

Abstract
The use of personalized medicine to treat rare monogenic diseases like lysosomal storage disorders (LSDs) is challenged by complex clinical trial designs, high costs, and low patient numbers. Hundreds of mutant alleles are implicated in most of the LSDs. The diseases are typically classified into 2 to 3 different clinical types according to severity. Moreover, molecular characterization of the genotype can help predict clinical outcomes and inform patient care. Therefore, we developed a simple cell culture assay based on HEK293H cells heterologously over-expressing the mutations identified in Fabry and Pompe disease. A similar assay has recently been introduced as a preclinical test to identify amenable mutations for Pharmacological Chaperone Therapy (PCT) in Fabry disease. This manuscript describes an amended cell culture assay which enables rapid phenotypic assessment of allelic variants in Fabry and Pompe disease to identify eligible patients for PCT and may aid in the development of novel pharmacochaperones.
Keywords: Medicine, Issue 130, Lysosomal storage disorder, phenotype, glycosidase, small molecule drugs, preclinical test, personalised therapy
Introduction
There are over a dozen lysosomal storage disorders (LSDs) related to glycosidase dysfunction as a result of primary gene mutations. In Fabry (OMIM #301500) and Pompe (OMIM #232300) disease, more than 500 and 200 missense mutations1,2,3 have been reported, respectively, which corresponds to about 60% of the total mutation count. Numerous new gene variants are still being identified, many of which have unknown significance. Extensive biochemical studies revealed that certain genotypes do not lead to a complete loss-of-function of the GLA gene (OMIM *300644) in Fabry disease, but cause the corresponding enzyme to fail to reach a thermodynamically favored folding state4. This results in ER retention and premature degradation of the otherwise functional enzyme. Similar conclusions have been drawn in other LSDs including Pompe disease5. Moreover, molecular characterization of enzyme variants can facilitate clinical interpretation of the mutations at the time of diagnosis6, suggesting that LSD progression is an individual process based on the nature of the mutation. Therefore, the conventional classification into typically 2 to 3 different clinical types should be reassessed in order to streamline clinical counselling and therapeutic decisions.
Enzyme Replacement Therapy (ERT) is available for both diseases. ERT, however, has limited efficacy in affected tissues/organs such as the brain and skeletal muscle. Furthermore, ERT can elicit an immunogenic response that jeopardizes its therapeutic benefits. Pharmacological Chaperones (PCs) are an attractive treatment alternative for patients with so-called responsive mutations. PCs serve as a molecular scaffold for correct protein folding and stabilization which in turn prevents endoplasmic reticulum (ER) retention and ER-associated degradation of the enzyme. Moreover, PCs can be administered orally and are potentially able to cross the blood brain barrier. Therefore, PCT might be a more viable option for treating patients with certain genotypes. For an extensive review on PC application in LSDs, refer to the excellent review by Parenti7.
The discovery of hundreds of disease causing mutant alleles challenges pre-clinical drug testing and necessitates a simple, fast, and highly standardized assessment of amenable patients for a personalized medicine approach. In order to assess the detrimental effects of LSD gene mutations and to test candidate mutations to predict amenable patients for PCT, a highly standardized over-expression system in HEK293H cells that allows for fast and reliable enzyme activity measurement was developed. Similar over-expression systems have been previously described for Fabry and Pompe disease using either COS-78,9,10,11, HeLa cells12, or HEK29313,14,15,16 cells for the glycosidase gene.
A very similar method has even been patented as a "Method to predict response to Pharmacological Chaperone treatment of diseases"17 indicating the relevance of a cell culture system capable of being integrated into clinical practice.
Protocol
1. Preparation of Mutant pcDNA3.1/GLA and pcDNA3.1/GAA Constructs
NOTE: The cloning strategies for the GLA and GAA coding sequences (cds) have been reported earlier15,18.
- Site-directed Mutagenesis Using Site-Directed Mutagenesis
- Use the reference sequences NM_000169.2 and NM_000152.4 as templates for the mutagenesis of GLA and GAA genes, respectively. Have a set of high purity salt free primers (25-37-mers) synthesized by a commercial provider, with sense and antisense primers carrying one of the respective sequence modifications central to their length to individually introduce the mutation. Use the free primer design tool to support the primer design19.
- For the reaction mixture, use the standard conditions for the reaction solution and PCR conditions provided by the manufacturer.
- Mix 5 µL of 10x reaction buffer, 10 ng of double-stranded template plasmid DNA (pcDNA3.1/GLA or pcDNA3.1/GAA), 125 ng of each primer, 1 µL of the provided dNTP mixture, 3 µL of DMSO reagent in an appropriate volume of deionized water (final reaction volume: 50 µL). Finally, add 2.5 U of DNA polymerase and mix by pipetting up and down. As a negative control, carry along a no-primer sample.
- Start the PCR using the following program: step 1: 95 °C for 1 min, step 2: 95 °C for 50 s, step 3: 60 °C for 50 s, step 4: 68 °C for 8 min, repeat step 2-4 18 times, and step 5: 68 °C for 10 min.
- Following PCR, add 1 µL of the DpnI restriction enzyme (10 U/µL) and further incubate the reaction vial at 37 °C for 1 h.
- Transformation and Screening for the Desired Clone
- Transform an aliquot of ultracompetent cells in accordance with the manufacturer's recommendations. Use SOC medium (tryptone 2% (w/v), yeast extract 0.5% (w/v), NaCl 10 mM, KCl 2.5 mM, sterilize at 121 °C, and then add sterile-filtered solutions of MgCl2 and glucose up to final concentrations of 10 and 20 mM, respectively) instead of the manufacturer's medium. After the procedure, plate 250 µL of the sample mutagenesis on an LB plate containing 100 µg/mL ampicillin and incubate at 37 °C for 18 h.
- Assure that the number of transformants is >10 and the reaction yields at least three times as many colonies as the no-primer control reaction, e.g., using a luminous plate to facilitate colony counting. Then pick 3 colonies and prepare 3 mL overnight cultures in LB Broth medium.
- The next day, carry out plasmid preparation with a standard kit and analyze whole sequence using T7 (5ʹ-TAA TAC GAC TCA CTA TAG GG-3ʹ) and BGHr (5ʹ-TAG AAG GCA CAG TCG AGG-3ʹ) primers via standard Sanger sequencing.
- Use a suitable molecular biology tool to analyze the sequence. When the desired mutation is detected and no further sequence abnormality compared to NM_000169.2 (α-galactosidase A) or NM_000152.4 (acid α-glucosidase) is seen, select the clone for transfection-grade plasmid purification.
- Determine the purity of the DNA by measuring the absorbance in a spectrophotometer. NOTE: Allow only preparations that yield a plasmid purity with a 260/280 absorbance ratio of >1.8 for cell culture experiments.
2. Cultivation of HEK293H Cells
Maintain HEK293H cells in high glucose (4.5 g/L) Dulbecco´s Modified Eagle Medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Keep the cells in a water-jacket incubator at 37 °C under a 5% CO2 atmosphere.
Cultivate the cells to a density of 80 - 90%.
Aspirate the medium and wash once using phosphate buffered saline (PBS) without Ca2+ and Mg2+.
Passage by adding 0.05% Trypsin-EDTA and incubate for 5 min at 37 °C and 5% CO2.
Split the cells 1:15 in fresh medium and seed into a new T75 flask to maintain the permanent culture. Do not use cells with more than 25 passages.
3. pcDNA3.1/GLA and pcDNA3.1/GAA Plasmid Transfection and Treatment of HEK293H
24 h prior to the transfection, wash the HEK293H cells in a T75 cell culture flask once with PBS with Ca2+, Mg2+. Harvest the cells with 0.05% Trypsin-EDTA as stated above and seed 1.5 x 105 cells in the cavities of a 24 well culture plate using 500 µL DMEM medium supplemented with 10% FBS without antibiotics.
Carry out a transfection protocol according to the manufacturer's manual. Typically, use a mixture of 1 µg of plasmid DNA and 2.5 µL of transfection reagent in 100 µL of serum-free DMEM. Incubate for 20 min at room temperature and add to the cells in a drop-wise manner thereafter.
Remove the medium containing the transfection reagent after a period of 4 h at 37 °C/5% CO2 and add 500 µL of fresh DMEM with 10% FBS/ 1% penicillin/streptomycin. NOTE: During this step, 1-Deoxygalactonojirimycin Hydrochloride (DGJ) or 1-Deoxynojirimycin Hydrochloride (DNJ) might be added to the culture medium where intended (use an aqueous stock solution of 10 mM in order to obtain a final concentration of 20 µM DGJ and DNJ). Fresh DGJ or DNJ was added 42 h after plasmid transfection.
4. Cell Harvest and α-galactosidase A or Acid α-glucosidase Activity Measurement
- Cell harvest
- On the day of the harvest, remove the cells from the incubator and aspirate the medium. Carefully wash the cells 2 times with PBS with Ca2+ and Mg2+. NOTE: This step is critical because DGJ and DNJ are potent inhibitors of α-galactosidase and α-glucosidase, respectively, and any leftover would invalidate the test.
- Add 200 µL of deionized water directly on top of the cells. Rinse the cells from the plate and transfer them to a 1.5 mL reaction tube.
- Homogenization by freezing and thawing
- Put the samples in an appropriate foam rack and vortex for 5 s to make the lysis more efficient. Put the samples alternating in liquid nitrogen for 10 s and in a room temperature water bath until the thawing was complete (5 min).
- Repeat this procedure 5 times and then spin the samples for 5 min at 10,000 x g. Retain the supernatant and pipette in a new reaction tube.
- Protein concentration determination using bicinchoninic acid (BCA) assay
- Prepare a fresh tube for each sample containing 40 µL of deionized H2O and add 10 µL of sample. Mix solution by vortexing briefly and transfer 10 µL into a cavity of a 96 well plate (each sample in triplicate). Dilute a 2 mg/mL bovine serum albumin (BSA) stock solution in deionized H2O as follows to obtain a standard curve: 50 µL H2O/50 µL BSA; 60 µL H2O/40 µL BSA; 70 µL H2O/30 µL BSA; 80 µL H2O/20 µL BSA; 90 µL H2O/10 µL BSA; 100 µL H2O.
- Start the reaction by adding 200 µL of BCA reagent (Reagent A and reagent B mixed at a 50:1 ratio) and incubate for 1 h in the dark at 37 °C under slight agitation on an orbital shaker (300 rpm). Measure the absorbance at 560 nm in a plate reader. NOTE: The samples typically contain between 1 and 1.5 µg protein per µL.
- Enzyme activity measurement with artificial 4-Methylumbelliferyl substrates (4-MUG)
- Dilute the calculated amount of each sample and pipet into fresh 1.5 mL reaction tubes to obtain 0.05 (for α-galactosidase A) or 0.5 (for acid α-glucosidase) µg protein/µL solutions. Vortex the samples for 5 s again and pipet 10 µL of this dilution into a 96 well plate (each sample in duplicate).
- Start the reaction by adding 20 µL of the respective substrate solution: For α-galactosidase A: 2 mM 4-Methylumbelliferyl-α-D-galactopyranoside (4-MU-gal) in 0.06 M phosphate citrate buffer, pH 4.7. For acid α-glucosidase: 2 mM 4-methylumbelliferyl α-D-glucopyranoside (4-MU-glu) in 0.025 M sodium acetate, pH 4.0.
- Incubate the enzyme reactions for 1 h in the dark at 37 °C under slight agitation on an orbital shaker (300 rpm). Terminate the reaction by the addition of 200 µL of 1.0 M, pH 10.5 adjusted glycine-NaOH buffer.
- Prepare a standard curve of 4-methylumbelliferone (4-MU) from a 0.01 mg/mL stock as follows: 100 µL H2O/ no 4-MU; 80 µL H2O/ 20 µL 4-MU; 60 µL H2O/ 40 µL 4-MU; 40 µL H2O/ 60 µL 4-MU; 20 µL H2O/ 80 µL 4-MU; no H2O/ 100 µL 4-MU, pipet 10 µL of each dilution into the 96 well plate (in duplicates) and add 200 µL of the 1.0 M glycine-NaOH buffer to each well in order to adjust the volume and pH.
- Measure the enzyme activity in a fluorescence reader equipped with the appropriate filter set and analyze the data using the appropriate software for the fluorescence reader device. NOTE: Both 4-MUG substrates are reduced to 4-MU during exposure to α-galactosidase A or acid α-glucosidase. Released 4-MU is a fluorochrome, which can be measured at 360 and 465 nm as the excitation and emission wavelengths, respectively, using a microplate fluorescence reader.
Representative Results
The mutagenesis procedure To assess the efficiency of GLA gene mutagenesis, the mutations were classified into one of the following categories. This approach to generate mutations revealed that about 66.5% of the GLA mutations were obtained in the first attempt. A further 25% could be obtained after a slightly modified second PCR.
Category 1: The mutagenesis PCR was effective at first attempt.
Category 2: First mutagenesis PCR failed (no colonies on the plate, no inserted mutation); repetition using the same primer set by increasing the annealing temperature up to 68 °C was effective.
Category 3: More effort had to be undertaken to yield the desired clone (e.g., typically one or more new sets of primers were designed).
α-galactosidase A and acid α-glucosidase enzyme activity measurement Enzyme activity of the different mutant enzymes was recorded after previous incubation of the transiently transfected cells in the presence or absence of a PC. Table 1 refers to the results for 3 α-galactosidase A and 3 acid α-glucosidase mutations. Data are displayed as (1) absolute values for the substrate turnover (nmol 4-MU* mg protein-1* h-1) and as (2) relative values normalized to the wild type enzyme. Both expressions are useful, since total substrate turnover illustrates the efficiency of the reaction and the sensitivity of the system, while the normalized values can give important hints for the likelihood of the malignancy of the mutation on the one hand and the efficiency of the applied PC treatment on the other hand. For the experimental phase, the work flow depicted in Figure 1 was set up, which scheduled a 60 h incubation period with the compound (due to technical reasons, e.g. fast HEK293H cell growth under the introduced conditions). Even though it has been stated that 10 µM was the approximate maximum achievable plasma concentration for DGJ14, the current protocol uses 20 µM as a reasonable concentration for the purpose of a screening for PC responsiveness as supported by numerous earlier works4,20,21,22,23. Moreover, it has been postulated that higher plasma levels can be reached24.
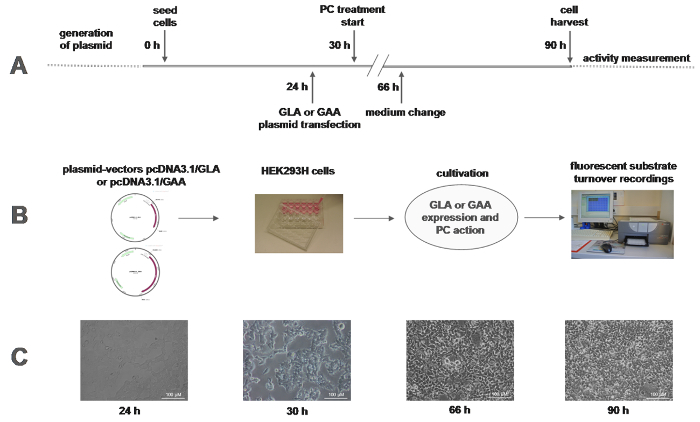
Figure 1:Work flow of the in vitro enzyme activity measurement. (A) The timeline of the experiment shows a 90 h cell culture effort with relatively little hands-on-time required at the indicated time points. (B) Representative plasmid vectors pcDNA3.1 containing the wild type cds of GLA and GAA are shown. GLA and GAA wild type plasmids and plasmids including the respective inserted mutation of interest were used to transiently transfect HEK293H cells plated in 24 well format. After a period to allow the cells to synthesize the respective gene products and allow for enzyme processing, lysosomal transport, and PC action, the cells were lysed and measured in a fluorescence plate reader and analyzed using the respective software. C: Cell morphology and growth status of the HEK293H cells are shown throughout the time course of the experiment. Scale bars = 100 µm Please click here to view a larger version of this figure.
| in vitro enzyme activity | |||||
| GLA | native | 20 µM DGJ | |||
| mutation (AA) | nmol 4-MUG-gal /mg/h (mean) | % mean (±SD, N) | nmol 4-MUG-gal /mg/h (mean) | % mean (±SD, N) | significance |
| p.A143T | 2384.7 | 29.2 (±2.6, 5) | 4223.0 | 52.0 (±4.4, 5) | *** |
| p.A156V | 379.2 | 4.3 (±.8, 5) | 1437.5 | 16.3 (±1.3, 5) | **** |
| p.R301Q | 777.4 | 8.8 (±1.1, 5) | 3002.6 | 44.2 (±3.6, 5) | **** |
| GAA | native | 20 µM DNJ | |||
| mutation (AA) | nmol 4-MUG-glu /mg/h (mean) | % mean (±SD, N) | nmol 4-MUG-glu /mg/h (mean) | % mean (±SD, N) | significance |
| p.Y455F | 41.1 | 5.4 (±.4, 5) | 246.6 | 31.4 (±2.8, 5) | *** |
| p.P545L | 65.7 | 6.6 (±.4, 5) | 166.5 | 16.7 (±1.5, 5) | ** |
| p.L552P | 0.0 | 0.0 (±.2, 5) | 104.3 | 10.4 (±1.4, 5) | *** |
Table 1: Enzyme activity of α-galactosidase A and acid α-glucosidase. The table shows representative results for 3 α-galactosidase A and 3 acid α-glucosidase mutants with and without the PCs DGJ or DNJ. Absolute enzyme activity data was corrected for endogenous enzyme activity of the HEK293H cells. To evaluate endogenous enzyme activity, the cells were transfected with a pcDNA3.1 empty vector. The values were 97.5 nmol 4-MU* mg protein-1* h-1 for α-galactosidase A and 41.6 nmol 4-MU* mg protein-1* h-1 for acid α-glucosidase, respectively; enzyme activity values were normalized to wild type enzyme from corresponding experiments, which explains the deviations of relative values between the different mutants. After 5 independent cell culture experiments (N = 5), each carried out in technical duplicates, the standard deviation was not allowed to be >15% of the mean. Ratio T-Tests were used to calculate the difference between untreated and PC-treated enzyme. *p <0.05, **p <0.01, ***p <0.005, ****p <0.001
Discussion
The protocol described herein delivers robust results for enzyme damage assessment in hereditary lysosomal diseases of metabolism. This manuscript is an amendment to the protocol published earlier15. The most crucial modifications involve stringency (i.e., in the process of mutant vector construct preparation), standardization of the cell culture protocol (i.e., HEK293H cell maintenance and transfection conditions), and the high number of experimental repetitions (at least 5), which highly contributed to the reproducibility of the results. Altogether, both target genes have been easily accessible for mutagenesis and a multiplicity of mutations can be assembled in parallel. A relatively high signal/noise ratio was achieved considering that endogenous enzyme activity within the HEK293H cells was a negligible 1/50 (α-galactosidase A) and 1/20 (acid α-glucosidase) of the over-expressed wild type gene. Thus, there is an excellent resolution between wild type and mutant enzyme. It should be noted that a different amount of total protein was applied for the activity assay of both enzymes (0.5 and 5 µg, respectively), because acid α-glucosidase activity was an order of magnitude lower than that of α-galactosidase A.
As stated above, assay parameters are often controversial, since many laboratories have developed their own assays to investigate lysosomal glycosidases. The systems can differ in cell type, plasmid-vector design, cultivation conditions, and period of drug exposure just to name a few of the numerous parameters. A recently published meta-analysis for Fabry disease demonstrated that enzyme activity recordings (with and without PC) of transfected cell models (e.g., HEK293, COS-7) was largely in accordance with the results obtained from patient-derived cells (mostly lymphocytes or fibroblasts) with regard to the question, whether a mutation is responsive to the PC24. Earlier reports directly comparing patient-derived cells and transfection models demonstrated that over-expression systems reflect the situation in patient cells without compromising the conclusion13,14. It can therefore be concluded that, regardless of the particular protocol, the results obtained by different authors was not changed by the different cellular systems, even though responder definitions can be divergent. The current definition for a responding mutation is 20% relative enzyme activity increase and 5% absolute enzyme activity increase compared to the wild type enzyme after incubation with 20 µM DGJ for 60 h. A comprehensive database, FabryCEP, permits the comparison of results obtained by different experimental approaches for hundreds of α-galactosidase mutants25.
Whether these criteria are sufficient to predict clinical benefits of DGJ and DNJ remains unclear, since the level of activity necessary to prevent symptomatic disease is controversial. A former study suggested that 10 to 15% of residual activity might be sufficient for the system to work properly26. However, in a recently published report from a phase 2 clinical study for DGJ, a 1% activity increase was deemed potentially beneficial for the patient, when the baseline activity was less than 1% of normal27. Recent clinical results suggest that patients predicted to respond to PCs by the responder definition of ≥20% relative increase and ≥3% absolute increase of wild type α-galactosidase A activity in HEK293H cells after incubation with 10 µM DGJ actually derived clinical benefit with disease stabilization or even improvement by renal and cardiac parameters28. Whereas DGJ has already been approved by the FDA and the European Commission as a monotherapy for Fabry disease, the course of PCs such as DNJ and the derivative N-Butyl-DNJ in Pompe disease appears to be different. A monotherapeutical approach with DNJ has been terminated during a Phase II clinical trial due to severe adverse events in some of the patients29. However, PC treatment in combination with approved ERT showed significantly improved results with regards to disease substrate (glycogen) reduction compared to the ERT monotherapy30.
We suggest that the presented approach can be extended to other LSDs and other PCs such as Gaucher, Krabbe, Tay-Sachs/Sandhoff, and GM1 gangliosidosis, but this might not work in specific cases, where for example the signal/noise ratio of the system is insufficient. Care should be taken to select a suitable cell disruption method to prepare a cell lysate for enzyme activity determination, because the addition of detergents into the lysis buffer might be indicative of membrane-bound enzymes such as glucocerebrosidase in Gaucher disease while similar amounts of detergent may harm other enzymes. For α-galactosidase A, a sonication-based cell disruption method should be avoided.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
The authors would like to acknowledge Mandy Loebert and Tina Czajka for excellent technical support. We thank Flora Luo (Harvard Medical School, Boston, MA, USA) for language editing help.
References
- Cardiff University. 2017. http://www.hgmd.cf.ac.uk/ac/index.
- Meiji Pharmaceutical University. 2017. http://fabry-database.org.
- Erasmus Medical Center. Mutations In Human Acid Alpha-Glucosidase. 2017. http://cluster15.erasmusmc.nl/klgn/pompe/mutations.html?lang=en.
- Ishii S, et al. Mutant alpha-galactosidase A enzymes identified in Fabry disease patients with residual enzyme activity: biochemical characterization and restoration of normal intracellular processing by 1-deoxygalactonojirimycin. Biochem J. 2007;406:285–295. doi: 10.1042/BJ20070479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tajima Y. Structural and biochemical studies on Pompe disease and a "pseudodeficiency of acid alpha-glucosidase". J Hum Genet. 2007;52:898–906. doi: 10.1007/s10038-007-0191-9. [DOI] [PubMed] [Google Scholar]
- Lukas J. Functional and Clinical Consequences of Novel α-Galactosidase A Mutations in Fabry Disease. Hum Mutat. 2016;37:43–51. doi: 10.1002/humu.22910. [DOI] [PubMed] [Google Scholar]
- Parenti G. Treating lysosomal storage diseases with pharmacological chaperones: from concept to clinics. EMBO Mol Med. 2009;1:268–279. doi: 10.1002/emmm.200900036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yasuda M. Fabry disease: characterization of alpha-galactosidase A double mutations and the D313Y plasma enzyme pseudodeficiency allele. Hum Mutat. 2003;22:486–492. doi: 10.1002/humu.10275. [DOI] [PubMed] [Google Scholar]
- Shimotori M, Maruyama H, Nakamura G, Suyama T, Sakamoto F, Itoh M, Miyabayashi S, Ohnishi T. Novel mutations of the GLA gene in Japanese patients with Fabry disease and their functional characterization by active site specific chaperone. Hum Mutat. 2008;29:331. doi: 10.1002/humu.9520. [DOI] [PubMed] [Google Scholar]
- Andreotti G, et al. Therapy of Fabry disease with pharmacological chaperones: from in silico predictions to in vitro tests. Orphanet J Rare Dis. 2011;6:66. doi: 10.1186/1750-1172-6-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna R. The pharmacological chaperone AT2220 increases the specific activity and lysosomal delivery of mutant acid alpha-glucosidase, and promotes glycogen reduction in a transgenic mouse model of Pompe disease. PLoS One. 2014;9:e102092. doi: 10.1371/journal.pone.0102092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siekierska A. α-Galactosidase aggregation is a determinant of pharmacological chaperone efficacy on Fabry disease mutants. J Biol Chem. 2012;287:28386–28397. doi: 10.1074/jbc.M112.351056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti G. Pharmacological enhancement of mutated alpha-glucosidase activity in fibroblasts from patients with Pompe disease. Mol Ther. 2007;15:508–514. doi: 10.1038/sj.mt.6300074. [DOI] [PubMed] [Google Scholar]
- Wu X. A pharmacogenetic approach to identify mutant forms of α-galactosidase A that respond to a pharmacological chaperone for Fabry disease. Hum Mutat. 2011;32:965–977. doi: 10.1002/humu.21530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukas J. Functional characterisation of alpha-galactosidase a mutations as a basis for a new classification system in fabry disease. PLoS Genet. 2013;9:e1003632. doi: 10.1371/journal.pgen.1003632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andreotti G, Citro V, Correra A, Cubellis MV. A thermodynamic assay to test pharmacological chaperones for Fabry disease. Biochim Biophys Acta. 2014;1840:1214–1224. doi: 10.1016/j.bbagen.2013.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2017. https://www.google.ch/patents/US9095584.
- Lukas J, et al. Enzyme enhancers for the treatment of Fabry and Pompe disease. Mol Ther. 2015;23:456–4564. doi: 10.1038/mt.2014.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2017. http://www.genomics.agilent.com/primerDesignProgram.jsp.
- Yam GH, Bosshard N, Zuber C, Steinmann B, Roth J. Pharmacological chaperone corrects lysosomal storage in Fabry disease caused by trafficking-incompetent variants. Am J Physiol Cell Physiol. 2006;290:C1076–C1082. doi: 10.1152/ajpcell.00426.2005. [DOI] [PubMed] [Google Scholar]
- Shin SH, et al. Screening for pharmacological chaperones in Fabry disease. Biochem Biophys Res Commun. 2007;359:168–173. doi: 10.1016/j.bbrc.2007.05.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin SH, et al. Prediction of response of mutated alpha-galactosidase A to a pharmacological chaperone. Pharmacogenet Genomics. 2008;18:773–7780. doi: 10.1097/FPC.0b013e32830500f4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filoni C, et al. Functional studies of new GLA gene mutations leading to conformational Fabry disease. Biochim Biophys Acta. 2010;1802:247–252. doi: 10.1016/j.bbadis.2009.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Citro V. The Large Phenotypic Spectrum of Fabry Disease Requires Graduated Diagnosis and Personalized Therapy: A Meta-Analysis Can Help to Differentiate Missense Mutations. Int J Mol Sci. 2016;17 doi: 10.3390/ijms17122010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cammisa M, Correra A, Andreotti G, Cubellis MV. Fabry_CEP: a tool to identify Fabry mutations responsive to pharmacological chaperones. Orphanet J Rare Dis. 2013;8:111. doi: 10.1186/1750-1172-8-111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinekugel P, Michel S, Conzelmann E, Sandhoff K. Quantitative correlation between the residual activity of beta-hexosaminidase A and arylsulfatase A and the severity of the resulting lysosomal storage disease. Hum Genet. 1992;88:513–523. doi: 10.1007/BF00219337. [DOI] [PubMed] [Google Scholar]
- Germain DP. Safety and pharmacodynamic effects of a pharmacological chaperone on α-galactosidase A activity and globotriaosylceramide clearance in Fabry disease: report from two phase 2 clinical studies. Orphanet J Rare Dis. 2012;7:91. doi: 10.1186/1750-1172-7-91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes DA, et al. Oral pharmacological chaperone migalastat compared with enzyme replacement therapy in Fabry disease: 18-month results from the randomised phase III ATTRACT study. J Med Genet. 2017;54:288–296. doi: 10.1136/jmedgenet-2016-104178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti G, Andria G, Valenzano KJ. Pharmacological Chaperone Therapy: Preclinical Development, Clinical Translation, and Prospects for the Treatment of Lysosomal Storage Disorders. Mol Ther. 2015;23:1138–1148. doi: 10.1038/mt.2015.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parenti G, et al. A Chaperone Enhances Blood α-Glucosidase Activity in Pompe Disease Patients Treated With Enzyme Replacement Therapy. Mol Ther. 2014;22:2004–2012. doi: 10.1038/mt.2014.138. [DOI] [PMC free article] [PubMed] [Google Scholar]