

Abstract
This article describes the steps for construction of a DNA library from soil, preparation and use of the nanopore flow cell, and analysis of the DNA sequences identified using computer software. Nanopore DNA sequencing is a flexible technique that allows for rapid microbial genome sequencing to identify bacterial and viral species, to characterize bacterial strains, and to detect genetic mutations that confer resistance to antibiotics. The advantages of nanopore sequencing (NS) for life sciences include its low complexity, reduced cost, and rapid real-time sequencing of purified genomic DNA, PCR amplicons, cDNA samples, or RNA. NS is an example of "strand sequencing" which involves sequencing DNA by guiding a single stranded DNA molecule through a nanopore that is inserted into a synthetic polymer membrane. The membrane has an electrical current applied across it, so as the individual bases pass through the nanopore the electrical current is disrupted to varying degrees by the four nucleotide bases. The identification of each nucleotide occurs by detecting the characteristic modulation of the electrical current by the different bases as they pass through the nanopore. The NS system consists of a handheld, USB powered portable device and a disposable flow cell that contains a nanopore array. The portable device plugs into a standard laptop computer that reads and records the DNA sequence using computer software.
Keywords: Environmental Sciences, Issue 130, Nanopore, DNA, metagenomics, sequencing, genomics, soil
Introduction
The goal of this procedure is to demonstrate the steps required for preparation of an environmental DNA library for sequencing, utilization of a nanopore flow cell sequencing device, and to perform analysis of the generated DNA sequences using system software and the National Center for Biotechnology Information (NCBI) bioinformatics tools to identify microbial species in soil. Currently, most DNA sequencing platforms require a major investment in technical training and complex instrumentation, which is not feasible in resource poor environments or in field applications. The nanopore sequencing (NS) platform eliminates these issues with a cost effective, simple to use library preparation protocol, and a portable device to sequence and analyze a variety of different types of nucleic acids1,3. We have incorporated the NS platform into several lab classes for master's degree students.
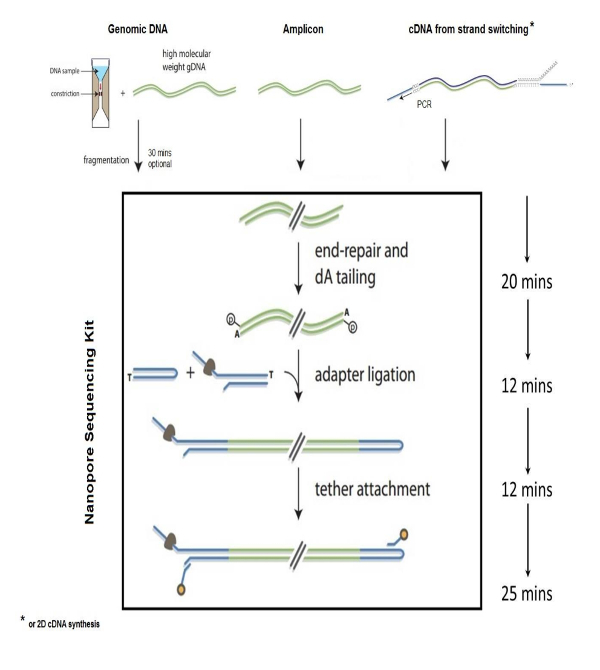
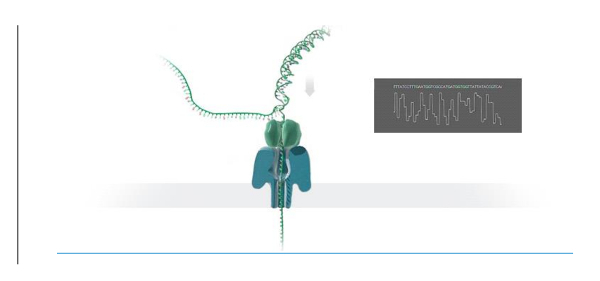
Nanopore technology for sequencing biomolecules has demonstrated wide applications in the life sciences, including identification of bacterial and viral pathogens1,2,6, environmental biodiversity studies, food safety monitoring, genomic analysis3,5, and characterization of bacterial antibiotic resistance4. NS is a fast and accurate method to sequence nucleic acids that is based on the principle of "strand sequencing" by detecting electrical disruption by individual nucleotide bases when single stranded DNA passes through a nanopore inserted into an electrified synthetic polymer membrane. The steps involved in preparing DNA for NS include genome fragmentation, end repair and 3' dA-tailing of genomic fragments, adaptor and tether annealing to the DNA, DNA library purification, and loading the library into the nanopore flow cell device. Fragmentation of the genome into ~8 kb sizes is accomplished by centrifuging 1 - 2 µg of genomic DNA through a g-tube fragmentation tube. The fragmented genomic ends are then repaired and tailed with poly dA using a commercially available kit. Single stranded adapter sequences, which are compatible with the nanopore motor protein, are added to DNA ends which are used to guide the DNA sequence through the nanopore (Figure 1). The tether sequences are required for DNA purification and for localizing the DNA molecules to the pore membrane. The hairpin is generated by ligating a hairpin adapter to one end of the dA tailed library. The hairpin structures at the DNA ends allows reading of the sense and antisense strands as the DNA passes through the nanopore (Figure 2). The prepared genomic library is then purified from the reaction by using streptavidin beads using a magnetic field, followed by loading the sample into the nanopore flow cell for analysis.
The sequenced DNA is assessed for quality and sequencing reads that are acceptable for analysis are then subjected to several bioinformatics tools to identify microbes. The sequences are "translated" into a FASTQ from a FAST5 format. In the FASTQ format, the sequences can then be used in BLAST analysis.
Protocol
Note: Metagenomic DNA is purified from soil (Baltimore County, Maryland) using a commercially available soil genomic isolation kit (see Table of Materials). Using an UV spectrophotometer (see Table of Materials), the purified genomic DNA should have a 260/280 (nm) ratio >1.8 and a 260/230 ratio between 2.0 - 2.2 to assure that the sample is free of contaminants. The amount of genomic DNA required for NS ranges from 200 ng to 2 µg.
1. Rapid Library Preparation Method (Short Protocol)
NOTE: This is a short protocol. See Table of Materials for the rapid sequencing kit.
In a 0.2 mL thin-walled tube (see Table of Materials) add 200 ng of high molecular weight DNA in a final of volume of 7.5 µL of distilled water.
Add 2.5 µL of fragmentation mix (FM) from the rapid library preparation kit and mix gently by inversion. Pulse centrifuge to spin down (1,000 x g for 5 s).
Place the sample in a thermocycler (see Table of Materials) for one round at 30 °C for 1 min followed by 75 °C for 1 min. Remove the tube and pulse centrifuge to spin down the sample.
Add 1 µL of rapid adapter and 0.2 µL of blunt/TA ligase master mix, and then store the adapted and tethered library on ice. NOTE: If using the short protocol proceed to step 8
2. Ligation Sequencing Protocol (Long Protocol): Metagenomic DNA Fragmentation
Note: See Table of Materials for the ligation sequencing kit.
Dilute 1 µg of purified genomic DNA to a final volume of 46 µL in deionized water.
Transfer the sample to a DNA fragmentation tube (see Table of Materials) and centrifuge for 1 min at room temperature at 8,000 x g using a microcentrifuge (see Table of Materials) to produce ~8 kb genomic fragments. Make sure all the liquid has passed into the collection tube.
Invert the fragmentation tube, return to the centrifuge, and centrifuge for 1 min to collect the fragmented genomic DNA into the lower chamber.
Transfer the fragmented genomic DNA to a sterile low-DNA binding 1.5 mL tube, and analyze a portion on a low percentage agarose gel (0.6%) to confirm fragmentation to >30 kb.
3. Fragmented Genomic DNA End-preparation
Using 800 ng of fragmented genomic DNA in 45 µL of deionized water, add 7 µL end-prep reaction buffer (see Table of Materials), 3 µL enzyme mix (see Table of Materials), and 5 µL of nuclease free water.
Mix by inversion and pulse centrifuge (1,000 x g for 5 s).
Transfer sample to a 0.2 mL thin-walled tube, and incubate sample for 5 min at 20 °C, followed by incubation at 65 °C for 5 min.
Pulse centrifuge to bring contents to the bottom of the tube.
Transfer sample to a 1.5 mL low-DNA binding microcentrifuge tube.
Prepare magnetic beads (see table of materials). Add 60 µL of resuspended beads to the end prep reaction from step 3.5 and mix by pipetting.
Incubate on a rotator mixer at 100 rpm (see Table of Materials) for 5 min at room temperature.
Pulse centrifuge the sample to pellet the beads, then place the sample next to magnet.
Once the beads are adhered to the magnet, pipette off supernatant and discard.
Remove the tube from the magnet and wash beads with 200 µL of freshly prepared 70% ethanol by gently pipetting the sample up-and-down.
Pulse centrifuge to pellet the beads and return to the magnet.
Remove the supernatant and discard. Repeat the washing step again using 200 µL of 70% ethanol.
Pulse centrifuge the tube, return to the magnet and remove all the 70% ethanol wash.
With the microcentrifuge tube lid open, allow the beads to air dry for 5 min at room temperature.
Remove the tube from magnet and suspend the pellet in 31 µL of sterile, nuclease-free water. Incubate for 2 min at room temperature.
Return the tube to the magnet until all the beads have pelleted and the eluate is clear.
Transfer the eluate to a 1.5 mL microcentrifuge tube. Using 1 µL of sample, quantify the end-prepped DNA using a UV spectrophotometer at 260 nm (see Table of Materials). Note: The percent recovery should be ~70% (700 ng) from a starting concentration of 1 µg of genomic DNA.
4. Adapter and Tether Addition to End-prepped Genomic DNA Fragments
Mix all the blunt/TA ligase master mix tubes by inversion, and then pulse centrifuge to bring contents to the bottom.
Using 30 µL of end-prepped DNA, add 20 µL of adapter mix, and 50 µL of blunt/TA ligation master mix.
Mix by inversion, pulse centrifuge, and incubate for 10 min at room temperature.
5. Magnetic Bead Preparation
Vortex the beads and then transfer 50 µL of suspended beads to a 1.5 mL microfuge tube.
Place the tube on the magnet to pellet the beads until the eluate clears.
Remove the supernatant and discard.
Add 100 µL of bead binding buffer (see Table of Materials for bead kit) to the beads, vortex to resuspend the beads, pellet the beads on the magnet, remove the supernatant and discard. Repeat the washing with 100 µL of binding buffer.
Add 100 µL of bead binding buffer to the washed beads. Vortex to resuspend the beads.
6. Library Purification
Using a P200 micropipette, add 40 µL of beads from the previous step to the adapted/tethered genomic DNA. Mix the sample carefully by pipetting.
Incubate the sample on a rotator mixer at 100 rpm for 5 min at room temperature.
Place the sample on the magnet and allow the beads to settle. Pipette off the supernatant and discard.
Resuspend the beads in 140 µL of bead binding buffer by pipetting. Place the sample against the magnet, discard the supernatant, and wash again with another 140 µL of bead binding buffer.
Pulse centrifuge the tube, place the tube on the magnet for 2 min. Remove the remaining bead binding buffer from the pellet.
7. Elution of Library from Magnet Beads
Resuspend the magnetic beads in 15 µL of elution buffer by pipetting. Incubate the sample for 10 min at room temperature.
Place the tube against the magnet to pellet the beads. When the solution clears, remove 15 µL of eluate and transfer to a 1.5 mL microfuge tube, and place the sample on ice.
Quantify the library using an UV spectrophotometer. NOTE: The percent recovery should be ~25% (250 ng) from a starting concentration of 1 µg of genomic DNA.
8. Starting a Run/Quality Control
Remove the flow cell from packaging and attach the flow cell to the portable, real-time sequencer (see Table of Materials).
Attach the sequencer to a computer via USB cable and start the sequencing software (see Table of Materials).
Click "Connect" device through the sequencing software, select "NC_Platform_QC.py", and click "Start".
The quality control (QC) takes approximately 6 - 7 min to complete; look for a 'sea of green' (large areas of output) on the QC readout to confirm that there are enough active pores (>800) for DNA sequencing.
9. Starting a Sequencing Run
Open the sequencing program; a dialog box will appear. In the Sample ID box, name the sample.
Click on the "Flow Cell ID" box and enter the code found on the sticker on the top of the flow cell.
Open the flow cell lid and then slide the sample port cover to the right (clockwise) so that the sample port is visible. Once the port is open, check for a small bubble. Remove a few microliters of buffer including the bubble.
Check to be sure that there is buffer across the sensor array.Make the priming buffer by mixing 480 µL of running buffer with fuel mix (RBF-1, see the Table of Materials) (be sure to mix thoroughly first) with 520 µL nuclease-free water.
Add 800 µL of the priming buffer to the priming port. Carefully lift the "SpotOn" cover to make it accessible.
After 5 min, add an additional 200 µL of priming buffer to the priming port.
10. Loading the Library
Before preparing the library place the following reagents on ice: RBF-1 (from the sequencing kit), adapted and tethered library, and library loading beads (LLB; from the sequencing kit).
To a 0.2 mL centrifuge tube, add 25.5 µL of RBF and 12 µL of nuclease-free water kept at room temperature.
Mix the LLB by pipetting up-and-down. Add 26.5 µL of the LLB to the 0.2 µL tube. Mix the reagents by pipetting up-and-down. NOTE: The LLB tends to settle out of the mixture.
Add 11 µL of the adapted and tethered library to the 0.2 µL tube. Mix by inversion and spin down by pulse centrifugation.
Add 75 µL of sample from step 9.4 to the "SpotOn" port dropwise. Make sure that each drop flows into the port before adding the next.
Carefully replace the sample port cover so that the bung enters the port, and replace the device lid.
11. Starting a Sequencing Software Protocol Script
Open the dropdown menu under 'Select Program' and select "48 hour sequencing."
Click on the "Execute" button to start the script.
- Check the results of the Mux scan reported in the notification stream for the number of active pores in the MUX scan. NOTE: This number may differ from the number of pores in the QC run and a large difference may indicate a problem.
- If there is a significant reduction of active pores from the QC run, restart the software, and if there is still a significant difference, reboot the computer.
Check that the heatsink temperature is 34 °C as reported by the sequencing software script. NOTE: If the temperature is not at 34 °C then there will not be enough active pores for the run.
Check the histogram of read lengths to determine if the sequence lengths are expected and appropriate for the experimental design.
Close the desktop agent using 'close x.' Quit the sequencing software by closing down the web GUI. Disconnect the device from the computer.
12. Analysis of Run and Results
Note: During the sequencing run, the data can be monitored using the "VIEW REPORT" program using the desktop agent. During or after the run is complete, files are available in a FAST5 file format in the data → reads → pass folder (default). If this folder is open while sequencing is active, the computer may freeze.
To view and manipulate sequences, download a "HDFViewer" .
To view the sequencing files, directly open the HDF viewer → open Data folder → select All file types → select C: drive (default) → select data → select reads → select pass.
Find and select the FASTQ file in the left dropdown menu; files will open and can be saved in FASTQ format. NOTE: For the purpose of the student experiment, sequences of 350 nucleotides or longer were selected for further analysis. Output from the base calling allows the user to sort sequence reads based on size. FASTQ files can be analyzed via BLAST (NCBI) or other bioinformatics programs.
Representative Results
The experimental design and nanopore technology provided a fast and inexpensive method for students to sequence soil DNA. The representative run passed the quality control parameters with more than 800 pores available for sequencing. The run resulted in over 125,000 reads available for study with a median sequence length of 5.38 kb. Sequences are given a quality score and only those sequences with acceptable scores were then analyzed. As the output of the sequencing reaction is in FAST5 format, which is not accepted by programs such as BLAST (NCBI), the sequences were viewed in the HDF viewer which converts the sequences to FASTQ format, which is compatible for BLAST analysis. In future iterations of the software, the data will be available as a FASTQ format, eliminating the need for the HDF viewer. See Supplementary Figures 1, 2, and 3.
Fifty sequences of over 350 nucleotides in length were subject to analysis by BLASTN (nucleotides against nucleotide database) or BLASTX (nucleotides translated into amino acid sequence) (NCBI) to identify organisms in the soil sample. Given the time constraints of the class and that the focus of the course is on laboratory methods and not bioinformatics, we have the students analyze sequences within these parameters. Our bioinformatics classes can use the data for development of pipeline analysis tools. This level of bioinformatics analysis is not covered in the laboratory based class. Table 1 is a list of the organisms that were identified with e values of less than 4 e-04. Generally, e-values less than 4 e-04 indicate strong similarity between the input sequence (query) and the matched sequence. The organisms identified by BLASTN (against the non-redundant (nr) database) are from a wide variety of species and are available for further genomic and protein analyses. This sample of soil, which came from a garden in Baltimore County, MD had never been tested, so there were no previous data from which to determine what organisms might be in the soil. BLASTN analysis (against the nr database) analysis also indicated there were sequences with no similarities in the nr database. To determine if these are truly novel sequences, further study is needed. Analysis of several sequences by BLASTX revealed several proteins from organisms not represented in the BLASTN analysis. Table 2 lists several possible proteins including endopeptidases, and ATPases-like proteins. Using this library of sequences, students have the opportunity to use more sophisticated bioinformatics tools to perform further analysis, if directed by the instructor.
Figure 1: Genomic library preparation. Steps for preparation of genomic DNA library for sequencing using nanopore technology. This figure has been modified with the necessary permissions. Please click here to view a larger version of this figure.
Figure 2. Schematic of DNA sequencing using a nanopore. DNA passing through a nanopore membrane with electrical signal generation and base identification. This figure has been modified with the necessary permissions. Please click here to view a larger version of this figure.
| Organism | e value |
| Streptomyces sp. | 3.00E-06 |
| Nocardoides sp. | 5.00E-04 |
| Gordonia sp. | 5.00E-04 |
| Hyphomicrobium nitrativorens | 1.00E-139 |
| Starkeya novella | 1.00E-31 |
| Hyphomicrobium denitrificans | 1.00E-30 |
| Fibomicrobium sp. | 5.00E-17 |
| Pseudomonas sp. | 2.00E-15 |
| Rhodothemus marinus | 1.00E-17 |
| Turneiella parva | 2.00E-24 |
| E. coli | 0.00E+00 |
| Bradyrhizobium sp. | 3.00E-30 |
| Gemmatirosa kalamazoonesis | 1.00E-20 |
| Burkholderia sp. | 8.00E-10 |
| Sphingomonas sp. | 8.00E-10 |
| Cellumonas sp. | 6.00E-11 |
Table 1: Selected results of BLASTN analysis. Soil metagenomics DNA sequences subjected to BLASTN similarity search against the NCBI non-redundant nucleotide database. The data reported have e-values of 4 x e-4 or less.
| Protein | Organism |
| Hypothetical protein | Acidobacter bacterium |
| SAM dependent methyltransferase | H. denitrificans |
| ATPase | Jannaschia sp. |
| Endopeptidase | Shigella sonnei |
| Lysis protein | E. coli |
Table 2: Selected results of BLASTX analysis. Soil metagenomics DNA sequences subjected to BLASTX similarity search against the NCBI non-redundant protein database.
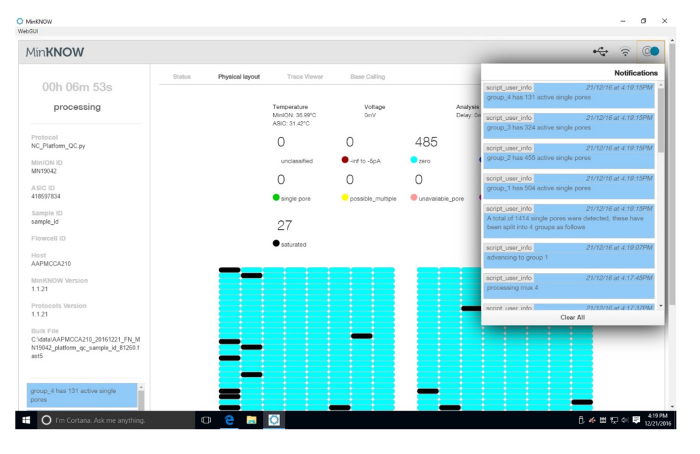 Supplemental Figure 1: Platform QC results. The results of the platform QC are presented. In the upper right corner are the results of the QC for active pores. Each quadrant of the flow cell is tested for active pores. The main page is dynamic and changes as each quadrant is tested. In this run, 1,414 single pores were detected. Please click here to view a larger version of this figure.
Supplemental Figure 1: Platform QC results. The results of the platform QC are presented. In the upper right corner are the results of the QC for active pores. Each quadrant of the flow cell is tested for active pores. The main page is dynamic and changes as each quadrant is tested. In this run, 1,414 single pores were detected. Please click here to view a larger version of this figure.
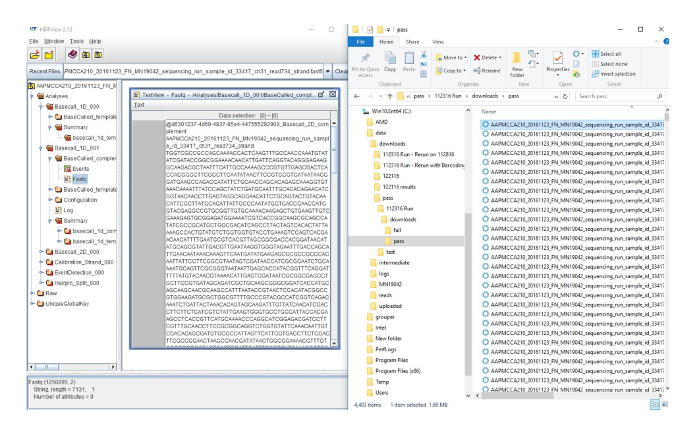 Supplemental Figure 2: Converting FAST5 files to FASTQ files. Here on the right side, is the output of the data from the sequencing run. Each line represents an individual sequence. The left side of the figure shows the highlighted sequence from the right side, in the HDF viewer that converts the sequence to a FASTQ file which can be used for further analysis. Please click here to view a larger version of this figure.
Supplemental Figure 2: Converting FAST5 files to FASTQ files. Here on the right side, is the output of the data from the sequencing run. Each line represents an individual sequence. The left side of the figure shows the highlighted sequence from the right side, in the HDF viewer that converts the sequence to a FASTQ file which can be used for further analysis. Please click here to view a larger version of this figure.
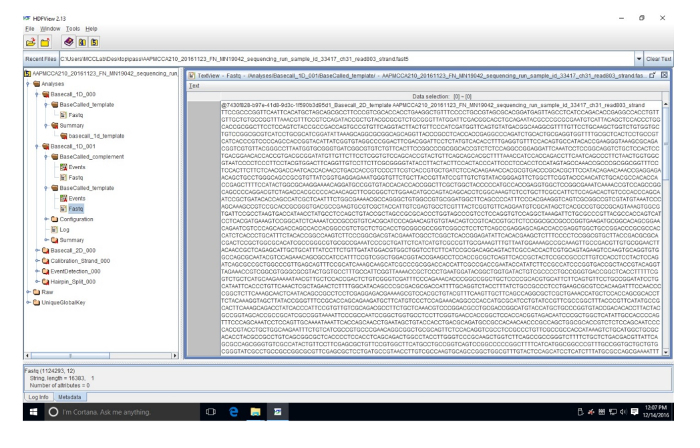 Supplemental Figure 3: Example of FASTQ sequence in HDF viewer. This sequence (read 803) is in a FASTQ file which converts the FAST5 data into nucleotides. Please click here to view a larger version of this figure.
Supplemental Figure 3: Example of FASTQ sequence in HDF viewer. This sequence (read 803) is in a FASTQ file which converts the FAST5 data into nucleotides. Please click here to view a larger version of this figure.
Discussion
Many next generation sequencing methods have been generated and each depends on sequencing by synthesis but the detection platforms for identifying nucleotides differ. NS, the most recent addition to the marketplace, uses a different method entirely, which does not require a sequencing reaction or labeled nucleotides. This method takes advantage of charge differential of already incorporated nucleotides as they pass through an electrified pore. The identification of each nucleotide occurs by modulation of the electrical current by the different bases as they pass through the nanopore. This multiplexed system allows the user to sequence many fragments at a time. By sequencing both strands of the DNA, the accuracy of the sequence is significantly increased and the sequencing software, which can be loaded onto a laptop computer, processes the signals and provides the sequence information that can be analyzed.
In pyrosequencing, a sequencing by synthesis (SBS) method, detection of the specific base incorporated into the template depends on the luciferase assay and the generation of chemiluminescent signals8. In ion semiconductor sequencing, the released hydrogen ion, which decreases the pH is detected by an ion sensor9. Single molecule real-time sequencing depends on the zero mode wave guide (ZMW)10, which illuminates for detection a florescent molecule tagged to the incorporated nucleotide. SBS uses a unique method to amplify the target DNA such that clusters of unique sequences are generated13. Detection of the added nucleotide is achieved when the fluorescence of the tagged nucleotide is recorded. NS on the other hand has unique advantage over other methods in that it requires limited technical resources, is portable, produces long sequencing reads, requires no prior DNA amplification, and can be operated at a reduced cost compared to other methods. Our students found the newer, rapid library prep protocol to be straightforward and amenable to a three hour lab class. Some of the issues we encountered were bubbles in the flow cell which were difficult to remove, it required significant computer power (one terabyte of storage), the current output of data is in a FAST5 file, and the sequencing flow cell has a limited shelf life before it deteriorates. In addition, other disadvantages of the NS Ligation sequencing protocol (long protocol) is that it requires several library preparation steps, requires expertise in molecular biology techniques, and generates reduced sequencing fidelity when compared to some sequencing methodologies3. However, recent advances with the new rapid library preparation kit requires only 10 min for library preparation and has demonstrated a reduced sequencing error rate. The new library preparation method was very amenable for use in a lab class.
There are several critical steps in the protocol, particularly in the QC of the flow cell. This includes performing an initial QC within five days of the receipt of the flow cell and using them within 8 weeks. Although we have used flow cells that were beyond 8 weeks, the number of open/active pores is greatly reduced. It is important that experiments are planned to fit a timeline where maximum use of the flow cells is achieved. We have used the cleaning protocol and reused the flow cells with success.
Investigating the metagenomics of soil represent an untapped genetic reservoir of microbial diversity. For example, one gram of soil is estimated to contain between 107 - 109 prokaryotic cells14. Moreover, soil organisms are a main source of novel natural products, enzymes, and antibiotics. Thus, soil metagenomics DNA sequence analysis represents a valuable instructional tool for students at every level of education. The NS technology's ease of use and low cost make this system a very effective teaching tool. Students can sequence environmental samples, and upon completion of the sequence use available bioinformatics tools to identify and characterize microbes and metagenomics sequences in test samples. Using the NS technology, students have true hands-on experience, which has until now been out of reach for use in laboratory courses because of the advanced technical expertise and high reagent, equipment, and maintenance costs in other sequencing platforms. Recently, one of our students (J. Harrison, personal communication) reported the use of this technology in an environmental monitoring project of farm soils. We expect that there will be many more applications for this technology in the education space.
Disclosures
The authors have no disclosures.
Acknowledgments
This project was support in part by Johns Hopkins University, Office of the Provost through the Gateway Science Initiative.
References
- Quick J, et al. Real-time, portable genome sequencing for Ebola surveillance. Nature. 2016;530(7589):228–232. doi: 10.1038/nature16996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greninger AL, et al. Rapid metagenomics identification of viral pathogens in clinical samples by real-time nanopore sequencing analysis. Genome Med. 2015;7:99. doi: 10.1186/s13073-015-0220-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quick J, et al. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biology. 2015;16:114. doi: 10.1186/s13059-015-0677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashton PM, et al. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat Biotechnol. 2015;33:296–300. doi: 10.1038/nbt.3103. [DOI] [PubMed] [Google Scholar]
- Loman NJ, Quick J, Simpson JT. A complete bacterial genome assembled de novo. using only nanopore sequencing data. Nat Method. 2015;12:733–735. doi: 10.1038/nmeth.3444. [DOI] [PubMed] [Google Scholar]
- Hoenen T, et al. Nanopore Sequencing as a Rapidly Deployable Outbreak Tool. Emerg Infect Dis. 2016;22(2):331–334. doi: 10.3201/eid2202.151796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrosino JF, Highlander S, Luna RA, Gibbs RA, Versalovic J. Metagenomic pyrosequencing and microbial identification. Clin Chem. 2009;55(5):856–866. doi: 10.1373/clinchem.2008.107565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cummings PJ, Ahmed R, Durocher JA, Jessen A, Vardi T, Obom KM. Pyrosequencing for Microbial Identification and Characterization. J. Vis. Exp. 2013. p. e50405. [DOI] [PMC free article] [PubMed]
- Rothberg JW, et al. An integrated semi-conductor device that enabling non-optical genome sequencing. Nature. 2011;475:348–352. doi: 10.1038/nature10242. [DOI] [PubMed] [Google Scholar]
- Levene MJ, Korlach J, Turner SW, Foquet M, Craighead HG, Webb WW. Zero-mode Waveguides for Single-Molecule Analysis at high concentrations. Science. 2003;299:682–686. doi: 10.1126/science.1079700. [DOI] [PubMed] [Google Scholar]
- Eid J, et al. Real-Time DNA Sequencing from Single Polymerase Molecules. Science. 2009;323:133–138. doi: 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- Stoddart D, Heron AJ, Mikhailova E, Maglia G, Bayley H. Single-nucleotide discrimination in immobilized DNA oligonucleotides with a biological nanopore. PNAS. 2009;106:7702–7707. doi: 10.1073/pnas.0901054106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shendure J, Hanlee J. Next generation DNA sequencing. Nat Biotechnol. 2008;26:1135–1145. doi: 10.1038/nbt1486. [DOI] [PubMed] [Google Scholar]
- Daniel R. The Metagenomics of Soil. Nat Rev Microbiol. 2005;3:470–478. doi: 10.1038/nrmicro1160. [DOI] [PubMed] [Google Scholar]