

Abstract
Classification and nomenclature of genes in a family can significantly contribute to the description of the diversity of encoded proteins and to the prediction of family functions based on several features, such as the presence of sequence motifs or of particular sites for post-translational modification and the expression profile of family members in different conditions. This work describes a detailed protocol for gene family characterization. Here, the procedure is applied to the characterization of the Arabidopsis Tóxicos in Levadura (ATL) E3 ubiquitin ligase family in grapevine. The methods include the genome-wide identification of family members, the characterization of gene localization, structure, and duplication, the analysis of conserved protein motifs, the prediction of protein localization and phosphorylation sites as well as gene expression profiling across the family in different datasets. Such procedure, which could be extended to further analyses depending on experimental purposes, could be applied to any gene family in any plant species for which genomic data are available, and it provides valuable information to identify interesting candidates for functional studies, giving insights into the molecular mechanisms of plant adaptation to their environment.
Keywords: Genetics, Issue 130, ATL E3 ubiquitin ligase, gene family, genome-wide, nomenclature, phylogeny, expression meta-analysis, gene duplication, grapevine
Introduction
During the last decade, much research has been carried out in grapevine genomics. Grapevine is a recognized economically relevant crop, which has become a model for research on fruit development and on the responses of woody plants to biotic and abiotic stresses. In this context, the release of the Vitis vinifera cv. PN40024 genome in 20071 and its updated version in 20112 led to a rapid accumulation of "Omics"-scale data and to a burst of high-throughput studies. Based on the published sequence data, the comprehensive analysis of a given gene family (generally composed of proteins sharing conserved motifs, structural and/or functional similarities and evolutionary relationships), can now be performed to uncover its molecular functions, evolution, and gene expression profiles. These analyses can contribute to understanding how gene families control physiological processes at a genome-wide level.
Many aspects of the plant life cycle are regulated by ubiquitin-mediated degradation of key proteins, which require a fine-tuned turnover to ensure regular cellular processes. Important components of the ubiquitin-mediated degradation process are the E3 ubiquitin ligases, which are responsible for system flexibility, thanks to the recruitment of specific targets3. Accordingly, these enzymes represent a huge gene family, with around 1,400 E3 ligase-encoding genes predicted in Arabidopsis thaliana genome4, each E3 ubiquitin ligase acting for the ubiquitination of specific target proteins. Despite the importance of substrate-specific ubiquitination in cellular regulation in plants, little is known about how the ubiquitination pathway is regulated and target proteins have been identified only in a few cases. The deciphering of such specificity and regulation mechanisms relies first on the identification and characterization of the different components of the system, in particular the E3 ligases. Among ubiquitin ligases, the ATL subfamily is characterized by 91 members identified in A. thaliana displaying a RING-H2 finger domain5,6, some of them playing a role in defense and hormone responses7.
The first crucial step to define the members of a new gene family is the precise definition of the family features, such as consensus motifs, key domains, and protein sequence characteristics. Indeed, the reliable retrieval of all gene family members based on BLAST analysis requires some mandatory sequence characteristics, in particular protein domains responsible for protein function/activity, serving as protein signature. This can be facilitated by previous characterization of the same gene family in other plant species or achieved by analyzing different genes putatively belonging to the same family in different plant species, to isolate common sequences. The family members can then be individually named following common rules settled by international consortia for a given plant species. In grapevine, for instance, such procedure is subjected to the recommendations of the Super-Nomenclature Committee for Grape Gene Annotation (sNCGGa), establishing the construction of a phylogenetic tree including V. vinifera and A. thaliana gene family members to allow gene annotation based on nucleotide sequences8.
Chromosome localization of family members and gene duplication survey allow highlighting the presence of whole-genome or tandem duplicated genes. Such information appears useful to unravel putative gene functions, since it might show functional redundancy or reveal different situations, i.e., non-functionalization, neo-functionalization, or sub-functionalization9. Both neo- and sub-functionalization are important events that create genetic novelty, providing new cellular components for plant adaptation to changing environments10. In particular, duplications of ancestral genes and production of new genes were very frequent during the evolution of the grapevine genome and newly formed genes originating from proximal and tandem duplications in grapevine were more likely to produce new functions11.
Another key factor in deciphering gene family function is the transcriptomic profile. The availability of public databases giving access to a huge amount of transcriptomic data can be thus exploited to assign putative functions to gene family members using large-scale in silico expression analyses. Indeed, the peculiar expression of some genes in specific plant organs or in response to certain stresses can give some hints regarding the putative roles of the corresponding proteins in defined conditions, and give support to hypotheses about possible sub-functionalization of duplicated genes to respond to different challenges. For that purpose, it is important to consider several datasets: these can be already available gene expression matrixes, such as the genome-wide transcriptomic atlas of grapevine organs and developmental stages12, or can be built ad hoc by retrieving transcriptomic datasets for the particular plant species subjected to defined stresses. Moreover, a simple approach using two matrices, one with pairwise similarity data and the other one with pairwise co-expression coefficients can be applied to evaluate the relationships between sequence similarity and expression patterns within a gene family.
The aim of this work is to provide a global approach, defining gene structure, conserved protein motifs, chromosomal location, gene duplications, and expression patterns, as well the prediction of protein localization and phosphorylation sites, to attain an exhaustive characterization of a gene family in plants. Such a comprehensive approach is applied here to the characterization of the ATL E3 ubiquitin ligase family in grapevine. According to the emerging role of ATL subfamily members in regulating key cellular processes7, this work can well assist the identification of strong candidates for functional studies, and eventually unravel the molecular mechanisms governing the adaptation of this important crop to its environment.
Protocol
1. Identification of Putative ATL Gene Family Member(s)
- PSI-BLAST web version
- Open the BLAST web page13 and click on the protein BLAST section.
- In the "Enter Query sequence" field, enter the amino acid sequence of the protein (here VIT_05s0077g01970) that will be used as the probe to identify the other family members. NOTE: A good representative protein should be used (a protein displaying all the important features that characterize the family).
- In the field "Choose search set", select the "Reference protein" database (refseq_protein) and the organism of interest (V. vinifera - taxid:29760).
- In the field "Program selection", select PSI-BLAST algorithm and click the BLAST button to run the analysis. NOTE: By clicking on the "Algorithm parameters" it is possible to adjust some advanced parameters (Max target sequences, Scoring matrix, PSI-BLAST threshold, etc.).
- The first BLAST round retrieves all the sequences displaying relevant matches with the query (e-value above the selected threshold - by default 0.005; 0.001 in this experiment). Unselect all the entries, which clearly do not belong to the family under examination by clicking on the tick in the "select for PSI-BLAST" column and run the second PSI-BLAST iteration by clicking the BLAST button as in step 1.1.4.
- Newly identified sequences are highlighted in yellow. Unselect the clearly wrong retrieved hits and uncover further iterations as described in step 1.1.5.
- Continue with iterations until the algorithm does not find any relevant entry or it reaches convergence (no new entries are found). Download the list of putative gene family members for further analyses. Visually inspect the retrieved hits in each iteration to avoid the presence of false positives.
- PSI-BLAST standalone version
- Download the standalone version of BLAST by clicking the "download BLAST" button on the BLAST home page13. NOTE: The standalone BLAST software is a command line version of the web interface described before. It enables executing the PSI-BLAST search against a custom local or remote database. Moreover, it allows searching with a pre-defined Position Specific Score Matrix (PSSM).
2. Manual Inspection of the PSI-BLAST-identified Family Members
- Multiple alignment
- Collect the amino acidic sequences previously identified in a FASTA-formatted file and upload it into the MEGA software14 to proceed with the multiple alignment.
- Open the MEGA software, click the "Align" button, click "Edit/Build Alignment", click "Create a new alignment", click "Protein".
- Click "Edit" from the alignment menu and "Insert Sequence from File". Browse for the FASTA file created before and confirm the upload of all the surveyed sequences.
- Click "Alignment" from the alignment menu and "Align by MUSCLE". Use default parameters, click "Compute" button, and wait for the completion of the multiple alignment.
- Visually inspect the multiple alignment to exclude incorrectly predicted family members. The canonical CxxC(13x)PxCxHxxHxxCxxxW(7x)CxxCW motif, (in particular the presence of the proline residue before the third cysteine), is the key feature required to define the ATL family members.
- Analysis of specific LOGO
- Submit the definitive list of family members (96 grapevine sequences fulfill the requirements to be considered ATL) to the Multiple Em for Motif Elicitation (MEME)15 to define conserved motifs across the family.
- From the MEME home page, click the "MEME" button, and complete the "Data Submission Form" with particular information regarding the family of interest.
- Use MEME analysis to confirm the presence of the two expected motifs within the grapevine ATL family members, i.e., the RING-H2 and the GLD motifs.
- Alternatively, perform steps 2.1 and 2.2 simultaneously using the bioinformatics software suite (see Table of Materials).
- Upload FASTA file (see step 2.1.1) into the suite. Select "File" from the menu, then "Import" and click "From file". Browse the FASTA file and click "Open".
- Select all the imported sequences in the list and click on "Align/Assemble" button in the toolbar, then click "Pairwise Multiple Alignment". Select "Muscle alignment" and click "OK" to launch the alignment using default parameters.
- To visualize the LOGO of the alignment, click on "Graphs" → "options" and select "Sequence Logo".
3. Analysis of Protein Physical Parameters and Domains
- As the definition of the different physical parameters of the surveyed family members is important to have a comprehensive description of the family, submit the list of family members to specific web tools.
- For isoelectric point (pI) and molecular weight (kDa), use the ProtParam tool16 on the Expasy website with default parameters.
- For protein subcellular localization, use different tools to obtain a more reliable prediction such as ngLOC v1.017 with default settings, targetP v1.118 with default settings, and protein prowler subcellular localization v1.219 with a cut-off of probability of 0.5. For phosphorylation sites, use the MUsite v1.0 web tool20 with default parameters.
- Investigate additional protein domains in family members.
- Open the Pfam database webpage21, select "Sequence search" tool, submit protein sequences in the query box, and click "Go" to run the analysis. NOTE: Each protein sequence is analyzed individually. An e-value of 1.0 in the default setting allows discriminating between significant and non-significant hits.
- Open the TMHMM Server22 from the Center for Biological Sequence Analysis to investigate the presence of putative transmembrane regions. Paste all protein sequences simultaneously in the query box (or alternatively upload a text file including all protein sequences in FASTA format) and click "Submit" to run the analysis.
- Analyze proteins lacking predicted transmembrane domains, according to TMHMM (step 3.2.2), with ProtScale tool to identify putative hydrophobic regions. Open ProtScale webpage23. Paste each protein sequence in the query box and select "Hphob. / Kyte & Doolittle" as amino acid scale. Click "Submit" to run the analysis.
4. Chromosomal Distribution, Duplications, and Exon-intron Organization
- Map the ATL family members on the chromosomes based on the information retrieved from the Grapevine Genome CRIBI Biotech Center website24.
- Browse the PhenoGram website homepage25. Write the "Input File" as a tab-delimited text file with the specific features of the genes to be mapped on the chromosomes, according to the exhaustive guidelines and examples regarding the compilation of the provided file following the path "Phenogram" → "Documentation" → "Options" → "Input file".
- Write the "Title" of the work. Select the genome to be drawn. For genomes not implemented in the software, such as the grapevine genome, select "other" in the drop-down menu. Write the genome file according to the guidelines and examples provided, following the path "Phenogram" → "Documentation" → "Options" → "Genome", and upload it.
- Use default parameters of "Phenotype spacing", "Phenotype color", "Image format", or select alternatives in the respective menus, and click "Plot" to obtain the visualization of the genes on the chromosomes.
- Define the duplication state of the family members using the MCScanX software26.
- Download and unzip a copy of MCscanX on a local machine running command lines 1 (Supplementary File 1). Enter the MCscanX folder and create the required executables running command lines 2 (Supplementary File 1). NOTE: Installation of MCscanX is known to fail on some Linux 64 bit machines due to an issue regarding the function chdir. If an error message is returned related to this function upon the make command execution, the command lines 3 (Supplementary File 1) should be run and the command "make" should be attempted afterwards.
- Download the V. vinifera proteins and the annotation file running command lines 4 (Supplementary File 1). NOTE: The grapevine annotation file needs to be unzipped and the single chromosomes information cat in a unique file by running command lines 5 (Supplementary File 1).
- Run an "all versus all" blastp search using the V. vinifera protein file as both the query and the subject.
- Create a searchable blast database using the V. vinifera protein file running command lines 6 (Supplementary File 1). Perform the blastp search by using the V. vinifera proteins file as a query against the database created previously by running command lines 7 (Supplementary File 1).
- Convert the annotation file in a suitable format for MCScanX. Run command lines 8 (Supplementary File 1) to download the custom perl script parseMSCanXgff.pl. Perform the analysis running command lines 9 (Supplementary File 1). NOTE: A file vitis.gff is generated that holds gene coordinates in the following format: sp# gene starting position ending position where "sp" is a two-letter code for the species (Vv for grapevine) whereas "#" is the name of the scaffold. Note that the provided custom perl script is suitable for most conversion, although some code modification may be required in some specific cases due to the diversity of the information provided in the available annotation file.
- Launch MCScanX running command lines 10 (Supplementary File 1). NOTE: The "vitis" is the prefix of both the annotation and the blast output file. This represents a compulsory requirement for the software to run.
- Analyze MCScanX results. MCScanX produces one text file "vitis.collinearity", which contains collinear blocks. Such a file can be inspected by any text editor (see example output 1 Supplementary File 1). NOTE: A "mcscaxOutput.html" directory is generated that contains html files featuring multiple alignments of collinear blocks against each reference chromosome. These files can be inspected through a web browser.
- Classify paralogous genes based on their relative positions in chromosomes running command lines 11 (Supplementary File 1). NOTE: Paralogous gene classification is described in Supplementary Table II. The generated output file "vitis.gene_type" contains all origin information with a simple tab delimited format.
- Perform enrichment analysis to evaluate whether the gene family has prevalently originated by a specific mechanism running command lines 12 (Supplementary File 1). NOTE: File "vitis.gene_type" is generated at step 4.2.8, whereas file "gene_family_file" represents a one line text file in which the name of the family (e.g., ATL_genes) is followed by the locus names for the all the genes belonging to the family separated by a tab. The applied statistical test for enrichment is a Fisher exact test and the p-values of different origins are stored in the file "outputFile.txt".
- Visualize the exon-intron organization of the genes using Interactive Tree Of Life (iTOL)27, an on-line tool for the display, annotation, and management of phylogenetic trees.
- Upload a phylogenetic tree in the "Upload" section of the iTOL website. The tree is built according to Section 5 below. For each family member gene, retrieve gene structure prediction from the V1 annotation of the grapevine genome (CRIBI website cited above). Calculate the length (in bp) of putative exons, introns, and untranslated regions (UTRs).
- Use the "Protein domains" dataset for graphical visualization of the exon-intron pattern. Write a plain text file including calculated lengths according to the specifications provided following the path "Help" → "Help pages" → "Dataset types" → "Protein domains" in the iTOL website27. Using "Protein domains" dataset, the "rectangle (RE)" and the "rectangle gap (GP)" shapes represent the exon and the UTRs, respectively.
5. Phylogenetic Analysis and Nomenclature
- Analyze the relationships among ATL family members through the construction of a high quality phylogenetic tree and the definition of a family nomenclature.
- For a grapevine gene family, follow the rules established by the Grapevine Super Nomenclature Committee8.
- Write a FASTA file including all nucleotide sequences of grapevine and A. thaliana gene family members to be included in the phylogenetic analysis. The nucleotide sequences allow the maximum of variability among family members (compared to protein sequences).
- Phylogenetic tree NOTE: The use of the Phylogeny.fr 29 pipeline is recommended to get a high quality phylogenetic tree, but not mandatory.
- Browse the Phylogeny.fr homepage29, and select the "Phylogeny analysis" pipeline. NOTE: "One Click" is suitable in most of the cases, but if needed it is possible to select specific advanced settings ("Advanced") or even a fully customized analysis ("A la Carte"; see step 5.2.5).
- Write the "Name of the analysis", upload the FASTA file created previously (step 5.2.1, and click "Submit" to run the analysis.
- Alternatively, if the procedure described above (steps 5.2.1, 5.2.2) results in an error message, complete each step of the Phylogeny suite pipeline individually, as follows.
- From the MUSCLE software homepage30, upload the FASTA file in "STEP 1", select "Pearson/FASTA" as "Output format" in "STEP 2", and click "Submit" in "STEP 3" to align query sequences.
- Click "Download alignment file" and save as FASTA file for further steps.
- Process the alignment FASTA file to eliminate poorly aligned positions using Gblocks Server tool31. Upload the alignment FASTA file, select "DNA" as "Type of sequence" and chose the option(s) of stringency that best fits with the analysis (e.g., for grapevine ATL gene family select all the three options proposed for "less stringent selection" because of high sequence divergence). Click "Get blocks" to run the analysis.
- Click "Resulting alignment" at the bottom of the output page and save the results as a new FASTA file.
- From the Phylogeny.fr homepage29, select "A la Carte" as "Phylogeny analysis" pipeline. Then, deselect "Multiple alignment" and "Alignment curation". Click "Create workflow", upload the Gblocks-curated FASTA file (step 5.2.5.4), select "Bootstrapping procedure" with default parameters in "Settings", and click "Submit" to run the analysis.
- Collapse poorly supported branches (i.e., bootstrap values < 70%) by clicking "Collapse branches" in the "Select and action" section and download the final results in the Newick format to further analyses.
- Assign a gene name based on the phylogeny.
- Review the phylogenetic tree to evaluate the reliability of the tree structure by uploading it into the iTOL suite cited above (section 4.3).
- Assign manually a gene name to each family member. In the case of one-to-one orthologues, assign the Arabidopsis-like name (e.g., AtATL3 → VviATL3). Differentiate grapevine genes (two or more) deriving from a single Arabidopsis homolog with the same phylogenetic distance using numbers, or letters if the Arabidopsis gene ends with a number (e.g., AtATL23 → VviATL23a, VviATL23b).
- In the case of one-to-many or many-to-many orthologues, assign a new gene name composed of the Arabidopsis-like name (here, "ATL") paired with a number higher than the highest number already used for both V. vinifera and Arabidopsis (e.g., VviATL83).
- Complete the nomenclature of the newly defined family descending from the top to the bottom of the phylogenetic tree.
6. Grapevine Organ and Stage Expression Profiling
- Generate the working data matrix containing expression data for the family members.
- Download the V. vinifera cv. Corvina gene expression Atlas datamatrix from the link distributed on the ResearchGate platform32. This file contains the RMA normalized expression values to be used in following steps.
- Extract the expression values for each family gene from the Atlas datamatrix and write a "working datamatrix" containing the same header row as the Atlas datamatrix. Save the "working datamatrix" as a tab-delimited text file.
- Perform the hierarchical bi-clustered analysis using Multi Experiment Viewer (MeV) software.
- Download and install MeV software33.
- Upload the "working datamatrix" (step 6.1.2) following the path "File" → "Load Data" → "Browse" and select the text file. Select "Single-color Array" and remove the tick from "Load Annotation" when an automatic annotation is not provided. Select the upper-leftmost expression value of the expression table preview and click the "Load" button.
- Adjust the data applying Log2 transformation ("Adjust Data" → "Log Transformations" → "Log2 Transform") and Gene/Row normalization ("Adjust Data" → "Gene/Row Adjustments" → "Median Center Gene/Row"). Set the proper scale limit ("Display" → "Set Color Scale Limits").
- Calculate the Hierarchical Clustering following the path "Analysis" → "Clustering" → "HCL". Select "Optimize Gene Leaf Order" and "Optimize Sample Leaf Order" in "Ordering Optimization field", "Pearson Correlation" in the "Distance Matrix Selection" field, and "Average linkage clustering" in the "Linkage Method Selection" field. Then, click "OK" to run the analysis.
- View the results in the "Analysis Results" → "HCL" menu on the left panel of the window. Export the heat map by clicking "Save Image" in the "File" menu.
7. Expression Profiling in Response to Biotic and Abiotic Stresses
Repeat step 6.1 with the GSE accession ID obtained from respective publications and studies investigating biotic and abiotic stress on grapevine. For example, experiments providing the transcriptome profile of grapevine berries infected with the fungal pathogen Botrytis cinerea using the NimbleGen Grape Whole-genome microarray can be browsed with GSE ID of GSE52586. Repeat steps 6.1.1 and 6.1.2.
- Search the NCBI Sequence Reads Archive34 with the SRA/BioProject ID (e.g., SRP055458 or PRJNA275778 for "grapevine flower shading" experiments) and download all associated raw sequence reads. RNA-seq datasets from many different studies are processed using a single pipeline for consistency.
- Perform differential gene expression (re-)analysis in R38 with limma39 libraries for RMA-normalized matrices and DESeq240 libraries for count matrix tables obtained from steps 7.1.1 and 7.2.1, respectively.
- Perform a standard "two-group" comparison (i.e., "treatment"/"control"). Ensure that the design matrix/groupings of "controls" and "treatment" conditions are properly specified. NOTE: A typical design for microarray differential expression analysis (GSE52586) to compare EL-33 berries infected with Botrytis cinerea against control (healthy) berries at the same development stage with limma running command lines 13 is shown in Supplementary File 1. A typical design for RNA-seq differential expression analysis (SRP055458 or PRJNA275778) to compare flower (at 7 days after cap-fall) under shade treatment against the control with DESeq2 running command lines 14 is shown in Supplementary File 1.
- Obtain the lists of differentially expressed genes (DEG) in each contrast, for limma, use the functions lmFit(), followed by eBayes(), and then by topTable()functions, while for DESeq2, use the DESeqDataSetFromMatrix(), DESeq(), and results() functions. Below, a typical workflow to be followed.
- For microarray differential expression analysis, see command lines 15 (Supplementary File 1). For RNA-seq differential expression analysis see command lines 16 (Supplementary File 1). Repeat the above steps for all other contrasts with different appropriate design schema (See examples in step 7.3.1)
From the lists of DEGs generated, extract all rows that do not correspond to ATL V1 accession, retain columns containing the log2 Fold Change (Treatment/Control) > |0.5| and adjusted p-values (FDR) < 0.05, and merge them accordingly into a matrix table, whether a study falls into "abiotic" or "biotic/pathogen interaction" compendia.
Construct the hierarchical clustered heatmaps (abiotic and biotic compendia) in R using the libraries gplots. NOTE: Calling the heatmap.2 function constructs the heatmap along with row dendrograms from the respective matrix tables. Additional arguments using cellnote function helps to distinguish differentially expressed (log2FC > 0.5, FDR < 0.05) ATL genes in each comparison across a large range of experimental conditions by a * symbol. Apply the typical workflow in R running command lines 17 (Supplementary File 1) or alternatively, repeat steps 6.2.2 to 6.2.5 to construct the heatmaps using MeV software.
8. Analysis of the Relationships Between Paralogous Sequence Divergence and Gene Co-expression
- Construct the matrix containing pairwise similarity. The elements of the similarity matrix are the values of sequence similarity calculated from the pairwise protein alignments.
- Use the EMBOSS needle web server41 with default settings to make pairwise sequence alignments and save as text file. Open the output text file and remove all comment lines, together with column and row names to generate a file called "similarityTable.txt". NOTE: Such a table features a line for each ATL gene reporting the similarity values calculated in each of the pairwise alignment. The order of the loci in rows and columns is the same so that a symmetric matrix is generated with respect of the diagonal values.
- Construct the matrix with co-expression data by calculating the Pearson correlation coefficient. The following procedure requires R and the perl module PDL.
- Download the expression values for the 96 ATL genes running command lines 18 (Supplementary File 1) within a terminal. Perform a co-expression analysis by using a custom perl script that can be downloaded by running command lines 19 (Supplementary File 1). Such script will calculate the Pearson correlation coefficient between pairs of ATL loci as previously reported.
- Launch the script running command lines 20 (Supplementary File 1) and follow the output instructions. The script will produce an output file (namely "coexpressionTable.txt") containing a co-expression matrix featuring the same locus names order of matrix obtained in step 8.1 (this ordering is essential to run the Mantel test, see below).
- Perform a Mantel test between the data matrices obtained in steps 8.1 and 8.2. After entering the R environment (run command "R" from within a terminal), load the ade4 library using the following command: library(ade4)
- Run the Mantel test by loading the two data matrices and performing the statistics running command lines 21 (Supplementary File 1), with "nrep" representing the number of permutations. The test consists of calculating the correlation between the elements of these matrices, permuting the matrices and then calculating the same test statistic again. NOTE: All the obtained values of the statistic test are used to build a reference distribution of the statistic test, which will be used to calculate a p-value to test for significance. The number of permutations defines the precision with which the p-value can be obtained.
Representative Results
The VIT_05s0077g01970 gene, identified as the most similar to A. thaliana ATL2 (At3g16720) through a BLASTp search, was used as probe to survey the ATL family members in the grapevine genome (V. vinifera cv Pinot Noir PN40024). The PSI-BLAST analysis converged after a few cycles revealing a list of putative genes belonging to the grapevine ATL gene family (Figure 1A). The presence of the canonical RING-H2 domain for each candidate was evaluated by the visual inspection of the MUSCLE alignment of all the entries identified in the analysis (Figure 1B). Only those genes containing the correctly spaced conserved amino acids, the two histidine residues, as well as the proline residue before the third cysteine were considered as ATLs according to the original ATL definition in Arabidopsis5. A total of 96 grapevine genes fulfilled the requirements and were considered for further characterization. Each ATL family member was analyzed to define the specific characteristics of the gene and the corresponding encoded protein, i.e., the presence of other known domain(s) in addition to the RING-H2, transmembrane or hydrophobic rich regions, subcellular localization, and putative phosphorylation sites (Table 1 and Table 2).
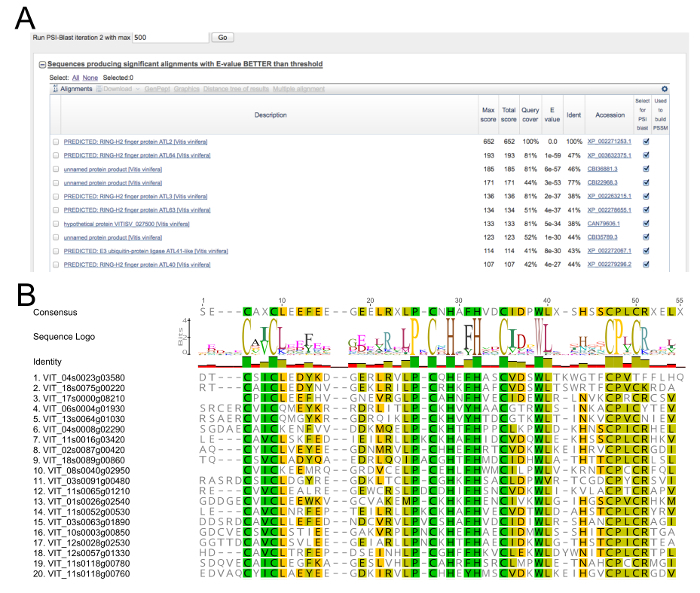
Figure 1: PSI-BLAST survey and alignment of putative grapevine ATLs. (A) Screenshot of the top 10 hits of the first PSI-BLAST iteration search using the protein sequence VIT_05s0077g01970 as bait. (B) Portion of the alignment of the 96 selected grapevine putative ATLs showing their RING-H2 domain and the corresponding LOGO obtained using a suite of molecular biology (see Table of Materials). Reproduced from Ariani et al. licensed under a Creative Commons Attribution 4.0 International License42. Please click here to view a larger version of this figure.
| Name | Gene ID | Gene length (bp) | Intron number | UniProt ID | Protein length (aa) | RING-H2 motif | TM/H domain number | Other domains |
| VviATL3 | VIT_09s0002g00220 | 1245 | 0 | F6HXK6 | 304 | PxC | 1 | |
| VviATL4[VviRHX1A] | VIT_15s0021g00890 | 1827 | 3 | D7SM36 | 203 | PxC | 0 | |
| VviATL18 | VIT_11s0118g00780 | 1113 | 2 | F6HCI8 | 193 | PC | 0 | |
| VviATL23a | VIT_18s0001g01060 | 935 | 0 | F6H0E4 | 114 | PxC | 0.5 | |
| VviATL23b | VIT_18s0001g01050 | 399 | 0 | E0CQX3 | 132 | PxC | 1 | |
| VviATL24 | VIT_17s0000g06460 | 4466 | 4 | D7SI89 | 217 | PxC | 1 | |
| VviATL27 | VIT_00s0264g00020 | 2554 | 4 | D7T1R5 | 235 | PxC | 1 | |
| VviATL43 | VIT_11s0052g00530 | 1576 | 2 | D7SQD9 | 457 | PxC | 3 | |
| VviATL54a | VIT_18s0001g06640 | 3221 | 1 | F6H0Y5 | 405 | PxC | 1 | |
| VviATL54b | VIT_03s0017g00670 | 2774 | 1 | F6HTI0 | 427 | PxC | 1 | |
| VviATL55[VviRING1] | VIT_07s0191g00230 | 1844 | 0 | F6HRP9 | 372 | PxC | 1 | |
| VviATL63 | VIT_06s0004g06930 | 804 | 0 | D7SJU6 | 267 | PxC | 1 | |
| VviATL65 | VIT_03s0063g01890 | 2068 | 0 | F6HQI8 | 396 | PxC | 1 | |
| VviATL82 | VIT_01s0026g02540 | 820 | 0 | F6HPQ9 | 233 | PC | 0.5 | |
| VviATL83 | VIT_17s0000g08400 | 1887 | 0 | F6GSQ4 | 143 | PC | 0 | |
| VviATL84 | VIT_06s0004g00120 | 1853 | 0 | F6GUP5 | 368 | PC | 0.5 | zf-RING_3 |
| VviATL85 | VIT_12s0034g01400 | 786 | 0 | F6H965 | 261 | PC | 0.5 | |
| VviATL86 | VIT_12s0034g01390 | 1434 | 1 | D7T016 | 451 | PC | 0.5 | |
| VviATL87 | VIT_18s0001g03270 | 1002 | 0 | F6H0T2 | 333 | PC | 0.5 | zf-RING_3 |
| VviATL88 | VIT_08s0040g00590 | 1320 | 0 | F6HQR2 | 314 | PC | 0 | zf-RING_3 |
Table 1: First 20 VviATL genes and sequence characteristics of the corresponding proteins. TM: transmembrane; H: hydrophobic; 0.5 indicates the presence of one or more hydrophobic regions. Reproduced from Ariani et al. licensed under a Creative Commons Attribution 4.0 International License42.
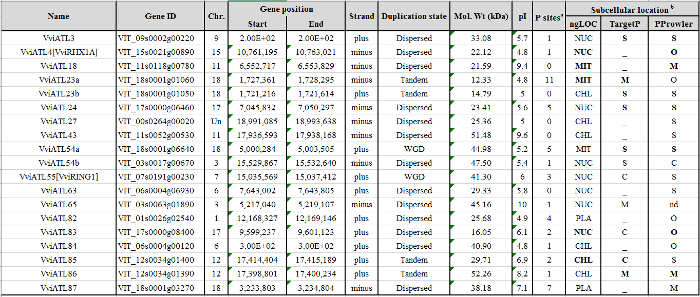 Table 2: Details on the first 20 VviATL gene position in V. vinifera genome, duplication state, and ATL protein physico-chemical characteristics and location. (a) Number of phosphorylation sites predicted by Musite; (b) similar predictions obtained with at least two software are highlighted in bold; ngLOC was used with default settings, whereas TargetP v1.1 and Protein Prowler Subcellular Localization were used with a cut-off of probability of 0.5. NUC, nucleus; MIT, mitochondria; CHL, chloroplast; PLA, plasma membrane; S, secretory pathway (presence of a signal peptide); M, mitochondria; C, chloroplast; O or -, other locations; nd, not determined (i.e., value below the threshold). Reproduced from Ariani et al. licensed under a Creative Commons Attribution 4.0 International License42. Please click here to download this file.
Table 2: Details on the first 20 VviATL gene position in V. vinifera genome, duplication state, and ATL protein physico-chemical characteristics and location. (a) Number of phosphorylation sites predicted by Musite; (b) similar predictions obtained with at least two software are highlighted in bold; ngLOC was used with default settings, whereas TargetP v1.1 and Protein Prowler Subcellular Localization were used with a cut-off of probability of 0.5. NUC, nucleus; MIT, mitochondria; CHL, chloroplast; PLA, plasma membrane; S, secretory pathway (presence of a signal peptide); M, mitochondria; C, chloroplast; O or -, other locations; nd, not determined (i.e., value below the threshold). Reproduced from Ariani et al. licensed under a Creative Commons Attribution 4.0 International License42. Please click here to download this file.
A phylogenetic analysis including the nucleotide sequences of identified grapevine ATL-encoding genes together with the sequences of the reference A. thaliana ATL gene family was used for grapevine ATL nomenclature, according to the guidelines of the sNCGGa8. Ninety-six and 83 nucleotide sequences from V. vinifera and A. thaliana, respectively, were subjected to the Phylogeny.fr pipeline to obtain a reliable phylogenetic tree. The latter sequences were later used to annotate and name grapevine genes on the basis of solid relationships (Figure 2). Following this approach, 13 out of 96 grapevine ATLs received a specific identifier considering their one-to-one orthology with an A. thaliana ATL. The names of the other 83 genes were assigned based on the phylogenetic tree, with a progressive numbering from top to bottom, starting from an ATL gene number higher than the highest number used in A. thaliana.
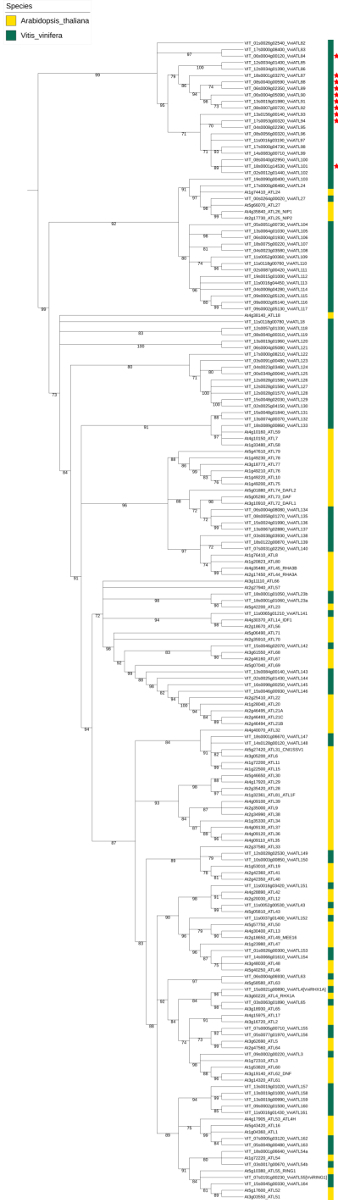
Figure 2: Phylogenetic tree of V. vinifera and A. thaliana ATL E3 ubiquitin ligase-encoding genes. The unrooted tree was generated with the Phylogeny.fr suite (V. vinifera (in green) and the 83 ATL genes of A. thaliana reported in the UniProt database (in yellow). Branch support values were obtained from 100 bootstrap replicates. The red stars indicate the presence of a BCA2 zinc finger (BZF) domain in the corresponding proteins. Reproduced from Ariani et al. licensed under a Creative Commons Attribution 4.0 International License42. Please click here to view a larger version of this figure.
Mapping ATL-encoding genes to the grapevine chromosomes showed a wide distribution throughout the genome, suggesting whole-genome duplication as the major evolutionary force in the expansion of ATL gene family in grapevine. Indeed, 31 ATLs were found in homologous chromosomal regions potentially originating from segmental or whole genome duplication events. Moreover, the same analysis highlighted 13 tandemly duplicated genes, one proximal duplicate, and 51 dispersed duplicates (Figure 3). Considering the very large number of duplicated genes in the ATL family, we performed an enrichment test (Fisher's exact test) to check the preferential retention of the duplicated genes during the genome fractionation. With a p-value < 0.001, this test confirmed the hypothesis that duplicated ATL genes were retained more than randomly expected, suggesting a role for the ATL gene family during grapevine adaptation and evolution.
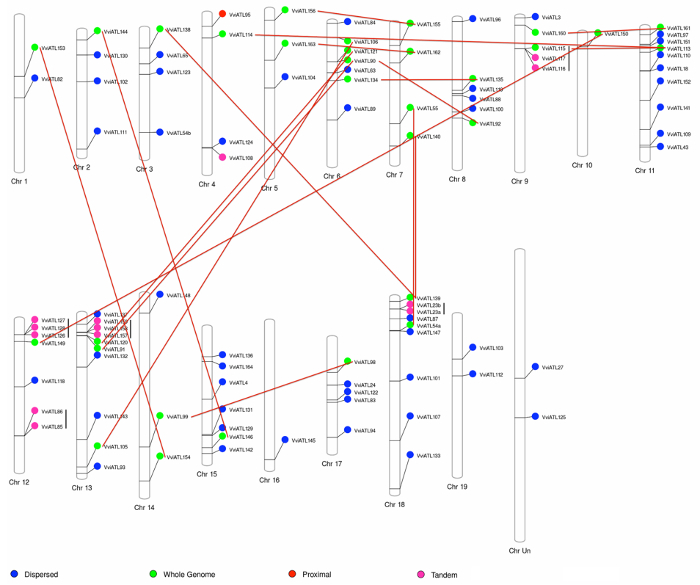
Figure 3: Grapevine ATL-encoding gene distribution on V. vinifera chromosomes and duplication state. The 96 grapevine ATL genes with exact chromosomal information available in the database were mapped to the 19 V. vinifera chromosomes. The colors indicate the original duplication event. Vertical black lines and red lines identify pairs derived from tandem duplications and whole genome duplications, respectively. Reproduced from Ariani et al. licensed under a Creative Commons Attribution 4.0 International License42. Please click here to view a larger version of this figure.
To further investigate the putative biological functions of the ATLs in grapevine, a meta-analysis was carried out on the V. vinifera cv. Corvina global gene expression Atlas12. The dataset includes whole-genome expression values of 54 different grapevine organs and developmental stages and was used to perform a hierarchical bi-clustered analysis. Results not only confirmed that all the 96 ATLs were expressed in at least one of the 54 tissues/stages, but also pointed out the presence of five main clusters of expression profiles (Figure 4A). Briefly, clusters A and E showed opposite behaviors, in particular the first is characterized by a general downregulation of ATL genes in juvenile samples, including early berry stages, young leaf, tendrils, inflorescence, and most of the bud stages. On the other hand, in the same cluster A, mature samples such as berries at ripening and post-harvest withering stages, woody tissues, and late stages of seed development ATL genes showed a predominant upregulation. Genes in Cluster C were mainly downregulated in most of the samples, while ATL genes in cluster D were often upregulated at late stages of berry development. Finally, cluster B did not show any relevant variation in the expression profiles.
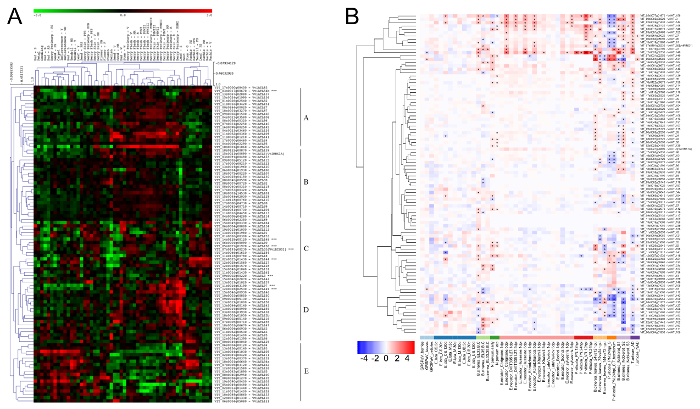
A similar approach was applied to study the expression of grapevine ATL family members in response to biotic and abiotic stresses, using specific datasets built for this purpose. A huge amount of expression data deriving from microarray and RNA-seq experiments are available from public access databases such as Gene Expression Omnibus (GEO) and ArrayExpress. Once collected and conveniently normalized, the information was exploited for further insights into the potential function of ATLs in plant response to stresses. Analyzing the expression profiles of grapevine ATLs in response to biotic stresses revealed that 62 out of 96 transcripts showed a significant modulation (log2 fold-change (FC) >|0.5|) in at least two conditions, with a false discovery rate (FDR) < 0.05 (Figure 4B). The number increases to 81 considering only the FDR threshold in a single condition. These results strongly suggested a direct involvement of the ATL gene family in the response to pathogens also in grapevine. In particular, a group of 12 genes (VviATL3-27-54b-55-90-97-123-144-148-149-156) were strongly upregulated in response to most pathogens, including biotrophic and necrotrophic fungi and herbivores, and thus, deserve attention for further functional analyses.
Figure 4: Hierarchical clusteringof ATL gene expression in grapevine Atlas and in grapevine biotic stress-related dataset. (A) The log transformed expression values of grapevine ATL genes in the grapevine Atlas12 were used for hierarchical cluster analysis based on Pearson's distance metric. The color scale represents higher (red) or lower (green) expression levels with respect to the median transcript abundance of each gene across all samples. Letters A to E on the right side indicate the different clusters identified. AB: after burst; B: burst; bud-W: winter bud; F: flowering; FB: flowering begins; FS: fruit set; G: green; MR: mid-ripening; PFS: post-fruit set; PHWI-II-III: post-harvest withering 1, 2 and 3 months; R: ripening; S: senescent; stem-W: woody stem; V: veraison; WD: well developed; Y: young. (B) The color scale represents increased (red) or decreased (blue) fold changes of grapevine ATL gene expression in infected samples compared to controls for each condition. Asterisks indicate the significant differential expression (FDR < 0.05) of each ATL under the corresponding conditions. Reproduced from Ariani et al. licensed under a Creative Commons Attribution 4.0 International License42. Please click here to view a larger version of this figure.
Supplementary Table 1: ATL genes candidates for alternative splicing. (a) ATL gene ID according to the V1 grape gene prediction and annotation, (b) ATL gene ID according to the V2 grape gene prediction and annotation43, (c) number of putative ATL alternative splicing variants, (d) information on coding sequence of each putative ATL variant. Please click here to download this file.
Supplementary Table 2: Please click here to download this file.
Supplementary File 1: Please click here to download this file.
Discussion
In the genomic era, many gene families have been deeply characterized in several plant species. This information is preliminary to functional studies and provide a frame to investigate further the role of different members in a family. In this context, there is also a need for a nomenclature system allowing to uniquely identify each member in a family, avoiding the redundancy and confusions that may arise when names are assigned independently to different genes by different research groups.
After thoughtful consideration, the grapevine scientific community agreed to name grapevine genes in a family based on similarities with Arabidopsis genes and established a series of rules that must be applied to describe new gene families in grapevine, basically starting from the phylogenetic comparison of nucleotide sequences between grapevine and Arabidopsis family members8. Therefore, only genes that are already annotated and named properly in Arabidopsis can be used in the grapevine nomenclature. The procedure applied for the identification of grapevine ATL orthologues in Arabidopsis described here was therefore carried out solely to fulfill the requirement of assigning the correct grapevine gene family nomenclature. Nevertheless, for other plant species, alternative approaches could be an option. For instance, orthology could be inferred using a bidirectional BLAST hits (BBH), where orthologues are defined as pairs of genes in two species that are more similar (i.e., with highest alignment score) to one another than to any other gene in the other species44. However, this method could miss many orthologues in the case of high rate of gene duplication, such as in plants and animals45. Moreover, in the case of ATL-encoding genes, BBH may retrieve genes lacking the precise ATL-type RING-H2 structure (including the proline residue) or genes that are not annotated and named as ATLs in Arabidopsis. Although from an evolutionary perspective this search may be relevant, the retrieval of orthologues that are not annotated would not have fulfilled the scope of grapevine ATL gene family annotation and nomenclature, and orthologues that are not annotated as ATLs cannot be used to name grapevine family members. Another possibility is to infer orthology based on amino acid instead of nucleotide sequences using InParanoid46, or the most recent Hieranoid 247, albeit such workflows are not expressly recommended by the scientific community.
Expression meta-analysis, which can be defined as a systematic approach to study and combine different publicly available dataset repositories of expression data, allows highlighting shared and different molecular mechanisms in a variety of conditions. Thus, the integration of gene expression information from multiple large-scale transcriptomic experiments can improve the characterization of a gene family, by defining the expression profiles of the family members across experiments, thus minimizing the impact of experiment-specific factors and supporting a more robust assumption of putative gene function in particular processes. However, the use of microarray data requires the integration of expression data obtained with different platforms, considering their own limitations. For instance, in the grapevine Nimblegen microarray platform, a significant proportion of probesets for corresponding genes represented on the array (~ 13,000 genes) have potentially cross-hybridization issues48. In the case of the grapevine ATL family, 15 genes may be affected by such phenomenon. Nevertheless, as discussed by Cramer et al.48, the cross-identification of highly similar gene family members by the same probe could provide interesting information regarding the expression, in specific conditions, not only of a single gene but of two to more genes sharing high sequence similarities, and thus potentially sharing targets and functions. Another potential issue related to microarray datasets is the expression detection limit of microarray platforms, which are not very sensitive. To solve both concerns, i.e., cross-hybridization and signal sensitivity, a possible solution could be to consider only RNAseq expression datasets. However, the meta-analysis of RNAseq data of very large datasets from many different studies can become highly time-consuming and may require many computational resources and high expertise.
Though the approach presented here aims to be exhaustive, it can be certainly further complemented with other analyses. First, to achieve further insights into the molecular evolution and phylogenetic relationship among gene family members in plants, the phylogenetic analysis could be extended building a phylogenetic tree using multiple sequence alignments of family members from several plant species. It is also possible to calculate the evolutionary time of family genes, an estimation of their synonymous and non-synonymous substitution rates during evolution, by determining the values Ks (number of synonymous substitutions per synonymous site in a given period of time) and Ka (number of nonsynonymous substitutions per non-synonymous site in the same period). The Ka/Ks ratio is used to infer the mechanisms of gene duplication events after divergence from their ancestors. A value of Ka/Ks = 1 suggests neutral selection, a Ka/Ks value of < 1 suggests purifying selection, and a Ka/Ks value of > 1 suggests positive selection49. Moreover, if gene structure analysis reveals the presence of introns, the gene family characterization can be further extended to the detection of alternative splicing variants. Indeed, based on a deep survey of RNA-seq data from different tissues, stress conditions and genotypes43, 21 (of the 96) ATLs are strong candidates for alternative splicing events, with potential number of isoforms ranging from 2 to 16 for these ATLs (see Supplementary Table 1). Alternative transcripts frequently produce protein isoforms that vary in amino acid sequences and these changes may alter the cellular properties of proteins and may cause alterations from subtle modulation to loss of function of the gene product. For that reason, alternative splicing events have been involved in important plant functions, including stress response, disease resistance, photosynthesis, and flowering50,51. Integration of ATL gene promoter information that contains putative cis-regulatory elements52 or finding molecules (e.g., microRNA and long non-coding RNA) potentially targeting ATLs53 can also be supplemented to reveal system insights into the complex molecular regulation and interaction of grapevine ATLs.
In conclusion, the choice of the analyses to be performed as well as the procedures to be applied to characterize a new gene family in a plant species are mainly driven by scientific community rules as well as by the scope of gene family identification. It is important to keep in mind the possible subsequent investigation steps, which will exploit the set of information, among which includes gene evolution among plant species, genome structure description, or reliable candidates for selection in functional studies.
Disclosures
The authors have nothing to disclose.
Acknowledgments
The work was supported by the University of Verona within the frame of Joint Project 2014 (Characterization of the ATL gene family in grapevine and of its involvement in resistance to Plasmopara viticola).
References
- Jaillon O, et al. The grapevine genome sequence suggests ancestral hexaploidization in major angiosperm phyla. Nature. 2007;449(7161):463–467. doi: 10.1038/nature06148. [DOI] [PubMed] [Google Scholar]
- Adam-Blondon A-F, et al. Genetics, Genomics, and Breeding of Grapes. Science Publishers; 2011. pp. 211–234. [Google Scholar]
- Chen L, Hellmann H. Plant E3 Ligases: Flexible Enzymes in a Sessile World. Mol. Plant. 2013;6(5):1388–1404. doi: 10.1093/mp/sst005. [DOI] [PubMed] [Google Scholar]
- Vierstra RD. The ubiquitin-26S proteasome system at the nexus of plant biology. Nat. Rev. Mol. Cell Biol. 2009;10(6):385–397. doi: 10.1038/nrm2688. [DOI] [PubMed] [Google Scholar]
- Serrano M, Parra S, Alcaraz LD, Guzmán P. The ATL Gene Family from Arabidopsis thaliana and Oryza sativa Comprises a Large Number of Putative Ubiquitin Ligases of the RING-H2 Type. J. Mol. Evol. 2006;62(4):434–445. doi: 10.1007/s00239-005-0038-y. [DOI] [PubMed] [Google Scholar]
- Aguilar-Hernández V, Aguilar-Henonin L, Guzmán P. Diversity in the Architecture of ATLs, a Family of Plant Ubiquitin-Ligases, Leads to Recognition and Targeting of Substrates in Different Cellular Environments. PLoS One. 2011;6(8):e23934. doi: 10.1371/journal.pone.0023934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzmán P. The prolific ATL family of RING-H2 ubiquitin ligases. Plant Signal Behav. 2012;7(8):1014–1021. doi: 10.4161/psb.20851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimplet J, et al. The grapevine gene nomenclature system. BMC Genomics. 2014;15:1077. doi: 10.1186/1471-2164-15-1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prince VE, Pickett FB. Splitting pairs: the diverging fates of duplicated genes. Nat. Rev. Genet. 2002;3(11):827–837. doi: 10.1038/nrg928. [DOI] [PubMed] [Google Scholar]
- Magadum S, Nerjee U, Murugan P, Gangapur D, Ravikesavan R. Gene duplication as a major force in evolution. J. Gen. 2013;92(1):155–161. doi: 10.1007/s12041-013-0212-8. [DOI] [PubMed] [Google Scholar]
- Wang N. Patterns of Gene Duplication and Their Contribution to Expansion of Gene Families in Grapevine. Plant Mol. Biol. Rep. 2013;31(4):852–861. [Google Scholar]
- Fasoli M. The Grapevine Expression Atlas Reveals a Deep Transcriptome Shift Driving the Entire Plant into a Maturation Program. Plant Cell. 2012;24(9):3489–3505. doi: 10.1105/tpc.112.100230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLAST. BLAST2.6.0. 2016. Available from: https://blast.ncbi.nlm.nih.gov/Blast.cgi.
- MEGA. MEGA7.0.25 build 7170412. 2017. Available from: http://www.megasoftware.net/
- MEME. MEME Suite Version 4.11.4. 2017. Available from: http://meme-suite.org/
- ProtParam. ExPASy Server. 2005. Available from: http://web.expasy.org/protparam/
- ngLOC v1.0. 2007. Available from: http://genome.unmc.edu/ngLOC/index.html.
- TargetP v1.1 Server. 2000. Available from: http://www.cbs.dtu.dk/services/TargetP/
- Prowler v1.2. 2005. Available from: http://bioinf.scmb.uq.edu.au:8080/pprowler_webapp_1-2/
- MuSite v1.0. 2010. Available from: http://musite.sourceforge.net/
- Pfam. Pfam version 31.0. 2016. Available from: http://pfam.xfam.org/
- TMHMM v2.0c. 2007. Available from: http://www.cbs.dtu.dk/services/TMHMM/
- ExPASy. ProtScale. 2005. Available from: http://web.expasy.org/protscale/
- CRIBI. Grape genome database. 2012. Available from: http://genomes.cribi.unipd.it/grape/
- PhenoGram. 2012. Available from: http://visualization.ritchielab.psu.edu/phenograms/plot.
- ScanX v0.8. 2013. Available from: http://chibba.pgml.uga.edu/mcscan2/
- Interactive Tree Of Life (iTOL) Version3.5.3. 2016. Available from: http://itol.embl.de/
- UniProt. 2016. Available from: http://www.uniprot.org/
- Phylogeny.fr. 2008. Available from: http://www.phylogeny.fr/index.cgi.
- MUSCLE. 2017. Available from: http://www.ebi.ac.uk/Tools/msa/muscle/
- Gblocks Server. Version 0.91b. 2002. Available from: http://molevol.cmima.csic.es/castresana/Gblocks_server.html.
- Vitis vinifera cv. Corvina gene expression Atlas. 2015. Available from: https://www.researchgate.net/publication/273383414_54sample_datamatrix_geneIDs_Fasoli2012.
- Multiple Experiment Viewer (MeV) Version 4.8.1. 2017. Available from: http://mev.tm4.org/
- Sequence Read Archive (SRA) 2017. Available from: https://www.ncbi.nlm.nih.gov/sra.
- Bolger AM, Lohse M, Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL. Fast gapped-read alignment with Bowtie 2. Nat Meth. 2012;9(4):357–359. doi: 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq-a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R. Version 3.4.1. 2017. Available from: https://www.r-project.org/
- Ritchie ME. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43(7):e47. doi: 10.1093/nar/gkv007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- EMBL-EBI. EMBOSS Needle. 2017. Available from: http://www.ebi.ac.uk/Tools/psa/emboss_needle/
- Ariani P. Genome-wide characterisation and expression profile of the grapevine ATL ubiquitin ligase family reveal biotic and abiotic stress-responsive and development-related members. Sci. Rep. 2016;6:38260. doi: 10.1038/srep38260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitulo N, et al. A deep survey of alternative splicing in grape reveals changes in the splicing machinery related to tissue, stress condition and genotype. BMC Plant Biol. 2014;14(1):99. doi: 10.1186/1471-2229-14-99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Fonstein M, D'Souza M, Pusch GD, Maltsev N. The use of gene clusters to infer functional coupling. Proc. Natl. Acad. Sci. USA. 1999;96(6):2896–2901. doi: 10.1073/pnas.96.6.2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalquen DA, Dessimoz C. Bidirectional Best Hits Miss Many Orthologs in Duplication-Rich Clades such as Plants and Animals. Genome Biol. Evol. 2013;5(10):1800–1806. doi: 10.1093/gbe/evt132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remm M, Storm CEV, Sonnhammer ELL. Automatic clustering of orthologs and in-paralogs from pairwise species comparisons1. J. Mol. Biol. 2001;314(5):1041–1052. doi: 10.1006/jmbi.2000.5197. [DOI] [PubMed] [Google Scholar]
- Kaduk M, Sonnhammer E. Improved orthology inference with Hieranoid 2. Bioinformatics. 2017;33(8) doi: 10.1093/bioinformatics/btw774. [DOI] [PubMed] [Google Scholar]
- Cramer GR, et al. Transcriptomic analysis of the late stages of grapevine (Vitis vinifera cv. Cabernet Sauvignon) berry ripening reveals significant induction of ethylene signaling and flavor pathways in the skin. BMC Plant Biol. 2014;14:370. doi: 10.1186/s12870-014-0370-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juretic N, Hoen DR, Huynh ML, Harrison PM, Bureau TE. The evolutionary fate of MULE-mediated duplications of host gene fragments in rice. Genome Res. 2005;15(9):1292–1297. doi: 10.1101/gr.4064205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filichkin SA. Genome-wide mapping of alternative splicing in Arabidopsis thaliana. Genome Res. 2010;20(1):45–58. doi: 10.1101/gr.093302.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quesada V, Macknight R, Dean C, Simpson GG. Autoregulation of FCA pre-mRNA processing controls Arabidopsis flowering time. EMBO J. 2003;22(12):3142–3152. doi: 10.1093/emboj/cdg305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DCJ, Gutierrez RL, Gambetta GA, Castellarin SD. Genome-wide analysis of cis-regulatory element structure and discovery of motif-driven gene co-expression networks in grapevine. DNA Res. 2017;24(3):311–326. doi: 10.1093/dnares/dsw061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong DCJ, Matus JT. Constructing Integrated Networks for Identifying New Secondary Metabolic Pathway Regulators in Grapevine: Recent Applications and Future Opportunities. Front. Plant Sci. 2017;8:505. doi: 10.3389/fpls.2017.00505. [DOI] [PMC free article] [PubMed] [Google Scholar]