

Abstract
The quantification of small molecules in single cells raises new potentials for better understanding the basic processes that underlie embryonic development. To enable single-cell investigations directly in live embryos, new analytical approaches are needed, particularly those that are sensitive, selective, quantitative, robust, and scalable to different cell sizes. Here, we present a protocol that enables the in situ analysis of metabolism in single cells in freely developing embryos of the South African clawed frog (Xenopus laevis), a powerful model in cell and developmental biology. This approach uses a capillary microprobe to aspirate a defined portion from single identified cells in the embryo, leaving neighboring cells intact for subsequent analysis. The collected cell content is analyzed by a microscale capillary electrophoresis electrospray ionization (CE-ESI) interface coupled to a high-resolution tandem mass spectrometer. This approach is scalable to various cell sizes and compatible with the complex three-dimensional structure of the developing embryo. As an example, we demonstrate that microprobe single-cell CE-ESI-MS enables the elucidation of metabolic cell heterogeneity that unfolds as a progenitor cell gives rise to descendants during development of the embryo. Besides cell and developmental biology, the single-cell analysis protocols described here are amenable to other cell sizes, cell types, or animal models.
Keywords: Chemistry, Issue 130, Capillary electrophoresis, electrospray ionization, mass spectrometry, microsampling, metabolomics, single cell, Xenopus, frog, embryogenesis
Introduction
A comprehensive understanding of embryonic development requires characterization of all molecular changes that unfold in every cell of the developing organism. While Next-Generation Sequencing with molecular amplification enables deep measurement of single-cell transcriptomes1 in developing systems2,3, considerably less is known about the suite of smaller molecules produced in single embryonic cells, including proteins and, especially, metabolites (molecular mass <~1,500 Da). With a fast and dynamic response to intrinsic and extrinsic events, the metabolome serves as a powerful descriptor of a cell's molecular state. The single-cell metabolome, therefore, raises the potential to track the spatial and temporal development of cell heterogeneity in the early embryo and to identify new molecules for functional studies. However, without molecular amplification available for these molecules, detection of the metabolome demands exceptional sensitivity using mass spectrometry (MS), which is the technology of choice for metabolite analysis.
Single-cell MS is a collection of technologies with sufficient sensitivity to measure metabolites in single cells (see reviews 4,5,6,7,8,9,10,11,12,13,14,15). Reproducible sampling of cells and efficient extraction of metabolites are essential to the successful detection of metabolites in single cells. Whole-cell dissection of identified cells from Xenopus embryos has enabled the characterization of small molecules and peptides16. Other approaches employ micropipettes to sample individual live cells followed by detection using electrospray ionization (ESI) MS. For example, metabolites were measured in plant or mammalian cells by single-cell video MS17, pressure probe18, single probe19, and fluidic force microscopy20, among other techniques21,22,23,24. Additionally, incorporation of chemical separation prior to ionization into the single-cell MS workflow efficiently simplifies the metabolome, thus alleviating potential interferences during ion generation before detection. Importantly, separation also provides compound-specific information to assist in molecular identifications. Capillary electrophoresis (CE) has been used to detect metabolites in single dissected25,26 or microsampled27neurons, capturing small-molecule differences between neuron phenotypes. We recently adapted CE to ESI tandem MS to enable the trace-level detection of hundreds of metabolites in individual cells that were dissected from early embryos of Xenopus laevis16,28. These studies revealed surprising metabolic differences between embryonic cells at an early stage of development and led to the discovery of metabolites with previously unknown developmental impacts16.
Here we provide a protocol that enabled the detection of metabolites in single cells directly in a live vertebrate embryo using microprobe single-cell CE-ESI-MS29,30. The model organism chosen is the 8-to-32-cell X. laevis embryo, although the approach is also applicable to later stages of development and other types of model organisms. This protocol uses sharpened capillaries with multi-axis translational control under guidance by a high-resolution imaging system to aspirate an ~10 nL portion of identified cells in situ in the morphologically complex developing embryo. This microprobe is scalable to smaller cells and operates within seconds, which is sufficiently fast to track cell lineages in the embryo. After extracting polar or apolar small molecules, such as metabolites and peptides, from the collected sample in ~4-5 µL extraction solution, a ~10 nL of the resulting extract is analyzed in a custom-built CE platform hyphenated to an ESI mass spectrometer. Construction and operation of the CE-ESI-MS platform builds on protocols described elsewhere.31,32 The co-axial CE-ESI interface is constructed as described elsewhere.31 This platform is maintained in the cone-jet spraying regime to achieve trace-level sensitivity with a capability for quantification over a 4-5 log-order dynamic range (relative28,29,30 or absolute16). The CE-ESI-MS platform offers a 60-amol lower limit of detection with 8% relative standard deviation (RSD) in quantitation over a tested range of 10 nM to 1 µM for small molecules16, which are sufficient to characterize endogenous metabolites in X. laevis cells. Microprobed cells continue to divide as the embryo progresses through development30, allowing for temporally and spatially resolved analysis of cellular metabolism. Indeed, single-cell CE-ESI-MS can be used to find metabolic differences between cells that occupy the dorsal-ventral16,29, animal-vegetal16, and left-right28 developmental axes as well as cells that form the neural-tissue fated dorsal lineage from a common progenitor cell in X. laevis30. Besides querying metabolic differences between individual embryonic cells at different developmental stages of the X. laevis embryo30, we anticipate that the protocols described here are applicable to a broad array of biomolecules and single cells microsampled from different stages of embryonic development as well as other types of cells and model organisms. Additionally, the microprobe could be used for microsampling while a different platform compatible with miniscule samples could be used for separation and/or characterization of biomolecules.
Protocol
All protocols related to the maintenance and handling of Xenopus laevis were approved by the Institutional Animal Care and Use Committee at the George Washington University (IACUC no. A311).
1. Preparation of Sampling Instruments, Media, Solvents, and Sampling Dishes
Prepare 1x Steinberg's solution (SS) by dissolving the following salts in ultrapure water (~18.2 MΩ.cm at 25 °C) in the following order and at the indicated concentrations following a standard protocol33: Sodium chloride (58.2 mM), potassium chloride (0.67 mM), calcium nitrate (0.34 mM), magnesium sulfate (0.83 mM), Tris-hydrochloride (4.19 mM), and Tris base (0.66 mM). Make 0.5x SS by two-fold, and 0.1x SS by ten-fold dilution, of the 1x SS using ultrapure water.
Prepare sampling dishes by first making 2% agarose in 1x SS. Autoclave at 120 °C for 20 min to dissolve. While still liquid, coat the bottom of 60 mm Petri dishes with the solution. Once the agarose gel has cooled and solidified, flame the end of a six-inch Pasteur pipette until it forms a ball, and lightly touch the heated end to imprint 5-10 wells, ~1 mm deep, into the agarose. NOTE: These wells are used to immobilize the embryos during sampling.
Prepare the metabolite extraction solvent. Tailor the physicochemical properties of the solvent (e.g., polarity and pH) to the classes of molecules that are of interest in the study. NOTE: For example, we use 40% methanol and 40% acetonitrile in LC-MS-grade water as a discovery approach to target mainly polar metabolites, and a few apolar metabolites and peptides28.
Make hair loops using clean hair and a Pasteur pipette as described elsewhere33 for gently moving embryos in the Petri dishes with minimal perturbation.
- Fabricate tapered-tip micropipettes as shown in Figure 1a.
- First, pull borosilicate capillaries (1,000/500 µm outer/inner diameter) in a Flaming-Brown-type capillary puller with the following settings: heat = 355; pull = 65, velocity = 80; time = 150.
- Next, break-off the tip of the pulled micropipette using a pair of fine sharp forceps to obtain a capillary tip outer diameter of ~20 µm. Perform this step under a stereomicroscope to aid precision and reproducibility. NOTE: Capillaries with a small tip are prone to clogging during aspiration of the viscous cytoplasm. While capillaries with a larger tip certainly help avoid clogging and aspirate more of the cellular content, large-bore capillaries may pose challenges during the sampling of smaller cells and possibly damage the cell for subsequent sampling. Careful adjustment of the pressure and time of aspiration may partially alleviate these challenges. We find micropipettes with ~20 µm outer diameter ideal for the work presented here.
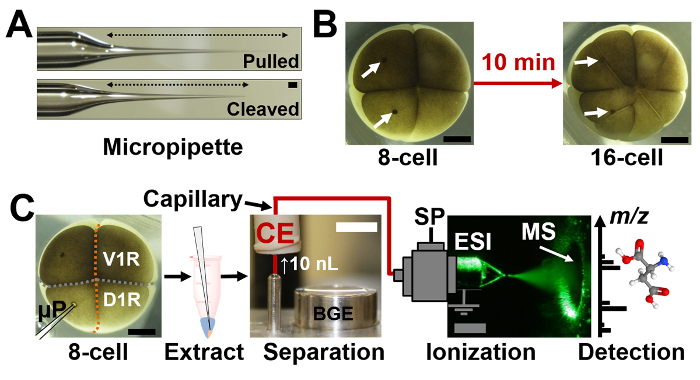
2. Microsampling Single Cells and Metabolite Extraction
Obtain embryos (fertilized eggs) through gonadotropin-induced natural mating of adult Xenopus laevis or via in vitro fertilization as described in protocols elsewhere33,34. NOTE: Natural mating ensures that the embryonic developmental stages are staggered while embryos obtained by in vitro fertilization are more reliable in supply. However, in vitro fertilization requires sacrificing the adult male frog.
Freshly prepare 2% cysteine dejellying solution by dissolving 4 g of cysteine into 200 mL of ultrapure water and dropwise adjust the solution to pH 8 using 10 N sodium hydroxide solution.
Remove the jelly coats surrounding the embryos as they begin to cleave into the 2-cell stage as follows: let embryos rest in the dejellying solution for 2 min, then gently swirl them for an additional 2 min to prevent embryos from adhering to the surface of the collection dish.
Gently pour dish contents into a clean beaker and quickly decant the dejellying solution from the beaker. Immediately cover the eggs with 0.1x SS to rinse off the remaining dejellying solution, gently swirl, and then decant the solution. Repeat this step four times to thoroughly wash the embryos. NOTE: Limit exposure of the embryos to the dejellying solution to 4 min to ensure viability. Comprehensive protocols to remove the jelly coats are available elsewhere33.
Transfer dejellied embryos into 1x SS in a Petri dish. To minimize crowding within the plates, place ~100 embryos per 100 mm dish33. NOTE: Dishes containing embryos can be stored between 14-18 °C to slow down development and obtain embryos at staggered developmental stages from the same parents. Further guidelines on temperature dependence of growth and development are published on Xenbase and elsewhere33,35,36,37.
- Sort cleaving embryos at the 2-cell stage into a separate dish in which stereotypical pigmentation confidently marks the dorsal-ventral axis, with reference to established cell fate maps38,39,40.
- Identify correctly cleaving embryos by ensuring that the first cleavage furrow, which demarks the midsagittal plane, bisects the darkly (ventral) and lightly (dorsal) pigmented animal pole such that the two halves are mirror images41.
Mount a fabricated micropipette on a multi-axis micromanipulator (manual or remotely controlled). Connect the micropipette to a microinjector.
Use a plastic transfer pipette to aspirate ~5 of the 8-cell embryos and transfer them into the sampling dish containing 0.5x SS. Using a hair loop, position the embryo to be sampled into an individual well, and ensure that the correctly identified cell of interest is facing the microprobe at a ~90° angle. NOTE: Identify the cells based on pigmentation and position in the embryo, with reference to cell fate maps38,39,40.
While working under the stereomicroscope (at 20-30X magnification), guide the tip of the micropipette into the identified single cell within the live embryo as shown in Figure 1 (see sampling points on cells in panel B and the apparatus in panel C).
Withdraw a desired portion of the cell's content by applying negative pressure pulses to the microcapillary using the microinjector ('fill mode'). NOTE: For example, we routinely aspirate ~10-15 nL volume from the cell by applying ~3 pulses of -30 psi to the capillary. This whole step lasts ~5 s for aspiration30.
- Gently retract the microprobe from the cell and transfer its tip into 4 µL of the metabolite extraction solvent chilled (4 °C) in a micro-sized vial. Next, apply a pressure surge of +80 psi for 1 s to the capillary using the microinjector ('clear mode') to expel the aspirate into the solvent.
- Tightly close the vial to prevent evaporation and place the vial back into the 4 °C ice bucket until sampling is complete. Discard the used micropipette into a sharps container to prevent a needlestick hazard. NOTE: To determine the volume of the aspirated cell content, inject the aspirate into mineral oil, where it obtains a spherical shape. The diameter of this sphere can be measured using a microscope. Calculate the aspirated volume: V = 4/3 π r3, where V is the volume, and r is the radius of the sphere.
To sample the same cell again or other cells of the embryo, repeat steps 2.7-2.11 (Figure 1c). To avoid potential carry over between consecutive samplings, use a fresh micropipette for the analysis of each different cell.
Once sampling is completed, vortex-mix the sample-containing microcentrifuge vials for ~1 min to expedite extraction of metabolites, then centrifuge at 8,000 × g for 5 min at 4 °C to pellet cellular debris and other particulates.
Store the cell extracts together with the cell debris and precipitated materials at -20 °C for a day or at -80 °C for up to 1 month until measurement by CE-ESI-MS.
3. CE-ESI-MS Measurement
- Preparation of Standards and Solutions for CE-ESI-MS
- Prepare the background electrolyte (BGE) consisting of 1% formic acid in LC-MS grade water.
- Prepare the sheath solution to contain 50% methanol in LC-MS grade water and 0.1% formic acid.
- Prepare 50 nM acetylcholine solution in the sheath solution for daily evaluation of the performance of the CE-ESI-MS system.
- Prepare 150 mM sodium chloride solution as mass-calibration standard for the low m/z range in the positive ion mode. A mass (m/z) accuracy of <10 ppm is recommended. Follow the mass spectrometer vendor's instructions to carry out this step. NOTE: Alternatively, other standards with known m/z values can be used to mass-calibrate the mass spectrometer.
- Construction of the CE-ESI Platform
- Construct a CE injection platform capable of rapid vertical translation of a stage holding the BGE vial and the sample loading microvial. For construction and operation of the platform, refer to details in Reference31.
- Assemble the CE-ESI interface (Figure 1c) as follows. Mount the electrospray metal emitter (130/260 µm inner/outer diameter and ~35 mm length) into a 3-port T-union. Feed the separation CE capillary (40/105 µm inner/outer diameter and ~100 cm length) through the electrospray emitter allowing it to protrude ~40-100 µm beyond the tip of the emitter. Work under a stereomicroscope to aid accuracy.
- Connect the sheath solution capillary (75/360 µm inner/outer diameter and ~100 cm length) to the remaining port to supply the electrospray solution. Use appropriate sleeves and finger-tighten connections for leak-free operation of the CE-ESI interface. Refer to previous protocols31,32 for details on the assembly and trouble-shooting of this interface. NOTE: Capillary dimensions affect the signal-to-noise ratio (S/N) and the duration of separation. For example, narrow-bore and short capillaries facilitate fast separations using higher separation voltages42,43. Additionally, depending on the types of molecules that are of interest in a study, separation capillaries may be coated to minimize/avoid unwanted molecule-capillary wall interactions44.
- Using a plate-holder, mount the CE-ESI interface onto a three-axis translation stage and position the electrospray emitter tip ~2 mm from the mass spectrometer orifice (Figure 1c).
- To clean components of the interface, rinse by supplying the electrospray sheath solution through the electrospray emitter at 1 µL/min and the BGE through the CE separation capillary. Use syringe pumps to feed the solvents at a steady rate.
- Flush the CE separation capillary before each measurement by connecting a syringe to the capillary inlet end. Use sufficiently large syringes to minimize refilling and priming of solvent supply lines. NOTE: Experiments typically use 1 mL gas-tight syringes to supply the electrospray sheath solvent and a 500 µL syringe to flush the separation capillary.
- Validation of CE-ESI-MS Platform and Measurement of Metabolites NOTE: The goal of this step is to confirm the analytical sensitivity of the CE-ESI-MS instrument daily before analyzing single-cell extracts. Using the CE-ESI platform described here, we can accomplish a lower limit of detection at 60 amol using a quadrupole orthogonal-acceleration time-of-flight mass spectrometer16.
- After rinsing the separation capillary for ~5 min, transfer its inlet into the BGE solution located in a stainless-steel vial.
- Position the electrospray emitter tip ~2 mm from the mass spectrometer orifice and fine-adjust this distance using a translation stage to generate electrospray in the stable cone-jet regime while monitoring the spray using a stereomicroscope (see references31,45). Monitor the stability of the total ion current (TIC) for ~30-45 min to ensure stable operation.
- Apply ~20 kV to the BGE vial by gradually ramping up the potential over ~15 s, typically generating ~7.5 µA current across the separation capillary using 1% formic acid as the BGE. Before each measurement, ensure system stability by monitoring the TIC profile for ~5-10 min, and then step-wise lower the separation (CE) potential to 0 V (ground). NOTE: To semi-automate this process, we use a custom-written software to remotely control the CE high voltage power supply31. If the CE-ESI-MS platform is unstable, carefully evaluate the source of instability by testing the CE-ESI-MS platform first in ESI-only mode and then in the CE-ESI operational mode as recommended elsewhere31. Briefly, to test the platform in the ESI-only mode, turn off the CE high voltage and monitor the TIC for ~30 min in the cone-jet spraying regime. If necessary, proceed as follows to address errors: (i) inspect connections for leaks; (ii) clean the electrospray emitter with water, isopropanol, water, and methanol; (iii) de-gas solvents; (iv) flush the emitter for ~25 min before testing again. If the CE-ESI platform is found stable in ESI-only mode but becomes unstable during CE separation, inspect the system for Joule heating and/or electrolysis: (i) flush the separation capillary with BGE for ~25 min and repeat the experiment; (ii) use lower separation potentials to maintain a linear Ohmic response (i.e., linear CE current vs. separation voltage curve); (iii) inspect the CE capillary for potential damages, such as cracks, and replace the capillary if necessary.
- Analyze ~6 nL of the sample as follows:
- Pipette 1 µL of the acetylcholine standard solution into the injection vial.
- Transfer the separation capillary from the BGE vial into the injection vial.
- Lift the CE injection stage 15 cm in 1s.
- Hold the stage elevated for 60 s to hydrodynamically inject ~6 nL of the sample into the separation capillary.
- Subsequently, translate the stage back to starting levels (in line with the capillary outlet).
- Gently move the capillary inlet end into the BGE.
- Immediately after, ramp up the CE voltage to start electrophoretic separation.
- Start MS data acquisition. NOTE: System performance may be characterized using any chemical standard. The single-cell CE-ESI-MS delivers lower limits of detection at ~10 nM (~60 amol) for acetylcholine, methionine, and histidine16. The volume injected (Vinj), into the capillary in nL, depends on the height difference (H, cm) during injection, density (ρ, g cm-3) and viscosity (η, kg m-1s-1) of the BGE, length (L, m) and radius (r, µm) of the CE capillary, and injection duration (tinj, s). This relationship is expressed by the following formula, where g (m s-2) is the gravitational acceleration:
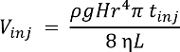
- Once the standard has been detected, stop data acquisition, lower the separation voltage stepwise to 0 V (ground), then retrieve the emitter to 2 cm from the orifice. Flush the separation capillary for 5 min before analyzing the cell extract.
- Measure 10 nL of the single-cell extract by repeating Steps 3.3.1-3.3.5 using 90 s to hydrodynamically inject the sample.
- Data Processing NOTE: The goal of data processing is to identify and quantify compounds between single cells. The single-cell CE-ESI-MS protocol generates narrow electropherographic peaks with typical base widths of a few seconds. By performing data analysis semi-manually, it is possible to find molecular features (unique m/z values with unique migration times) using the following steps. Representative separation is shown for select identified metabolites in Figure 2a.
- Mass-calibrate raw data files post data acquisition. NOTE: We use signals from sodium formate clusters that are generated during the separation of abundant sodium ions from the sample, which are natively present in the cells or extracted from the embryo culture media. The goal of this step is to enhance metabolite identification in later steps by ensuring a high mass (m/z) accuracy, preferably <5 mDa, or <10 ppm, between m/z 50-1,000. Here, post data acquisition calibration makes it possible to routinely obtain mass accuracies <1 mDa, or <2 ppm for m/z 50-500.
- Using a processing script, search for molecular features across the detected mass range. Average mass spectra across each peak to determine the accurate mass, and note down their corresponding migration times. For the identification of low-mass metabolites in the m/z 50-500 range, use a 500 mDa step window to monitor molecular features with S/N > 3.
- Integrate the peak area-under-the-curve for each molecular feature manually or automatically. The resulting area values are used as a measure of metabolite abundance.
- Identify molecular features of interest with high confidence as follows (see Fig. 2b).
- Next, evaluate these mass matches by comparing their tandem mass spectrum obtained from the cell extracts with data available in the metabolite databases or the tandem mass spectrum measured for the corresponding chemical standard.
- Last, validate these assignments by comparing the migration time of molecular features recorded in the cell extracts with related chemical standards analyzed by the same CE-ESI-MS instrument. NOTE: To enhance experimental throughput, we typically identify only molecular features that are statistically significantly different between experimental conditions or cell types. Representative identifications are shown in Figure 2. To identify metabolites with high mass accuracy, we recommend externally calibrating the mass spectrometer daily, performing real-time recalibration during each measurement using an internal standard, and/or externally mass-calibrating each measurement file post-data acquisition (e.g., for sodium formate clusters here) are recommended.
- To compare small-molecule production between single cells, use peak area-under-the-curve in selected ion electropherograms (from Step 3.4.3) as relative measures of compound abundance. NOTE: In our experiments, online software platforms47 were used to perform all steps of subsequent data analysis, including the following steps: i) filtering of molecular features with occurrence in at least 50% of each sample-set (e.g., cell type); ii) normalization of the data; iii) statistical (e.g., t-test) and multivariate data analysis, such as principal component analysis (PCA) and hierarchical cluster analysis (HCA). We use p < 0.05 (Student's t-test) to mark statistical significance and fold-change ≥ 1.5 to note biological significance.
- To determine the absolute concentration of a small molecule in a single cell, implement classical methods of calibration (e.g., external calibration or standard addition) using peak area-under-the-curve of the compound of interest16.

Representative Results
We recently employed microprobe single-cell CE-ESI-MS to characterize metabolites in individual identified cells in freely developing Xenopus laevis embryos29,30. The microprobe enables fast (~5 sec/cell), in situ aspiration of ~10 nL from an individual cell, multiple aspirations of the same cell, or several different cells within the same or later stages of development of the live embryo (Figure 1b). The aspirated cellular content is extracted using a suitable solvent and metabolites are then separated and ionized using the custom-built CE-ESI interface and detected by MS. Typically, the approach yields ~300 nonredundant metabolite signals (after removing isotopic and cluster peaks) from each cell. Separation is exemplified for select molecules in Figure 2a. We identified 70 different signals with high confidence as small polar metabolites, based on migration time and MS/MS fragmentation patterns of cell extract signals, compared to those from a standard or from tandem mass spectral libraries. For example, Figure 2b shows the confident identification of histidine based on the combination of these orthogonal pieces of information.
The microprobe CE-ESI-MS allows for the quantification of metabolic differences between single cells. CE-ESI-MS has a quantitative repeatability of 8% RSD based on under-the-curve peak areas in selected-ion electropherograms16,30. With microprobe sampling, this technical reproducibility is 14% RSD30, which is sufficient to query metabolic activity differences between cells to a statistical and biological significance. As an example, Figure 3 shows that microbe CE-ESI-MS enabled in situ micro-sampling of three dorsal cell-types (Figure 3a). Furthermore, PCA of the quantitative metadata uncovered metabolic changes as an identified progenitor cell of the 8-cell embryo (see D1R cell) divided to form a cell clone in the 16-cell (see D11R cell) and 32-cell (D111R cell) embryo (Figure 3b). Representative trends are shown for select metabolites in Figure 3c. Metabolites such as acetylcholine decreased in abundance with cell division, whereas trolamine and Ser-Arg showed opposing trends in these cell types. Trolamine decreased in abundance from the 8- to the 16-cell embryo and then became enriched in the 32-cell embryo, while Ser-Arg depicted an opposite trend by increasing to the 16-cell embryo before decreasing in the 32-cell embryo. Arginine did not significantly change in abundance between these cell stages. These trends are expected to shed more light on the fundamental processes that underlie the early developmental stages of X. laevis.
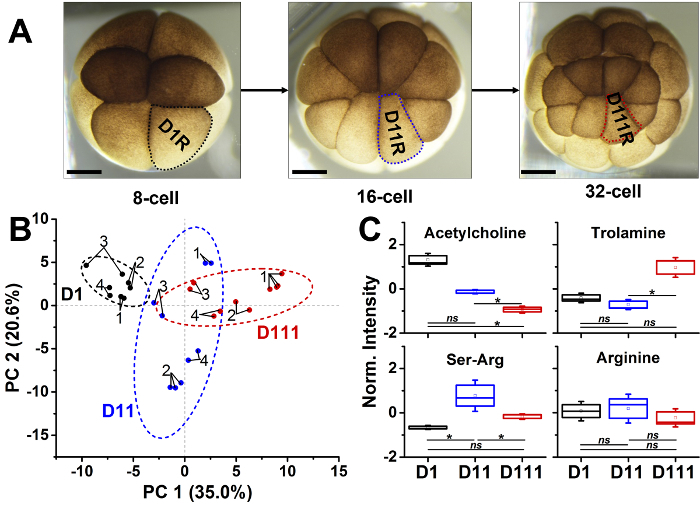
Figure 1: Experimental workflow for in situ microprobe single-cell capillary electrophoresis (CE) mass spectrometry (MS). (A) Fabricated micropipettes (top) before and (bottom) after cleaving the tip to 20 µm outer diameter for use in microsampling. (B) Microsampled cells continued to divide as the 8-cell embryo developed to the 16-cell stage, opening the stage for in vivo studies. (C) Identified single cells were sampled using a microprobe (µP) in the 8-cell Xenopus laevis embryo (see V1R and D1R cells), the cell metabolites extracted and analyzed using CE-MS. Scale bars = 250 µm (dark/gray); 10 mm (white). Key: SP, syringe pump. (Figures adapted with permission from references29,30.) Please click here to view a larger version of this figure.
Figure 2: Microprobe single-cell CE-MS enables confident identification of metabolites. (A) Representative electropherogram for select identified metabolites. (B) Identification of histidine based on a comparison of accurate mass, migration time, and fragmentation data (collision-induced dissociation) against the metabolite standard. Key: 1, methyl histidine; 2, homolysine; SAM, S-adenosylmethionine; Orn, ornithine; TEA, triethylamine; Car, carnitine; AcCho, acetylcholine; AcCar, acetylcarnitine; HPX, hypoxanthine; GSH, glutathione. Please click here to view a larger version of this figure.
Figure 3: Quantitative metabolic differences between single cells that form a dorsal lineage in X. laevis. (A) Optical images of the progenitor D1R cell of the 8-cell embryo, and its descendants, the D11R cell in the 16-cell embryo and the D111R cell in the 32-cell embryo. (B) Principal component analysis (PCA) of metabolites quantified in these three cell types uncovering different metabolic profiles. Each data point denotes a different cell (see numbers) that was measured in technical replicates (connected points). Ellipses mark 95% confidence. (C) Analysis of variance (ANOVA) and post-hoc Fisher's least significant discriminant analysis reveals complex metabolic trends across the cell stages. For the box plots, square is mean, box is 1 × standard error of the mean (SEM), and whisker is 1.5 × SEM. Key: *p < 0.05; ns, no significant difference. Scale bars = 250 µm. (Figures adapted with permission from reference30.) Please click here to view a larger version of this figure.
Discussion
Microprobe CE-ESI-MS enables the direct characterization of metabolites in single cells in live, freely developing embryos. At the heart of the approach are two technical subcomponents, namely in situ capillary microsampling and high-sensitivity CE-ESI-MS. Compared to whole-cell dissection, capillary microsampling has the advantage of fast operation (few seconds vs. 5 min/cell by dissection), compatibility with the complex three-dimensional morphology of embryos, and scalability to smaller cells that form at later stages of development. Unlike dissection, microprobe sampling leaves other cells in the embryo intact for subsequent analysis. Intriguingly, microsampled cells can continue to divide, raising the possibility for long-term studies of cell differentiation. The ability to control the tip size of the microprobe also allows microprobe CE-ESI-MS to serve as a versatile tool for studying the role of metabolism in cells and developmental processes using other model organisms, including zebrafish and mouse. To assure credible results, it is critical that the cells of interest are precisely, accurately, and consistently identified and sampled. Experimental parameters of aspiration (e.g., capillary dimensions, pressure, and time) can be tailored to sample a desired portion of a cell. Additionally, it is critical to immobilize the embryos to enhance precision in targeting the cell of interest, which in turn minimizes damage to the embryo. In this protocol, we created wells within agarose-coated Petri dishes to immobilize the embryos during sampling. In our experience, capillaries with beveled tips ensure minimal damage to the cell compared to blunt tips. Further improvements in the throughput of sampling and chemical analysis will enable the characterization of large populations of cells, thus empowering statistical analysis to identify important metabolites underlying cell and developmental processes.
The purpose of CE-ESI-MS is to enable the characterization of the metabolome in trace-level sensitivity. CE offers complementary advantages for single-cell studies over other popular separation techniques, such as nano-flow liquid chromatography or gas chromatography. CE provides exquisite separation efficiency to address the complex metabolome. The physical dimensions of CE are compatible with the volume-limited samples that result from single cells. Additionally, various enrichment techniques exist in CE (e.g., field-amplified sample stacking or dynamic pH junction) to preconcentrate the sample on column, thus boosting detection sensitivity. For successful studies, the CE-ESI-MS instrument should be characterized and validated daily. For example, we require a S/N of at least 3 for 60 amol of acetylcholine standard (6 nL of 50 nM standard injected) and a reproducibility of <5% RSD in separation (migration) time and <25% RSD in quantification based on peak area-under-the-curve in selected-ion electropherograms for the standard. Trace-sensitive detection and dynamic quantification facilitate the measurement of diverse types of metabolites (and peptides) spanning a broad range of concentration in single cells using CE-ESI-MS6,16,25,31. While we routinely detect ~300 different positively charged metabolite signals, CE-MS is also compatible with negative ionization mode48, thus enhancing the depth of the detectable portion of the single-cell metabolome.
Though X. laevis offered several advantages in the creation of this technology for studying development, microprobe CE-ESI-MS may also be adaptable to other types of model organisms. In Xenopus, pigmentation and size differences between cells along with reproducible cell fate maps49 make it possible to identify specific cells with known tissue fates. This, in turn, opens the door to discovery or targeted metabolic experiments of entire cell lineages as cells solidify different developmental programs toward differentiation. To eliminate differences arising due to varying cell-cycles, embryos should be allowed to fully divide to the next stage before sampling, as demonstrated elsewhere50. Microprobe CE-ESI-MS is adaptable to classical embryological manipulations or molecular tools (e.g., gene knock out/down) to test the developmental significance of small molecules with significant dysregulation between select cells or cell types. For example, the combination of single-cell metabolomics with cell fate tracking via the expression of green fluorescent protein led to the discovery of metabolites with previously unknown developmental impacts16.
Single-cell metabolomics studies call for careful attention to analytical details, particularly in the case of quantification; our technology is no exception. Ideally, each step of the sample preparation workflow should be carefully evaluated to minimize potential degradation and/or loss of endogenous metabolites or contamination to the samples. Use of high-purity denaturing solvents (e.g., LC/MS grade), acids or bases, and low temperatures (~4 °C) are recommended to quench the activity of enzymes and alleviate/avoid metabolic changes to samples prior to instrumental analysis6,51. Simple organic solutions based on methanol and acetonitrile allow efficient extraction of small polar metabolites28. Of course, experiments targeted to specific classes of metabolites (e.g., apolar compounds, lipids) benefit from tailoring the composition of the extraction solvent beforehand. For example, we recently demonstrated that the use of multiple types of extraction solvents enhances the coverage of the single-cell metabolome28. Quantification of these metabolites can be facilitated by internal standards, such as isotopically labeled metabolites, which may be spiked into the extracts during metabolite extraction. The internal standards are also beneficial in quality assurance for the extraction and measurement steps, thus facilitating the evaluation of technical and system reproducibility and the recovery of sample analytes. Last, we advocate for biological replicates (different cells aspirated from different embryos) with each cell analyzed in technical replicates (same extract measured multiple times) to aid statistical power and the interpretation of results.
Disclosures
The authors have nothing to disclose.
Acknowledgments
This work was supported by National Institutes of Health Grants GM114854 (to P.N.) and CA211635 (to P.N.), the Arnold and Mabel Beckman Foundation Beckman Young Investigator grant (to P.N.), the DuPont Young Professor award (to P.N.), the American Society for Mass Spectrometry Research Award (to P.N.), and COSMOS Club Foundation fellowships (to R.M.O. and E.P.P.). The opinions and conclusions expressed in this publication are solely those of the authors and do not necessarily represent the official views of the funding sources.
References
- Tang FC, Lao KQ, Surani MA. Development and applications of single-cell transcriptome analysis. Nat. Methods. 2011;8(4):S6–S11. doi: 10.1038/nmeth.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veselovska L, et al. Deep sequencing and de novo assembly of the mouse oocyte transcriptome define the contribution of transcription to the DNA methylation landscape. Genome Biol. 2015;16(209) doi: 10.1186/s13059-015-0769-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tran DA, Bai AY, Singh P, Wu XW, Szabo PE. Characterization of the imprinting signature of mouse embryo fibroblasts by RNA deep sequencing. Nucleic Acids Res. 2014;42(3):1772–1783. doi: 10.1093/nar/gkt1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang DJ, Bodovitz S. Single cell analysis: the new frontier in 'omics'. Trends Biotechnol. 2010;28(6):281–290. doi: 10.1016/j.tibtech.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Svatos A. Single-cell metabolomics comes of age: new developments in mass spectrometry profiling and imaging. Anal. Chem. 2011;83(13):5037–5044. doi: 10.1021/ac2003592. [DOI] [PubMed] [Google Scholar]
- Rubakhin SS, Romanova EV, Nemes P, Sweedler JV. Profiling metabolites and peptides in single cells. Nat. Methods. 2011;8(4):S20–S29. doi: 10.1038/nmeth.1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bodenmiller B, et al. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat. Biotechnol. 2012;30(9):858–889. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubakhin SS, Lanni EJ, Sweedler JV. Progress toward single cell metabolomics. Curr. Opin. Biotechnol. 2013;24(1):95–104. doi: 10.1016/j.copbio.2012.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleparnik K, Foret F. Recent advances in the development of single cell analysis: A review. Anal. Chim. Acta. 2013;800:12–21. doi: 10.1016/j.aca.2013.09.004. [DOI] [PubMed] [Google Scholar]
- Zenobi R. Single-cell metabolomics: Analytical and biological perspectives. Science. 2013;342(6163):1243259. doi: 10.1126/science.1243259. [DOI] [PubMed] [Google Scholar]
- Gholipour Y, Erra-Balsells R, Nonami H. In situ pressure probe sampling and UV-MALDI MS for profiling metabolites in living single cells. Mass Spectrom (Tokyo) 2012;1(1):A0003. doi: 10.5702/massspectrometry.A0003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comi TJ, Do TD, Rubakhin SS, Sweedler JV. Categorizing cells on the basis of their chemical profiles: progress in single-cell mass spectrometry. J. Am. Chem. Soc. 2017;139(11):3920–3929. doi: 10.1021/jacs.6b12822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard-Banek C, Portero EP, Onjiko RM, Nemes P. New-generation mass spectrometry expands the toolbox of cell and developmental biology. Genesis. 2017;55:e23012. doi: 10.1002/dvg.23012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YY, et al. Single-cell analysis by ambient mass spectrometry. Trac-Trends Anal. Chem. 2017;90:14–26. [Google Scholar]
- Lanni EJ, Rubakhin SS, Sweedler JV. Mass spectrometry imaging and profiling of single cells. J. Proteomics. 2012;75(16):5036–5051. doi: 10.1016/j.jprot.2012.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onjiko RM, Moody SA, Nemes P. Single-cell mass spectrometry reveals small molecules that affect cell fates in the 16-cell embryo. Proc. Natl. Acad. Sci. U. S. A. 2015;112(21):6545–6550. doi: 10.1073/pnas.1423682112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizuno H, Tsuyama N, Harada T, Masujima T. Live single-cell video-mass spectrometry for cellular and subcellular molecular detection and cell classification. J. Mass Spectrom. 2008;43(12):1692–1700. doi: 10.1002/jms.1460. [DOI] [PubMed] [Google Scholar]
- Nakashima T, et al. Single-cell metabolite profiling of stalk and glandular cells of intact trichomes with internal electrode capillary pressure probe electrospray ionization mass spectrometry. Anal. Chem. 2016;88(6):3049–3057. doi: 10.1021/acs.analchem.5b03366. [DOI] [PubMed] [Google Scholar]
- Pan N, et al. The single-probe: A miniaturized multifunctional device for single cell mass spectrometry analysis. Anal. Chem. 2014;86(19):9376–9380. doi: 10.1021/ac5029038. [DOI] [PubMed] [Google Scholar]
- Guillaume-Gentil O, et al. Single-cell mass spectrometry of metabolites extracted from live cells by fluidic force microscopy. Anal. Chem. 2017;89(9):5017–5023. doi: 10.1021/acs.analchem.7b00367. [DOI] [PubMed] [Google Scholar]
- Saha-Shah A, Green CM, Abraham DH, Baker LA. Segmented flow sampling with push-pull theta pipettes. Analyst. 2016;141(6):1958–1965. doi: 10.1039/c6an00028b. [DOI] [PubMed] [Google Scholar]
- Hu J, et al. Synchronized polarization induced electrospray: Comprehensively profiling biomolecules in single cells by combining both positive-ion and negative-ion mass spectra. Anal. Chem. 2016;88(14):7245–7251. doi: 10.1021/acs.analchem.6b01490. [DOI] [PubMed] [Google Scholar]
- Zhang LW, Vertes A. Energy charge, redox state, and metabolite turnover in single human hepatocytes revealed by capillary microsampling mass spectrometry. Anal. Chem. 2015;87(20):10397–10405. doi: 10.1021/acs.analchem.5b02502. [DOI] [PubMed] [Google Scholar]
- Zhang LW, et al. In Situ metabolic analysis of single plant cells by capillary microsampling and electrospray ionization mass spectrometry with ion mobility separation. Analyst. 2014;139(20):5079–5085. doi: 10.1039/c4an01018c. [DOI] [PubMed] [Google Scholar]
- Lapainis T, Rubakhin SS, Sweedler JV. Capillary electrophoresis with electrospray ionization mass spectrometric detection for single-cell metabolomics. Anal. Chem. 2009;81(14):5858–5864. doi: 10.1021/ac900936g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemes P, Knolhoff AM, Rubakhin SS, Sweedler JV. Metabolic differentiation of neuronal phenotypes by single-cell capillary electrophoresis electrospray ionization mass spectrometry. Anal. Chem. 2011;83(17):6810–6817. doi: 10.1021/ac2015855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aerts JT, et al. Patch clamp electrophysiology and capillary electrophoresis mass spectrometry metabolomics for single cell characterization. Anal. Chem. 2014;86(6):3203–3208. doi: 10.1021/ac500168d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onjiko RM, Morris SE, Moody SA, Nemes P. Single-cell mass spectrometry with multi-solvent extraction identifies metabolic differences between left and right blastomeres in the 8-cell frog (Xenopus) embryo. Analyst. 2016;141(12):3648–3656. doi: 10.1039/c6an00200e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Onjiko RM, Plotnick DO, Moody SA, Nemes P. Metabolic comparison of dorsal versus ventral cells directly in the live 8-cell frog embryo by microprobe single-cell CE-ESI-MS. Anal. Methods. 2017. [DOI] [PMC free article] [PubMed]
- Onjiko RM, Portero EP, Moody SA, Nemes P. In situ microprobe single-cell capillary electrophoresis mass spectrometry: Metabolic reorganization in single differentiating cells in the live vertebrate (Xenopus laevis) embryo. Anal. Chem. 2017;89:7069–7076. doi: 10.1021/acs.analchem.7b00880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemes P, Rubakhin SS, Aerts JT, Sweedler JV. Qualitative and quantitative metabolomic investigation of single neurons by capillary electrophoresis electrospray ionization mass spectrometry. Nat. Protoc. 2013;8(4):783–799. doi: 10.1038/nprot.2013.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knolhoff AM, Nemes P, Rubakhin SS, Sweedler JV. Wevers R, Lutz N, Sweedler JV, editors. Methodologies for Metabolomics. 2013. pp. 119–139.
- Sive HL, Grainger RM, Harland RM. Early development of Xenopus laevis: a laboratory manual. Cold Spring Harbor Laboratory Press; 2000. [Google Scholar]
- Moody SA. Cell lineage analysis in Xenopus embryos. Methods Mol Biol. 2000;135:331–347. doi: 10.1385/1-59259-685-1:331. [DOI] [PubMed] [Google Scholar]
- Bowes JB, et al. Xenbase: a Xenopus biology and genomics resource. Nucleic Acids Res. 2008;36:D761–D767. doi: 10.1093/nar/gkm826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpinka JB, et al. Xenbase, the Xenopus model organism database; new virtualized system, data types and genomes. Nucleic Acids Res. 2015;43(D1):D756–D763. doi: 10.1093/nar/gku956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James-Zorn C, et al. Xenbase: expansion and updates of the Xenopus model organism database. Nucleic Acids Res. 2013;41(D1):D865–D870. doi: 10.1093/nar/gks1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moody SA. Fates of the blastomeres of the 16-cell stage Xenopus embryo. Dev. Biol. 1987;119(2):560–578. doi: 10.1016/0012-1606(87)90059-5. [DOI] [PubMed] [Google Scholar]
- Moody SA. Fates of the blastomeres of the 32-cell-stage Xenopus embryo. Dev. Biol. 1987;122(2):300–319. doi: 10.1016/0012-1606(87)90296-x. [DOI] [PubMed] [Google Scholar]
- Dale L, Slack JMW. Fate map for the 32-cell stage of Xenopus laevis. Development. 1987;99(4):527–551. doi: 10.1242/dev.99.4.527. [DOI] [PubMed] [Google Scholar]
- Klein SL. The first cleavage furrow demarcates the dorsal-ventral axis in Xenopus embryos. Dev. Biol. 1987;120(1):299–304. doi: 10.1016/0012-1606(87)90127-8. [DOI] [PubMed] [Google Scholar]
- Rollman CM, Moini M. Ultrafast capillary electrophoresis/mass spectrometry of controlled substances with optical isomer separation in about a minute. Rapid Commun. Mass Spectrom. 2016;30(18):2070–2076. doi: 10.1002/rcm.7691. [DOI] [PubMed] [Google Scholar]
- Moini M, Martinez B. Ultrafast capillary electrophoresis/mass spectrometry with adjustable porous tip for a rapid analysis of protein digest in about a minute. Rapid Commun. Mass Spectrom. 2014;28(3):305–310. doi: 10.1002/rcm.6786. [DOI] [PubMed] [Google Scholar]
- Huhn C, Ramautar R, Wuhrer M, Somsen GW. Relevance and use of capillary coatings in capillary electrophoresis-mass spectrometry. Anal. Bioanal. Chem. 2010;396(1):297–314. doi: 10.1007/s00216-009-3193-y. [DOI] [PubMed] [Google Scholar]
- Nemes P, Marginean I, Vertes A. Spraying mode effect on droplet formation and ion chemistry in electrosprays. Anal. Chem. 2007;79(8):3105–3116. doi: 10.1021/ac062382i. [DOI] [PubMed] [Google Scholar]
- Zhu ZJ, et al. Liquid chromatography quadrupole time-of-flight mass spectrometry characterization of metabolites guided by the METLIN database. Nat. Protoc. 2013;8(3):451–460. doi: 10.1038/nprot.2013.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wishart DS, et al. HMDB 3.0 The Human Metabolome Database in 2013. Nucleic Acids Res. 2013;41(D1):D801–D807. doi: 10.1093/nar/gks1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu JX, Aerts JT, Rubakhin SS, Zhang XX, Sweedler JV. Analysis of endogenous nucleotides by single cell capillary electrophoresis-mass spectrometry. Analyst. 2014;139(22):5835–5842. doi: 10.1039/c4an01133c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubrecht L, Nieuwkoop PD, Faber J. Normal table of Xenopus laevis (Daudin). A systematical and chronological survey of the development from the fertilized egg till the end of metamorphosis. North-Holland Pub. Co; 1967. [Google Scholar]
- Grant PA, Herold MB, Moody SA. Blastomere explants to test for cell fate commitment during embryonic development. J. Vis. Exp. 2013. [DOI] [PMC free article] [PubMed]
- Sellick CA, Hansen R, Stephens GM, Goodacre R, Dickson AJ. Metabolite extraction from suspension-cultured mammalian cells for global metabolite profiling. Nat. Protoc. 2011;6(8):1241–1249. doi: 10.1038/nprot.2011.366. [DOI] [PubMed] [Google Scholar]