

Abstract
Lentiviral vectors are an ideal choice for delivering gene-editing components to cells due to their capacity for stably transducing a broad range of cells and mediating high levels of gene expression. However, their ability to integrate into the host cell genome enhances the risk of insertional mutagenicity and thus raises safety concerns and limits their usage in clinical settings. Further, the persistent expression of gene-editing components delivered by these integration-competent lentiviral vectors (ICLVs) increases the probability of promiscuous gene targeting. As an alternative, a new generation of integrase-deficient lentiviral vectors (IDLVs) has been developed that addresses many of these concerns. Here the production protocol of a new and improved IDLV platform for CRISPR-mediated gene editing and list the steps involved in the purification and concentration of such vectors is described and their transduction and gene-editing efficiency using HEK-293T cells was demonstrated. This protocol is easily scalable and can be used to generate high titer IDLVs that are capable of transducing cells in vitro and in vivo. Moreover, this protocol can be easily adapted for the production of ICLVs.
Keywords: Genetics, Issue 130, Viral-mediated gene transfer, HEK-293T cells, integrase-deficient lentiviral vectors, CRISPR/Cas9 gene editing system, p24 ELISA, sucrose-gradient ultracentrifugation, FACS
Introduction
Precise gene editing forms the cornerstone of major biomedical advances that involve the development of novel strategies to tackle genetic diseases. At the forefront of gene-editing technologies is the method relying on the usage of the clustered regularly-interspaced short palindromic repeats (CRISPR)/Cas9 system that was initially identified as a component of bacterial immunity against the invasion of viral genetic material (reviewed in references1,2). A major advantage of the CRISPR/Cas9 system over other gene-editing tools, such as zinc-finger nucleases (ZFNs) and transcription activator-like effector nucleases (TALENs) (reviewed in reference3), is the relative simplicity of plasmid design and construction of CRISPR components — a feature that has powered the expansion of gene-editing from a few specialized laboratories to a much wider research community. Additionally, the simplicity of CRISPR/Cas9 programming and its capacity for multiplexed target recognition have further fueled its popularity as a cost-effective and easy-to-use technology. Among the various methods available to researchers to deliver such gene-editing components to cells, viral vectors remain by far the most popular and efficient system.
Lentiviral vectors (LVs) have emerged as the vehicle of choice to deliver the components of CRISPR/Cas9 system in vivo for diverse applications4,5,6,7. Several key features make LVs a popular choice for this process including their ability to infect both dividing and non-dividing cells, low immunogenicity, and minimal cellular toxicity (reviewed in reference8). As a result, LV-mediated gene therapy has been employed in treatments of infectious diseases, such as HIV-1, HBV, and HSV-1, as well as in the correction of defects underlying human hereditary diseases, such as cystic fibrosis and neo-vascular macular degeneration4,5,7,9,10,11. Moreover, LVs have been effectively modified to perform multiplex gene editing at distinct genomic loci using a single vector system12.
However, the inherent property of LVs to integrate into the host genome can be mutagenic and often handicaps their utility as transgene delivery vehicles, especially in clinical settings. Moreover, since stably-integrated LVs express their transgenes at sustainably high levels, this system is ill-suited for the delivery of gene-editing components such as CRISPR/Cas9; overexpression of Cas9-guide RNA (gRNA), and similar proteins such as ZFNs, are associated with elevated levels of off-target effects, which include undesirable mutations13,14,15,16,17 and can potentially enhance cytotoxicity18. Therefore, to achieve precise gene-editing with minimal off-target effects, it is imperative to design systems that allow for the transient expression of gene editing components.
In recent years, a variety of delivery platforms have been developed to transiently express CRISPR/Cas9 in cells16,19,20,21 (reviewed in reference22). These include methods that rely on directly introducing purified Cas9 along with the appropriate guide RNAs into cells, which was shown to be more effective at targeted gene-editing in comparison to plasmid-mediated transfection16. Studies have demonstrated that ribonucleoprotein (RNP) complexes consisting of guide RNA/Cas9 particles are rapidly turned over after mediating DNA cleavage at their targets, suggesting that short-term expression of these components is sufficient to achieve robust gene editing16. Conceivably, non-integrating viral vector platforms such as adeno-associated viral vectors (AAVs) can provide a viable alternative to deliver gene-editing machinery to cells. Unfortunately, AAV capsids possess significantly lower packaging capability than LVs (<5kb), which severely impedes their ability to package the multi-component CRISPR toolkit within a single vector (reviewed in reference8). It is worth noting that addition of compounds that inhibit histone deacetylases (e.g., sodium butyrate23) or impede the cell cycle (e.g., caffeine24) have been shown to increase lentiviral titers. Despite the recent progress, the transient expression systems developed so far are still impeded by several shortcomings, such as lower production efficiency, which lead to reduced viral titers, and low transduction efficiency of the viruses generated through such approaches25.
Integrase-deficient lentiviral vectors (IDLVs) represent a major advancement in the development of gene-delivery vehicles, as they combine the packaging capability of LVs with the added benefit of AAV-like episomal maintenance in cells. These features help IDLVs largely circumvent the major issues associated with integrating vectors, vis-à-vis continuous overexpression of potentially genotoxic elements and integration-mediated mutagenicity. It was previously demonstrated that IDLVs can be successfully modified to enhance episomal gene expression26,27. With regards to IDLV-mediated CRISPR/Cas9 delivery, low production titers and lower expression of episome-borne genomes relative to integrase-proficient lentiviral systems limits their utility as bona fide tools for delivering genome-editing transgenic constructs. We recently demonstrated that both transgene expression and viral titers associated with IDLV production are significantly enhanced by the inclusion of binding sites for the transcription factor Sp1 within the viral expression cassette28. The modified IDLVs robustly supported CRISPR-mediated gene editing both in vitro (in HEK-293T cells) and in vivo (in post-mitotic brain neurons), while inducing minimal off-target mutations compared to the corresponding ICLV-mediated systems28. Overall, we developed a novel, compact, all-in-one CRISPR toolkit carried on an IDLV platform and outlined the various advantages of using such a delivery vehicle for enhanced gene editing.
Here, the production protocol of the IDLV-CRISPR/Cas9 system is described, including the various steps involved in the assembly, purification, concentration, and titration of IDLVs, as well as strategies to validate the gene-editing efficacy of these vectors. This protocol is easily scalable to meet the needs of different investigators and is designed to successfully generate LV vectors with titers in the range of 1 x 1010 transducing units (TU)/mL. The vectors generated through this protocol can be utilized to efficiently infect several different cell types, including difficult-to-transduce embryonic stem cells, hematopoietic cells (T-cells and macrophages), and cultured and in vivo-injected neurons. Furthermore, the protocol is equally well-suited for the production of integrase-competent lentiviral vectors in similar quantities.
Figure 1: IDLV packaging.(a) Schematic of the wild type integrase protein (b) The modified plasmid was derived from psPAX2 (see Methods, plasmid construction for details). Representative agarose gel image of clones screened for mutated integrase clones. DNA samples prepared using a standard plasmid DNA isolation mini-kit were analyzed by digestion with EcoRV and SphI. The correctly-digested clone (number 5, dashed red box) was further verified by direct (Sanger) sequencing for the D64E substitution in INT. The integrase-deficient packaging cassette was named pBK43. (c) Schematic of the transient transfection protocol employed to generate IDLV-CRISPR/Cas9 vectors, showing 293T cells transfected with VSV-G, packaging, and transgene cassettes (Sp1-CRISPR/Cas9 all-in-one plasmid). Viral particles that bud out from the cell membrane contain the full-length RNA of the vector (expressed from the transgene cassette). The second generation of the IDLV-packaging system was used, which includes the regulatory proteins Tat and Rev. Rev expression is further supplemented from a separate cassette (RSV-REV-plasmid). Abbrev: LTR-Long-terminal repeat, VSV-G, vesicular stomatitis virus G-protein, pCMV-cytomegalovirus promoter; Rous sarcoma virus (RSV) promoter; RRE- (Rev Response Element). Other regulatory elements on the expression cassette include Sp1-binding sites, Rev Response element (RRE), Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element (WPRE), a core-elongation factor 1α promoter (EFS-NC), the vector packaging element ψ (psi), human Cytomegalovirus (hCMV) promoter, and human U6 promoter. Please click here to view a larger version of this figure.
Protocol
1. Culturing HEK-293T Cells and Seeding Cells for Transfection
NOTE: Human Embryonic Kidney 293T (HEK-293T) cells are grown in DMEM, high glucose media supplemented with 10% bovine calf serum supplemented with iron and growth promoters, and 1x antibiotic-antimycotic solution (100x solution contains 10,000 units penicillin, 10 mg streptomycin and 25 µg amphotericin B per mL). The media is also supplemented with 1x sodium pyruvate, 1x non-essential amino acid mix, and 2 mM L-Glutamine (stock 200 mM L-alanyl-L-glutamine dipeptide in 0.85% NaCl). Cells are cultured in 100 mm tissue culture plates (approximate growth surface area is 55 cm²). A sub-cultivation ratio of 1:10 is used with sub-culturing every 2 - 3 days. Trypsin-EDTA 0.05% is used for dissociation of cells between passages. To maintain consistency between experiments, we recommend testing calf sera when switching to a different lot/batch and monitor any changes in cell growth, transfection efficiency, and vector production.
Start a new culture by using low passage cells (it is recommended to not use cells after passage 15 or if growth slows down) by seeding into a 10-cm tissue culture plate. Use DMEM supplemented with 10% serum for cell growth. Grow cells at 37 °C with 5% CO2 in a standard tissue-culture incubator. Use a standard hemocytometer to count cells for all subsequent steps.
Once the cells reach 90 - 95% confluent growth, reseed into 15 cm tissue culture plates (below, Steps 1.3 - 1.5).
To reseed, aspirate media from the confluent plate and gently rinse with sterile 1x PBS. Incubate cells with 2 mL of a dissociation reagent (e.g., Trypsin-EDTA) at 37 °C for 3 - 5 min. Add 8 mL of media containing serum to inactivate the dissociation reagent, and triturate 10 - 15 times with a 10 mL serological pipette to create a single cell suspension. Resuspend cells in the culture media to obtain a cell density of approximately 4 x 106 cells/mL.
To enhance adherence to the substrate, pre-coat the 15 cm plates with 0.2% gelatin, adding 8 mL of gelatin per plate. Spread evenly across the surface of the plate, incubate at room temperature for 10 min, and siphon off the liquid.
Bring the total volume of each plate to 25 mL with warm (37 °C) HEK-293 T media (see Note under Step 1) and seed the plates by adding 2.5 mL of cells (total ~1 x 107 cells/plate). Incubate plates at 37 °C with 5% CO2 overnight or until 70 - 80% confluency is reached. NOTE: Up to six 15 cm plates can be used for production. Adjust volume of media per plate to ~20 - 22 mL when using more than four plates (refer to Step 5 of the protocol for rationale).
2. Transfecting HEK-293T Cells Using a Calcium Phosphate-based Protocol
- Transfection reagents
- To prepare 2x BES-buffered solution BBS (50 mM BES, 280 mM NaCl, 1.5 mM Na2HPO4), combine 16.36 g of NaCl, 10.65 g of BES (N, N-bis (2-hydroxyethyl)-2-amino-ethanesulfonic acid), and 0.21 g of Na2HPO4. Add double-distilled H2O (dd-H2O) up to 900 mL. Dissolve, titrate to pH 6.95 with 1M NaOH, and bring volume to 1 L. Filter via 0.22 µM filter unit. Store at -20 °C.
- Prepare 1M CaCl2. Filter solution via a 0.22 µM filter. Store at 4 °C.
Observe the plates that were seeded a day prior. Cells are ready for transfection once they reach 70 - 80% confluency.
Aspirate old media from the plates and gently add freshly-prepared media without serum. NOTE: See Supplementary File 1-Plasmids for details about the chosen plasmids and their preparation.
Prepare the plasmid mix by aliquoting the four plasmids into a 15-mL conical tube. For a single 15-cm dish preparation, use 37.5 µg of the CRISPR/Cas9-transfer vector (pBK198 or pBK189), 25 µg of pBK43 (psPAX2-D64E), 12.5 µg pMD2.G, and 6.25 µg of pREV (Figure 1c).
Add 312.5 µL 1M CaCl2 to the plasmid mix. To this, add up to 1.25 mL of sterile dd-H20.
Slowly (drop-wise) add 1.25 mL of 2x BBS solution while vortexing the mix. Incubate for 30 min at room temperature.
Add the transfection mixture dropwise to each 15-cm plate (2.5 mL per plate). Swirl the plates gently and incubate at 37 °C with 5% CO2 for 2 - 3 h. Thereafter, add 2.5 mL (10%) serum per plate and continue incubating overnight (12 - 18 h). NOTE: Some labs use 3% CO2 incubators to stabilize media pH. However, we do not observe any difference in transfection efficiency between 3% and 5% CO2. Also, the size of CaPO4 precipitates is crucial for transfection efficiency; the transfection mix has to be clear prior to its addition onto the cells. If the mix becomes cloudy during incubation, prepare fresh 2x BBS (pH = 6.95).
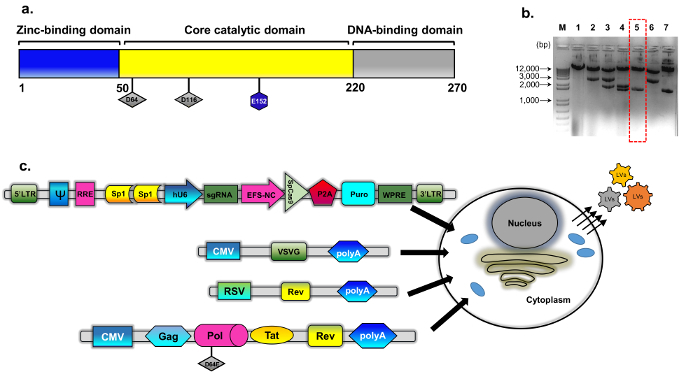
Figure 2: Concentration of the viral particles using double-sucrose gradient protocol. Viral particles collected from the supernatant (SN) are loaded onto gradient sucrose gradient. 70%, 60%, 30%, and 20% sucrose solutions are used to create the gradient. Following centrifugation, the particles collected from the 30 - 60% sucrose fractions are further loaded onto a 20% sucrose cushion and precipitated. The final pellet containing purified viral particles is resuspended in 1x PBS for further usage (see text for further details). Please click here to view a larger version of this figure.
3. Day After Transfection
Observe the cells to ensure that they are nearing 100% confluency at this point with little to no cell death. Replace media by adding 25 mL of fresh DMEM + 10% serum to each plate. Continue incubating at 37 °C with 5% CO2 for an additional 48 h. NOTE: Adjust volume of media in step 3.1 if using more than six 15 cm plates (see Step 5.2, Note).
4. Harvesting Virus
Carefully collect the supernatant from all the tissue culture plates containing the transfected cells using a sterile 10 mL tissue culture pipette and pool in 50 mL centrifuge tubes.
Clear the suspension by centrifugation at 400 - 450 x g for 10 min using a tabletop centrifuge. Filter the supernatant through a 0.45 µm vacuum filter unit. NOTE: The filtered supernatant can be stored at 4 °C for up to 4 days before proceeding with concentration, or aliquoted and stored at -80 °C. The expected titers of IDLV-Sp1-CRISPR/Cas9 (non-concentrated) viral preparations should be ~2 x 107 TU/mL (refer to Step 6 for titer determination). However, avoid subjecting the viral preparations to multiple freeze-thaw cycles, as each round of freezing and thawing results in a 10 - 20% loss in functional titers.
5. Concentration of Viral Particles by Ultracentrifugation
NOTE: We utilize a double-sucrose method of purification that involves two steps: a sucrose gradient step and a sucrose cushion step (Figure 2).
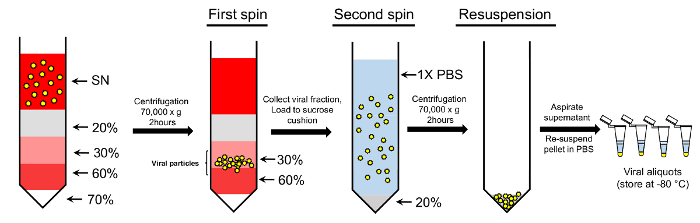
Load the conical ultracentrifugation tubes in the following order to create a sucrose gradient: 0.5 mL 70% sucrose (dissolved 1x PBS), 0.5 mL 60% sucrose (dissolved in DMEM), 1 mL 30% sucrose (dissolved in DMEM), and 2 mL 20% sucrose (dissolved in 1x PBS).
- Add the virus-containing supernatant carefully to the gradient. As the total volume of supernatant from four 15 cm plates is 100 mL, use six ultracentrifugation tubes per spin to process the full volume of the virus supernatant.
- Each ultracentrifugation tube used for this step has a volume capacity of up to 30 mL (including the volume of sucrose), distribute viral supernatant equally among tubes, leaving at least 10% headspace to prevent spillage.
- Adjust the culture volume to ~20 - 22 mL per plate when using more than four 15 cm plates for each experiment, so that the final volume of the pooled viral supernatant can easily be accommodated within six ultracentrifugation tubes.
- Fill ultracentrifugation tubes to at least three-fourths their total volume capacity, otherwise breakage of tubes can occur during centrifugation, resulting in possible loss of sample and/or equipment damage.
Balance the tubes with 1x PBS and centrifuge samples at 70,000 x g for 2 h at 17 °C (see Table of Materials for rotor details). NOTE: To prevent disruption of the sucrose layer during acceleration, set the ultracentrifuge to slowly accelerate the rotor to 200 rpm during the first 3 min of the spin. Similarly, set the ultracentrifuge to slowly decelerate the rotor from 200 rpm to 0 rpm over 3 min at the end of the spin.
Carefully collect 30 - 60% sucrose fractions into clean tubes (Figure 2). Add cold 1x PBS to the pooled fractions and bring up the volume to 100 mL; mix by pipetting up and down several times.
Proceed to the sucrose cushion step by carefully layering the viral preparation on a sucrose cushion. For this, add 4 mL of 20% sucrose (in 1x PBS) to the tube, followed by ~20 - 25 mL of the viral solution per tube. If tubes are less than three-fourths full, top up with sterile 1x PBS.
Carefully balance and centrifuge the samples at 70,000 x g for 2 h at 17 °C, as before. Pour off the supernatant and allow the remaining liquid to drain by inverting tubes on paper towels.
Aspirate remaining droplets in order to remove all liquid from the pellet. At this step, the virus-containing pellets should be barely visible as small translucent spots.
Resuspend pellets by adding 70 µL of 1x PBS to the first tube and thoroughly pipetting the suspension, subsequently transferring the suspension to the next tube and mixing as before, continuing until all the pellets are resuspended.
Rinse the tubes with an additional 50 µL of cold 1x PBS and mix as before. Ensure that the combined volume of the final suspension is ~120 µL, and appears slightly milky; clear it by centrifugation at 10,000 x g for 30 s on a tabletop microcentrifuge.
Transfer the supernatant to a fresh microfuge tube, make 10 µL aliquots, and store them at -80 °C. NOTE: Avoid performing repeated freeze-thaw cycles on the lentiviral samples. Except when centrifugation is required, efforts should be made to carry out the remaining steps in tissue culture hoods, or designated tissue-culture rooms using appropriate biosafety measures (see Discussion).
6. Estimation of Viral Titers
- p24 -enzyme-linked immunosorbent assay (ELISA) method NOTE: The assay is carried out using high-binding 96-well plates as per the instructions of the NIH AIDS Vaccine Program for HIV-1 p24 Antigen Capture Assay Kit (see Table of Materials) with modifications29.
- The next day, wash the wells three times with 200 µL 0.05% Tween 20 in cold PBS (PBS-T solution). Coat the plate with 100 µL of monoclonal anti-p24 antibody at a dilution of 1:1500 in 1x PBS and incubate overnight at 4 °C.
- To eliminate non-specific binding, block the plate with 200 µL 1% BSA in PBS; wash three times with 200 µL 0.05% Tween 20 in cold PBS (PBS-T solution) for at least 1 h at room temperature.
- Prepare samples: For concentrated vector preparations, dilute 1 µL of the sample 100-fold by adding 89 µL of dd-H20 and 10 µL of Triton X-100 (final concentration of 10%). For non-concentrated preparations, prepare ten-fold diluted samples (add 80 µL of dd-H20 and 10 µL of Triton X-100 (final concentration of 10%) to 10 µL of the sample). NOTE: Samples can be stored at -20 °C at this step for an extended period of time for later usage.
- Prepare HIV-1 standards by applying a 2-fold serial dilution (with a starting concentration 5 ng/mL).
- Dilute concentrated samples (from 1:100 pre-diluted stocks) in RPMI 1640 supplemented with 0.2% Tween 20 and 1% BSA to establish 1:10,000, 1:50,000, and 1:250,000 dilutions. Dilute non-concentrated samples (from 1:10 pre-diluted stocks) in RPMI 1640 supplemented with 0.2% Tween 20 and 1% BSA to establish 1:500, 1:2500, and 1:12,500 dilutions.
- Apply samples on the plate in triplicates and incubate overnight at 4 °C.
- The next day, wash the wells six times and incubate at 37 °C for 4 h with 100 µL polyclonal rabbit anti-p24 antibody, diluted 1:1000 in RPMI 1640, 10% FBS, 0.25% BSA, and 2% normal mouse serum (NMS).
- Wash six times as above and incubate at 37 °C for 1 h with goat anti-rabbit horseradish peroxidase IgG diluted 1:10,000 in RPMI 1640 supplemented with 5% normal goat serum, 2% NMS, 0.25% BSA, and 0.01% Tween 20.
- Wash the plate, as above, and incubate with TMB peroxidase substrate at room temperature for 15 min.
- Stop the reaction by adding 100 µL of 1 N HCL. Measure sample absorbance at 450 nm using the absorbance plate reader.
- Measurement of fluorescent reporter intensity
- FACS Method NOTE: The extent of GFP signal depletion in cells can be accurately estimated by measuring mean fluorescence intensity of the transduced cells via flow cytometry. Please refer to the recent paper 28 for FACS data analysis, presentation, and interpretation. The protocol is described as follows.
- Make a ten-fold serial dilution of the preparation (from 10-1 to 10-5) of the viral preparation in 1x PBS.
- Seed approximately 5 x 105 293T cells in each well of a 6-well plate in a final volume of 2 mL per well. Add 10 µL of each viral dilution to the cells and incubate cells at 37 °C for 48 h.
- Harvest cells for FACS analysis as follows: add 200 µL of 0.05% Trypsin-EDTA solution and incubate cells at 37 °C for 5 min. Add 2 mL of a complete DMEM media and collect samples into 15 mL conical tubes.
- Pellet cells by centrifugation at 400 x g at 4 °C, and resuspend the pellet in 500 µL of cold 1x PBS.
- For fixation, add an equal volume of 4% formaldehyde solution to this suspension and incubate for 10 min at room temperature.
- Pellet fixed cells and resuspend in 1 mL of 1x PBS. Analyze GFP expression using a FACS instrument, as described in Ortinski et al.28 Briefly, use the following formula to determine the virus' functional titer:
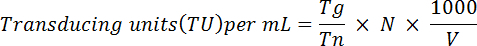 NOTE: Here Tg = number of GFP-positive cells counted; Tn = total number of cells counted; N = total number of cells transduced; V = volume (in µL) used for transduction. For example: If 1 x 106 cells were transduced with 10 µL of virus, 2 x 104 cells were counted and 5 x 103 were GFP-positive, based on the above equation the functional titer would be:
NOTE: Here Tg = number of GFP-positive cells counted; Tn = total number of cells counted; N = total number of cells transduced; V = volume (in µL) used for transduction. For example: If 1 x 106 cells were transduced with 10 µL of virus, 2 x 104 cells were counted and 5 x 103 were GFP-positive, based on the above equation the functional titer would be: 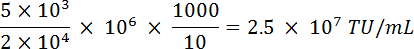
- Counting GFP-positive cells
- Calculate the multiplicity of infection (MOI) used for transduction. Test a broad range of MOIs (1 - 10), with increasing MOIs resulting in a higher transduction efficiency.
- Prior to transfection, seed a 6-well plate with approximately 3 - 4 x 105 cells per well. Once the cells reach >90% confluency (usually within 24 h), transduce with the purified virus at pre-determined MOIs.
- Incubate plate at 37 °C with 5% CO2 in a standard tissue culture, and monitor cells at regular intervals for 1 - 7 days for changes in GFP signal.
- Count the number of GFP-positive cells with a fluorescent microscope (PLAN 4X objective, 0.1 N.A, 40X magnification) fitted with a GFP filter set (excitation wavelength-470 nm, emission wavelength-525 nm), using naïve (un-transduced) cells to set the population of GFP-negative and positive cells.
- Estimate the final titer by adjusting for the dilution factor and the volume, using the following formula:
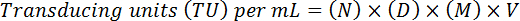 NOTE: Here, N = number of GFP-positive cells, D = dilution factor, M = magnification factor (usually 20X), V = volume of virus used for transduction. For example, for 20 GFP-positive cells (N) counted at a dilution of 10-4 (1:10,000) in a 10 µL sample (V) at 20X magnification (M) (D) would result in a functional titer of (20 x 104) x (20) x (10) x (100*) = 4 x 108 TU/mL. (*to adjust to per mL)
NOTE: Here, N = number of GFP-positive cells, D = dilution factor, M = magnification factor (usually 20X), V = volume of virus used for transduction. For example, for 20 GFP-positive cells (N) counted at a dilution of 10-4 (1:10,000) in a 10 µL sample (V) at 20X magnification (M) (D) would result in a functional titer of (20 x 104) x (20) x (10) x (100*) = 4 x 108 TU/mL. (*to adjust to per mL)
Representative Results
Validation of the knockout-efficiency of IDLV-CRISPR/Cas9 vectors We used GFP-expressing 293T cells as a model to validate the efficiency of CRISPR/Cas9-mediated gene knockout. GFP+ cells were generated by transduction of HEK-293T cells with pLenti-GFP (vBK201a) at an MOI of 0.5 (Figure 3b, "no-virus" panel). The sgRNA-to-GFP/Cas9 all-in-one vector cassette was packaged into IDLV or ICLV particles and the production efficiency was assessed by p24 ELISA (see above; and Figure 3a). The titers of the vectors containing Sp1-binding sites (following concentration) were found to be in the range of 1010 TU/mL. We then assessed the efficiency of GFP knockout using fluorescent microscopy (Figure 3b). To this end, 5 x 105 GFP-expressing 293T cells were seeded into a 6-well plate and we transduced them 24 h later with IDLV- or ICLV-CRISPR/Cas9 at MOIs of 1 and 5 (Figure 3b). The cells were incubated for 24 h, after which the culture medium was replaced and cells were incubated for an additional 48 h, before reseeding the plates for subsequent days of the experiment.
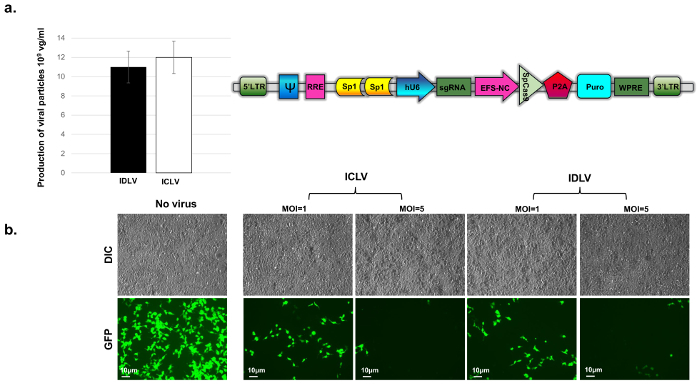
In a recent study, we measured the mean fluorescence intensity of GFP+ cells transduced with ICLV/CRISPR or IDLV/CRISPR components via flow cytometry28. We saw comparable GFP-depletion in both samples 14 days post-transduction (pt) (2 - 4% signal depletion), and nearly-identical GFP depletion 21 days pt (>99% signal depletion)28. In concordance with previous observations, we observed a ~five-fold reduction in the number of GFP-positive cells as early as 7 days pt (Figure 3), with a nearly complete signal depletion observed by 14 days pt (data not shown) following transduction with both ICLV- and IDLV-CRISPR/Cas9 systems. The signal loss was evaluated as the ratio of the cells that remained GFP-positive after treatment and the total number of naïve GFP-positive cells. These results clearly demonstrate that CRISPR/Cas9 constructs delivered by IDLVs are comparable with those delivered by their integrating counterparts in their ability to mediate rapid, robust, and sustained gene editing in dividing cells.
Figure 3: Evaluation of viral titers. (a) by p24-ELISA assay. Titers for the concentrated Sp1-IDLV-CRISPR/Cas9 (black bar) and Sp1-ICLV-CRISPR/Cas9 (white bar) were evaluated. The results are recorded in copy numbers per mL, where 1 ng p24-gag = 1 x 104 viral particles. Bar graph data represents mean ± SD from triplicate experiments. (b) Evaluation of CRISPR-mediated GFP-knockout efficiency. Depletion of GFP signal was compared between ICLV-CRISPR/Cas9- and IDLV-CRISPR/Cas9-transduced 293T GFP+ cells at MOIs of 1 and 5. Un-transduced ("no virus" panel in figure) GFP-positive cells were used as controls. Images were acquired using a fluorescence microscope at 40X magnification at 7 days post transduction. Abbrev: RRE: Rev Response Element, EFS-NC: core-elongation factor 1α promoter, ψ (psi): vector packaging element, hU6: human U6 promoter, Puro: Puromycin-resistance cassette, WPRE: Woodchuck Hepatitis Virus Posttranscriptional Regulatory Element, LTR: Long-terminal repeat. Please click here to view a larger version of this figure.
Supplementary File 1: Plasmids Please click here to download this file.
Discussion
IDLVs have begun to emerge as the vehicle of choice for in vivo gene-editing, especially in the context of genetic diseases, owing largely to the low risk of mutagenesis associated with these vectors compared to integrating delivery platforms22,28. In the current manuscript, we sought to detail the protocol associated with production of the improved all-in-one IDLV-CRISPR/Cas9 system that was recently developed in our laboratory28.
Modifications to existing platforms With this method, we were able to generate IDLV-CRISPR/Cas9 and ICLV-CRISPR/Cas9 vectors at titers in the range of 1 x 1010 TU/mL (Figure 3a). This enhancement in production efficiency can be attributed to the addition of Sp1-binding site into all-in-one CRISPR/Cas9 vector cassette. Indeed, we recently reported that inclusion of Sp1 results in a ~2.5-fold increase in the packaging efficiency of IDLV- and ICLV-CRISPR/Cas9 vectors and a ~7-fold increase in the overall functional titers. These results are in agreement with earlier work from various groups highlighting Sp1 as a key regulator of wild-type HIV-130,31,32,33,34,35.
Critical steps and troubleshooting As pointed out above, Sp1-IDLVs carrying CRISPR/Cas9 transgenes were capable of generating titers in the vicinity of 1 x 1010 TU/mL per ~5 x 107 producer cells. Titers lower than these would be indicative of errors in the production process, in which case the following crucial points should be considered for titer improvement: 1) It is recommended that the producer cells preferably be of low passage number, with replacement after ≥15 passages and/or when slower growth is observed. 2) The choice of cell media components is important in terms of production efficiency. For instance, in place of the commonly-used fetal bovine serum, we find that the usage of Cosmic Calf Serum consistently improved cell growth and viral production, while being cost-effective at the same time. 3) The production fitness of the different HEK lines should be carefully evaluated. For example, we found a ~threefold difference in viral production yield between 293T cells and 293-FT (see Table of Materials) cells (data not shown). 4) It is recommended that cells be transfected when they are roughly 70 - 80% confluent, with lower cell densities resulting in premature cell death due to viral toxicity, and higher densities resulting in a marked drop in production efficiency. As a rule of thumb, we suggest accounting for a cell density that allows cells to undergo one additional round of cell division post-transfection. 5) Lastly, the efficiency of transfection is highly dependent on the pH of 2x BBS, which is to be maintained at exactly 6.95 for ideal transfection. It is therefore highly recommended that each new batch of 2x BBS be checked on a pilot-transfection scale.
Vector handling and safety There are several important safety considerations during the production of IDLV and ICLV vectors with this protocol. First, working with lentiviruses requires Bio-Safety Level II containment. Despite the safety features afforded by SIN vectors, residual transcriptional activity from SIN vectors has been reported36. Furthermore, previous work has demonstrated that IDLV- and ICLV-genomes can be productively rescued by HIV-127. Therefore, it is strongly recommended that replication competence assays (RCA) be performed, especially when concentrated lentiviruses are being used37. For safety procedures regarding handling of lentiviral vector preparations, see Biosafety in Microbiological and Biomedical Laboratories, 4th edition, published by the Centers for Disease Control (CDC), which can be found online38. On the same note, third generation packaging systems39 with enhanced biosafety features can be used to package IDLVs and ICLVs, albeit with lower efficiency than that of the second-generation system.
Significance and future directions Overall, the production protocol for IDLVs for CRISPR/Cas9- mediated gene editing described here represents the first evaluation of the efficiency of this system in rapidly-dividing cells. Using GFP-positive HEK-293T cells, we demonstrated that Sp1-CRISPR/Cas9 delivered by IDLVs can efficiently and quickly edit GFP in these cells, with similar kinetics as ICLVs. These observations are broadly consistent with the results from previous work using ribonucleoprotein complexes (Cas9 RNPs) for gene editing through non-viral transfection methods. This novel platform further enriches the ever-expanding toolbox for the delivery of gene-editing components and other molecular cargos to cells.
As discussed earlier, despite the recent progress in developing lentiviral-based gene-delivery systems, there are few to no options for platforms that allow the sustainable production of high-titer viral vectors for rapid, transient, and targeted gene manipulation. Through the incorporation of binding motifs for a highly-expressed transcription factor in the transgene expression cassette, we were able to simultaneously address several of these issues. This simple yet critical manipulation opens up a number of avenues for developing similar delivery platforms. It would be particularly useful to test if transgene expression can be enhanced through either the addition of numerous copies of the same binding motif, or by multiplexing the Sp1 motif with binding sites for other transcription factors. With such tools at our disposal, it is tempting to speculate that expression from relatively weak tissue-specific promoters, such as those for human Synapsin I (hSyn) and mouse calcium/calmodulin-dependent protein kinase II (CaMKII), could be significantly improved both in vitro and in vivo.
While the current study was limited to testing the gene knockdown capability of IDLV-delivered CRISPR/Cas9, in principle, the versatility of our system would be amenable to diverse gene-editing applications, such as those relying on the catalytically-inactive dCas940. Finally, combined with smaller endonucleases, such as SaCas941 and Cpf142, the platform described in this study can be adopted toward establishing highly-efficient AAV-based gene-delivery systems in a relatively short span of time, which would provide the added advantage of low immunogenicity compared to lentiviral vectors. Needless to say, such approaches would be a positive step towards the development of novel, efficient, and clinically safe viral vectors.
Disclosures
Patent USC-499-P (1175) was filed by the University of South Carolina in relation to the work described in this manuscript.
Acknowledgments
We would like to thank the Department of Neurobiology, Duke University School of Medicine and Dean's Office for Basic Science, Duke University. We also thank members of the Duke Viral Vector Core for comments on the manuscript. Plasmid pLenti CRISPRv2 was gift from Feng Zhang (Broad Institute). The LV-packaging system including the plasmids psPAX2, VSV-G, pMD2.G and pRSV-Rev was a kind gift from Didier Trono (EPFL, Switzerland). Financial support for this work was provided by the University Of South Carolina School Of Medicine, grant RDF18080-E202 (B.K).
References
- Horvath P, Barrangou R. CRISPR/Cas, the immune system of bacteria and archaea. Science. 2010;327(5962):167–170. doi: 10.1126/science.1179555. [DOI] [PubMed] [Google Scholar]
- Marraffini LA, Sontheimer EJ. CRISPR interference: RNA-directed adaptive immunity in bacteria and archaea. Nat Rev Genet. 2010;11(3):181–190. doi: 10.1038/nrg2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim H, Kim JS. A guide to genome engineering with programmable nucleases. Nat Rev Genet. 2014;15(5):321–334. doi: 10.1038/nrg3686. [DOI] [PubMed] [Google Scholar]
- Bellec J, et al. CFTR inactivation by lentiviral vector-mediated RNA interference and CRISPR-Cas9 genome editing in human airway epithelial cells. Curr Gene Ther. 2015;15(5):447–459. doi: 10.2174/1566523215666150812115939. [DOI] [PubMed] [Google Scholar]
- Kennedy EM, et al. Suppression of hepatitis B virus DNA accumulation in chronically infected cells using a bacterial CRISPR/Cas RNA-guided DNA endonuclease. Virology. 2015;476:196–205. doi: 10.1016/j.virol.2014.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roehm PC, et al. Inhibition of HSV-1 Replication by Gene Editing Strategy. Sci Rep. 2016;6:23146. doi: 10.1038/srep23146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, et al. CCR5 gene disruption via lentiviral vectors expressing Cas9 and single guided RNA renders cells resistant to HIV-1 infection. PLoS One. 2014;9(12):e115987. doi: 10.1371/journal.pone.0115987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor B, Bailey RM, Wimberly K, Kalburgi SN, Gray SJ. Methods for gene transfer to the central nervous system. Adv Genet. 2014;87:125–197. doi: 10.1016/B978-0-12-800149-3.00003-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebbink RJ, et al. A combinational CRISPR/Cas9 gene-editing approach can halt HIV replication and prevent viral escape. Sci Rep. 2017;7:41968. doi: 10.1038/srep41968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, et al. CRISPR/Cas9-Mediated CCR5 Ablation in Human Hematopoietic Stem/Progenitor Cells Confers HIV-1 Resistance In Vivo. Mol Ther. 2017. [DOI] [PMC free article] [PubMed]
- Yiu G, Tieu E, Nguyen AT, Wong B, Smit-McBride Z. Genomic Disruption of VEGF-A Expression in Human Retinal Pigment Epithelial Cells Using CRISPR-Cas9 Endonuclease. Invest Ophthalmol Vis Sci. 2016;57(13):5490–5497. doi: 10.1167/iovs.16-20296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabadi AM, Ousterout DG, Hilton IB, Gersbach CA. Multiplex CRISPR/Cas9-based genome engineering from a single lentiviral vector. Nucleic Acids Res. 2014;42(19):e147. doi: 10.1093/nar/gku749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y, Sander JD, Reyon D, Cascio VM, Joung JK. Improving CRISPR-Cas nuclease specificity using truncated guide RNAs. Nat Biotechnol. 2014;32(3):279–284. doi: 10.1038/nbt.2808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsu PD, et al. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat Biotechnol. 2013;31(9):827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Kim D, Cho SW, Kim J, Kim JS. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24(6):1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pattanayak V, et al. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat Biotechnol. 2013;31(9):839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalem O, et al. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014;343(6166):84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaefer KA, et al. Unexpected mutations after CRISPR-Cas9 editing in vivo. Nat Methods. 2017;14(6):547–548. doi: 10.1038/nmeth.4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JG, et al. Lentivirus pre-packed with Cas9 protein for safer gene editing. Gene Ther. 2016;23(7):627–633. doi: 10.1038/gt.2016.27. [DOI] [PubMed] [Google Scholar]
- Platt RJ, et al. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159(2):440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truong DJ, et al. Development of an intein-mediated split-Cas9 system for gene therapy. Nucleic Acids Res. 2015;43(13):6450–6458. doi: 10.1093/nar/gkv601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson CE, Gersbach CA. Engineering Delivery Vehicles for Genome Editing. Annu Rev Chem Biomol Eng. 2016;7:637–662. doi: 10.1146/annurev-chembioeng-080615-034711. [DOI] [PubMed] [Google Scholar]
- Jaalouk DE, Crosato M, Brodt P, Galipeau J. Inhibition of histone deacetylation in 293GPG packaging cell line improves the production of self-inactivating MLV-derived retroviral vectors. Virol J. 2006;3:27. doi: 10.1186/1743-422X-3-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellis BL, Potts PR, Porteus MH. Creating higher titer lentivirus with caffeine. Hum Gene Ther. 2011;22(1):93–100. doi: 10.1089/hum.2010.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoban MD, et al. Delivery of Genome Editing Reagents to Hematopoietic Stem/Progenitor Cells. Curr Protoc Stem Cell Biol. 2016;36:1–10. doi: 10.1002/9780470151808.sc05b04s36. [DOI] [PubMed] [Google Scholar]
- Bayer M, et al. A large U3 deletion causes increased in vivo expression from a nonintegrating lentiviral vector. Mol Ther. 2008;16(12):1968–1976. doi: 10.1038/mt.2008.199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor B, Ma H, Webster-Cyriaque J, Monahan PE, Kafri T. Epigenetic activation of unintegrated HIV-1 genomes by gut-associated short chain fatty acids and its implications for HIV infection. Proc Natl Acad Sci U S A. 2009;106(44):18786–18791. doi: 10.1073/pnas.0905859106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortinski PI, O'Donovan B, Dong X, Kantor B. Integrase-Deficient Lentiviral Vector as an All-in-One Platform for Highly Efficient CRISPR/Cas9-Mediated Gene Editing. Mol Ther Methods Clin Dev. 2017;5:153–164. doi: 10.1016/j.omtm.2017.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor B, et al. Notable reduction in illegitimate integration mediated by a PPT-deleted, nonintegrating lentiviral vector. Mol Ther. 2011;19(3):547–556. doi: 10.1038/mt.2010.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berkhout B, Verhoef K, van Wamel JL, Back NK. Genetic instability of live, attenuated human immunodeficiency virus type 1 vaccine strains. J Virol. 1999;73(2):1138–1145. doi: 10.1128/jvi.73.2.1138-1145.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Gonzalo M, et al. The hepatitis B virus X protein induces HIV-1 replication and transcription in synergy with T-cell activation signals: functional roles of NF-kappaB/NF-AT and SP1-binding sites in the HIV-1 long terminal repeat promoter. J Biol Chem. 2001;276(38):35435–35443. doi: 10.1074/jbc.M103020200. [DOI] [PubMed] [Google Scholar]
- Kim YS, et al. Artificial zinc finger fusions targeting Sp1-binding sites and the trans-activator-responsive element potently repress transcription and replication of HIV-1. J Biol Chem. 2005;280(22):21545–21552. doi: 10.1074/jbc.M414136200. [DOI] [PubMed] [Google Scholar]
- Ortinski PI, Lu C, Takagaki K, Fu Z, Vicini S. Expression of distinct alpha subunits of GABAA receptor regulates inhibitory synaptic strength. J Neurophysiol. 2004;92(3):1718–1727. doi: 10.1152/jn.00243.2004. [DOI] [PubMed] [Google Scholar]
- Van Lint C, et al. Transcription factor binding sites downstream of the human immunodeficiency virus type 1 transcription start site are important for virus infectivity. J Virol. 1997;71(8):6113–6127. doi: 10.1128/jvi.71.8.6113-6127.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Lint C, Ghysdael J, Paras P, Burny A, Verdin E. A transcriptional regulatory element is associated with a nuclease-hypersensitive site in the pol gene of human immunodeficiency virus type 1. J Virol. 1994;68(4):2632–2648. doi: 10.1128/jvi.68.4.2632-2648.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu W, Russ JL, Eiden MV. Evaluation of residual promoter activity in gamma-retroviral self-inactivating (SIN) vectors. Mol Ther. 2012;20(1):84–90. doi: 10.1038/mt.2011.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Testing for Replication Competent Retrovirus (RCR)/Lentivirus (RCL) in Retroviral and Lentiviral Vector Based Gene Therapy Products - Revisiting Current FDA Recommendations. 2017. Available from: https://sites.duke.edu/dvvc/files/2016/05/FDA-recommendation-for-RCR-testing.pdf.
- CDC. Biosafety in Microbiological and Biomedical Laboratories. 2017. Available from: https://www.cdc.gov/biosafety/publications/bmbl5/bmbl.pdf.
- Dull T, et al. A third-generation lentivirus vector with a conditional packaging system. J Virol. 1998;72(11):8463–8471. doi: 10.1128/jvi.72.11.8463-8471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu XS, et al. Editing DNA Methylation in the Mammalian Genome. Cell. 2016;167(1):233–247. doi: 10.1016/j.cell.2016.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ran FA, et al. In vivo genome editing using Staphylococcus aureus Cas9. Nature. 2015;520(7546):186–191. doi: 10.1038/nature14299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zetsche B, et al. Cpf1 is a single RNA-guided endonuclease of a class 2 CRISPR-Cas system. Cell. 2015;163(3):759–771. doi: 10.1016/j.cell.2015.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]