

Abstract
Bacterial and plant A/B toxins exploit the natural trafficking pathways in eukaryotic cells to reach their intracellular target(s) in the cytosol and to ultimately kill. Such A/B toxins generally consist of an enzymatically active Asubunit (e.g., ricin toxin A (RTA)) and one or more cell binding Bsubunit(s), which are responsible for toxin binding to specific cell surface receptors. Our current knowledge of how A/B toxins are capable of efficiently intoxicating cells helped scientists to understand fundamental cellular mechanisms, like endocytosis and intracellular protein sorting in higher eukaryotic cells. From a medical point of view, it is likewise important to identify the major toxin trafficking routes to find adequate treatment solutions for patients or to eventually develop therapeutic toxin-based applications for cancer therapy.
Since genome-wide analyses of A/B toxin trafficking in mammalian cells is complex, time-consuming, and expensive, several studies on A/B toxin transport have been performed in the yeast model organism Saccharomyces cerevisiae. Despite being less complex, fundamental cellular processes in yeast and higher eukaryotic cells are similar and very often results obtained in yeast can be transferred to the mammalian situation.
Here, we describe a fast and easy to use reporter assay to analyze the intracellular trafficking of RTA in yeast. An essential advantage of the new assay is the opportunity to investigate not only RTA retro-translocation from the endoplasmic reticulum (ER) into the cytosol, but rather endocytosis and retrograde toxin transport from the plasma membrane into the ER. The assay makes use of a reporter plasmid that allows indirect measurement of RTA toxicity through fluorescence emission of the green fluorescent protein (GFP) after in vivo translation. Since RTA efficiently prevents the initiation of protein biosynthesis by 28S rRNA depurination, this assay allows the identification of host cell proteins involved in intracellular RTA transport through the detection of changes in fluorescence emission.
Keywords: Cellular Biology, Issue 130, S. cerevisiae, ricin toxin A chain (RTA), A/B toxin, endocytosis, retrograde protein transport, fluorescence-based reporter assay
Introduction
Patients suffering from infections by toxin producing bacteria represent a severe medical and financial burden for each social health care system, in particular since efficient therapeutic treatments are still largely missing. To develop new therapeutic strategies, the complex intoxication mechanisms of medically relevant A/B toxins such as cholera toxin, Shiga toxin, or ricin need to be fully understood at the molecular level based on novel powerful assays that have to be implemented.
In recent years, several studies attempted to analyze A/B toxin transport in yeast and mammalian cells by using time-consuming and cost-intensive methods such as radioactive toxin labeling1,2 as well as siRNA-based screening approaches3. In some cases, toxin trafficking has been visualized in vivo by fluorescence microscopy after chemical and/or genetic coupling of individual toxin subunits with fluorophores, quantum dots, or fluorescent proteins4,5. Unfortunately, such modifications often lead to inactive and/or altered natural properties of the toxins. Another elegant way to indirectly answer a wide variety of scientific questions is the use of reporter systems based on enzymes such as lacZ, luciferase, or fluorescent proteins (e.g. GFP or Discosoma sp. red fluorescent protein (dsRed)).
In this manuscript, a simple protocol is described which identifies cellular components required for the intracellular transport of extracellularly applied RTA in S. cerevisiae. Thereby, a fluorescence-reporter plasmid containing an N-terminal ER-import signal followed by GFP acts as a protein biosynthesis sensor, which indirectly measures RTA-mediated protein translation inhibition by GFP fluorescence emission after in vivo translation6. In case that RTA endocytosis and/or intracellular trafficking is negatively (or positively) affected in a particular yeast deletion mutant compared to wild-type, this can be detected through an increase (or decrease) in GFP fluorescence emission6.
So far, all methods analyzing RTA transport in yeast cells were restricted to the ER-to-cytosol retro-translocation process of RTA. In such an artificial system, RTA containing an ER import signal is expressed from an inducible promoter resulting in a suicidal phenotype1,7. Although the cell binding B-subunit of ricin is likewise missing in the experimental setup described in this manuscript and, thus, does not fully represent the natural situation of ricin holotoxin intoxication8, toxin transport from the plasma membrane through the Golgi apparatus to the ER can be closely mimicked with this novel assay. Interestingly, the preliminary results obtained in the pilot study indicate that the trafficking pathways used by RTA reveal striking similarities with the intoxication route of ricin holotoxin.
In summary, the described method can be used to determine the specific role of selected cellular proteins in RTA endocytosis and trafficking in yeast. Furthermore, this experimental setup might be easily adapted to other ribosome inactivating toxins produced and secreted from various yeast and bacterial species such as zymocin or Shiga toxin.
Protocol
NOTE: An overview of the general experimental workflow is depicted in Figure 1.
Caution: RTA is highly toxic for humans. Safety lab permission S2 (biosafety level 2 equivalent) is needed. Please wear gloves during the entire experiment.
1. Heterologous Expression of His-tagged RTA in Escherichia coli
Transform E. coli cells with the expression plasmid pET24-RTA(His)6 or the empty vector pET24a(+) using standard electroporation protocols9,10. Cells containing the empty plasmid serve as a negative control.
After selection of positive clones on kanamycin-containing (100 µg/mL) LB plates, inoculate cells containing the RTA expression plasmid or the empty vector in 5 mL LBkan medium (LB medium with 100 µg/mL of kanamycin) and incubate at 37 °C and 220 rpm for 24 h.
Supplement 1 L LBkan medium with 5 mL pre-culture and incubate cells at 37 °C and 220 rpm until cells have reached an OD600 of 0.8-1.0 (approximately 3-4 h). Thereafter, reduce culture temperature to 28 °C and prepare a 1 M of isopropyl-β-D-1-thiogalactopyranoside (IPTG) stock solution in H2O.
Induce RTA expression of the E. coli by adding IPTG to a final concentration of 1 mM.
After 3.5 h at 28 °C and 220 rpm, harvest cells by centrifugation at 10,000 x g and 4 °C for 10 min, wash the pellet two times with 5 mL of binding buffer (500 mM NaCl, 10 mM imidazole, and 20 mM KH2PO4, pH = 7.4) at 10,000 x g and 4 °C for 10 min, and resuspend pellet in 5 mL binding buffer. NOTE: Protocol can be paused at this stage and cells can be stored at 80 °C for several days.
2. Purification of His-tagged RTA via Affinity Chromatography
Sonicate cells on ice using the following protocol: 15 s pulse (20 microns), 30 s pause. Repeat this step five times.
Centrifuge cell lysate at 21,000 x g and 4 °C for 15 min and filter supernatant using a sterile syringe filter system (0.2 µm pore size). NOTE: Cell pellets of successfully sonicated samples show transparent borders.
- Use an automated purification system equipped with a 5 mL Ni2+-based affinity column to purify the His-tagged RTA fraction from the sterile-filtered E. coli supernatant. In general, use an elution speed of 1 mL/min and cool the whole purification system to prevent non-efficient toxin binding and loss of toxin activity. NOTE: Parameters for efficient RTA purification are listed in Table 1. See also Becker et al. for further information9.
- Briefly, equilibrate the affinity column with 20 mL of binding buffer to remove the storage buffer. Apply sterile-filtered supernatant onto the affinity column using a syringe.
- Wash the column with 25-35 mL of binding buffer to remove the unbound proteins from the column. Perform the washing step until UV absorbance at 280 nm is close to the initial UV value.
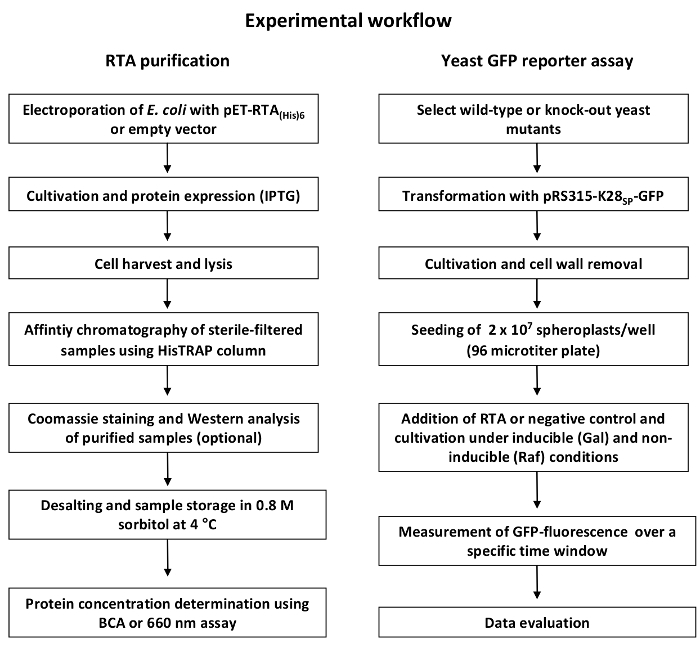
- Elute bound RTA fraction in 20-35 mL of elution buffer (500 mM imidazole, 500 mM NaCl, 20 mM KH2PO4, pH = 7.2) and keep the sample on ice (Figure 2A and Figure 2B). NOTE: Elution of the RTA fraction is marked by an increase in UV absorption. Please note to exclusively collect this fraction to prevent contamination with nonspecific bound proteins.
- Desalt eluted RTA fraction on a 5 mL desalting column and equilibrate sample in 0.8 M sorbitol.
- Replace the affinity column of the purification system by the desalting column and first equilibrate the column with 20 mL of 0.8 M sorbitol.
- Apply the eluted RTA fraction to the column via a syringe. Wash the column with 100 mL of 0.8 M sorbitol and elute the desalted RTA fraction in a 15 mL tube as soon as the UV absorption starts to increase (Figure 2C).
- Store the eluted RTA fraction at 4 °C. NOTE: Directly stop RTA fraction sampling when conductance increases to avoid salt contamination. Parameters for the desalting procedure are listed in Table 1.
Concentrate eluted RTA fraction at 10,000 x g and 4 °C for 30-180 min using a 10 kDa cut-off spin column to a final volume of 1-2 mL and store sample at 4 °C for 3-4 weeks. Caution: Do not freeze the sample since freezing leads to a complete loss of RTA activity.
Determine protein concentration using a conventional protein determination kit. Protein concentration should be in the range of 1-1.5 mg/mL. NOTE: RTA tends to precipitate if the protein concentration is too high (>5 mg/mL).
3. Yeast Transformation and Cell Wall Removal
Transform wild-type or selected yeast deletion mutants with the GFP reporter plasmid pRS315-K28SP-GFP6 using standard lithium acetate transformation methods14. Incubate cells on leucine drop-out (d/o) glucose plates (2% glucose, 1.5% agar, 0.5% ammonium sulfate, 0.17% Yeast Nitrogen Base (YNB), and 0.087% d/o mix without leucine) at 30 °C for 2-3 days for positive clone selection.
Pick 3 different yeast clones from each plate and inoculate the clones in 100 mL of leucine d/o raffinose medium (2% raffinose, 0.5% ammonium sulfate, 0.17% YNB, and 0.087% d/o mix without leucine) at 220 rpm and 30 °C to OD600 = 1.0-2.0 (2-4 x 107 cells/mL). NOTE: Cell growth of the different yeast deletion strains is different. Monitor OD600 via a spectrophotometer.
To calculate OD600 values, dilute samples to OD600 = 0.1-0.3 (1:5 to 1:10 dilutions) and measure OD600 in a spectrophotometer. As reference, use H2O supplemented with the corresponding amount of leucine d/o raffionose medium. Finally, resuspend OD600 = 125 in 5 mL of sterile water (5 x 108 cells/mL).
Centrifuge cells at 25 °C for 5 min at 10,000 x g, wash cells twice with 5 mL sterile water, and resuspend cells in 50 mL spheroplasting buffer (0.8 M sorbitol, 10 mM Tris-HCl (pH 7.5), 10 mM CaCl2, 2 mM dithiothreitol (DTT), and 200 µg/mL lytic enzyme).
Incubate the 50 mL culture at 100 rpm and 30 °C for 90 min. Optional: Monitor spheroplast formation every 15 min by phase contrast microscopy. Under the microscope, cells should have a dark translucent gray color. Bright (refractile) cells are insufficiently digested, whereas ghosts (pale gray cells with little, if any, internal structure) are over-digested.
- Check spheroplast preparation efficacy for each sample.
- After finishing step 3.5, mix 4 µL of the spheroplasted 50 mL culture with 496 µL spheroplasting buffer (approximately 2 × 106 spheroplasts) and centrifuge for 10 min at 400 x g.
- Resuspend pellet in 10 mL H2O distilled, vortex sample for 30 s, and plate out 10 µL of the sample on leucine d/o glucose plates (2% glucose, 1.5% agar, 0.5% ammonium sulfate, 0.17% YNB, and 0.087% d/o mix without leucine).
- Incubate cells for 3 days at 30 °C. Count the total number of grown cell colonies on the plate. For data evaluation, use only samples with efficiency higher than 98% (total cell colony number <40 colonies/plate).
Centrifuge cells from step 3.5 at 400 x g and 4 °C for 10 min and wash cells twice with 5 mL stabilized leucine d/o raffinose medium (0.8 M sorbitol, 2% raffinose, 0.5% ammonium sulfate, 0.17% YNB, and 0.087% drop-out mix without leucine). Resuspend cells in 5 mL stabilized leucine d/o raffinose medium (1 x 108 cells/mL) and directly use cells for the GFP reporter assay measurements (Section 4).
4. GFP Reporter Assay Measurement in 96-Well Plates
Seed out the yeast cell spheroplasts obtained in step 3.7 into 96-well plates (200 µL/well).
Add 70 µL stabilized leucine d/o raffinose medium containing negative control (eluate of a Ni2+-affinity purified cell lysate from IPTG-induced E. coli cells expressing the empty vector) or purified RTA in a final RTA concentration of 5 µM (corresponding to 160 g/L RTA) in each well.
Immediately add 30 µL stabilized galactose solution (30% galactose, 0.8 M sorbitol) to induce GFP expression and subsequently start the measurement. NOTE: Perform at least 3 technical replicates per experiment and 3 biological replicates per yeast strain.
After finishing sample preparation (steps 4.1-4.3), put the 96-well plate in a fluorescence reader and start the measurement. Use the 475/509 nm filter set required for GFP fluorescence detection. Perform measurements at 30 °C, 120 rpm, and with a shaking diameter of 1 mm over a time window of 20 h (measuring intervals of 10 min). NOTE: The GFP filter set is normally available in all reader systems. Temperature, time window, measuring intervals, and RTA concentration can be adjusted for own needs. Representative results are shown in Figure 4.
Optional: Use additional internal controls in the measurement for quality control. Prepare a negative control by adding 30 µL stabilized leucine d/o raffinose medium instead of 30% galactose (no GFP induction). In addition, add 70 µL of 0.8 M sorbitol stabilized G418 solution (300 µg/mL). The protein translation inhibitor G418 serves as positive control for protein inhibition as it prevents GFP expression in yeast.
- Calculate relative GFP fluorescence in percent for the 20 h time point according to the following equation (see Figure 4A):
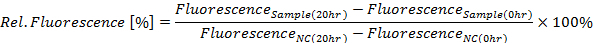 Where NC is the negative control
Where NC is the negative control
- Alternatively, determine the relative GFP fluorescence for each measuring point (in this case 10 min, see also step 4.4) using the above equation. As shown in Figure 4B, create a graph by blotting the GFP fluorescence intensities (y-axis) over time (x-axis).
Representative Results
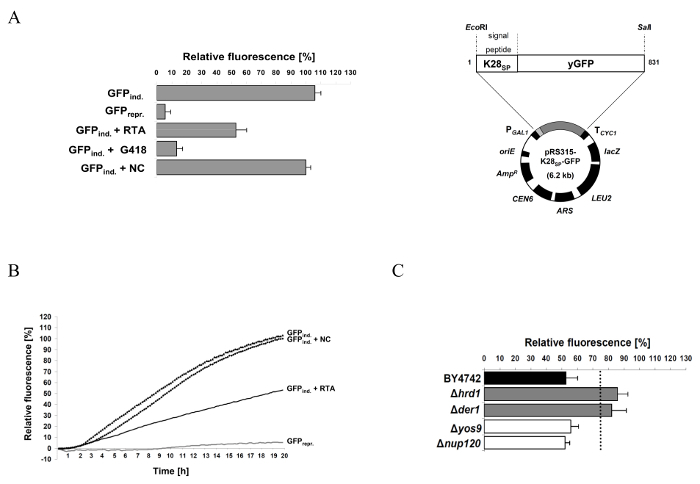
The general workflow of the protocol described in this manuscript is illustrated in Figure 1, roughly summarizing the single steps for successful RTA purification and the subsequent GFP reporter assay experiment. A more detailed description of each individual step can be found in the protocol. Figure 2 illustrates the expected result of a successful RTA purification by affinity chromatography (Figure 2A) and empty vector control (Figure 2B). Figure 2C shows a representative outcome of a desalting experiment illustrating the ideal separation of the protein peak (blue) and the salt-eluted peak (brown). An effective RTA purification is presented in form of a Coomassie-stained SDS-gel in Figure 3. Finally, Figure 4 summarizes some original results obtained by this simple and robust GFP-based reporter assay method6. Figure 4A illustrates that the established GFP reporter assay is able to produce reliable results and schematically depicts the GFP reporter plasmid used in this study. In the graph, the relative GFP fluorescence of yeast spheroplasts under RTA-treated and non-treated conditions is compared with each other. As expected, non-induced as well as G418-treated cells show almost no GFP fluorescence. In non-treated and negative control-treated (empty vector) cells, GFP expression is not affected and significant differences between both samples were not observed. In contrast, RTA treatment leads to the expected GFP fluorescence reduction mediated by its ribosome inhibiting activity. In Figure 4B, a time curve of RTA-induced GFP fluorescence inhibition is displayed. This kind of data presentation contains additional information about the timing of GFP expression (90 min after galactose induction) and the starting point of the translational inhibition by RTA (210 min after application in wild-type cells). The delay in RTA-mediated fluorescence decrease most likely reflects the uptake kinetics and intracellular transport of RTA to the cytosol. Figure 4C illustrates the relative GFP fluorescence obtained from selected yeast mutants after a 20 h RTA-treatment and compared to RTA-treatment and compared to RTA-treated wild-type cells. The ERAD components Hrd1p and Der1p were chosen as suitable positive controls because earlier studies already demonstrated that both proteins are involved in ER-to-cytosol retrotranslocation of RTA in yeast1. Therefore, both mutants show the expected increase in GFP fluorescence compared to wild-type cells. In contrast, relative GFP fluorescence values in Δyos9 and Δnup120 cells are not significantly different. It has already been described that neither a lack of the ER quality control lectin Yos9p nor a lack in the nuclear pore protein Nup20p affect RTA toxicity and transport1. A list of the specific affinity chromatography and desalting parameters can be found in Table 1.
Figure 1: General workflow of RTA purification and GFP-based reporter assay. The RTA purification (left) starts with transformation of E. coli with the expression or empty vector followed by cultivation and IPTG-induced RTA expression. After cell lysis and sterile-filtration, the supernatant is purified via Ni2+ affinity chromatography and sample purity is verified via Coomassie staining or Western blot. Before the eluted RTA fractions are ready to use in the GFP reporter assay experiment, fractions need to be desalted and concentrated, and protein concentration must be determined. The GFP reporter assay experiment (right) starts with the transformation of wild-type and/or selected yeast deletion mutants with the GFP expression construct. After cultivation, the cell wall is removed by enzymatic treatment to allow RTA in vivo uptake. Then, GFP fluorescence of yeast cell spheroplasts is measured under toxin-treated and non-treated conditions over time in a fluorescence reader (475/509 nm). Finally, relative GFP fluorescence is determined using the equation shown in step 4.6 and is displayed as illustrated in Figure 4. Please click here to view a larger version of this figure.
Figure 2: Representative affinity chromatography and desalting results of E. coli cells containing pET-RTA(His)6 or the empty vector control. (A) Typical purification graph of an E. coli cell lysate sample expressing RTA(His)6 from vector pET24-RTA(His)6. Blue curve represents the UV absorption at 280 nm caused by the aromatic rings of the amino acids Trp, Tyr, and Cys and serves as protein indicator. During column loading, unbound proteins are detected in the flow-through (0-10 mL). After removing unbound proteins by washing with binding buffer, elution buffer concentration is increased from 0 to 100% (red curve) and bound proteins are immediately eluted from the column. The peak of the blue curve between 25 and 30 mL represents the fraction of bound His-tagged RTA (black box). (B) Typical purification graph of an E. coli lysate from cells expressing empty vector control. Same purification protocol as in (A). As shown in the black box, no proteins are eluted from the column in the negative control. (C) Desalting diagram of an E. coli pET24-RTA(His)6 sample after affinity purification. The diagram shows the separation of the RTA protein fraction (peak of 280 nm absorption between 20-30 mL) and the salt fraction (increasing conductance peak after 30 mL). Please click here to view a larger version of this figure.
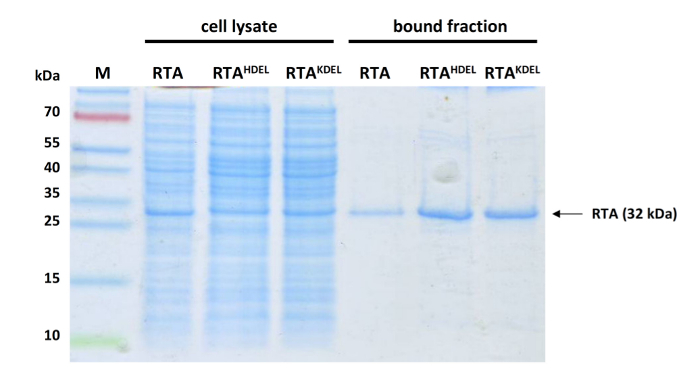
Figure 3: Representative Western analysis of different samples before and after affinity purification. 20 µL cell lysate (lanes 2-4) or purified sample (lanes 5-7) of different RTA variants (only unmodified RTA was used in this study) analyzed by SDS-PAGE and Coomassie blue staining. Lane 1, protein ladder; lane 2, unmodified RTA; lane 3, RTAHDEL; lane 4, RTAKDEL; lane 5, unmodified RTA; lane 6, RTAHDEL; lane 7, RTAKDEL. Please click here to view a larger version of this figure.
Figure 4: Representative results obtained by the reporter assay for RTA-treated wild-type and selected yeast deletion mutants. (A) Relative GFP fluorescence of wild-type yeast spheroplasts containing the reporter plasmid pRS315-K28SP-GFP (right) under induced (3% galactose, GFPind.) and non-induced (2% raffinose, GFPrepr.) conditions after 20 h GFP induction. Relative GFP fluorescence was also analyzed in the presence of RTA (5 µM), G418 (300 µg/mL) or the negative control (NC). The protein translation inhibitor G418 serves as positive control and prevents GFP expression in yeast. Mean values and standard deviation (SD) are indicated. (B) Relative GFP fluorescence from (A) displayed as time course over 20 h. This diagram provides additional information about the timing of GFP expression and translation inhibition by RTA; the curve behavior indirectly reflects the kinetics of RTA uptake and intracellular transport to the cytosol. (C) Representative result of selected yeast mutants defective in cellular proteins that are known to be involved (Hrd1p and Der1p) or not involved (Yos9p and Nup120p) in RTA trafficking in yeast1,6. The dotted line represents a threshold of significance which is based on the fluorescence emission obtained by the positive control strains (for further explanation see reference6). Mean values and standard deviation (SD) are indicated. The figure was modified from Becker et al.6 Please click here to view a larger version of this figure.
| Affinity chromotography parameter | ||
| Measuring parameters | UV (280 nm), Elution buffer concentration, Conductance | |
| Settings | Variable/Unit | Value |
| Flow rate | mL/min | 1 |
| Maximum column pressure | MPa | 0.35 |
| Wavelength | nm | 280 nm |
| Binding buffer | Pump position | A |
| Elution buffer | Pump position | B |
| Starting concentration elution buffer | % | 0 |
| System volume compensation | mL | 10 |
| Column equilibration | mL | 20 |
| Sample injection over superloop/syringe | mL | cell lysate volume |
| Washing step | mL | 20-35 |
| Elution step | mL | 20-35 |
| Concentration elution buffer during elution | % | 100 |
| Recalibration of column with 20% EtOH | mL | 50 |
| Desalting parameter | ||
| Measuring parameters | UV (280 nm), Conductance | |
| Settings | Variable/Unit | Value |
| Flow rate | mL/min | 4 |
| Maximum column pressure | MPa | 0.35 |
| Wavelength | nm | 280 nm |
| Binding buffer | Pump position | A |
| Elution buffer | Pump position | B |
| Starting concentration elution buffer | % | 0 |
| System volume compensation | mL | 10 |
| Column equilibration | mL | 20 |
| Sample injection over superloop/syringe | mL | cell lysate volume |
| Desalting buffer injection (0.8 M sorbitol) | mL | 100 |
| Recalibration of column with 20% EtOH | mL | 50 |
Table 1: Parameters used for column-based affinity purification and desalting. List of all relevant column settings (e.g., flow rate, elution buffer concentration, equilibration and washing step duration) and measured parameters (e.g., conductance or UV absorption).
Discussion
When performing the above protocol, we recommend the following suggestions to achieve a successful outcome of the experiment.
For heterologous protein expression, it is important to not exceed the IPTG concentration of 1 mM. IPTG concentrations >1 mM inhibit promoter-induced RTA expression and lead to lower toxin yields. Furthermore, cells should not be cultivated at temperatures higher than 28 °C to prevent inclusion body formation, inefficient folding, and toxin inactivation. RTA expression at lower temperature (e.g., 20-28 °C) is possible and also leads to the production of biological active toxin. However, the additional temperature reduction does not significantly improve RTA activity/folding and results in a lower total toxin yield compared to 28 °C (data not shown).
Purification parameters of the affinity chromatography (Table 1) are optimized for the use of an automated purification system equipped with 5 mL Ni2+ affinity column and should be adjusted to other home-made or commercial column systems if RTA purity and yield are sub-optimal. In case of sub-optimal RTA purity (indicated by the appearance of additional proteins in Coomassie-stained SDS-gels), an increase in the imidazole concentration of the binding buffer normally reduces unspecific binding of positively charged proteins to the column, thereby increasing overall RTA purity. In case of sub-optimal RTA yields, introduction of additional sonification steps (4-8 pulses), longer incubation times (5 h) or larger culture volumes (>1 L) can help to improve the overall toxin yield. However, there is also the opportunity to successfully purify RTA via spin column based systems (data not shown) if an automated system is not available in the lab.
During the desalting step, it is important to skip the sampling procedure as soon as the conductance starts to rise. Contamination with salt, especially imidazole, influences the protein concentration measurement, resulting in inaccurate RTA concentration determination which directly affects the RTA content used in the GFP reporter assay.
It is essential to store purified RTA at 4 °C and avoid freezing. Frozen samples show a reduced activity of RTA in XTT-based cell vitality assays on HeLa cells (data not shown). Purified samples can be kept for around 3-4 weeks before their biological activity is continuously diminished. In addition, high RTA concentrations (≥5 mg/mL) after spin column concentration are critical because RTA shows a tendency to precipitate in 0.8 M sorbitol.
A drawback of the method is the fact that RTA is normally not taken up by intact yeast cells. To achieve reliable results, the yeast cell wall needs to be completely removed by enzymatic treatment. Therefore, it is essential to check spheroplast formation efficiency of each yeast strain by the methods described in steps 3.4-3.5. In case of sub-optimal spheroplast preparation, the incubation time and lytic enzyme concentration should be optimized and monitored via phase contrast microscopy. Overdigestion (ghost formation) can be prevented by reducing lytic enzyme concentration and/or incubation time whereas the opposite approach is needed in case of insufficient digestion. Data from strains with insufficient cell wall removal should be excluded from the data evaluation.
It should also be mentioned that the experimental setup does not completely mimic the natural intoxication process of ricin. The cell binding B subunit of ricin (RTB) is missing which is normally responsible for efficient toxin binding to galactose residues at the cell surface of mammalian cells15. Theoretically, this limitation can be overcome by performing the experiment with purified ricin holotoxin containing its A and B subunit. Nevertheless, yeast cells do not possess galactose residues on their cell surface which could mediate binding of RTB8; this fact also explains why intact yeast cells are not sensitive against ricin. Thus, it is unclear if RTB would act as cell binding component in the yeast model. Interestingly, former studies already demonstrated that RTA is able to reach the cytosol of mammalian cells without its cell binding B subunit16. To understand this phenomenon, we decided to use only RTA in the experimental setup to identify cellular components in yeast which facilitate toxin trafficking from the plasma membrane into the cytosol. Surprisingly, the components involved in RTA trafficking in yeast show striking similarities to the components required for efficient ricin holotoxin transport in mammalian cells6. Based on these results, it can be speculated that RTB plays a minor role in intracellular ricin transport than previously supposed and is only important for the initial contact to the mammalian cell surface.
In contrast, siRNA-based systems in mammalian cells perfectly reflect the natural toxin situation, but are time-consuming and expensive3. Furthermore, the use of yeast deletion mutants bears the important advantage that the mutants show a complete knock-out of the protein of interest, whereas siRNA-based systems generally lead to a reduction of the protein level. In some cases, residual protein levels can be sufficient for toxin transport whereby no altered phenotype is observed and, thus, the involvement of the specific protein is not visible in siRNA-based systems.
Although only non-essential yeast deletion strains were used in the original study6, temperature-sensitive strains as well as decreased abundance by mRNA perturbation (DAmP) collection strains17 (reduced expression level of essential proteins) are likewise suitable for this kind of method in the future. In general, the role of essential proteins cannot be investigated with this assay because of their lack of viability, therefore represent a limitation of the assay system.
However, the new reporter assay represents an elegant and alternative strategy to existing methods which try to analyze RTA transport in the model organism S. cerevisiae. The existing studies used intracellular plasmid-driven RTA expression systems and are restricted to the analysis of the retro-translocation from the ER into the cytosol1,7. Successful implementation of the extracellular RTA application now opens the possibility to not only study ER retrotranslocation but also to indirectly analyze RTA transport from the PM though the Golgi to the ER. As the cellular function and localization of the majority of yeast genes is nowadays discovered and available in databases, the identified candidate proteins can be directly allocated to specific compartments and/or transport pathways.
Overall, the introduced reporter assay method represents an easy and robust approach to identify candidate proteins involved in intracellular trafficking of toxins or toxin subunits (e.g., RTA) which block protein translation in vivo. Relatively low standard deviations (1-10%) as well as the high data reproducibility in the original study confirms these statements. Due to the 96-well plate format, a fast screening of various yeast mutants is possible within a day after toxin purification.
In the future, there is the possibility to use the method for high-throughput screening of yeast deletion libraries. In the current setup, the applied RTA concentration (5 µM) is able to decrease the relative GFP fluorescence to 50%. Under these conditions, the method offers the advantage to identify deletion mutants with negative (increased GFP fluorescence) and positive (reduced GFP fluorescence) effects on RTA transport. For high throughput screenings, a stronger difference between treated and non-treated samples is sometimes beneficial. By using higher RTA concentrations and/or increasing the measure interval (e.g., 48 h), an even more pronounced block of GFP expression should be achievable.
It is to be emphasized that this method is not only restricted to analyze the intoxication process of extracellular applied RTA in yeast, but there is also the opportunity to analyze the ER import of RTA by expressing RTA via a plasmid in this GFP reporter strain (data not shown). Furthermore, the screening approach can likewise be adapted to study intracellular transport pathways of other protein biosynthesis inhibiting proteins such as zymocin18 in the future. In contrast to the well characterized killing mode of zymocin, relatively little is known about the trafficking pathways responsible for efficient toxin transport into the cytosol19,20. Thereby, zymocin represents an optimal candidate for future studies as zymocin is naturally taken up by yeast cells. Thus, the removal of the cell wall can be omitted which improves the overall performance of the GFP reporter assay (with respect to time and costs). With the help of this powerful reporter assay, it would be feasible to dissect the intracellular transport route(s) of zymocin in yeast.
Disclosures
The authors have nothing to disclose.
Acknowledgments
Parts of this study were kindly supported by a grant from the Deutsche Forschungsgemeinschaft (SFB 1027, A6).
References
- Li S, et al. Folding-competent and folding-defective forms of ricin A chain have different fates after retrotranslocation from the endoplasmic reticulum. Mol Biol Cell. 2010;21(15):2543–2554. doi: 10.1091/mbc.E09-08-0743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S, Spooner RA, Hampton RY, Lord JM, Roberts LM. Cytosolic entry of Shiga-like toxin a chain from the yeast endoplasmic reticulum requires catalytically active Hrd1p. PLoS One. 2012;7(7):e41119. doi: 10.1371/journal.pone.0041119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau D, et al. Genome-wide RNAi screens identify genes required for Ricin and PE intoxications. Dev Cell. 2011;21(2):231–244. doi: 10.1016/j.devcel.2011.06.014. [DOI] [PubMed] [Google Scholar]
- Giepmans BN, Adams SR, Ellisman MH, Tsien RY. The fluorescent toolbox for assessing protein location and function. Science. 2006;312(5771):217–224. doi: 10.1126/science.1124618. [DOI] [PubMed] [Google Scholar]
- Majoul IV, Bastiaens PI, Soling HD. Transport of an external Lys-Asp-Glu-Leu (KDEL) protein from the plasma membrane to the endoplasmic reticulum: studies with cholera toxin in Vero cells. J Cell Biol. 1996;133(4):777–789. doi: 10.1083/jcb.133.4.777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Becker B, Schnoder T, Schmitt MJ. Yeast Reporter Assay to Identify Cellular Components of Ricin Toxin A Chain Trafficking. Toxins (Basel) 2016;8(12) doi: 10.3390/toxins8120366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li XP, Baricevic M, Saidasan H, Tumer NE. Ribosome depurination is not sufficient for ricin-mediated cell death in Saccharomyces cerevisiae. Infect Immun. 2007;75(1):417–428. doi: 10.1128/IAI.01295-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lord JM, Roberts LM, Robertus JD. Ricin: structure, mode of action, and some current applications. Faseb J. 1994;8(2):201–208. [PubMed] [Google Scholar]
- Becker B, Schmitt MJ. Adapting yeast as model to study ricin toxin a uptake and trafficking. Toxins (Basel) 2011;3(7):834–847. doi: 10.3390/toxins3070834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidman CE, Struhl K, Sheen J, Jessen T. Introduction of plasmid DNA into cells. Curr Protoc Mol Biol. 2001. [DOI] [PubMed]
- Brunelle JL, Green R. Coomassie blue staining. Methods Enzymol. 2014;541:161–167. doi: 10.1016/B978-0-12-420119-4.00013-6. [DOI] [PubMed] [Google Scholar]
- Shapiro AL, Vinuela E, Maizel JV., Jr Molecular weight estimation of polypeptide chains by electrophoresis in SDS-polyacrylamide gels. Biochem Biophys Res Commun. 1967;28(5):815–820. doi: 10.1016/0006-291x(67)90391-9. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, Gordon J. Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: procedure and some applications. 1979. Biotechnology. 1992;24:145–149. [PubMed] [Google Scholar]
- Ito H, Fukuda Y, Murata K, Kimura A. Transformation of intact yeast cells treated with alkali cations. J Bacteriol. 1983;153(1):163–168. doi: 10.1128/jb.153.1.163-168.1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitetta ES, Yen N. Expression and functional properties of genetically engineered ricin B chain lacking galactose-binding activity. Biochim Biophys Acta. 1990;1049(2):151–157. doi: 10.1016/0167-4781(90)90035-z. [DOI] [PubMed] [Google Scholar]
- Wales R, Roberts LM, Lord JM. Addition of an endoplasmic reticulum retrieval sequence to ricin A chain significantly increases its cytotoxicity to mammalian cells. J Biol Chem. 1993;268(32):23986–23990. [PubMed] [Google Scholar]
- Breslow DK, et al. A comprehensive strategy enabling high-resolution functional analysis of the yeast genome. Nat Methods. 2008;5(8):711–718. doi: 10.1038/nmeth.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonowski D, Schaffrath R. Zymocin, a composite chitinase and tRNase killer toxin from yeast. Biochem Soc Trans. 2007;35(Pt 6):1533–1537. doi: 10.1042/BST0351533. [DOI] [PubMed] [Google Scholar]
- Jablonowski D, Schaffrath R. Saccharomyces cerevisiae RNA polymerase II is affected by Kluyveromyces lactis zymocin. J Biol Chem. 2002;277(29):26276–26280. doi: 10.1074/jbc.M203354200. [DOI] [PubMed] [Google Scholar]
- Jablonowski D, Frohloff F, Fichtner L, Stark MJ, Schaffrath R. Kluyveromyces lactis zymocin mode of action is linked to RNA polymerase II function via Elongator. Mol Microbiol. 2001;42(4):1095–1105. doi: 10.1046/j.1365-2958.2001.02705.x. [DOI] [PubMed] [Google Scholar]