

Abstract
N6-methyladenosine–sequencing (m6A-seq) is a critical tool to obtain an unbiased genome-wide picture of m6A sites of modification at high resolution. It allows the study of the impact of various perturbations on m6A modification distribution and the study of m6A functions. Herein, we describe the m6A-seq protocol, which entails RNA immunoprecipitation (RIP) performed on fragmented poly(A) RNA utilizing anti-m6A antibodies. The captured/enriched m6A positive RNA fragments are subsequently sequenced by RNA-seq in parallel with background control non-immunoprecipitated input RNA fragments. Analyses reveals peaks of m6A enrichment containing sites of modifications analogous to chromatin modification immunoprecipitation experiments.
Keywords: m6A-seq, epitranscriptome, N6-methyladenosine, genome-wide, METTL3, METTL14
1. Introduction
The emergence of observations that N6-methyl-adenosine (m6A) is a wide-spread reversible RNA chemical modification with proteins acting as “writers”, “readers” and “erasers” of m6A has led to a new field coined ‘epitranscriptomics’. Although m6A modifications have been recognized from yeast to humans since the 1970s, many aspects of m6A modification, including function(s), are only beginning to be understood [1]. For example, the m6A biogenesis machinery appears to be critical for developmental cell fate decisions from yeast to humans [2]. The development of m6A “location analyses” called m6A-seq has been a critical step in the field. In this protocol, RNA immunoprecipitation (RIP) is performed on fragmented poly(A) RNA utilizing anti-m6A antibodies followed by RNA-seq of the captured/enriched m6A positive RNA fragments with non-immunoprecipitated RNA fragments serving as input or background control. Utilizing m6A-seq, studies have revealed that m6A modification(s) sites occur on thousands of mRNAs and hundreds of non-coding RNAs in mouse and human cells [3,4]. Topologically along the transcriptome, m6A modifications exhibit enrichment near 3′ end of both coding (often near stop codon or 3′UTR) and non-coding RNAs as well as long internal exons. Subsets of m6A sites on RNAs appear to exhibit tissue and stimuli specificity, suggesting a regulated and dynamic m6A epitranscriptome [5]. Indeed, it has been shown for example that under conditions of stress such as ultraviolet radiation and heat shock that m6A sites accumulate in the 5′UTR of genes to promote 5′ cap independent translation [6]. The “writing” of m6A RNA modification is accomplished via an m6A methyltransferase complex, including two known catalytic mammalian components encoded by METTL3 and METTL14 [7]. Additional critical components required for m6A methyltranferase complex activity include WTAP and VIRILIZER although their exact functions and mechanism of action are unclear [8,9]. m6A modification(s) are reversible, based on the discovery that the protein encoded by fat mass and obesity gene, FTO, and a related protein ALKBH5, operate as m6A demethylases or “erasers” [10,11]. At the molecular level, m6A has been implicated in many aspects of RNA metabolism through the binding of so called YTH domain “reader” proteins including splicing, translational efficiency, cap-independent translation, RNA export and RNA structure [12].
m6A modifications location analyses by m6A-seq is a critical assay to probe the functions of m6A in normal physiology and in pathophysiological conditions. The basic schema of m6A-seq is shown in Figure 1 and allows for 50 nt resolution of sites of modifications. In short, poly(A) RNA is fragmented and subjected to anti-m6A RNA immunoprecipitation (RIP) utilizing a commercially available anti-m6A antibodies. The m6A positive enriched fragments are subsequently sequenced and compared to the distribution of input/non-immunoprecipitated fragmented RNA as a background control. Peaks of m6A enrichment over background/input are then computationally identified utilizing either custom computer scripts or programs such as HOMER [13]. An example of the results of the m6A-seq utilizing sequence alignments to the UCSC genome browser is shown in Figure 2.
Fig 1.
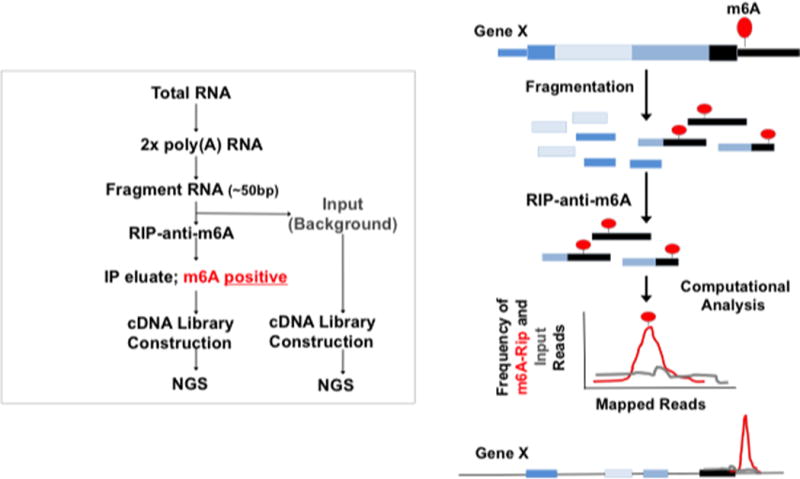
Work flow and schematic diagram of m6A-seq protocol
Fig 2.
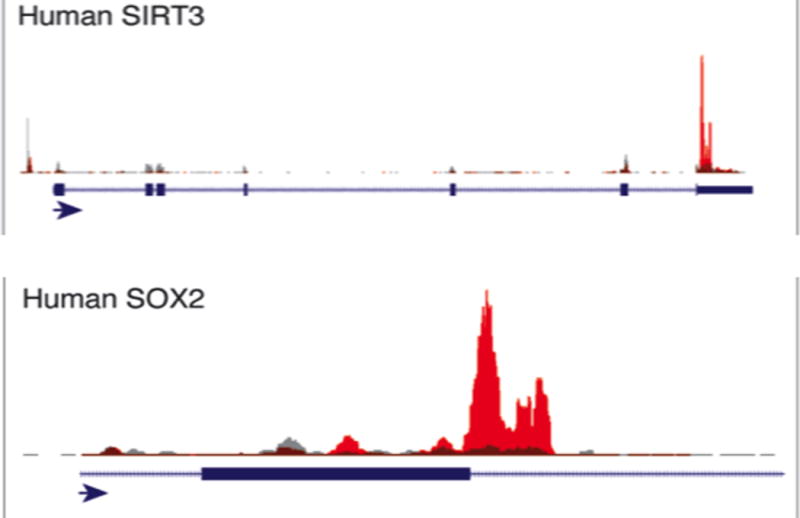
m6A-seq example data tracks. UCSC genome browser tracks of m6A-seq position analyses for two genes of our previously published H1-ESC data (red for m6A-RIP and gray for input)
2. Materials
Prepare all solutions using ultrapure autoclaved distilled water and analytical grade reagents that are RNase and DNase free. Use RNase/DNase free consumables and glassware. All pipetting is performed using sterile RNase/DNase free low retention filtered tips. Prior to be used, every stock solution, buffer solution, antibody batch, Dynabeads and chemicals are independently tested on total RNA extract. The RNA is then run on a BioAnalyzer to control for potential degradation due to RNase contamination.
2.1 Buffers
Fragmentation buffer: 10 mM ZnCl2, 10 mM Tris-HCl pH7.0
Stop Buffer: 0.5M EDTA
m6A Binding Buffer: 50 mM Tris-HCl pH7.4, 150 mM NaCl2, 1% NP-40, 2 mM EDTA; Add RNAse inhibitor at the manufacturer recommended concentration
Low Salt Buffer: 0.2X SSPE, 0.001 M EDTA, 0.05% Tween-20. Add RNAse inhibitor at the manufacturer recommended concentration
High Salt Buffer: 0.2X SSPE, 0.001 M EDTA, 0.05% Tween-20, 137.5 mM NaCl; Add RNAse inhibitor at the manufacturer recommended concentration
TET: 10 mM Tris-HCl pH8.0, 1 mM EDTA pH8.0, 0.05% Tween-20; Add RNAse inhibitor at the manufacturer recommended concentration
Elution Buffer: 0.02 M DTT, 0.150 M NaCl, 0.05 M Tris-HCl pH7.5, 0.001M EDTA, 0.10% SDS; Add RNAse inhibitor at the manufacturer recommended concentration
2.2 M6A RIP-seq
Antibody: Anti-m6A (see Note 1)
Dynabeads Antibody Coupling Kit: (see Note 2)
Acid-phenol:chloroform pH 4.5 (with IAA, 125:24:1)
Chloroform
Absolute Ethanol (200 proof)
SUPERase-in RNAse inhibitor (Ambion)
Ultra Pure Glycogen: 20 mg/mL
Magnets: (see Note 3)
mRNA Purification Kit: Ambion Dynabeads mRNA Purification Kit polyA isolation kit
Kapa library quantification kit: (see Note 4)
3. Methods
3.1. M6A RIP
Carry out all procedures at room temperature unless otherwise specified. All procedures should be done in a clean RNAse free environment.
Each biological replicate for m6A-seq starts by using 400 μg of total RNA yielding approximately 10 μg of double poly(A) selected RNA (see Note 5).
Resuspend each sample obtained from step 1 (10 μg of poly(A) RNA) in 50 μL of UltraPure H2O.
Add 250 μL of fragmentation buffer to the 50 μL of isolated 2x poly(A) RNA to a final volume of 300 μL.
3.2. Fragmentation Step of the RIP
3.3. RNA binding to m6A-Dynabeads
Add 150 μl of pre-equilibrated m6A-Dynabeads (see Notes 2 and 8) to the 350 μl of fragmented RNA from step 2 of section 3.2 to a final volume of 500 μl.
Allow the fragmented RNA to bind to the m6A-Dynabeads at room temperature while rotating (tail-over-head) at 7 rotations per minute for 1 hour.
Place the tubes containing the samples on a magnet allowing the bead complexes to cluster until the solution becomes clear (see Note 3).
Discard the 500 μL liquid phase or supernatant as this fraction represents the m6A negative fragments not captured by the anti-m6A antibody.
3.4 Washing of m6A-Dynabeads
The m6A positive fragments which are retained on the surface of the m6A-coupled Dynabeads are then subjected to a series of wash steps.
Resuspend m6A-Dynabeads-RNA complexes in 500 μL of m6A Binding Buffer, incubate for 3 minutes at room temperature and remove clear supernatant after placing the beads in the magnet.
Repeat step 1 with 500 μL of Low Salt Buffer.
Repeat step 1 with 500 μL of High Salt Buffer. Do not exceed 3-minute incubation time for this step to prevent release of the RNA from the beads.
Repeat step 1 twice with 500 μL of TET buffer.
3.5. Elution of m6A-positive RNA
Add 125 μL of 42°C pre-heated Elution Buffer to the m6A-Dynabead complexes from section 3.4, step 4 and incubate at 42°C for 5 minutes.
At the end of the 5 minutes, vortex the beads gently and place them on the magnet.
Collect the liquid phase and transfer to a fresh tube, kept on ice, as it represents the eluate fraction containing the m6A “enriched RNA”.
Add an additional 125 μL of pre-heated Elution Buffer to the beads and process as described in step 1-3 above for 3 additional times, for a total of 4 elutions.
Collect the liquid phase obtained at each elution step and pool with the previous ones. Keep sample on ice while working on the next elution. After the fourth round of elution, the final total eluate volume of the m6A positive RNA fraction is 500 μL.
3.6. Extraction and Cleanup Step of the RIP
Extract the 500 μl of m6A positive RNA collected in previous step by adding 500 μL of acid phenol-chloroform.
Centrifuged at 4°C at 10,000g for 7.5 minutes.
Carefully collected the upper phase making sure not to touch the inter-phase and transfer to a fresh 1.5 ml tube.
Add 500 μL of previously tested RNAse free chloroform to the fresh tube, vortex briefly and centrifuged at 4°C at 10,000g for 7.5 minutes.
Transfer the upper phase to a fresh 1.5 mL tube and proceed to RNA precipitation (see Note 9) overnight at −20°C
Centrifuged the sample at 4°C for 20 minutes at 16,000g.
Wash the pellet twice in 70% ethanol by centrifuging for 10 minutes at 4°C at 16,000g.
Dry the pellet at room temperature for 10 minutes prior to re-suspend it in the desired volume (typically 5-6 μl) of Ultra-Pure H20. (see Note 10)
3.7. Library Construction
We have generally utilized 100 ng of RNA (100 ng of input and 100 ng of post m6A-IP positive fraction) for library construction utilizing the Illumina TrueSeq Stranded mRNA Sample Preparation Guide.
Add 13 μL of Fragment, Prime, Finish Mix to the 5 μl m6A positive fragmented RNA obtained in step 13 (to final volume of 18 μL).
Skip the fragmentation step in the Illumina protocol given the RNA has already been fragmented, and proceed immediately to the synthesis of the First Strand cDNA.
Follow the Illumina protocol to the end.
Verify the fragment sizes of each individual library on an Agilent BioAnalyzer 2100 or equivalent using High Sensitivity DNA chip.
Quantify the library by qPCR on using the Kapa library quantification kit according to the manufacturer’s instructions (see Note 4).
Submit the libraries for high-throughput sequencing (Note 11-13)
Acknowledgments
This work was supported by MGH Start-up funds to Cosmas Giallourakis. We thank Yi Xing, PhD and Jinkai Wang, PhD at UCLA who have been our computational biologist collaborators on m6A related projects.
Footnotes
This protocol has been tested using Anti-M6A (N6-methyladenosine) antibody from Synaptic Systems (Cat. No 202 003).
This protocol was tested using Dynabeads Antibody Coupling Kit following exactly the manufacturer protocol. The anti-m6A antibody was coupled to the Dynabeads at ratio of 5 μg of anti-m6A antibody per 1 mg of Dynabeads, as suggested by the manufacturer (coupling range of 5-10 μg of antiboby per mg of Dynabeads). Based on the number of samples in the experiment, the amount of Dynabeads and antibody to be coupled has to be adjusted accordingly following the manufacturer recommendations.
-
-16× 1.5 mL tubes rack: Invitrogen, DYNAL Invitrogen based separations
-
-96 Wells plate: Ambion, Magnetic Stand-96.
We used KAPABIOSYSTEMS Cat.NO KK4824
To isolate 10 μg of double poly(A) RNA, we often start with approximately 400 μg of total RNA.
The conditions of fragmentation detailed in this protocol allow for approximately 50 bp fragments on multiple types of poly(A) RNA examined.
Individual users might need to optimize the fragmentation conditions based on their samples.
Preparation of equilibrated coupled m6A-Dynabeads. 50 μL of coupled m6A-Dynabeads are utilized per sample. The 50 μL m6A-Dynabeads are equilibrated by re-suspending them in 500 μL of m6A Binding Buffer for 5 minutes at room temperature and then placed on the magnet. The supernatant is discarded and this step is repeated a second time. The 50 μL of equilibrated m6A-Dynabeads is then re-suspended in 150μL of Binding Buffer and used in Step 1 of section 3.3.
NaCl/ethanol precipitation is carried out overnight at −20°C in the presence of 1μL Ultra Pure Glycogen. To this end, 1/10 volume of 3M NaCl and 2.5× volumes of absolute ethanol are added to the sample.
Following pellet resuspension you can choose to use 1μL of your resuspended RNA to check the quantity and size of your m6A immunoprecipated RNA by Nanodrop photometer and bioanalyzer respectively, or proceed to library construction directly.
The basic schema of m6A-seq is shown in Figure 1 and allows for 50 to 200 bp resolution of sites of modifications depending on fragmentation condition, depth of sequencing and whether paired end reads or single end reads are used. The current protocol has been optimized so that the m6A peaks are identified to approximately 50 nt resolution based on average length of fragmented RNA and paired end sequencing.
To date, the produced antibodies that recognize the m6A modification also appear to bind another structurally related modification N6,2′-O-dimethyladenosine called m6Am. This m6Am modification can be distinguished from m6A modifications as it is always in the 5′UTR and if present always on the first nucleotide of a transcript if it starts with adenosine. Utilizing single site resolution CLIP based m6A-seq, it was estimated that approximately 4-8% of sites identified by m6A-seq may actually be m6Am (14). As transcripts often have heterogeneous start sites, the peak intensity of potential m6Am sites identified by m6A-seq are often less then peak intensities of m6A found in long internal exons or in the 3′end of genes.
In terms of identifying the site(s) of modifications, the consensus m6A motif is RR(m6A)CH. We then search for this motif surrounding the point of maximal peak enrichment and this is inferred to be the site of modification. However, it is possible that there is more then one consensus site near the peak and further experiments such as SCARLET or CLIP based m6A-seq would be needed to determine if there are multiple m6A sites harbored within the m6A peak [14–16].
References
- 1.Meyer KD, Jaffrey SR. The dynamic epitranscriptome: N6-methyladenosine and gene expression control. Nat Rev Mol Cell Biol. 2014;15(5):313–326. doi: 10.1038/nrm3785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Batista PJ, Molinie B, Wang J, Qu K, Zhang J, Li L, Bouley DM, Lujan E, Haddad B, Daneshvar K, Carter AC, Flynn RA, Zhou C, Lim KS, Dedon P, Wernig M, Mullen AC, Xing Y, Giallourakis CC, Chang HY. m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell. 2014;15(6):707–719. doi: 10.1016/j.stem.2014.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dominissini D, Moshitch-Moshkovitz S, Schwartz S, Salmon-Divon M, Ungar L, Osenberg S, Cesarkas K, Jacob-Hirsch J, Amariglio N, Kupiec M, Sorek R, Rechavi G. Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature. 2012;485(7397):201–206. doi: 10.1038/nature11112. [DOI] [PubMed] [Google Scholar]
- 4.Meyer KD, Saletore Y, Zumbo P, Elemento O, Mason CE, Jaffrey SR. Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell. 2012;149(7):1635–1646. doi: 10.1016/j.cell.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz S, Agarwala SD, Mumbach MR, Jovanovic M, Mertins P, Shishkin A, Tabach Y, Mikkelsen TS, Satija R, Ruvkun G, Carr SA, Lander ES, Fink GR, Regev A. High-resolution mapping reveals a conserved, widespread, dynamic mRNA methylation program in yeast meiosis. Cell. 2013;155(6):1409–1421. doi: 10.1016/j.cell.2013.10.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Meyer KD, Patil DP, Zhou J, Zinoviev A, Skabkin MA, Elemento O, Pestova TV, Qian SB, Jaffrey SR. 5′ UTR m(6)A Promotes Cap-Independent Translation. Cell. 2015;163(4):999–1010. doi: 10.1016/j.cell.2015.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu J, Yue Y, Han D, Wang X, Fu Y, Zhang L, Jia G, Yu M, Lu Z, Deng X, Dai Q, Chen W, He C. A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat Chem Biol. 2014;10(2):93–95. doi: 10.1038/nchembio.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schwartz S, Mumbach MR, Jovanovic M, Wang T, Maciag K, Bushkin GG, Mertins P, Ter-Ovanesyan D, Habib N, Cacchiarelli D, Sanjana NE, Freinkman E, Pacold ME, Satija R, Mikkelsen TS, Hacohen N, Zhang F, Carr SA, Lander ES, Regev A. Perturbation of m6A writers reveals two distinct classes of mRNA methylation at internal and 5′ sites. Cell Rep. 2014;8(1):284–296. doi: 10.1016/j.celrep.2014.05.048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ping XL, Sun BF, Wang L, Xiao W, Yang X, Wang WJ, Adhikari S, Shi Y, Lv Y, Chen YS, Zhao X, Li A, Yang Y, Dahal U, Lou XM, Liu X, Huang J, Yuan WP, Zhu XF, Cheng T, Zhao YL, Wang X, Rendtlew Danielsen JM, Liu F, Yang YG. Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 2014;24(2):177–189. doi: 10.1038/cr.2014.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zheng G, Dahl JA, Niu Y, Fedorcsak P, Huang CM, Li CJ, Vagbo CB, Shi Y, Wang WL, Song SH, Lu Z, Bosmans RP, Dai Q, Hao YJ, Yang X, Zhao WM, Tong WM, Wang XJ, Bogdan F, Furu K, Fu Y, Jia G, Zhao X, Liu J, Krokan HE, Klungland A, Yang YG, He C. ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol Cell. 2013;49(1):18–29. doi: 10.1016/j.molcel.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jia G, Fu Y, Zhao X, Dai Q, Zheng G, Yang Y, Yi C, Lindahl T, Pan T, Yang YG, He C. N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat Chem Biol. 2011;7(12):885–887. doi: 10.1038/nchembio.687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Roundtree IA, He C. RNA epigenetics-chemical messages for posttranscriptional gene regulation. Curr Opin Chem Biol. 2016;30:46–51. doi: 10.1016/j.cbpa.2015.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Linder B, Grozhik AV, Olarerin-George AO, Meydan C, Mason CE, Jaffrey SR. Single-nucleotide-resolution mapping of m6A and m6Am throughout the transcriptome. Nat Methods. 2015;12(8):767–772. doi: 10.1038/nmeth.3453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ke S, Alemu EA, Mertens C, Gantman EC, Fak JJ, Mele A, Haripal B, Zucker-Scharff I, Moore MJ, Park CY, Vagbo CB, Kussnierczyk A, Klungland A, Darnell JE, Jr, Darnell RB. A majority of m6A residues are in the last exons, allowing the potential for 3′ UTR regulation. Genes Dev. 2015;29(19):2037–2053. doi: 10.1101/gad.269415.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu N, Parisien M, Dai Q, Zheng G, He C, Pan T. Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA. 2013;19(12):1848–1856. doi: 10.1261/rna.041178.113. [DOI] [PMC free article] [PubMed] [Google Scholar]