

Abstract
Translation of mRNA into proteins is a complex process involving several layers of regulation. It is often assumed that changes in mRNA transcription reflect changes in protein synthesis, but many exceptions have been observed. Recently, a technique called ribosome profiling (or Ribo-Seq) has emerged as a powerful method that allows identification, with high accuracy, which regions of mRNA are translated into proteins and quantification of translation at the genome-wide level. Here, we present a generalized protocol for genome-wide quantification of translation using Ribo-Seq in budding yeast. In addition, combining Ribo-Seq data with mRNA abundance measurements allows us to simultaneously quantify translation efficiency of thousands of mRNA transcripts in the same sample and compare changes in these parameters in response to experimental manipulations or in different physiological states. We describe a detailed protocol for generation of ribosome footprints using nuclease digestion, isolation of intact ribosome-footprint complexes via sucrose gradient fractionation, and preparation of DNA libraries for deep sequencing along with appropriate quality controls necessary to ensure accurate analysis of in vivo translation.
Keywords: Biochemistry, Issue 130, RNA, translation, ribosome profiling, next-generation sequencing, RNA-sequencing, Saccharomyces cerevisiae
Introduction
mRNA translation is one of the fundamental processes in the cell, which plays an important role in the regulation of protein expression. Therefore, mRNA translation is tightly controlled in response to different internal and external physiological stimuli 1,2. However, the mechanisms of translational regulation remain understudied. Here, we describe the protocol for the genome-wide quantification of translation in budding yeast by ribosome profiling. The overall goal of the ribosome profiling technique is to study and quantify the translation of specific mRNAs under different cellular conditions. This technique uses next-generation sequencing to quantitatively analyze ribosome occupancy throughout the genome and allows monitoring the rate of protein synthesis in vivo at the single codon resolution 3,4. Currently, this method provides the most advanced means of measuring the levels of protein translation, and has proven to be a useful discovery tool providing information that cannot be revealed by other currently available techniques, e.g. microarrays or translation state array analysis (TSAA) 5. As ribosome profiling reports on the combined changes in transcript levels and translational output, it also provides much greater sensitivity compared to other methods.
This approach is based on deep sequencing of ribosome-protected mRNA fragments 3. During protein translation, ribosomes protect ~ 28 nt portions of the mRNA (called footprints) 6. By determining the sequence of the ribosome-protected fragments, Ribo-Seq can map the position of ribosomes on the translated mRNA and identify which regions of mRNA are likely to be actively translated into protein 3,7. In addition, we can quantitatively measure the translation of mRNA by counting the number of footprints that align to a given mRNA transcript.
In order to isolate the ribosome-protected fragments, cell lysates are initially treated with a translation inhibitor to stall the ribosomes followed by ribonuclease digestion. Whereas free mRNA and portions of translated mRNAs not protected by ribosomes are degraded by ribonuclease, the ribosome-protected mRNA fragments can be recovered by purifying intact ribosome-footprint complexes. These mRNA footprints are then converted into cDNA library and analyzed by deep sequencing (Figure 1). In parallel to ribosome profiling, intact mRNA is extracted from the same sample and sequenced. By comparing the level of translation identified by Ribo-Seq with mRNA abundance measurements, we can identify genes that are specifically up- or down-regulated at the level of translation and calculate translation efficiency of mRNA at the genome-wide level. While the protocol described in this article is specific for yeast, it should be also useful for researchers who will try to establish the Ribo-Seq protocol in other systems.
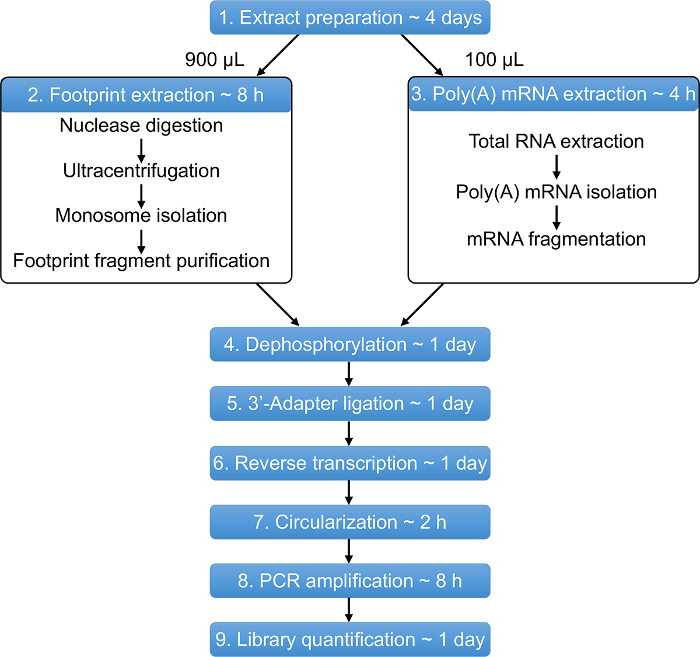
Protocol
1. Extract Preparation
Streak yeast strains from frozen stocks for single colonies on YPD plates (1% yeast extract, 2% peptone, 2% glucose, and 2% agar). Incubate the plates at 30 °C for 2 days.
Inoculate yeast from a YPD plate (use a single colony) into 15 mL of YPD medium (1% yeast extract, 2% peptone, 2% glucose) in a 50 mL conical centrifuge tube and grow overnight with shaking (200-250 rpm) at 30 °C.
Dilute culture into 500 mL of YPD medium in a 2 L sterile flask, so that the OD600 < 0.1. Grow yeast cells with shaking at 30 °C for 3-5 h until OD600 = 0.5 (mid-log phase).
Collect cells by filtering through 0.45 μm membrane filters using a glass holder filter assembly. Scrape the pellet with a spatula, flash freeze in liquid nitrogen, and store at -80 °C. The expected size of the frozen pellet is ~ 0.2 - 0.5 g. Caution: Liquid nitrogen is extremely low temperature; wear appropriate protection.
Prepare a fresh Lysis Buffer (20 mM Tris-HCl pH 8.0, 140 mM KCl, 5 mM MgCl2, 100 μg/mL cycloheximide, 0.5 mM DTT, 1% Triton X-100) immediately prior to use, keep on ice.
Put 3 chrome-steel beads (3.2 mm) into a 1.8 mL stainless steel tube. Pre-chill in liquid nitrogen and add frozen pellets, cover with a silicone rubber cap. Homogenize the sample by cryogrinding for 10 s at 4200 rpm; repeat 10 times. Chill the tube in liquid nitrogen for at least 10 s between each round. NOTE: It is important to always keep the sample frozen.
Add 1 mL of Lysis Buffer, mix well by pipetting. Transfer into a new 1.5 mL tube.
Centrifuge at 20,000 x g for 5 min at 4 °C. While working on ice, transfer the supernatant into a new 1.5 mL tube. Transfer 100 μL of lysate to a new 1.5 mL tube for poly(A) mRNA isolation. Proceed immediately to RNA isolation or flash freeze the tube in liquid nitrogen. Measure OD260 of the rest of the lysate, which will be used for Footprint extraction, and flash freeze the tubes in liquid nitrogen. Store the lysates at -80 °C.
2. Footprint Extraction
- Preparation of sucrose gradients
- Prepare solutions for 50%, 40%, 30%, 20%, and 10% sucrose in Gradient Buffer (20 mM Tris-HCl pH 8.0, 140 mM KCl, 5 mM MgCl2, 100 μg/mL cycloheximide, 0.5 mM DTT).
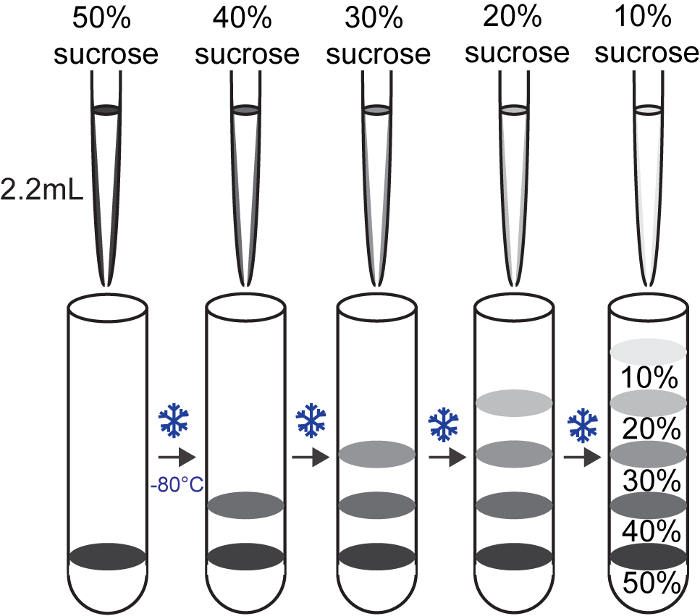
- Pipette 2.2 mL of the prepared 50% sucrose buffer to the bottom of the 13.2 mL thin wall polyallomer tube and freeze the solution for 10 min at -80 °C (Figure 2). Then layer 2.2 mL of 40% sucrose buffer on top of the frozen 50% sucrose buffer, and freeze for 10 min at -80 °C. Following the same instructions, layer 30%, then 20%, and finally 10% sucrose buffers atop of each other. Avoid making air bubbles while layering. Keep the gradients at -80 °C until use. The sucrose gradients should be prepared at least one day before the experiment and stored at -80 °C.
- Nuclease digestion
- Thaw the sucrose gradients the night before experiments at 4 °C.
- Thaw cell lysate (Footprint sample) on ice. Transfer an aliquot of cell lysate containing 50 OD260 units to a new 1.5 mL tube and add fresh Lysis Buffer up to 1 mL. Store the remaining lysate at -80 °C. Add 10 μL RNase I (100 U/μL) and incubate 1 h at room temperature with gentle rotation on a head-over-heels rotator.
- (Optional) Clarify the samples at 20,000 x g for 5 min at 4 °C and recover the supernatant into a new 1.5 mL tube.
- Ultracentrifugation
- Gently layer lysate samples to the top of a 10-50% sucrose gradient.
- Ultracentrifuge the polyallomer tubes at 210,000 x g (35,000 rpm) at 4 °C for 3 h using SW-41 Ti rotor.
- Gradient fractionation system set-up
- Turn on the components and allow the UV monitor to warm-up for at least 30 min.
- If using a digital recorder software, start the software on the computer, set the graph's scaling limits to -0.01 and 1.
- Fill the syringe with Chase Solution (20 mM Tris-HCl pH 8.0, 140 mM KCl, 5 mM MgCl2, 60% sucrose), remove any air bubbles inside the syringe and cannula.
- Install an ultracentrifuge tube filled with RNase-free water. Pierce the tube with the cannula until two black marks can be seen. Start the syringe pump at 1 mL/min.
- As the water passes through the flow cell, press "Auto Zero" button on the UV monitor to adjust baseline to 0. Set the sensitivity of the UV monitor to "2.0 AU". After a stable baseline has been set up, stop the syringe pump. Recover the Chase Solution and remove the ultracentrifuge tube.
- Monosome isolation
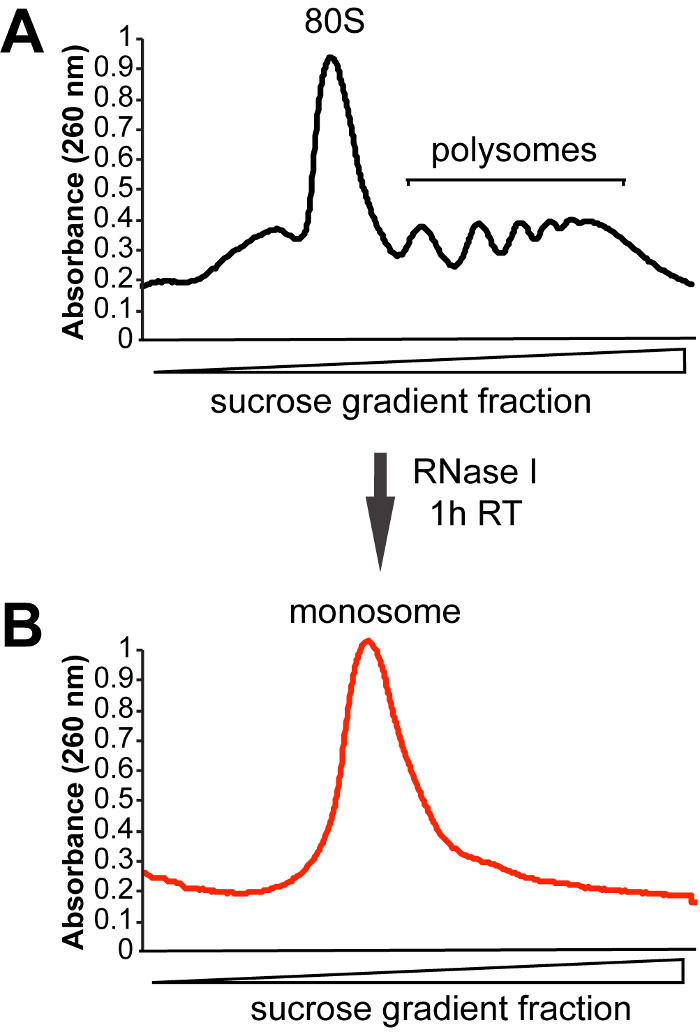
- Install and pierce the ultracentrifuge tube containing the sucrose gradient. Start the syringe pump at 1 mL/min, collect 1 mL fractions monitoring A254 values. Pool fractions representing the 80S monosome peak into one tube, keep on ice (Figure 3).
- Once Chase Solution reaches the flow cell, stop the syringe pump. Recover Chase Solution and remove the ultracentrifuge tube. Repeat fractionation for the rest of the samples starting at step 2.5.1. NOTE: When finished, clean the fractionation system with at least 30 mL of RNase-free water. Thoroughly wash tubing, syringe and all removable components with warm water.
- Filter the fractions through 0.5 mL centrifugal filters (100 kDa MWCO) at 12,000 x g for 10 min at 4 °C and discard the flow-through, repeat until volume is < 100 μL. Add 400 μL of Release Buffer (20 mM Tris-HCl pH 7.0, 2 mM EDTA, 40 U/mL RNase inhibitor). Mix by pipetting, incubate on ice 10 min and transfer the unit into a new collection tube. Centrifuge at 12,000 x g for 10 min at 4 °C and collect the flow-through.
- Footprint fragment purification
- Transfer the flow-through containing footprint RNA fragments to a new 1.5 mL tube, add 20 μL of 20% SDS (to a final concentration of 1%), mix by pipetting.
- Add 1 volume of Acid-Phenol:Chloroform (pH 4.5), vortex for 10 s. Caution: Acid-Phenol:Chloroform is toxic, avoid contact with skin and inhalation.
- Heat at 65 °C for 5 min, put on ice for 1 min. Centrifuge the sample at 12,000 x g for 5 min at 4 °C, transfer the aqueous phase (top) to a new 1.5 mL tube.
- Precipitate footprint RNA fragments by adding 1/10 volume of NaOAc (pH 5.5), 1/100 volume of glycogen (10 mg/mL), and 2.5 volumes of 100% ethanol. Incubate at -20 °C for at least 1 h.
3. Poly(A) mRNA Extraction
- Total RNA extraction
- Thaw a 100 μL aliquot of cell lysate (Total RNA sample) on ice, add 300 μL of 20 mM Tris-HCl pH 7.0 and 20 μL of 20% SDS (to a final concentration of 1%), mix by pipetting.
- Add 1 volume (400 μL) of Acid-Phenol:Chloroform (pH 4.5) and vortex for 10 s.
- Heat at 65 °C for 5 min, put on ice for 1 min. Centrifuge the sample at 12,000 x g for 5 min at 4 °C, transfer the aqueous phase (top) to a new 1.5 mL tube.
- Perform a second phenol extraction. Add 1 volume of Acid-Phenol:Chloroform (pH 4.5), vortex for 10 s. Heat at 65 °C for 5 min, put on ice for 1 min. Centrifuge the sample at 12,000 x g for 5 min at 4 °C, transfer the aqueous phase to a new 1.5 mL tube.
- Precipitate the RNA. For this add 1/10 volume of 3M NaOAc (pH 5.5), 1/100 volume of glycogen (10 mg/mL), and 2.5 volumes of 100% ethanol. Incubate at -20 °C for at least 1 h.
- Poly(A) mRNA isolation
- Centrifuge the RNA samples at 20,000 x g for 30 min at 4 °C, remove ethanol. Spin down 30 s at max speed, remove the rest of the ethanol with a gel-loading tip, and air dry the pellet for 5 min.
- Dissolve the pellet in 300 μL of Lysis/Binding buffer from the poly(A) mRNA isolation kit. Proceed to poly(A) mRNA extraction according to the poly(A) mRNA isolation kit manufacturer's protocol.
- Precipitate the mRNA samples by adding 1/10 volume of 3 M NaOAc (pH 5.5), 1/100 volume of glycogen (10 mg/mL), and 2.5 volumes of 100% ethanol. Incubate at -20 °C for at least 1 h.
- mRNA fragmentation
- Centrifuge the mRNA samples at 20,000 x g for 30 min at 4 °C, remove ethanol. Spin down 30 sec, remove the rest of the ethanol with a gel-loading tip and air dry the pellet for 5 min.
- Resuspend mRNA in 18 μL of RNase-free water, add 2 μL of 10x RNA fragmentation buffer. Incubate 5 min at 94°C. Transfer tube to ice.
- Precipitate by adding 1/10 volume of 3 M NaOAc (pH 5.5), 1/100 volume of glycogen (10 mg/mL), and 2.5 volumes of 100% ethanol. Incubate at -20 °C for at least 1 h.
4. Dephosphorylation
Treat Footprint and fragmented mRNA samples with T4 polynucleotide kinase. For this, centrifuge the samples at 20,000 x g for 30 min at 4 °C, remove ethanol. Spin down 30 s at max speed, remove the rest of the ethanol with a gel loading tip. Air dry the pellet for 5 min.
Resuspend the pellet in 7.75 µL of water, add 1 µL of 10x T4 polynucleotide kinase buffer, 1 µL of T4 polynucleotide kinase (10,000 U/mL), and 0.25 µL of RNase inhibitor (20 U/µL). Incubate 1 h at 37 °C.
(Optional) Pre-run a 15% TBE-urea gel at 180 V for 15 min using 1x TBE buffer.
Add 10 µL of 2X TBE-urea sample buffer to 10 µL of each Footprint and mRNA sample. Prepare a control sample containing 1 µL of 10 µM upper size marker oligonucleotide (32 nt), 1 µL of 10 µM lower size marker oligonucleotide (28 nt), 8 µL water, and 10 µL of 2x TBE-urea sample buffer for Footprint samples. Prepare a control sample containing 1 µL of 10 µM RNA marker oligonucleotide (63 nt), 9 µL water, and 10 µL of 2x TBE-urea sample buffer for mRNA samples.
Incubate the samples and controls 3 min at 75 °C, spin down, put on ice for 1 min. Load each sample into 2 wells of the 15% TBE-urea gel, separate RNA fragments by gel electrophoresis at 180 V for 1 h.
Stain the gel for 5 min with a nucleic acid gel stain (dilute 10,000x in water), protect from light.
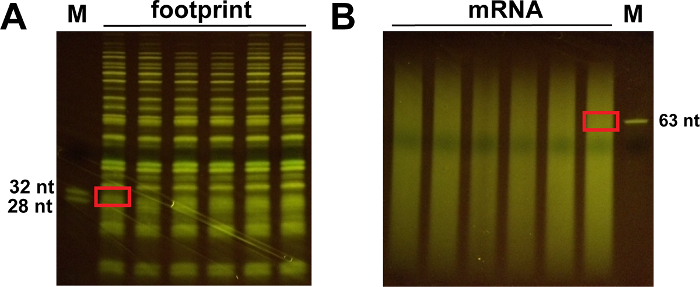
Using a blue light transilluminator, cut the proper band between 28 and 32 nt markers for footprint samples (Figure 4A) and around 50-70 nt for mRNA samples (Figure 4B) with a clean razor blade. Freeze the polyacrylamide gel pieces at -80 °C for at least 10 min.
Extract RNA from the polyacrylamide gel as follows. Heat the gel pieces at 70 °C for 2 min, grind the gel using disposable pellet pestles. Elute RNA with 300 µL elution buffer (20 mM Tris-HCl pH 7.0, 2 mM EDTA), add 1 µL RNase inhibitor (20 U/µL), and incubate at 37 °C for 3 h.
Centrifuge the sample at 12,000 x g for 5 min at 4 °C, collect the supernatant, and precipitate by adding 1/10 volume of NaOAc (pH 5.5), 1/100 volume of glycogen (10 mg/mL), and 2.5 volumes of 100% ethanol. Incubate at -20 °C for at least 1 h.
5. 3'-Adapter Ligation
Centrifuge the Footprint and mRNA samples at 20,000 x g for 30 min at 4 °C, remove ethanol. Spin down 30 s at max speed, remove the rest of the ethanol with a gel-loading tip. Air dry the pellet for 10 min.
Resuspend the pellet in 4.75 µL of nuclease-free water, 2 µL of 50% PEG8000, 1 µL of 10X T4 RNA ligase buffer, 1 µL of 3'-adapter (100 ng/µL), 0.25 µL of RNase inhibitor (20 U/µL), 1 µL of T4 RNA ligase 2 truncated KQ (200,000 U/mL). Incubate overnight at 16 °C.
The next morning, remove the excess of adapter by adding 0.5 µL of 5'-deadenylase (10 U/µL) and 0.5 µL of Rec J exonuclease (10 U/µL) directly to the ligation reaction. Incubate for 30 min at 30 °C.
Precipitate the samples. For this, add 30 µL of nuclease-free water, 1 µL glycogen (10 mg/mL), 1/10 volume of NaOAc (pH 5.5), and 2.5 volumes of 100% ethanol. Incubate at -20 °C for at least 1 h.
6. Reverse Transcription
Centrifuge the samples at 20,000 x g for 30 min at 4 °C, remove ethanol. Spin down 30 s at max speed, remove the rest of the ethanol with a gel-loading tip. Air dry the pellet for 10 min.
Resuspend the pellet in 11.5 µL of nuclease-free water, add 0.5 µL of 8 µM reverse transcription primer (RT primer) and 1 µL of dNTP mix (10 mM). Incubate for 5 min at 65 °C, place on ice.
Add 4 µL of 5X first strand buffer, 2 µL of 0.1 M DTT, 0.5 µL of RNase inhibitor (20 U/µL), 0.5 µL of reverse transcriptase (200 U/µL). Incubate for 30 min at 48 °C, 1 min at 65 °C, 5 min at 80 °C.
Do not precipitate and proceed immediately to the hydrolysis of RNA, by adding 0.8 µL of 2M NaOH, incubate 30 min at 98 °C. Add 0.8 µL 2M HCl to neutralize reaction.
Precipitate the samples. For this, add 20 µL of nuclease-free water, 1 µL glycogen (10 mg/mL), 1/10 volume of NaOAc (pH 5.5), and 2.5 volumes of 100% ethanol. Incubate at -20 °C for at least 1 h.
Centrifuge the samples at 20,000 x g for 30 min at 4 °C, remove ethanol. Spin down 30 s at max speed, remove the rest of the ethanol with a gel-loading tip. Air dry the pellet for 10 min.
Resuspend the pellet in 5 µL of nuclease-free water, add 5 µL of 2X TBE-urea sample buffer. Incubate 3 min at 75 °C, spin down, put on ice for 1 min.
(Optional) Pre-run a 10% TBE-urea gel at 180 V for 15 min using 1X TBE buffer.
Load the sample on 1 well of the 10% TBE-urea gel. Separate the products of the reverse transcription reaction by gel electrophoresis at 180 V for 50 min. As a control, prepare a sample containing 1 µL of 2.5 µM RT primer, 1 µL of 2.5 µM 128 nt marker oligonucleotide, 3 µL water, and 5 µL of 2X TBE-urea sample buffer, and run on a separate lane of the gel.
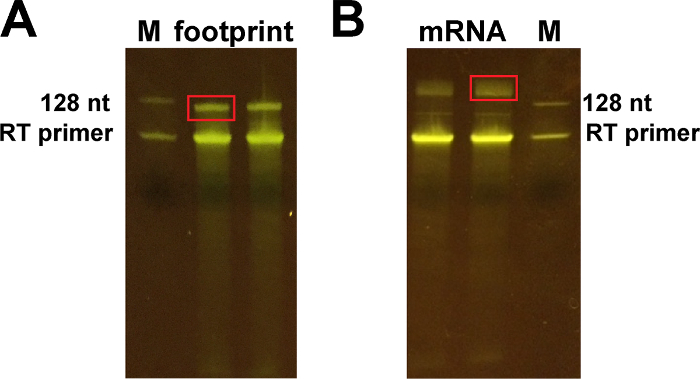
Stain the gel for 5 min with a nucleic acid gel stain (dilute 10,000x in water), protect from light. Using a blue light transilluminator, cut the band around 128 nt and higher (Figure 5) with a clean razor blade. Freeze the polyacrylamide gel pieces at -80 °C for at least 10 min.
Heat the gel pieces at 70 °C for 2min, grind the gel using disposable pellet pestles. Elute DNA with 300 µL of 20 mM Tris-HCl pH 7.0, and incubate at 37 °C for 3 h.
Centrifuge the sample at 12,000 x g, for 5 min at 4 °C, collect the supernatant, and precipitate by adding 1/10 volume of NaOAc (pH 5.5), 1/100 volume of glycogen (10 mg/mL), and 2.5 volumes of 100% ethanol. Incubate at -20 °C for at least 1 h.
7. Circularization
Centrifuge the samples at 20,000 x g for 30 min at 4 °C, remove ethanol. Spin down 30 s at max speed, remove the rest of the ethanol with a gel-loading tip, and air dry the pellet for 10 min.
Resuspend the pellet in 16.75 µL of nuclease-free water. Add 2 µL of 10x single-stranded DNA (ssDNA) ligase buffer, 1 µL of 50 mM MnCl2, 0.25 µL of ssDNA Ligase (100 U/µL). Incubate for 1 h at 60 °C, 10 min at 80 °C. Do not precipitate, and store the ssDNA ligation reaction product at -20 °C.
8. PCR Library Amplification
While working on ice, mix the following in a PCR tube: 146 µL of nuclease-free water, 2 µL of 20 µM Forward primer, 2 µL of 20 µM Indexed Reverse primer, 40 µL of 5x high-fidelity DNA plymerase buffer, 4 µL of dNTPs (10 mM), 4 µL of ssDNA ligation reaction product, and 2 µL of high-fidelity DNA polymerase. Choose a different Indexed Reverse primer for each sample that will be multiplexed for sequencing. Transfer a 50 µL aliquot of the PCR mixture into 4 new PCR tubes.
Perform the PCR amplification with varying number of cycles (8, 10, 12, 14) using the following settings: 1 min at 98 °C initial denaturation 8 to 14 cycles: 15 s at 94 °C 5 s at 55 °C 10 s at 65 °C 2 min at 65 °C final extension
Precipitate the PCR product by adding 1/10 volume of 3 M NaOAc (pH 5.5), 1/100 volume of glycogen (10 mg/mL), and 2.5 volumes of 100% ethanol. Incubate at -20 °C for at least 1 h.
Centrifuge the samples at 20,000 x g for 30 min at 4 °C, remove ethanol. Spin down 30 s at max speed, remove the rest of the ethanol with a gel-loading tip, and air dry the pellet.
Resuspend the pellet in 8 µL of nuclease-free water, add 2 µL of non-denaturing 5x nucleic acid sample buffer. Load the sample on 1 well of a non-denaturing 8% TBE gel. Separate the DNA products by gel electrophoresis at 150 V for 35 min. As a control, prepare a sample containing 0.5 µL of 10 bp DNA ladder (1 µg/µL), 7.5 µL water, and 2 µL of non-denaturing 5x nucleic acid sample buffer, and run on a separate lane of the gel.
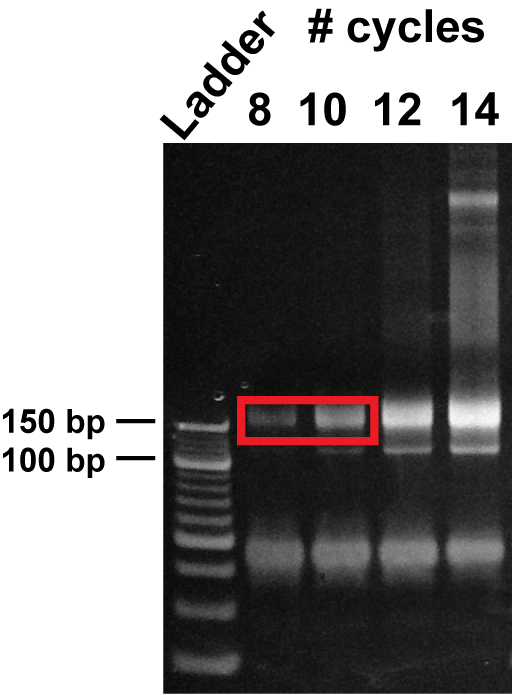
Stain the gel for 5 min with a nucleic acid gel stain (dilute 10,000x in water), protect from light. Using a blue light transilluminator, cut the band around 150 bp for footprint samples and around 180 bp for mRNA samples (Figure 6) with a clean razor blade. Store the gel pieces at -80 °C for at least 10 min.
Heat the gel pieces at 70 °C for 2 min, grind the gel using disposable pellet pestles. Elute DNA with 300 µL of 20 mM Tris-HCl pH 7.0, and incubate at 37 °C for 3 h.
Centrifuge the sample at 12,000 x g, for 5 min at 4 °C, collect the supernatant, and precipitate by adding 1/10 volume of NaOAc (pH 5.5), 1/100 volume of glycogen (10 mg/mL), and 2.5 volumes of 100% ethanol. Incubate at -20 °C for at least 1 h.
Centrifuge the samples at 20,000 x g for 30 min at 4 °C, remove ethanol. Spin down 30 s at max speed, remove the rest of the ethanol with a gel-loading tip, and air dry the pellet. Finally, resuspend the libraries in 20 µL of nuclease-free water and proceed to quantification, quality control and sequencing.
9. Library Quantification and High-throughput Sequencing
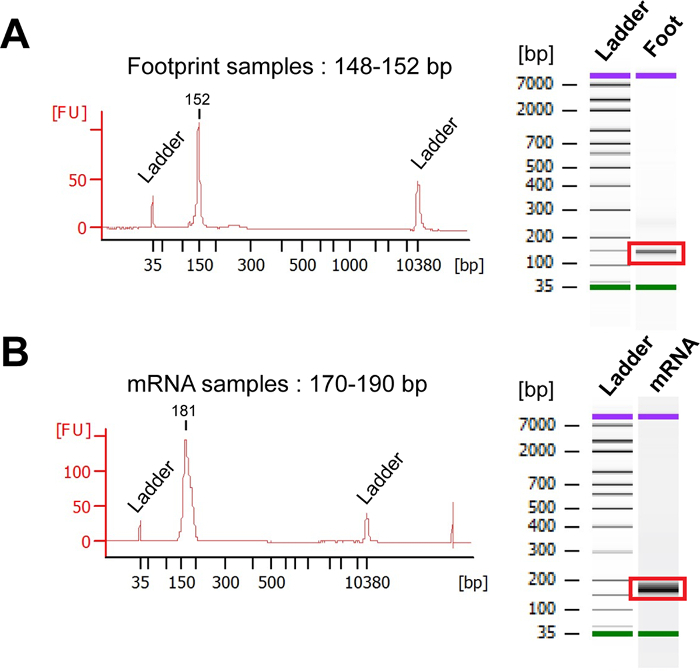
Before sending the samples for sequencing, determine the yield and quality of PCR-amplified libraries. Representative profiles for footprint and mRNA sequencing libraries are shown in Figure 7. The expected size of the PCR-amplified library is 148-152 bp for Footprint samples, and 170-190 bp for mRNA samples.
If several samples with different barcodes will be pooled for sequencing, perform accurate quantification of the libraries using a qPCR-based sequencing library quantification assay.
Pool the libraries in equimolar ratios. Calculate the total volume of the pool as 3 µL x total number of samples to be pooled. Determine the volume of each library that needs to be added so that libraries are mixed in the equimolar ratios to achieve 10 nM final concentration of the pool. Add the calculated volumes of each library into a new 1.5 mL tube, mix by pipetting. Add water to achieve the calculated total volume. Store pooled libraries at -20 °C.
Send an aliquot of the pooled library to be sequenced using 50 bp single-end sequencing run on an Illumina sequencing platform. We routinely multiplex 12 samples in a single sequencing run, which yields ~ 200 million reads per sequencing lane. NOTE: The final number of footprint reads collected per each sample depends on the quantity of the input material and the level of rRNA contamination.
Representative Results
Detailed pipelines for bioinformatic analysis of ribosome profiling data have been described previously 8,9. In addition, several research groups have developed bioinformatics tools for differential gene expression analysis and processing of sequencing data, which are specific for ribosome profiling method 10,11,12,13,14,15,16,17,18. The first step in the analysis of Ribo-Seq data is demultiplexing and trimming the 3' adapter sequence AGATCGGAAGAGCACACGTCT using Cutadapt software 19. Then the sequencing reads are aligned against non-coding RNAs, such as rRNA and tRNA, using Bowtie 20 to remove contaminating sequences, and reads that do not align are then mapped to the yeast genome. Read count per gene can then be assessed by HTseq-count software 21, and differentially expressed genes are identified using DESeq2 22.
Figure 8 shows representative results of quantitative analysis of translation in hbs1Δ 23,24 yeast deletion mutant that were obtained using our protocol. First, we assessed the number and proportion of reads that correspond to non-coding RNA (e.g. rRNAs) obtained after sequencing footprint and mRNA libraries generated for wild-type cells and the hbs1Δ mutant. We found that, even without any rRNA depletion steps (discussed below), from 50 to 60% of all sequenced reads in our footprint libraries correspond to ribosome-protected footprint fragments (Figure 8A). In contrast, only 3% of rRNA-derived fragments were observed in mRNA libraries demonstrating that poly(A) mRNA isolation allows effective elimination of rRNA reads. Together, we were able to obtain more than 10 million footprint reads for each of the footprint samples by sequencing 2 replicates. To assess the variability of library generation and reproducibility of the data, we usually analyze at least two independent biological replicates per each experimental condition. Both footprint and mRNA sample libraries show good reproducibility with Pearson correlation coefficient R ~ 0.99 between matched samples (Figure 8B).
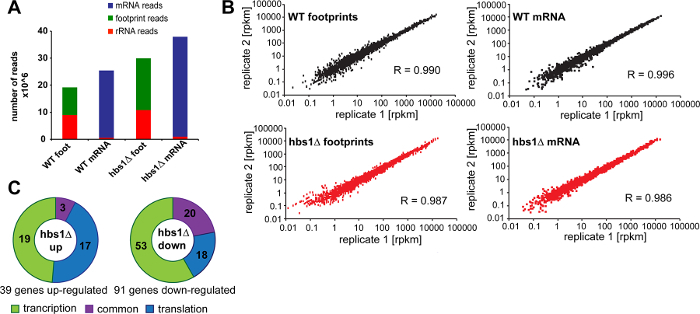
In addition to assessing the level of protein translation at the genome-wide level, ribosome profiling allows measuring changes in translation efficiency between experimental conditions 3. For this, an aliquot of the cell lysate (not treated with ribonuclease) is used for poly(A) mRNA isolation and preparation of an RNA-Seq library. Because both footprint library and RNA-Seq library are prepared under the same controlled conditions and can be traced to each individual replicate, Ribo-Seq and RNA-Seq datasets can be directly compared to identify genes that are regulated by the changes in mRNA transcription, translation efficiency, or by a combined effect. To identify genes that are up- or down-regulated specifically at the level of protein translation in the hbs1Δ mutant, we calculated changes in translation efficiency by dividing footprint rpkm values by mRNA rpkm for each of the genes (Figure 8C).
Figure 1: Overview of the Ribo-Seq protocol. The entire protocol can be performed in approximately 11 days. Estimated time for each step is shown. Please click here to view a larger version of this figure.
Figure 2: Preparation of sucrose gradients Please click here to view a larger version of this figure.
Figure 3: Representative sucrose gradient profiles.(A) Sucrose gradient profile obtained for control (not treated with RNase I) sample. (B) In order to extract ribosome-protected RNA fragments, cell lysates are treated with RNase I for 1 h at room temperature (RT). Fractions corresponding to the monosomal peak are then collected for footprint extraction. Please click here to view a larger version of this figure.
Figure 4: Representative images of the 15% polyacrylamide gels obtained after T4 polynucleotide kinase treatment.(A) The size of the excised gel slice around 28 and 32 nt is shown for footprint samples. (B) Cut the gel slice about 50-70 nt for mRNA samples. Please click here to view a larger version of this figure.
Figure 5: Representative images of the 10% polyacrylamide gels obtained after reverse transcription.(A) Cut the upper band ~ 128 nt for footprint samples, corresponding to the product of reverse transcription. (B) Cut the band around 150-170 nt for the mRNA samples (upper band). The lower bands correspond to the RT primer. Please click here to view a larger version of this figure.
Figure 6: Polyacrylamide gel purification of PCR-amplified libraries. PCR products obtained after 8, 10, 12, and 14 cycles of library amplification were resolved on a non-denaturing 8% TBE gel. The size of the full-length footprint libraries is ~ 150 bp, whereas the size of mRNA libraries is ~170-190 bp. Avoid the lower band that does not contain insert.
Figure 7: Bioanalyzer analysis.(A) Representative Bioanalyzer profiles of the PCR-amplified footprint library. The average size of the footprint library is expected to be from 148 to 152 nt. (B) Representative Bioanalyzer profiles of the sequencing library obtained for mRNA samples. The expected size of the mRNA library is 170-190 nt. Please click here to view a larger version of this figure.
Figure 8: Representative results.(A) Number of mRNA, footprint, and rRNA reads obtained for wild-type sample and the hbs1Δ mutant. Combined number of reads generated by sequencing two biological replicates are shown for wild-type cells and the hbs1Δ mutant. (B) Reproducibility of footprint and mRNA-abundance measurements between two replicates. Pearson correlation coefficients (R) are indicated. (C) Transcriptional and translational changes in the hbs1Δ mutant. Significantly up-regulated and down-regulated genes in hbs1Δ are grouped in accordance to whether they are affected by a change in mRNA transcription, translation efficiency, or by a combined effect. Please click here to view a larger version of this figure.
| Primers | Sequence | Index |
| 3' adapter (100 ng/µL) | /5rApp/AGATCGGAAGAGCACACGTCT/3ddC/ | |
| RT primer | pGATCGTCGGACTGTAGAACTCTGAACGTGTAGATCTCGGTGGTCGCCGTATCATT/iSp18/GTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | |
| Forward PCR primer | AATGATACGGCGACCACCGAGATCTACACGTTCAGAGTTCTACAGTCCGACG | |
| Index primer 1 | CAAGCAGAAGACGGCATACGAGATCGTGATGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | ATCACG |
| Index primer 2 | CAAGCAGAAGACGGCATACGAGATACATCGGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | CGATGT |
| Index primer 3 | CAAGCAGAAGACGGCATACGAGATGCCTAAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | TTAGGC |
| Index primer 4 | CAAGCAGAAGACGGCATACGAGATTGGTCAGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | TGACCA |
| Index primer 5 | CAAGCAGAAGACGGCATACGAGATCACTGTGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | ACAGTG |
| Index primer 6 | CAAGCAGAAGACGGCATACGAGATATTGGCGTGACTGGAGTTCAGACGTGTGCTCTTCCGATCT | GCCAAT |
Table 1: 3' Adapter and primer sequences.
Discussion
The Ribo-Seq approach has emerged as a powerful technology for the analysis of mRNA translation in vivo at the genome-wide level 3. Studies using this approach, which allows monitoring translation with single-codon resolution, has contributed to our understanding of translational regulation. Despite its advantages, Ribo-Seq has several limitations. Ribosomal RNA (rRNA) fragments are always co-purified during isolation of ribosome-protected footprints decreasing the yield of useful sequencing reads that can be obtained in Ribo-Seq experiments 9,25. Our data demonstrate that rRNA contamination can contribute to as many as 40-50% of all reads (Figure 8). One of the ways to overcome this limitation is to increase the sequencing depth. Additional rounds of sequencing should be performed until the required number of sequencing reads is achieved for each of the analyzed samples. Analysis of the sequencing libraries prepared using our protocol shows that more than 5 million footprint reads can be obtained for each footprint library, when 12 samples are multiplexed together in a standard 50-bp single-end run on Illumina HiSeq 2000 platform. However, if significant contamination with rRNA is observed, an optional rRNA removal step can be performed.
One of the strategies to remove rRNA contaminating fragments from footprint libraries is to use subtractive hybridization with the biotinylated oligonucleotides as described previously 8,26,27. Alternatively, rRNA contaminating reads can be removed by using commercially available rRNA depletion kits. For this, researchers can perform rRNA depletion on purified 3'-adapter-ligated footprint fragments from step 5.4. Following rRNA depletion, footprint samples can be precipitated and used directly for reverse transcription (step 6) as described in the protocol.
In addition to purification of monosomes using sucrose gradient fractionation described in our protocol, several methods have been developed that utilize a column-based purification 28 and ultracentrifugation through a sucrose cushion 8,29. While these methods can speed up the process of footprint library preparation and are less technically challenging compared to sucrose gradient fractionation, they do not allow qualitative analysis of the purified monosomes and efficiency of the nuclease digestion step. Recently, the choice of nuclease has been shown to affect the efficiency of purification of ribosome-protected RNA fragments in different species 30. While RNase I has robust activity in yeast cell lysates, its activity has been shown to affect the integrity of ribosomes in mouse tissues as well as in fruit flies. Therefore, the choice and concentration of nuclease should be optimized when adopting this protocol to other species.
Another important consideration is the use of the translation inhibitors. Most protocols for ribosome profiling typically involve treating cells with elongation inhibitors, such as cycloheximide or harringtonine, in order to prevent the run-off of ribosomes during cell harvesting 3. One of the limitations of the use of translation inhibitors is that the drugs diffuse progressively in the cell, inducing a progressive inhibition of the ribosomes 31. Moreover, the drugs do not inhibit translation initiation or termination. As a consequence, ribosomes disproportionately accumulate at the start codon and are depleted at the stop codon 8,27,31. In order to limit this artifact, we chose to flash freeze the cells in liquid nitrogen and treat with cycloheximide at the time of extraction in lysis buffer only.
Despite a relatively large number of reports that have utilized Ribo-Seq in various species, standardized protocols for performing Ribo-Seq analysis are lacking. As a consequence, Ribo-Seq data generated by different labs cannot be directly compared. For example, comparison of published ribosome profiling datasets revealed significant discrepancies in results among studies 32. This is in part due to continuous improvements that have been made as the protocol evolved over the past several years. In addition, many researchers modified the ribosome profiling protocol to adopt it to the specific needs of their study or model system 9,25. Further standardizing the ribosome profiling protocol as well as data analysis and normalization, would allow preventing some of the experimental biases and ensure accurate and reproducible quantification of in vivo translation.
Disclosures
The authors declare that they have no competing financial interests.
Acknowledgments
This work was supported by the National Institutes of Health grants AG040191 and AG054566 to VML. This research was conducted while VML was an AFAR Research Grant recipient from the American Federation for Aging Research.
References
- Holcik M, Sonenberg N. Translational control in stress and apoptosis. Nat Rev Mol Cell Biol. 2005;6(4):318–327. doi: 10.1038/nrm1618. [DOI] [PubMed] [Google Scholar]
- Tanenbaum ME, Stern-Ginossar N, Weissman JS, Vale RD. Regulation of mRNA translation during mitosis. Elife. 2015;4 doi: 10.7554/eLife.07957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 2009;324(5924):218–223. doi: 10.1126/science.1168978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT. Genome-wide translational profiling by ribosome footprinting. Methods Enzymol. 2010;470:119–142. doi: 10.1016/S0076-6879(10)70006-9. [DOI] [PubMed] [Google Scholar]
- Brar GA, Weissman JS. Ribosome profiling reveals the what, when, where and how of protein synthesis. Nat Rev Mol Cell Biol. 2015;16(11):651–664. doi: 10.1038/nrm4069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolin SL, Walter P. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 1988;7(11):3559–3569. doi: 10.1002/j.1460-2075.1988.tb03233.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Ingolia NT, Weissman JS, Bartel DP. Mammalian microRNAs predominantly act to decrease target mRNA levels. Nature. 2010;466(7308):835–840. doi: 10.1038/nature09267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 2012;7(8):1534–1550. doi: 10.1038/nprot.2012.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartholomaus A, Del Campo C, Ignatova Z. Mapping the non-standardized biases of ribosome profiling. Biol Chem. 2016;397(1):23–35. doi: 10.1515/hsz-2015-0197. [DOI] [PubMed] [Google Scholar]
- Larsson O, Sonenberg N, Nadon R. anota: Analysis of differential translation in genome-wide studies. Bioinformatics. 2011;27(10):1440–1441. doi: 10.1093/bioinformatics/btr146. [DOI] [PubMed] [Google Scholar]
- Zhong Y, et al. RiboDiff: detecting changes of mRNA translation efficiency from ribosome footprints. Bioinformatics. 2017;33(1):139–141. doi: 10.1093/bioinformatics/btw585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao Z, Zou Q, Liu Y, Yang X. Genome-wide assessment of differential translations with ribosome profiling data. Nat Commun. 2016;7:11194. doi: 10.1038/ncomms11194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung BY, et al. The use of duplex-specific nuclease in ribosome profiling and a user-friendly software package for Ribo-seq data analysis. RNA. 2015;21(10):1731–1745. doi: 10.1261/rna.052548.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa A, et al. RiboProfiling: a Bioconductor package for standard Ribo-seq pipeline processing. F1000Res. 2016;5:1309. doi: 10.12688/f1000research.8964.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn JG, Weissman JS. Plastid: nucleotide-resolution analysis of next-generation sequencing and genomics data. BMC Genomics. 2016;17(1):958. doi: 10.1186/s12864-016-3278-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baranov PV, Michel AM. Illuminating translation with ribosome profiling spectra. Nat Methods. 2016;13(2):123–124. doi: 10.1038/nmeth.3738. [DOI] [PubMed] [Google Scholar]
- Calviello L, et al. Detecting actively translated open reading frames in ribosome profiling data. Nat Methods. 2016;13(2):165–170. doi: 10.1038/nmeth.3688. [DOI] [PubMed] [Google Scholar]
- Michel AM, et al. RiboGalaxy: A browser based platform for the alignment, analysis and visualization of ribosome profiling data. RNA Biol. 2016;13(3):316–319. doi: 10.1080/15476286.2016.1141862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet J. 2011;17(1):10–12. [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10(3):R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S, Pyl PT, Huber W. HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics. 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014;15(12):550. doi: 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Elzen AM, Schuller A, Green R, Seraphin B. Dom34-Hbs1 mediated dissociation of inactive 80S ribosomes promotes restart of translation after stress. EMBO J. 2014;33(3):265–276. doi: 10.1002/embj.201386123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guydosh NR, Green R. Dom34 rescues ribosomes in 3' untranslated regions. Cell. 2014;156(5):950–962. doi: 10.1016/j.cell.2014.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT. Ribosome profiling: new views of translation, from single codons to genome scale. Nat Rev Genet. 2014;15(3):205–213. doi: 10.1038/nrg3645. [DOI] [PubMed] [Google Scholar]
- Gerashchenko MV, Lobanov AV, Gladyshev VN. Genome-wide ribosome profiling reveals complex translational regulation in response to oxidative stress. Proc Natl Acad Sci U S A. 2012;109(43):17394–17399. doi: 10.1073/pnas.1120799109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DE, et al. Improved ribosome-footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep. 2016;14(7):1787–1799. doi: 10.1016/j.celrep.2016.01.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JE, Yi H, Kim Y, Chang H, Kim VN. Regulation of poly(A) tail and translation during the somatic cell cycle. Mol Cell. 2016;62(3):462–471. doi: 10.1016/j.molcel.2016.04.007. [DOI] [PubMed] [Google Scholar]
- Howard MT, Carlson BA, Anderson CB, Hatfield DL. Translational redefinition of UGA codons is regulated by selenium availability. J Biol Chem. 2013;288(27):19401–19413. doi: 10.1074/jbc.M113.481051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerashchenko MV, Gladyshev VN. Ribonuclease selection for ribosome profiling. Nucleic Acids Res. 2017;45(2):e6. doi: 10.1093/nar/gkw822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerashchenko MV, Gladyshev VN. Translation inhibitors cause abnormalities in ribosome profiling experiments. Nucleic Acids Res. 2014;42(17):e134. doi: 10.1093/nar/gku671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor PB, Andreev DE, Baranov PV. Comparative survey of the relative impact of mRNA features on local ribosome profiling read density. Nat Commun. 2016;7:12915. doi: 10.1038/ncomms12915. [DOI] [PMC free article] [PubMed] [Google Scholar]