clean 96-well reservoir plate. An example for two rows: 50 µL x 2 = 100 µL/well.
- Using a multichannel pipette, add 48 µL of the 20% PEG/1 M NaCl magnetic bead solution into each row of samples in the Ligation plate from step 4.5.6 and mix up and down 10 times. Change tips between rows. Incubate at room temperature for 15 min. Continue with the bead clean-up procedure as outlined in steps 3.3.3-3.3.7.
- Take the plate off the plate magnet and using a multichannel pipette, add 45 µL EB (Table of Materials). Mix well by pipetting 10 times up and down. Change tips between rows. Incubate for at room temperature for 3 min to elute.
- After incubation, place the IP reaction plate back onto the plate magnet and incubate for 2 min. Using a multichannel pipette, without disturbing the beads, carefully transfer the supernatant into the corresponding wells of a new 96-well plate. Label the plate "Ligation + 1 BC".
- To each well add 5 µL of 10x PCR high fidelity buffer and mix well by pipetting up and down. Change tips between rows. Cover the plate with a plastic seal, quick spin, and keep it at room temperature.
- Bead clean-up #2 after adapter ligation
- Perform bead clean-up as described in section 4.6 with the following changes: add 60 µL of 20% PEG/1 M NaCl magnetic bead solution to each active well in the Ligation + 1 BC plate (step 4.6.7) and elute from the beads using 35 µL of EB buffer. Label the plate "Ligation + 2 BC" and keep it on ice.
- PCR amplification
- Retrieve from the -20 °C freezer all the reagents (except enzymes) required for the PCR reaction (Table 9), thaw them at room temperature then immediately place them on ice.
- Working on ice, follow Table 9 to set up the PCR Master Mix in a sterile, 1.5 mL microcentrifuge tube. Follow the general brew set-up instructions as outlined in steps 4.1.4-4.1.5. On ice, aliquot an appropriate volume of the PCR Master Mix into one row of a new 96-well reservoir plate. Keep it on ice. See step 4.5.4 for sample calculations.
- Working on ice, using a multichannel pipette, add 2 µL of each unique 12.5 µM PCR reverse indexing primer (Supplemental Table 2) into each well in the Ligation + 2 BC plate (step 4.7.1) and mix slowly up and down. Change tips between rows. Keep the plate on ice.
- Working on ice, using a multichannel pipette, add 23 µL of the PCR master mix into each row of samples from step 4.8.3 and mix 10 times by pipetting up and down. Seal the plate with a PCR cover, spin it down at 200 x g for 1 min, and incubate in a thermocycler (see Table 10 for PCR cycling conditions).
- Bead clean-up after PCR amplification
- After PCR amplification spin the plate at 200 x g for 1 min, 4 °C. Perform the bead clean-up as described in section 4.6 with the following changes: add 51 µL of 20% PEG/1 M NaCl magnetic bead solution to each PCR reaction and elute from the beads using 25 µL of EB buffer. Seal the plate with an aluminum cover and spin down at 200 x g for 1 min. Store the samples at -20 °C.
- To validate constructed libraries, perform DNA quantification using a fluorescence-based assay, visualize the final product using a chip-based capillary electrophoresis analyzer (high sensitivity assay), and run enrichment quantitative PCR (qPCR) (See Representative Results, Validation of ndChIP-seq library quality by qPCR).
Representative Results
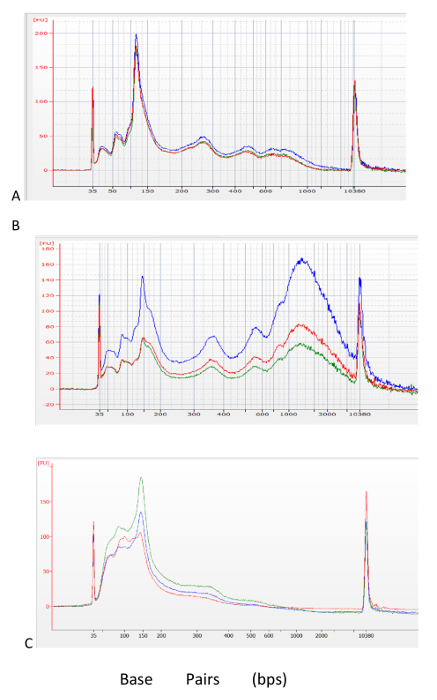
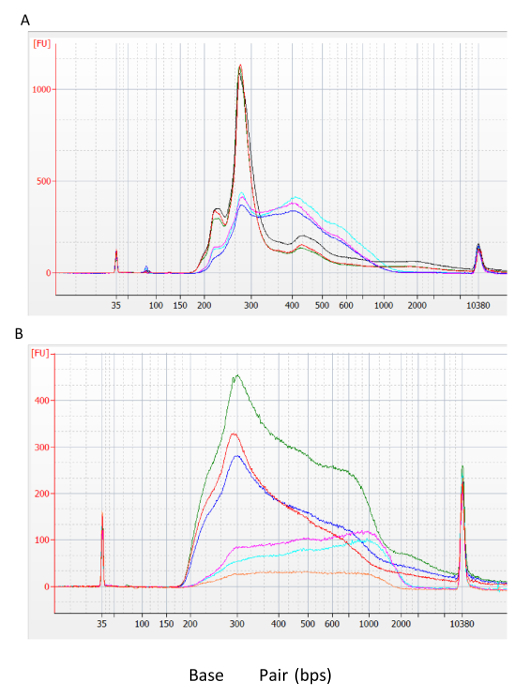
Chromatin Digestion Profiles Optimization of the MNase digestion is essential for the success of this protocol. It is crucial to generate a digestion profile dominated by single nucleosome fragment sizes, while not over-digested, to allow for recovery of higher order nucleosome fragments. An ideal digestion profile consists of a majority of single nucleosome fragments with a small fraction representing fragments smaller and larger than single nucleosomes. Figure 1 shows examples of an ideal, over-digested, and under-digested size distribution profiles. Note that sub-optimal digestion of chromatin will also be apparent in the profile of the sequencing library generated from the IP material (Figure 2).
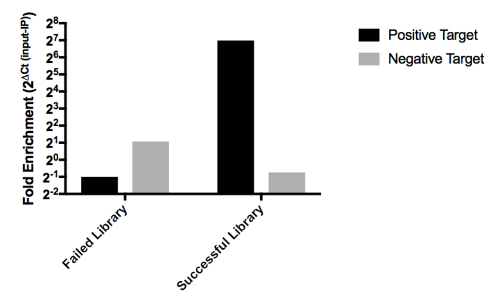
Validation of ndChIP-seq Library Quality by qPCR qPCR is a well-established method for assessment of the quality of ChIP18,19,20. When performing ndChIP-seq on 10,000 cells the yield of nucleic acid after IP will be below 1 ng. Therefore, it is essential to perform qPCR after library construction to assess the relative enrichment of target regions over background. To provide a background estimate, libraries constructed from the MNase digested chromatin (Input) are generated. For each IP library, two sets of primers are needed (see SupplementalTable 3 for a list of primers for commonly used histone marks). One primer set should be specific for a genomic region that is consistently associated with the histone modification of interest (positive target) and another region that is not marked with the histone modification of interest (negative target). The quality of the ChIP-seq library will be assessed as fold enrichment with respect to input library. Fold enrichment can be calculated using the following equation that assumes exponential amplification of the target genomic region: 2Ctinput- CtIP. Our custom made R statistical software package, qcQpcr_v1.2, is suitable for qPCR enrichment analysis of low input native ChIP-seq libraries (Supplemental Code Files). Figure 3 represents a qPCR result for successful and unsuccessful ChIP-seq libraries. The minimum expected fold enrichment value for good quality ndChIP-seq libraries are 16 for narrow marks, such as H3K4me3, and 7 for broad marks, for example H3K27me3.
Modeling MNase Accessibility Computational analysis of ChIP-seq is complex and unique for each experimental setting. A set of guidelines established by International Human Epigenomic Consortium (IHEC) and The Encyclopedia of DNA Elements (ENCODE) can be used to assess the quality of the ChIP-seq libraries21. It is important to note that the sequencing depth of the libraries impacts the detection and resolution of enriched regions20. The number of peaks detected increase and approaches a plateau as read depth increases. We recommend ndChIP-seq libraries to be sequenced in accordance with the IHEC recommendations of 50 million paired-reads (25 million fragments) for narrow marks (e.g., H3K4me3) and 100 million paired-reads (50 million fragments) for broad marks (e.g., H3K27me3) and input22. These sequencing depths provide sufficient sequence alignments for detection of the most significant peaks using widely used ChIP-seq peak callers, such as MACS2 and HOMER, without reaching saturation23,24. A high quality mammalian ndChIP-seq library has a PCR duplicate rate of <10% and reference genome alignment rate of > 90% (including duplicated reads). Successful ndChIP-seq libraries will contain highly correlated replicates with a significant portion (> 40%) of aligned reads within MACS222 identified enriched peaks and inspection of aligned reads on a genome browser should reveal visually detectable enrichments compared to the input library (Figure 4). In addition, ndChIP-seq can be used to assess nucleosome density by utilizing a Gaussian mixture distribution algorithm (w1 * n(x; μ1,σ1) + w2 * n(x; μ2,σ2) = 1) at MACS2 identified enriched regions to model nucleosome density as defined by MNase accessible boundaries. In this model, w1 represents mono-nucleosome distribution weight and w2 represents di-nucleosome distribution weight. Where w1 is greater than w2, there is dominance of mono-nucleosome fragments and vice versa. This analysis requires that libraries be sequenced in a paired-end fashion so that fragment sizes can be defined. In order to apply the Gaussian mixture distribution algorithm, statistically significant enriched regions are first identified. We suggest peak calling with MACS2 using Input as a control and with default settings for narrow marks and a q value cutoff of 0.01 for broad marks. A number of statistical packages employing a Gaussian mixture distribution algorithm are available from widely used statistical software packages. Utilizing average fragment size, determined by paired-end read boundaries of the IPed samples, distributions at MACS2 identified enriched promoters, and a Gaussian mixture distribution algorithm can be applied to each promoter using the R-statistical package Mclust version 3.025 to calculate a weighted distribution. In this application, we recommend eliminating promoters containing less than 30 aligned fragments because below this threshold the resulting weight estimates become unreliable. A good quality ndChIP-seq library generates a Gaussian mixture distribution that consist of two major components with mean values corresponding to mono-, di-nucleosome fragment lengths.
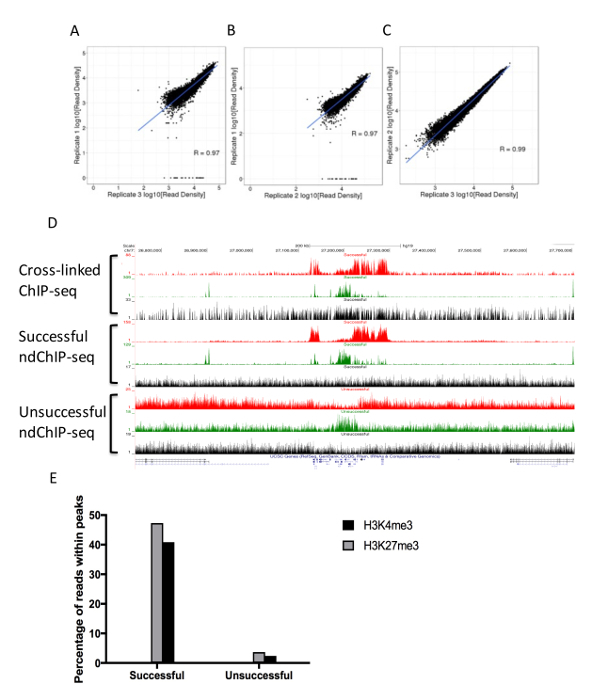
Figure 1: Assessment of MNase digestion before library generation. Chip-based capillary electrophoresis analyzer profiles of an optimal MNase digested (A), under-digested (B), and over-digested (C) chromatin. Biological replicates are shown as blue, red, and green traces. Please click here to view a larger version of this figure.
Figure 2: Assessment of MNase digestion after library generation. (A) Post library construction profiles of optimally digested input (biological replicates; red, green, black) and IP (biological replicates; cyan, purple, blue) and (B) sub-optimal input (biological replicates; red, green, blue) and IP (biological replicates; cyan, purple, orange) libraries. Please click here to view a larger version of this figure.
Figure 3: Post library construction quantitative PCR can be used to assess the quality of ndChIP-seq libraries. Fold enrichment of H3K4me3 IP libraries with respect to input libraries is calculated as 2(Ct of input - Ct of IP) for positive and negative targets using qcQpcr_v1.2. Please click here to view a larger version of this figure.
Figure 4: Representative ndChIP-seq library constructed from 10,000 primary CD34+ cord blood cells. Pearson correlation of H3K4me3 signal (reads per million mapped reads) calculated in the promoters (TSS+/-2Kb) between 3 biological replicates, (A) replicate 1 and 2, (B) replicate 1 and 3, (C) replicate 2 and 3. (D) UCSC browser view of the HOXA gene cluster of cross-linked ChIP-seq generated from 1 million cells per IP, successful ndChIP-seq from 10,000 cells per IP, and unsuccessful ndChIP-seq from 10,000 cells per IP. (red: H3K27me3, green: H3K4me3, and black: Input). (E) Fraction of mapped reads within MACS2 identified enriched regions of H3K4me3 (black) and H3K27me3 (grey). Please click here to view a larger version of this figure.
| Buffer Composition |
| A.1. Immunoprecipitation buffer (IP) |
| 20 mM Tris-HCl pH 7.5 |
| 2 mM EDTA |
| 150 mM NaCl |
| 0.1% Triton X-100 |
| 0.1% Deoxycholate |
| 10 mM Sodium Butyrate |
| A.2. Low Salt Wash buffer |
| 20 mM Tris-HCl pH 8.0 |
| 2 mM EDTA |
| 150 mM NaCl |
| 1% Triton X-100 |
| 0.1% SDS |
| A.3. High Salt Wash buffer |
| 20 mM Tris-HCl pH 8.0 |
| 2 mM EDTA |
| 500 mM NaCl |
| 1% Triton X-100 |
| 0.1% SDS |
| A.4. ChIP Elution buffer |
| 100 mM NaHCO3 |
| 1% SDS |
| A.5. 1x Lysis buffer – 1 mL |
| 0.1% Triton |
| 0.1% Deoxycholate |
| 10 mM Sodium Butyrate |
| A.6. Ab dilution buffer |
| 0.05% (w/v) Azide |
| 0.05% broad spectrum antimicrobial (e.g. ProClin 300) |
| in PBS |
| A.7. 30% PEG/1M NaCl Magnetic Bead Solution (reference16) |
| 30% PEG |
| 1 M NaCl |
| 10 mM Tris HCl pH 7.5 |
| 1 mM EDTA |
| 1 mL of washed super-paramagnetic beads |
| A.8. 20% PEG/1M NaCl Magnetic Bead Solution (reference16) |
| 30% PEG |
| 1 M NaCl |
| 10 mM Tris HCl pH 7.5 |
| 1 mM EDTA |
| 1 mL of washed super-paramagnetic beads |
Table 1: ndChIP-seq buffer composition.
| Histone Modification | Concentration (µg/µL) |
| H3K4me3 | 0.125 |
| H3K4me1 | 0.25 |
| H3K27me3 | 0.125 |
| H3K9me3 | 0.125 |
| H3K36me3 | 0.125 |
| H3K27ac | 0.125 |
Table 2: Antibody amount required for ndChIP-seq.
| Reganet | Volume (µL) |
| Ultra Pure Water | 478 |
| 1 M Tris-HCl pH 7.5 | 10 |
| 0.5 M EDTA | 10 |
| 5 M NaCl | 2 |
| Glycerol | 500 |
| Total Volume | 1,000 |
Table 3: Recipe for MNase dilution buffer.
| Reagent | Volume (µL) |
| Ultra Pure Water | 13 |
| 20 mM DTT | 1 |
| 10x MNase Buffer | 4 |
| 20 U/µl Mnase | 2 |
| Total Volume | 20 |
Table 4: Recipe for MNase Master Mix.
| Reagent | Volume (µL) |
| Elution Buffer | 30 |
| Buffer G2 | 8 |
| Protease | 2 |
| Total Volume | 40 |
Table 5: Recipe for DNA Purification Master Mix.
| Reagent | Volume (µL) |
| Ultra Pure Water | 3.3 |
| 10x Restriction Endonuclease Buffer (e.g. NEBuffer) | 5 |
| 25 mM ATP | 2 |
| 10 mM dNTPs | 2 |
| T4 Polynucleotide Kinase (10 U/µl) | 1 |
| T4 DNA polymerase (3 U/µl) | 1.5 |
| DNA polymerase I, Large (Klenow) Fragment (5 U/µl) | 0.2 |
| Total Volume | 15 |
Table 6: Recipe for End Repair Master Mix.
| Reagent | Volume (µL) |
| Ultra Pure Water | 8 |
| 10x Restriction Endonuclease Buffer (e.g. NEBuffer) | 5 |
| 10 mM dATP | 1 |
| Klenow (3'-5' exo-) | 1 |
| Total Volume | 15 |
Table 7: Recipe for A-Tailing Master Mix.
| Reagent | Volume (µL) |
| Ultra Pure Water | 4.3 |
| 5x Quick ligation buffer | 12 |
| T4 DNA ligase (2000 U/µl) | 6.7 |
| Total Volume | 23 |
Table 8: Recipe for Adapter Ligation Master Mix.
| Reagent | Volume (µL) |
| Ultra Pure Water | 7 |
| 25 uM PCR primer 1.0 | 2 |
| 5x HF buffer | 12 |
| DMSO | 1.5 |
| DNA Polymerase | 0.5 |
| Total Volume | 23 |
Table 9: Recipe for PCR Master Mix.
| Temprature (°C) | Duration (s) | Number of Cycles |
| 98 | 60 | 1 |
| 98 | 30 | |
| 65 | 15 | 10 |
| 72 | 15 | |
| 72 | 300 | 1 |
| 4 | hold | hold |
Table 10: PCR run method.
| Oligo | Sequence | Modification |
| PE_adapter 1 | 5’- /5Phos/GAT CGG AAG AGC GGT TCA GCA GGA ATG CCG AG -3’ | 5’ Modification: Phosphorylation |
| PE_adapter 2 | 5’ - ACA CTC TTT CCC TAC ACG ACG CTC TTC CGA TC*T - 3’ | 3’Modification: *T is a phosphorothioate bond |
Supplemental Table 1: Oligo sequences for generation of PE adapter.
| Primer Name | Sequence | Index | IndexRevC (To be used for sequencing) |
| PCR reverse indexing primer 1 | CAAGCAGAAGACGGCATACGAGATCGTGATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | CGTGAT | atcacg |
| PCR reverse indexing primer 2 | CAAGCAGAAGACGGCATACGAGATCTGATCCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | CTGATC | gatcag |
| PCR reverse indexing primer 3 | CAAGCAGAAGACGGCATACGAGATGGGGTTCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | GGGGTT | aacccc |
| PCR reverse indexing primer 4 | CAAGCAGAAGACGGCATACGAGATCTGGGTCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | CTGGGT | acccag |
| PCR reverse indexing primer 5 | CAAGCAGAAGACGGCATACGAGATAGCGCTCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | AGCGCT | agcgct |
| PCR reverse indexing primer 6 | CAAGCAGAAGACGGCATACGAGATCTTTTGCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | CTTTTG | caaaag |
| PCR reverse indexing primer 7 | CAAGCAGAAGACGGCATACGAGATTGTTGGCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | TGTTGG | ccaaca |
| PCR reverse indexing primer 8 | CAAGCAGAAGACGGCATACGAGATAGCTAGCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | AGCTAG | ctagct |
| PCR reverse indexing primer 9 | CAAGCAGAAGACGGCATACGAGATAGCATCCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | AGCATC | gatgct |
| PCR reverse indexing primer 10 | CAAGCAGAAGACGGCATACGAGATCGATTACGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | CGATTA | taatcg |
| PCR reverse indexing primer 11 | CAAGCAGAAGACGGCATACGAGATCATTCACGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | CATTCA | tgaatg |
| PCR reverse indexing primer 12 | CAAGCAGAAGACGGCATACGAGATGGAACTCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | GGAACT | agttcc |
| PCR reverse indexing primer 13 | CAAGCAGAAGACGGCATACGAGATACATCGCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | ACATCG | cgatgt |
| PCR reverse indexing primer 14 | CAAGCAGAAGACGGCATACGAGATAAGCTACGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | AAGCTA | tagctt |
| PCR reverse indexing primer 15 | CAAGCAGAAGACGGCATACGAGATCAAGTTCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | CAAGTT | aacttg |
| PCR reverse indexing primer 16 | CAAGCAGAAGACGGCATACGAGATGCCGGTCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | GCCGGT | accggc |
| PCR reverse indexing primer 17 | CAAGCAGAAGACGGCATACGAGATCGGCCTCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | CGGCCT | aggccg |
| PCR reverse indexing primer 18 | CAAGCAGAAGACGGCATACGAGATTAGTTGCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | TAGTTG | caacta |
| PCR reverse indexing primer 19 | CAAGCAGAAGACGGCATACGAGATGCGTGGCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | GCGTGG | ccacgc |
| PCR reverse indexing primer 20 | CAAGCAGAAGACGGCATACGAGATGTATAGCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | GTATAG | ctatac |
| PCR reverse indexing primer 21 | CAAGCAGAAGACGGCATACGAGATCCTTGCCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | CCTTGC | gcaagg |
| PCR reverse indexing primer 22 | CAAGCAGAAGACGGCATACGAGATGCTGTACGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | GCTGTA | tacagc |
| PCR reverse indexing primer 23 | CAAGCAGAAGACGGCATACGAGATATGGCACGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | ATGGCA | tgccat |
| PCR reverse indexing primer 24 | CAAGCAGAAGACGGCATACGAGATTGACATCGGTCTCGGCATTCCTGCTGAACCGCTCTTCCGATCT | TGACAT | atgtca |
Supplemental Table 2: PCR reverse indexing primer sequences.
| Primers | Sequence | |
| ZNF333_genic_H3K9me3_F | 5'-AGCCTTCAATCAGCCATCATCCCT-3' | |
| ZNF333_genic_H3K9me3_R | 5'-TCTGGTATGGGTTCGCAGATGTGT-3' | |
| HOXA9-10_F | 5'-ACTGAAGTAATGAAGGCAGTGTCGT-3' | |
| HOXA9-10_R | 5'-GCAGCAYCAGAACTGGTCGGTG-3' | |
| GAPDH_genic_H3K36me3_F | 5'-AGGCAACTAGGATGGTGTGG-3' | |
| GAPDH_genic_H3K36me3_R | 5'-TTGATTTTGGAGGGATCTCG-3' | |
| GAPDH-F | 5'-TACTAGCGGTTTTACGGGCG-3' | |
| GAPDH-R | 5'-TCGAACAGGAGGAGCAGAGAGCGA-3' | |
| Histone modification | Positive Target | Negative Target |
| H3K4me3 | GAPDH | HOXA9-10 |
| H3K4me1 | GAPDH_genic | ZNF333 |
| H3K27me3 | HOXA9-10 | ZNF333 |
| H3K27ac | GAPDH | ZNF333 |
| H3K9me3 | ZNF333 | HOXA9-10 |
| H3K36me3 | GAPDH_genic | ZNF333 |
Supplemental Table 3: A list of primers for commonly used histone marks (H3K4me3, H3K4me1, H3K27me3, H3K27ac, H3K9me3, and H3K36me3).
Supplemental File 1: ndChIP-seq WorkSheet. Please click here to download this file.
Supplemental Code Files: qcQpcr_v1.2. R statistical software package for qPCR enrichment analysis of low input native ChIP-seq libraries. Please click here to download this file.
Discussion
Given the combinatorial nature of chromatin modification and nucleosome positioning in transcriptional regulation, a method that enables simultaneous measurements of these features is likely to provide new insights into epigenetic regulation. The ndChIP-seq protocol presented here is a native ChIP-seq protocol optimized to enable simultaneous interrogation of histone modification and nucleosome density in rare primary cells9,10. ndChIP-seq utilizes enzymatic digestion of chromatin that, when coupled to paired-end massively parallel sequencing and a Gaussian mixture distribution model, allows for the investigation histone modifications at single nucleosome level and the deconvolution of epigenomic profiles driven by heterogeneity within a population. Using this protocol, we have previously reported a unique distribution of immuno-precipitated fragment sizes, determined by paired-end read boundaries, associated with specific chromatin states defined by ChromHMM10,24.
The quality of a ndChIP-seq library depends on multiple factors, such as antibody specificity and sensitivity, optimal MNase digestion conditions, and quality of the chromatin. The specificity of the antibodies used is crucial in producing successful ndChIP-seq library. An ideal antibody shows high affinity against the epitope of interest with little cross reactivity with other epitopes. It is equally important to choose magnetic beads with the highest affinity for the antibody of choice.
MNase digestion is a critical and time- and concentration-sensitive reaction in this protocol. Therefore, when processing multiple samples, it is important that each reaction is incubated for an equivalent amount of time (see step 2.2). The quality of the chromatin is another factor that significantly effects the outcome of ndChIP-seq. Fragmented chromatin leads to a sub-optimal MNase digestion profile and results in libraries with a low signal to noise ratio. Primary samples with low cell viability or degraded chromatin, such as formalin-fixed paraffin-embedded (FFPE) tissue are not recommended for this protocol.
The addition of a PIC during chromatin extraction reduces undesired (i.e., random) chromatin fragmentation and preserves integrity of histone tails. As such, PIC needs to be added to lysis buffer and immunoprecipitation buffer just prior to use. While selecting cells via flow cytometry, select for viable cells and ensure cells are sorted at a low flow rate to increase the accuracy of the cell number estimate and viability of cells. Avoid sorting cells directly into lysis buffer. The sheath buffer will dilute the lysis buffer and prevents effective permeabilization of the cell membrane to MNase. Depending on a type of cells or organism, titration of MNase may be required to obtain optimal digestion.
ndChIP-seq on mammalian cells requires a minimum sequencing depth of 50 million paired-reads (25 million fragments) for narrow marks and 100 million paired-reads (50 million fragments) for broad marks and input. The Gaussian mixture distribution algorithm will not perform optimally on libraries that have been sequenced to a depth significantly below this recommendation. ndChIP-seq will not classify promoters with little separation between the weighted distribution value for mono- and di-nucleosome fragment lengths into mono- or di-nucleosome dominated promoters. Therefore, these promoters must be removed in the subsequent analysis. Biological replicates can be generated to increase confidence in predicted distributions and identify technical variability in the MNase digestion and library construction.
Unlike previous iterations of native ChIP-seq protocols, ndChIP-seq provides the means to investigate combinatorial effect of chromatin structure and histone modification by utilizing fragment size post immunoprecipitation to integrate nucleosome density, determined by MNase accessibility, with histone modification measurements. Application of ndChIP-seq to primary cells and tissues will provide novel insights into the integrative nature of epigenetic regulation and permit identification of epigenetic signatures due to heterogeneity within the population.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
A.L. was supported by a Canadian Graduate Scholarship from the Canadian Institutes of Health Research. This work was supported by grants from Genome British Columbia and the Canadian Institutes of Health Research (CIHR-120589) as part of the Canadian Epigenetics, Environment and Health Research Consortium Initiative and by the Terry Fox Research Institute Program Project Grant #TFF-122869 to M.H. and a Terry Fox Research Institute New Investigator Award (Grant # 1039) to M.H.
References
- Simpson RT. Nucleosome Positioning: Occurrence, Mechanisms, and Functional Consequences. Prog. Nucleic Acid Res. Mol. Biol. 1991;40:143–184. doi: 10.1016/s0079-6603(08)60841-7. Issue C. [DOI] [PubMed] [Google Scholar]
- Kornberg RD, Lorch Y. Twenty-five years of the nucleosome, fundmamental particle of the eukaryotic chromosome. Cell. 1999;98:285–294. doi: 10.1016/s0092-8674(00)81958-3. [DOI] [PubMed] [Google Scholar]
- Schones DE, et al. Dynamic regulation of nucleosome positioning in the human genome. Cell. 2008;132(5):887–898. doi: 10.1016/j.cell.2008.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal E, et al. A genomic code for nucleosome positioning. Nature. 2006;442(7104):772–778. doi: 10.1038/nature04979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiio Y, Eisenman RN. Histone sumoylation is associated with transcriptional repression. PNAS. 2003;100(23):13225–13230. doi: 10.1073/pnas.1735528100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Lorzadeh A, Hirst M. Regulatory variation: An emerging vantage point for cancer biology. Wiley Interdiscip. Rev. Syst. Biol. Med. 2014;6(1):37–59. doi: 10.1002/wsbm.1250. [DOI] [PubMed] [Google Scholar]
- Jiang J, et al. Investigation of the acetylation mechanism by GCN5 histone acetyltransferase. PLoS ONE. 2012;7(5) doi: 10.1371/journal.pone.0036660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kouzarides T. Histone methylation in transcriptional control. Curr. Opin. Genet. Dev. 2002;12(2):198–209. doi: 10.1016/s0959-437x(02)00287-3. [DOI] [PubMed] [Google Scholar]
- Brind'Amour J, Liu S, Hudson M, Chen C, Karimi MM, Lorincz MC. An ultra-low-input native ChIP-seq protocol for genome-wide profiling of rare cell populations. Nat. Commun. 2015;6:6033. doi: 10.1038/ncomms7033. [DOI] [PubMed] [Google Scholar]
- Lorzadeh A, et al. Nucleosome Density ChIP-Seq Identifies Distinct Chromatin Modification Signatures Associated with MNase Accessibility. Cell Rep. 2016;17(8):2112–2124. doi: 10.1016/j.celrep.2016.10.055. [DOI] [PubMed] [Google Scholar]
- Noll M, Kornberg RD. Action of micrococcal nuclease on chromatin and the location of histone H1. J. Mol. Biol. 1977;109(3):393–404. doi: 10.1016/s0022-2836(77)80019-3. [DOI] [PubMed] [Google Scholar]
- Skene PJ, Henikoff S. A simple method for generating high-resolution maps of genome wide protein binding. eLife. 2015;4:09225. doi: 10.7554/eLife.09225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brogaard K, Xi L, Wang J-P, Widom J. A map of nucleosome positions in yeast at base-pair resolution. Nature. 2012. [DOI] [PMC free article] [PubMed]
- Cui K, Zhao K. Genome-wide approaches to determining nucleosome occupancy in metazoans using MNase-Seq. Methods Mol. Biol. 2012;833:413–419. doi: 10.1007/978-1-61779-477-3_24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mieczkowski J, et al. MNase titration reveals differences between nucleosome occupancy and chromatin accessibility. Nat. Commun. 2016;7:11485. doi: 10.1038/ncomms11485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu S, Campbell HM, Dittel BN, Ray A. Purification of Specific Cell Population by Fluorescence Activated Cell Sorting (FACS) JOVE. 2010. [DOI] [PMC free article] [PubMed]
- Rohland N, Reich D. Cost-effective, high-throughput DNA sequencing libraries for multiplexed target capture. Genome Res. 2012;22(5):939–946. doi: 10.1101/gr.128124.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Josefowicz SZ, Kas A, Chu TT, Gavin MA, Rudensky AY. Genome-wide analysis of Foxp3 target genes in developing and mature regulatory T cells. Nature. 2007;445(7130):936–940. doi: 10.1038/nature05563. [DOI] [PubMed] [Google Scholar]
- Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, Kraus WL. Reciprocal Binding of PARP-1 and Histone H1 at Promoters Specifies Transcriptional Outcomes. Science. 2008;319(5864):819–821. doi: 10.1126/science.1149250. [DOI] [PubMed] [Google Scholar]
- Mendenhall EM, et al. Locus-specific editing of histone modifications at endogenous enhancers. Nat. Biotechnol. 2013;31(12):1133–1136. doi: 10.1038/nbt.2701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landt SG, et al. ChIP-seq guidelines and practices of the ENCODE and modENCODE consortia. Genome Res. 2012;22(9):1813–1831. doi: 10.1101/gr.136184.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reference Epigenome Standards - IHEC. 2017. Available from: http://ihec-epigenomes.org/research/reference-epigenome-standards/
- Heinz S, et al. Simple Combinations of Lineage-Determining Transcription Factors Prime cis-Regulatory Elements Required for Macrophage and B Cell Identities. Mol. Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng J, Liu T, Qin B, Zhang Y, Liu XS. Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 2012;7(9):1728–1740. doi: 10.1038/nprot.2012.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraley C, Raftery AE. MCLUST: Software for Model-Based Cluster Analysis. J. Classif. 1999;16(2):297–306. [Google Scholar]