

Abstract
Ligand–protein binding kinetic rates are growing in importance as parameters to consider in drug discovery and lead optimization. In this study we analysed using surface plasmon resonance (SPR) the transition state (TS) properties of a set of six adenosine A2A receptor inhibitors, belonging to both the xanthine and the triazolo-triazine scaffolds. SPR highlighted interesting differences among the ligands in the enthalpic and entropic components of the TS energy barriers for the binding and unbinding events. To better understand at a molecular level these differences, we developed suMetaD, a novel molecular dynamics (MD)—based approach combining supervised MD and metadynamics. This method allows simulation of the ligand unbinding and binding events. It also provides the system conformation corresponding to the highest energy barrier the ligand is required to overcome to reach the final state. For the six ligands evaluated in this study their TS thermodynamic properties were linked in particular to the role of water molecules in solvating/desolvating the pocket and the small molecules. suMetaD identified kinetic bottleneck conformations near the bound state position or in the vestibule area. In the first case the barrier is mainly enthalpic, requiring the breaking of strong interactions with the protein. In the vestibule TS location the kinetic bottleneck is instead mainly of entropic nature, linked to the solvent behaviour.
Electronic supplementary material
The online version of this article (10.1007/s40203-017-0037-x) contains supplementary material, which is available to authorized users.
Keywords: Metadynamics, Supervised molecular dynamics, Ligand binding kinetics, SPR, Biacore, Molecular dynamics
Introduction
The importance of the pharmacology of adenosine receptors (ARs) is daily experienced by millions of coffee drinkers worldwide. Indeed, it is well established that caffeine is able to non-selectively inhibit AR subtypes (Rivera-Oliver and Díaz-Ríos 2014) (A1, A2A, A2B and A3) leading to a range of different biological responses and suggesting the potential usefulness of AR agonists or blocking agents (Jacobson and Gao 2006; Polosa and Blackburn 2009; Stone et al. 2009). Increasing attention is being addressed to the A2A AR and its modulation due to its emerging value in multiple disease states: Parkinson’s disease (Richardson et al. 1997), mainly attributed to the heterodimerization with the dopamine receptor D2 in the central nervous system (CNS) striatum (Fink et al. 1992), attention deficit hyperactivity disorder (ADHD) and immuno-oncology. The A2A AR represents a good starting point for structure-based drug design (SBDD) among all the G protein-coupled receptors (GPCRs) superfamily members. Despite the intrinsic difficulties of GPCR crystallography (Ghosh et al. 2015), to date more than 30 X-ray structures of the A2A AR have already been published, in complex with both agonists (Lebon et al. 2011, 2015; Xu et al. 2011), including a recent structure bound to an engineered G protein (Carpenter et al. 2016), and antagonists (Congreve et al. 2012; Doré et al. 2011; Hino et al. 2012; Jaakola et al. 2008; Liu et al. 2012; Segala et al. 2016).
Compared to the thermodynamic dissociation constant KD, binding kinetic rate constants k on and k off, are gaining in importance as parameters to consider in drug discovery and lead optimization. In fact, data obtained from in vitro steady state conditions are not always predictive for the in vivo biological environment, where the concentration of a ligand in proximity of its endogenous target is governed by pharmacokinetics (PK). As a consequence, increasing efforts are being addressed to the development of reliable structure-kinetic relationships (SKR), able to drive improvements in the kinetic profile of potential drug candidates. Indeed, compounds from a chemical series may show very similar affinities but dissimilar kinetic behaviour (Guo et al. 2017). The dynamic properties of binding equilibria allow the thermodynamic constant KD to be related to the kinetic rate constants k on and k off, as shown in Eq. (1)
| 1 |
Drug-like compounds have k on values generally in the range of 103–109 M−1 s−1 (the latter is approximately the rate limit of free diffusion in solution), with k off values ranging from about 10−7 s−1 to approximately 1 s−1 (Copeland 2015). Interestingly, super-fast binders (e.g. characterized by k on larger than 109 M−1s−1) have been evolutionary selected as effectors of physiologic processes that need instant regulation, like acetylcholine on acetylcholinesterase (AChE) (Radić et al. 1997) in the central nervous system (CNS). Nowadays it is common to refer to the kinetic concept of residence time (t r), first introduced in 2006 (Copeland et al. 2006) and defined as the reciprocal of the k off value (e.g. t r = 1/k off); t r is related to the in vivo biological effects triggered by ligands (Copeland 2015; Hothersall et al. 2016). The value of t r, especially when longer than the pharmacokinetic elimination lifetime, is generally associated with a favourable pharmacodynamic profile (Dahl and Akerud 2013) and may be important for tuning the agonist signalling bias (Kenakin and Christopoulos 2012), an emerging concept in GPCRs pharmacology. It is defined as the ability of ligands to preferentially signal through different effectors, triggering a distinct functional effect. Nevertheless, it is necessary to consider that also adverse effects can be linked to high t r values (Vauquelin et al. 2012). The k on has a crucial role in the setup of protocols for binding measurements (Hulme and Trevethick 2010), being the kinetic on rate of ligands involved in the experimental procedures; it critically drives the time needed to achieve the required equilibrium conditions.
From a mechanistic point of view, there is a range of driving forces that determine the free energy change during molecular binding and unbinding transitions. These include desolvation phenomena (Dror et al. 2011; Pan et al. 2013), conformational entropy loss (Frederick et al. 2007) and favourable and unfavourable intermolecular interactions (Radić et al. 1997; Schmidtke et al. 2011). The first extracellular vestibules that ligands encounter are the extracellular loops (ELs), excluding ligands able to reach the GPCR orthosteric binding site by diffusing from the membrane bilayer [as described by Stanley et al. (2016)]. These structural elements can modulate kinetic rates (Guo et al. 2017; Segala et al. 2016) and selectivity profiles (Nguyen et al. 2016; Seibt et al. 2013), mainly due to their intrinsic flexibility and high degree of structural variability. Molecular dynamics (MD) simulations represent the best computational approach for modeling events that are deeply influenced by flexibility and water molecules. Indeed, MD—based “enhanced sampling” methods are extensively used to simulate transitions of chemical systems between energy minima that are separated by high energy barriers and therefore associated with slow kinetic rates. This is the case for ligand unbinding, where the time scale reaches up to several hours or days, and is thus too computationally expensive for a single unbiased MD simulation starting from the ligand bound conformation; nowadays it is possible to reach the millisecond time scale on specialized machines (Shaw et al. 2009). Kinetic descriptions of binding and unbinding have been addressed by several different approaches, including, but not limited to, methods introducing an energy bias as a scalar in the potential energy equation of the system (Fukunishi et al. 2002; Hamelberg et al. 2004; Luitz and Zacharias 2014; Mollica et al. 2015, 2016; Pierce et al. 2012; Sinko et al. 2013; Wang et al. 2013) and methods requiring a preliminary definition of a set of collective variables (CVs) to be biased during the simulation (Barducci et al. 2011; Bui et al. 2003; Gervasio et al. 2005; Guo et al. 2016; Isralewitz et al. 2001; Laio and Parrinello 2002; Laio et al. 2005; Li 2005; Patel et al. 2014; Torrie and Valleau 1977; Yu et al. 2016). CVs can be for example intermolecular or interatomic distances, angles formed by atoms or group of atoms, coordination numbers, degree of solvation and they are used in order to drive the binding/unbinding transition and to map the corresponding energy profile. Among them, metadynamics (Barducci et al. 2011; Gervasio et al. 2005; Laio and Parrinello 2002; Laio et al. 2005) is probably one of the most used methods. During a metadynamics simulation, a history-dependent energetic term (centred along the pre-defined set of CVs) is added at discrete time intervals, decreasing the probability that the system will revisit that specific configuration and increasing the probability of a transition from one trough to another one (e.g. the ligand in the bound and unbound states, respectively) (Barducci et al. 2011). More recently, alternative metadynamics approaches like adaptive Gaussian (Branduardi et al. 2012) and well tempered metadynamics (WT-metaD) (Barducci et al. 2008, 2011) have been introduced.
Computational chemists are constantly working on new tools to allow evaluation of ligand kinetics and rationalize experimental data. From this perspective, the aMetaD protocol (Bortolato et al. 2015) was recently developed and tested on three GPCR systems. This approach combines adiabatic-bias molecular dynamics (ABMD) (Marchi and Ballone 1999) and WT-metaD in order to simulate ligand–protein unbinding events and provide an energy estimation of predicted transition states. The output from aMetaD allows rapid ranking of structurally related ligands according to predicted unbinding energetics (e.g. slow off and fast off compounds), as well as insights into the water dynamics during the dissociation. However, the need for a more complete ligand kinetics evaluation inspired us to develop a new protocol, capable of reconstructing an energy profile associated to both the binding and unbinding events: starting from a docked intermolecular complex. This MD—based sampling method is able to consecutively simulate ligand unbinding and binding, using a supervised MD (SuMD) approach (Cuzzolin et al. 2016; Sabbadin and Moro 2014) and keeping track of the energy required for the transition by mean of metadynamics. The supervised metadynamics (suMetaD) algorithm was tested on a set of A2A AR antagonists (Fig. 1), belonging to both the xanthine (XAC, DPCPX, KW3902) and the [1,2,4]triazolo[1,5-a][1,3,5]triazine [ZM241385, Z48 (Federico et al. 2011), Z80 (Federico et al. 2016)] scaffolds, whose transition state thermodynamics were experimentally determined using the surface plasmon resonance (SPR) technique (Fig. 2). The past 10 years have seen a significant surge in the application of SPR technology to study small molecule interactions; it uses a protein in real time without labelling (Du et al. 2016; Rich and Myszka 2009). Engineering stabilized GPCRs allows SPR techniques to be applied to this class of membrane receptors with promising results (Rich et al. 2011; Shepherd et al. 2014). Using SPR to measure the affinity of interaction K D at a series of temperatures allows the enthalpy ΔH° and entropy ΔS° of interaction to be calculated using the van’t Hoff equation (here in its integrated form)
where R is the universal gas constant. This approach has been used in a number of studies (Borea et al. 2004; Roos et al. 1998; Sahlan et al. 2010).
Fig. 1.
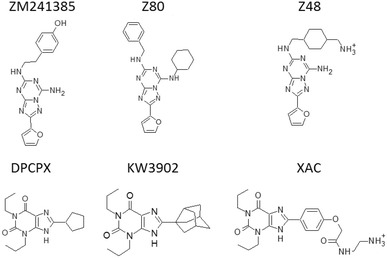
Chemical structures of the A2A AR ligands considered for the suMetaD test. ZM241385, Z80 and Z48 are [1,2,4]triazolo[1,5-a][1,3,5]triazine inhibitors; DPCPX, KW3902 and XAC are xanthine inhibitors
Fig. 2.
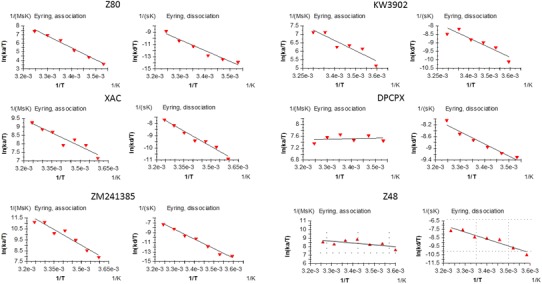
Eyring equation plots for the ligand binding association (left) and dissociation (right) from the SPR analysis for the 6 ligands included in this study. The values for ΔH‡ and ΔS‡ can be determined from kinetic data plotting ln(t/T) vs 1/T. In the resulting linear interpolation equation the slope corresponds to ΔH‡/R and the ΔS‡ can be calculated from the y-intercept
Likewise, measuring the rate constant k of association or dissociation as a function of temperature enables a crude approximation of the enthalpy ΔH°‡ and entropy ΔS°‡ of the transition state formation using the Eyring equation (here in its integrated form) to be obtained.
where R is the universal gas constant, k B is Boltzmann constant, and h is Planck’s constant.
Studying the mechanism of transition state formation can provide important additional information helping to understand why interactions with similar affinities can have different kinetics.
From this standpoint, the suMetaD computational protocol allows to obtain insights into kinetic bottlenecks along the simulated pathways that offer a rationale for understanding experimental transition state (TS) thermodynamic data and allow generation of working hypotheses on the role of enthalpy and entropy during the binding and unbinding rate limiting steps.
Using SPR we evaluated the ligand binding and unbinding event to the A2A AR stabilized receptor (StaR™) for the 6 ligands shown in Fig. 1. Transition state thermodynamics was evaluated using association and dissociation rate constants measured at temperatures between 5 and 35 °C at 5 °C intervals. A series of five twofold dilutions of the test compounds was injected and the obtained sensorgrams were fitted to a 1:1 interaction model to obtain the rate constants. The temperature dependence of the rate constants was fitted to Eyring equation using Biacore T200 evaluation software to obtain enthalpy and entropy of TS formation (Fig. 2).
Results
The obtained experimental transition state thermodynamics results are summarized in Table 1. It is interesting to note that the spread in the TS free energy for the test set is about 2.5 kcal/mol for both the association and dissociation events. The enthalpic and entropic components however have stark differences in the energy barriers: ΔH and TΔS cover a range of more than 20 kcal/mol for the binding event and more than 30 kcal/mol for the unbinding event. The smaller changes in ΔG‡ among the ligands is the result of enthalpy–entropy compensation effects: higher ΔH‡ energy barrier correspond to lower − TΔS‡ and vice versa.
Table 1.
Transition state thermodynamic results obtained from the SPR analysis of the association and dissociation of the six ligands considered in this study to the A2A AR
| Association (kcal/mol) | Dissociation (kcal/mol) | |||||
|---|---|---|---|---|---|---|
| ΔH‡ | − TΔS‡ | ΔG‡ | ΔH‡ | − TΔS‡ | ΔG‡ | |
| ZM241385 | 19.1 ± 2.2 | − 11.2 ± 2.2 | 7.9 | 40.6 ± 2.2 | − 19.8 ± 2.2 | 20.8 |
| XAC | 10.5 ± 1.7 | − 1.5 ± 1.7 | 9.0 | 16.7 ± 1.4 | 2.4 ± 1.4 | 19.1 |
| DPCPX | − 0.4 ± 1.0 | 10.0 ± 1.0 | 9.6 | 8.4 ± 0.9 | 11.0 ± 0.9 | 19.4 |
| KW3902 | 12.2 ± 2.4 | − 2.2 ± 2.4 | 10.0 | 11.2 ± 2.3 | 7.7 ± 2.3 | 18.9 |
| Z80 | 22.7 ± 1.8 | − 12.4 ± 1.8 | 10.3 | 19.6 ± 1.8 | − 1.4 ± 1.8 | 18.2 |
| Z48 | 5.5 ± 1.4 | 3.6 ± 1.4 | 9.1 | 16.5 ± 2.5 | 2.3 ± 2.5 | 18.8 |
Association and dissociation rate constants were measured at temperatures between 5 and 35 °C at 5 °C intervals
To support the analysis of the experimental data we developed a novel computational protocol based on MD to study putative ligand unbinding and binding events. It is based on a supervised algorithm (Sabbadin and Moro 2014), that drives the exploration only of ligand–protein conformations starting from a provided bound state conformation (SI Fig. 2) that are compatible with paths linking the orthosteric site to the extracellular bulk solvent. It evaluates if the ligand is moving in the right direction, calculating the root mean square deviation (RMSD) of the ligand coordinates during the MD from the unbound (unbinding path) or bound (binding path) ligand target conformation. At the same time the relative free energy of the unbinding/binding paths are estimated using metadynamics (Barducci et al. 2011). During the metadynamics simulation a history dependent bias is added to the potential energy landscape representing the unbinding and binding events. The analysis of the resulting energy profile (SI Fig. 3) allows the estimation of a representative protein–ligand conformation corresponding to the highest energy barrier the ligand has to overcome during its path toward the target positions. These conformations can be useful to understand at a molecular level the on and off rate TS energy barriers linked to the ligand binding/unbinding kinetic bottlenecks. Movies with the simulations of the unbinding/binding event for each ligand are available as Supplementary Material (videos S1–S6).
Visual inspection of the representative kinetic bottlenecks shows different location of the ligands in the predicted TS complex conformation (Figs. 3, 4). During the binding event the ligand faces ordered water molecules in the orthosteric site, some creating stable favourable interactions with the protein (Fig. 3). When the energy barrier is predicted to be located near the vestibule region of the receptor, the ligand generally does not need to displace tightly protein-bound waters. This is in agreement with the resulting low enthalpic energy barrier. In parallel, the solvent is trapped in the orthosteric site by the ligand position in the vestibule area, resulting in lower probability of exchange with bulk solvent and a high entropic barrier to the binding event. For some other ligands the energy barrier was predicted to correspond to a ligand location deep in the pocket, close to the final bound state. A high enthalpic barrier in this case is expected to be linked to the required displacement of water molecules in the orthosteric site tightly bounded to the protein. Their release to bulk results in a favourable entropic gain linked to the ligand binding event.
Fig. 3.
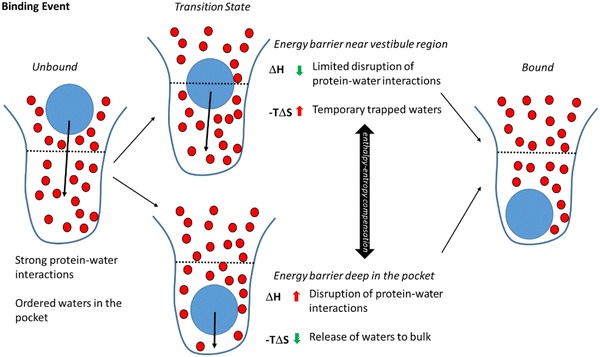
Schematic overview of the two alternative transition state locations detected by the suMetaD protocol for the ligand binding event. The ligand is represented by a blue circle, waters by smaller red circles, the pocket by a blue line divided by a black dotted line in the orthosteric site (bottom half) and the vestibule region (top half). Starting from the unbound state (left) with strong protein–water interactions and ordered waters the ligand reaches the bound state (right). Two alternative ligand locations corresponding to the transition state have been detected, the upper transition state showing the energy barrier is near the vestibule region and the lower one where it is deep in the orthosteric site. They are characterized by opposite enthalpic and entropic components related to the desolvation of the binding site
Fig. 4.
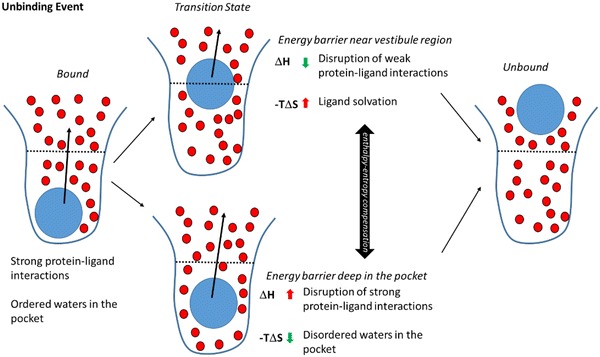
Schematic overview of the two alternative transition state locations detected by the suMetaD protocol for the ligand unbinding event. As in Fig. 3, the ligand is represented by a blue circle, waters by smaller red circles, the pocket by a blue line divided by a black dotted line in the orthosteric site (bottom half) and the vestibule region (top half). Starting from the bound state (left) with strong protein–ligand interactions and ordered waters the ligand reaches the unbound state (right). Two alternative ligand locations corresponding to the transition state have been detected: on the top, the energy barrier is near the vestibule region; on the bottom is deep in the orthosteric site. They are characterized by opposite enthalpic and entropic components related to the solvation of the binding site and of the small molecule
During the unbinding event the ligand faces a cap of ordered water molecules in the orthosteric site and it has to disrupt strong protein–ligand interactions (Fig. 4). Similar to the analysis of the binding event, we predicted two possible ligand locations corresponding to the unbinding transition states: near the vestibule region or close to the bound state. A kinetic bottleneck near the vestibule area and the extracellular loops is characterized by a high entropic barrier mainly linked to the solvation of non-polar ligand atoms. In contrast, a low enthalpic barrier is the result of weaker interactions with the protein in this region compared to when the ligand is located deep in the orthosteric site. An unbinding transition state close to the bound state conformation is characterized by a high enthalpic barrier related to strong interactions with the receptor. In this case the unbinding event starts from a complex conformation where the waters are tightly bound to the protein and/or to the small molecule, evolving into a nearby TS conformation where they are more disordered (resulting in a low entropic energy barrier).
Analysis of the TS thermodynamic properties of ligand binding shows a high enthalpic energy barrier for Z80 and ZM241385, counterbalanced by favourable entropic gains. Visual inspection of the kinetic bottlenecks obtained using the suMetaD protocol for these two small molecules (Fig. 5a, b) shows ligand positions deep in the orthosteric site. The high enthalpic barrier is linked to the displacement of most of the water molecules, with stable interactions with the protein orthosteric site then created. At the same time their release to bulk results in an entropically favourable binding event. For KW3902 and XAC, the enthalpic TS binding energy barriers decrease together with the entropic balancing effect. This is agreement with their kinetic bottlenecks predicted close to the vestibule region (Fig. 5c, d), resulting in the release to bulk of only part of the stable waters in the site. SPR data shows Z48 binding TS barrier is characterized by both enthalpic and entropic components, while for DPCPX is only entropic. Visual inspection of their kinetic bottlenecks (Fig. 5e, f) shows binding positions in the vestibule region creating weak interactions with the receptor. These interactions can explain the low enthalpic component of the binding energy. Their location near the loops results in a large structured water network in the pocket that needs to be disrupted to allow the small molecule to reach the final bound state, causing a higher entropic barrier.
Fig. 5.
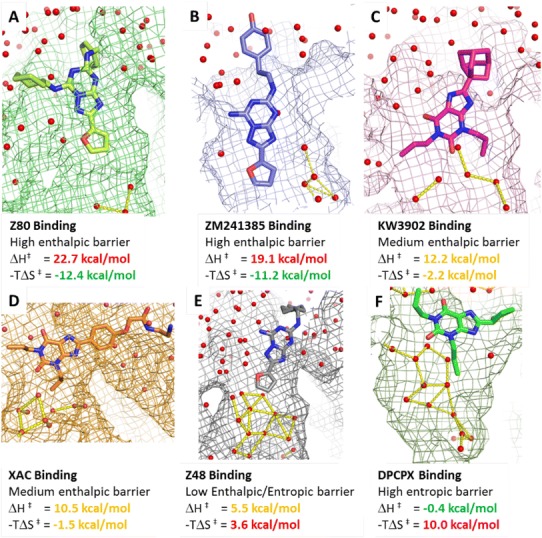
Protein–ligand locations/conformations corresponding to the binding kinetic bottlenecks detected by the suMetaD protocol for the 6 small molecules considered in this study. The ligand is shown in stick representation, the pocket as a mesh surface and waters as small spheres. Interactions among waters in the orthosteric site are shown as yellow dotted lines. The experimental energy of the enthalpic (ΔH‡) and entropic (−TΔS‡) components of the TS is reported
Analysis of the TS thermodynamic properties of ligand unbinding shows a high enthalpic energy barrier for ZM241385 counterbalanced by a favourable entropic gain. The predicted position/conformation (Fig. 6a) for its unbinding kinetic bottleneck shows the ligand still tightly bound in the orthosteric site. In this pose it creates good interactions with the receptor, linked to the high experimental enthalpic barrier. The counterbalancing entropic component can be related to the flexible 4-ethylphenol tail sitting on top of the binding site and less ordered waters (compared to the bound conformation) at the interface between the protein and the small molecule. For the other ligands, solvation of non-polar hydrophobic saturated ring or aliphatic tails results in increasing unbinding entropic energy barriers. In Z80, XAC and Z48 transition states (Fig. 6b–d) the water network starts to act more like a lid on the extracellular side. The solvent molecules create a complex web of hydrogen bond interactions hindering ligand unbinding. In KW3902 (Fig. 6e), the bulky and hydrophobic saturated ring system touching the solvent increases further the unbinding entropic barrier. The predicted kinetic bottleneck positions for these ligands are between the bound state and the loops creating interactions with the receptor not as strong as ZM241385 in its TS position. In particular, the predicted TS pose for DPCPX (Fig. 6f) is in the loop region close to several solvent molecules, creating a cage of H-bonds, resulting in a high entropic energy barrier for ligand unbinding.
Fig. 6.
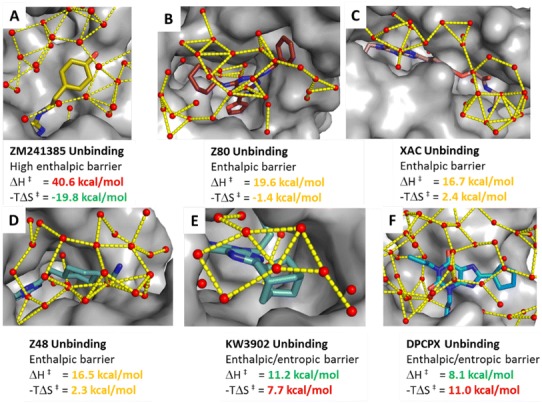
Protein–ligand positions/conformations corresponding to the unbinding kinetic bottlenecks detected by the suMetaD protocol for the 6 small molecules considered in this study. The ligand is shown in stick representation, the pocket as solid grey surface and waters as small spheres. Interactions among waters in the vestibule region near the ligand and among the extracellular loops are shown as yellow dotted lines. The experimental energy of the enthalpic (ΔH‡) and entropic (−TΔS‡) components of the TS is reported
Discussion
It is still challenging to understand ligand binding and unbinding kinetic properties at a molecular level. In general, transition state energy barriers correspond to kinetic bottleneck positions and conformations the ligand needs to overcome to reach the final state. Their enthalpic and entropic components determine ligand on and off rates. TS enthalpy barriers are mainly linked to polar interactions among the ligand, the protein and the waters, while the entropic component is strongly related to changes in protein/ligand flexibility and solvation/desolvation effects. For the ligands considered in this study, the binding entropy barrier was found to be correlated to the number of temporary trapped waters in the orthosteric site in the predicted energy barrier position/conformation (Fig. 7). For the unbinding event the TS entropic barrier was related to the number of waters in the extracellular side of the receptor at less than 4 Å from the ligand aliphatic carbon atoms (Fig. 7).
Fig. 7.
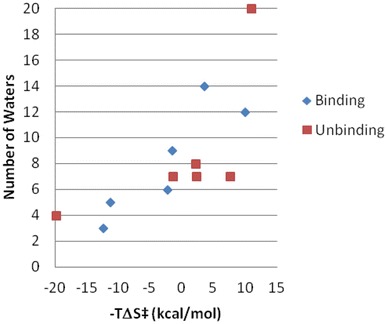
Role of the solvent in the transition state entropic energy barrier. The experimental binding (blue) and unbinding (red) TS entropic barrier for the 6 ligands considered in this study is plotted on the X-axis. The Y-axis shows for the binding event (blue) the corresponding number of temporary trapped waters in the orthosteric site in the representative TS conformation. For the unbinding event (red), the Y-axis includes the number of waters in the extracellular side of the receptor at less than 4 Å from the ligand aliphatic carbon atoms in the representative TS conformation
Using a molecular dynamics-based approach we have been able to simulate the ligand binding and unbinding events to extract possible low energy pathways linking the docked ligand location to the extracellular side of the receptor. Using a supervised MD algorithm the simulation exploration is optimized to consider only directions compatible with the desired binding or unbinding events. In parallel we enhanced the conformational space sampling and we evaluated the energy of the obtained hypothetical pathway using metadynamics. Metadynamics allows the extraction of a representative conformation of the system corresponding to the high energy barrier the ligand has to overcome to reach the target state. The resulting molecular details of the kinetic bottlenecks have been useful to generate testable working hypothesis to understand the key aspects of the ligand structure determining the on and off rates.
For the six ligands evaluated in this study their transition thermodynamic properties were linked in particular to the role of water molecules. During the binding event the ligand has to face a complex ordered water network in the pocket. Its ability to disrupt those interactions, resulting in the expulsion of waters into bulk solvent, determines if the TS conformation will be near the final bound state position or in the vestibule area, close to the extracellular loops. In the first case the barrier is mainly enthalpic, requiring the breaking of stable protein–waters interactions. In the second case it will be largely entropic, due to temporarily trapped waters in the pocket. These opposite energetic components result in the enthalpy–entropy compensation effect we see experimentally for the six ligands.
It is possible to reach a similar conclusion from the analysis of the unbinding events. TS positions/conformations can be near the starting bound state position or in the vestibule area. In the former case the barrier is mainly enthalpic, requiring the breaking of strong interactions with the protein. In the latter case with a vestibule TS location, the kinetic bottleneck is instead mainly of an entropic nature, linked to the solvation of the ligand and the binding site.
In conclusion, several different aspects can play important roles affecting the transition state energy barrier that the small molecule has to overcome to reach the final state. The suMetaD method we developed and presented in this paper provides a useful tool to improve our understanding of the TS molecular details. It can help both to interpret structure-kinetic relationships and to make predictions for new molecules. The promising results obtained for the test set presented herein need now to be extended to a bigger transition state thermodynamic datasets to fully prove the general applicability of this approach.
Methods
Expression of A2AR in insect cells
A2A AR StaR 2 carrying a C-terminal deca His tag was expressed in Sf21 cells grown in ESF921 medium supplemented with 10% (v/v) FBS and 1% (v/v) Penicillin/Streptomycin using the FastBac expression system (Invitrogen). Cells were infected at a density of 2.5 × 106 cells/ml with baculovirus at an approximate multiplicity of infection of 1. Cultures were grown at 27 °C and harvested 48 h post infection.
Membrane preparation and protein purification
All subsequent purification steps were carried out at 4 °C. To prepare membranes, 2 l of cells were re-suspended in PBS buffer supplemented with cOmplete Protease Inhibitor™ tablets (Roche) and 5 mM EDTA. Cells were disrupted by micro-fluidizer at 60 PSI and membranes collected by ultracentrifugation at 204.7 k×g for 1 h. Membranes were washed with PBS buffer supplemented with protease inhibitor tablets and 500 mM NaCl, collected by ultracentrifugation and re-suspended in 40 mM HEPES pH 7.5, 250 mM NaCl and stored at − 80 °C. Just prior to solubilization membranes were thawed, homogenized, supplemented with 10 µM ZM24134 and incubated on a roller mixer for 60 min. Membranes were solubilized with 1.5% (w/v) DM for 1 h, insoluble material was removed by ultra-centrifugation and the solubilized lysate batch bound to 5 ml of Ni–NTA Superflow resin (Qiagen) for 3 h in the presence of 10 mM imidazole. Resin was washed with a gradient of 10–50 mM imidazole in 40 mM HEPES pH 7.5, 250 mM NaCl, 0.15% (w/v) DM, and 10 µM ZM24134 over 35 column volumes before bound material was eluted in a step with 245 mM imidazole. Receptor was further purified by gel filtration (SEC) in 40 mM HEPES pH 7.5, 150 mM NaCl, 0.15% (w/v) DM, and 10 µM ZM24134. Receptor purity was analyzed using SDS-PAGE and LC–MS, and receptor monodispersity was assayed by analytical SEC. Protein concentration was determined using the receptor’s calculated extinction coefficient at 280 nm [ε280, calc = 47,780 (mg/ml × cm)−1] and confirmed by quantitative amino acid analysis.
Assay of binding thermodynamics by SPR
SPR experiments were carried out on a Biacore T200 instrument with a sensor chip NTA (GE Healthcare). The running buffer was 10 mM phosphate, pH 7.4, 2.7 mM KCl, 137 mM NaCl, 0.05 mM EDTA, 5% DMSO. A2A AR was injected over Ni-loaded chip NTA at 200 nM for 10 min at 10 °C to obtain about 5000 resonance units (RU) of immobilised receptor. Transition state thermodynamics was evaluated using association and dissociation rate constants measured at temperatures between 5 and 35 °C at 5 °C intervals. A series of five twofold dilutions of the test compounds was injected and the obtained sensorgrams were fitted to 1:1 interaction model to obtain the rate constants. The temperature dependence of the rate constants was fitted to Eyring equation using Biacore T200 evaluation software to obtain enthalpy and entropy of transition state formation.
suMetaD
For the ligands ZM241385, Z48 and Z80, the A2A AR conformation was based on PDB:4EIY (Liu et al. 2012), while for the other ligands PDB:3REY (Doré et al. 2011) was used. The fusion protein was removed from 4EIY and the protein sequence was modified to correspond to the construct used in the SPR experiments using Prime (Jacobson et al. 2004) (Schrödinger Release 2016-3). Receptors were prepared with the Protein Preparation Wizard in Maestro (Schrödinger Release 2016-3): the H-bond network has been optimized through an exhaustive sampling of hydroxyl and thiol moieties, tautomeric and ionic state of His and 180° rotations of the terminal dihedral angle of amide groups of Asp and Gln. His264 has been considered to be protonated. The starting docking poses for Z48 and Z80 were based on ZM241385 and refined using Glide (Friesner et al. 2004; Halgren et al. 2004). For Z80 the rotameric state of H264 and E169 were modified to be comparable to the conformation of the corresponding residues in the PDB 3PWH (Doré et al. 2011) A2A AR crystal structure. The docking poses of DPCPX and KW3902 were based on XAC bound crystallographic conformation and refined using Glide (Friesner et al. 2004; Halgren et al. 2004). For these two ligands Y2717.36 rotameric state was changed to be comparable to its conformation in 4EIY.
The supervised metadynamics protocol (included in a single python script, suMetaD.py) uses as input the protein PDB, and the bound conformations of the ligands (as SDF) and reasonable target ligand unbound positions near the extracellular side (also as SDF), at about 20 Å from the bound conformation. aMetaD.py protocol can be divided in the two steps: (1) system preparation and equilibration; (2) supervised metadynamics (suMetaD).
System preparation and equilibration
Every ligand-receptor complex is aligned to a reference (+Z corresponds to the extracellular side, −Z to the intracellular side, the membrane is in the XY plane). The system is equilibrated using the following molecular dynamics protocol. The AMBER99SB force field (ff) parameters (Lindorff-Larsen et al. 2010) were used for the protein and the GAFF ff (Wang et al. 2004) for the ligands using AM1-BCC partial charges (Jakalian et al. 2002). The system has been embedded in a triclinic box including an equilibrated membrane consisting of 256 DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) lipids (Jämbeck and Lyubartsev 2012) and 24,513 waters using g_membed (Wolf et al. 2010) in Gromacs. The SPC water model was used and ions were added to neutralize the system (final concentration 0.01 M). An energy minimization protocol based on 1000 steps steepest-descent algorithm has been applied to the system. The membrane has been equilibrated using 0.5 ns MD simulation with a time step of 2.5 fs, using LINCS on all bonds and keeping the protein and ligand restrained applying a force of 100 kJ mol−1 nm−1. Lennard-Jones and Coulomb interactions were treated with a cut-off of 1.069 nm with particle-mesh Ewald electrostatics (PME) (Darden et al. 1993). The MD has been executed in the NPT ensemble using v-rescale (Bussi et al. 2007) (tau_t = 0.5 ps) for the temperature coupling to maintain the temperature of 298 K and using Parrinello–Rahman (Parrinello and Rahman 1981) (tau_p = 10.0 ps) for the semi-isotropic pressure coupling to maintain the pressure of 1.013 bar. Without applying any positional restraints, the system has been minimized for 200 steps using the steepest-descent algorithm and equilibrated using MD using the same settings described above, but with a time step of 2 fs and increasing the temperature from 29.8 to 298 K in 10 steps (9 steps of 30 ps and the last one of 300 ps).
Supervised metadynamics (suMetaD)
The metadynamics (Barducci et al. 2011) protocol exploits a generic path collective variable (CV) (Branduardi et al. 2007) generated using the RMSD between the starting ligand bound state and a ligand position corresponding to the original starting ligand location translated 3 Å on the X-axis. Two path CVs have been considered: one defining the RMSD position on this path (s) and the other the RMSD distance from the path (z) using lambda = 20. The same MD settings used during the final system equilibration at 298 K are used for the suMetaD protocol. For the metadynamics algorithm the following settings have been used: initial energy bias Gaussian height of 0.25 kcal/mol with a deposition frequency of 1 ps. The width of the Gaussians was 0.01 Å. The suMetaD protocol is divided in two consecutive parts:
The ligand unbinding event is simulated first using a maximum of 200 metadynamics steps of 50 ps each (always writing to the same COLVAR file using the RESTART keyword) for a total of maximum 10 ns. The supervised algorithm is implemented in the following way: after every step if the RMSD from the target ligand position is decreased the next step starts from the end of the previous step, otherwise from the beginning of the previous step assigning new random atom velocities. If the ligand reaches a distance of 15 Å from the bound position the simulation is stopped.
The final output coordinates from the ligand unbinding simulation is used as a starting conformation for ligand binding simulation. As before, a maximum of 200 metadynamics steps of 50 ps each are simulated. A comparable supervised algorithm is used, but the RMSD from the target bound ligand position is used. If the ligand reaches a distance of 3 Å from the bound position the simulation is stopped.
The final results included for the analysis are: (1) the binding/unbinding trajectories; (2) the metadynamics energy profile as function of the bound state RMSD; (3) the conformation corresponding to the highest energy barrier (based on the metadynamics bias energy deposition) for the unbinding and binding event.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Footnotes
Electronic supplementary material
The online version of this article (10.1007/s40203-017-0037-x) contains supplementary material, which is available to authorized users.
References
- Barducci A, Bussi G, Parrinello M. Well-tempered metadynamics: a smoothly converging and tunable free-energy method. Phys Rev Lett. 2008;100:020603. doi: 10.1103/PhysRevLett.100.020603. [DOI] [PubMed] [Google Scholar]
- Barducci A, Bonomi M, Parrinello M. Metadynamics: metadynamics. Wiley Interdiscip Rev Comput Mol Sci. 2011;1:826–843. doi: 10.1002/wcms.31. [DOI] [Google Scholar]
- Borea PA, Varani K, Gessi S, Merighi S, Dal Piaz A, Gilli P, et al. Receptor binding thermodynamics at the neuronal nicotinic receptor. Curr Top Med Chem. 2004;4:361–368. doi: 10.2174/1568026043451410. [DOI] [PubMed] [Google Scholar]
- Bortolato A, Deflorian F, Weiss DR, Mason JS. Decoding the role of water dynamics in ligand-Protein unbinding: CRF 1 R as a test case. J Chem Inf Model. 2015;55:1857–1866. doi: 10.1021/acs.jcim.5b00440. [DOI] [PubMed] [Google Scholar]
- Branduardi D, Gervasio FL, Parrinello M. From A to B in free energy space. J Chem Phys. 2007;126:54103. doi: 10.1063/1.2432340. [DOI] [PubMed] [Google Scholar]
- Branduardi D, Bussi G, Parrinello M. Metadynamics with adaptive Gaussians. J Chem Theory Comput. 2012;8:2247–2254. doi: 10.1021/ct3002464. [DOI] [PubMed] [Google Scholar]
- Bui JM, Henchman RH, McCammon JA. The dynamics of ligand barrier crossing inside the acetylcholinesterase gorge. Biophys J. 2003;85:2267–2272. doi: 10.1016/S0006-3495(03)74651-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bussi G, Donadio D, Parrinello M. Canonical sampling through velocity rescaling. J Chem Phys. 2007;126:14101. doi: 10.1063/1.2408420. [DOI] [PubMed] [Google Scholar]
- Carpenter B, Nehmé R, Warne T, Leslie AGW, Tate CG. Structure of the adenosine A2A receptor bound to an engineered G protein. Nature. 2016;536:104–107. doi: 10.1038/nature18966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Congreve M, Andrews SP, Doré AS, Hollenstein K, Hurrell E, Langmead CJ, et al. Discovery of 1,2,4-triazine derivatives as adenosine A2A antagonists using structure based drug design. J Med Chem. 2012;55:1898–1903. doi: 10.1021/jm201376w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland RA. The drug–target residence time model: a 10-year retrospective. Nat Rev Drug Discov. 2015;15:87–95. doi: 10.1038/nrd.2015.18. [DOI] [PubMed] [Google Scholar]
- Copeland RA, Pompliano DL, Meek TD. Drug–target residence time and its implications for lead optimization. Nat Rev Drug Discov. 2006;5:730–739. doi: 10.1038/nrd2082. [DOI] [PubMed] [Google Scholar]
- Cuzzolin A, Sturlese M, Deganutti G, Salmaso V, Sabbadin D, Ciancetta A, et al. Deciphering the complexity of ligand-protein recognition pathways using supervised molecular dynamics (SuMD) simulations. J Chem Inf Model. 2016;56:687–705. doi: 10.1021/acs.jcim.5b00702. [DOI] [PubMed] [Google Scholar]
- Dahl G, Akerud T. Pharmacokinetics and the drug–target residence time concept. Drug Discov Today. 2013;18:697–707. doi: 10.1016/j.drudis.2013.02.010. [DOI] [PubMed] [Google Scholar]
- Darden T, York D, Pedersen L. Particle mesh Ewald: an N log(N) method for Ewald sums in large systems. J Chem Phys. 1993;98:10089–10092. doi: 10.1063/1.464397. [DOI] [Google Scholar]
- Doré AS, Robertson N, Errey JC, Ng I, Hollenstein K, Tehan B, et al. Structure of the adenosine A2A receptor in complex with ZM241385 and the xanthines XAC and caffeine. Structure. 2011;19:1283–1293. doi: 10.1016/j.str.2011.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror RO, Pan AC, Arlow DH, Borhani DW, Maragakis P, Shan Y, et al. Pathway and mechanism of drug binding to G-protein-coupled receptors. Proc Natl Acad Sci. 2011;108:13118–13123. doi: 10.1073/pnas.1104614108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du X, Li Y, Xia Y-L, Ai S-M, Liang J, Sang P, et al. Insights into protein-ligand interactions: mechanisms, models, and methods. Int J Mol Sci. 2016;17:144. doi: 10.3390/ijms17020144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federico S, Paoletta S, Cheong SL, Pastorin G, Cacciari B, Stragliotto S, et al. Synthesis and biological evaluation of a new series of 1,2,4-triazolo[1,5-a]-1,3,5-triazines as human A2A adenosine receptor antagonists with improved water solubility. J Med Chem. 2011;54:877–889. doi: 10.1021/jm101349u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Federico S, Ciancetta A, Porta N, Redenti S, Pastorin G, Cacciari B, et al. 5,7-Disubstituted-[1,2,4]triazolo[1,5- a][1,3,5]triazines as pharmacological tools to explore the antagonist selectivity profiles toward adenosine receptors. Eur J Med Chem. 2016;108:529–541. doi: 10.1016/j.ejmech.2015.12.019. [DOI] [PubMed] [Google Scholar]
- Fink JS, Weaver DR, Rivkees SA, Peterfreund RA, Pollack AE, Adler EM, et al. Molecular cloning of the rat A2 adenosine receptor: selective co-expression with D2 dopamine receptors in rat striatum. Brain Res Mol Brain Res. 1992;14:186–195. doi: 10.1016/0169-328X(92)90173-9. [DOI] [PubMed] [Google Scholar]
- Frederick KK, Marlow MS, Valentine KG, Wand AJ. Conformational entropy in molecular recognition by proteins. Nature. 2007;448:325–329. doi: 10.1038/nature05959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, et al. Glide: a new approach for rapid, accurate docking and scoring 1 method and assessment of docking accuracy. J Med Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Fukunishi H, Watanabe O, Takada S. On the Hamiltonian replica exchange method for efficient sampling of biomolecular systems: application to protein structure prediction. J Chem Phys. 2002;116:9058–9067. doi: 10.1063/1.1472510. [DOI] [Google Scholar]
- Gervasio FL, Laio A, Parrinello M. Flexible docking in solution using metadynamics. J Am Chem Soc. 2005;127:2600–2607. doi: 10.1021/ja0445950. [DOI] [PubMed] [Google Scholar]
- Ghosh E, Kumari P, Jaiman D, Shukla AK. Methodological advances: the unsung heroes of the GPCR structural revolution. Nat Rev Mol Cell Biol. 2015;16:69–81. doi: 10.1038/nrm3933. [DOI] [PubMed] [Google Scholar]
- Guo D, Pan AC, Dror RO, Mocking T, Liu R, Heitman LH, et al. Molecular basis of ligand dissociation from the adenosine A2A receptor. Mol Pharmacol. 2016;89:485–491. doi: 10.1124/mol.115.102657. [DOI] [PubMed] [Google Scholar]
- Guo D, Heitman LH, IJzerman AP. Kinetic aspects of the interaction between ligand and G protein-coupled receptor: the case of the adenosine receptors. Chem Rev. 2017;117:38–66. doi: 10.1021/acs.chemrev.6b00025. [DOI] [PubMed] [Google Scholar]
- Halgren TA, Murphy RB, Friesner RA, Beard HS, Frye LL, Pollard WT, et al. Glide: a new approach for rapid, accurate docking and scoring 2 enrichment factors in database screening. J Med Chem. 2004;47:1750–1759. doi: 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Hamelberg D, Mongan J, McCammon JA. Accelerated molecular dynamics: a promising and efficient simulation method for biomolecules. J Chem Phys. 2004;120:11919–11929. doi: 10.1063/1.1755656. [DOI] [PubMed] [Google Scholar]
- Hino T, Arakawa T, Iwanari H, Yurugi-Kobayashi T, Ikeda-Suno C, Nakada-Nakura Y, et al. G-protein-coupled receptor inactivation by an allosteric inverse-agonist antibody. Nature. 2012 doi: 10.1038/nature10750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hothersall JD, Brown AJ, Dale I, Rawlins P. Can residence time offer a useful strategy to target agonist drugs for sustained GPCR responses? Drug Discov Today. 2016;21:90–96. doi: 10.1016/j.drudis.2015.07.015. [DOI] [PubMed] [Google Scholar]
- Hulme EC, Trevethick MA. Ligand binding assays at equilibrium: validation and interpretation: equilibrium binding assays. Br J Pharmacol. 2010;161:1219–1237. doi: 10.1111/j.1476-5381.2009.00604.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isralewitz B, Gao M, Schulten K. Steered molecular dynamics and mechanical functions of proteins. Curr Opin Struct Biol. 2001;11:224–230. doi: 10.1016/S0959-440X(00)00194-9. [DOI] [PubMed] [Google Scholar]
- Jaakola V-P, Griffith MT, Hanson MA, Cherezov V, Chien EYT, Lane JR, et al. The 26 angstrom crystal structure of a human A2A adenosine receptor bound to an antagonist. Science. 2008;322:1211–1217. doi: 10.1126/science.1164772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson KA, Gao Z-G. Adenosine receptors as therapeutic targets. Nat Rev Drug Discov. 2006;5:247–264. doi: 10.1038/nrd1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson MP, Pincus DL, Rapp CS, Day TJF, Honig B, Shaw DE, et al. A hierarchical approach to all-atom protein loop prediction. Proteins Struct Funct Bioinform. 2004;55:351–367. doi: 10.1002/prot.10613. [DOI] [PubMed] [Google Scholar]
- Jakalian A, Jack DB, Bayly CI. Fast, efficient generation of high-quality atomic charges AM1-BCC model: II parameterization and validation. J Comput Chem. 2002;23:1623–1641. doi: 10.1002/jcc.10128. [DOI] [PubMed] [Google Scholar]
- Jämbeck JPM, Lyubartsev AP. Derivation and systematic validation of a refined all-atom force field for phosphatidylcholine lipids. J Phys Chem B. 2012;116:3164–3179. doi: 10.1021/jp212503e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenakin T, Christopoulos A. Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat Rev Drug Discov. 2012;12:205–216. doi: 10.1038/nrd3954. [DOI] [PubMed] [Google Scholar]
- Laio A, Parrinello M. Escaping free-energy minima. Proc Natl Acad Sci. 2002;99:12562–12566. doi: 10.1073/pnas.202427399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laio A, Rodriguez-Fortea A, Gervasio FL, Ceccarelli M, Parrinello M. Assessing the accuracy of metadynamics. J Phys Chem B. 2005;109:6714–6721. doi: 10.1021/jp045424k. [DOI] [PubMed] [Google Scholar]
- Lebon G, Warne T, Edwards PC, Bennett K, Langmead CJ, Leslie AGW, et al. Agonist-bound adenosine A2A receptor structures reveal common features of GPCR activation. Nature. 2011;474:521–525. doi: 10.1038/nature10136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lebon G, Edwards PC, Leslie AGW, Tate CG. Molecular determinants of CGS21680 binding to the human adenosine A2A receptor. Mol Pharmacol. 2015;87:907–915. doi: 10.1124/mol.114.097360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W. Possible pathway(s) of testosterone egress from the active site of cytochrome P450 2B1: a steered molecular dynamics simulation. Drug Metab Dispos. 2005;33:910–919. doi: 10.1124/dmd.105.004200. [DOI] [PubMed] [Google Scholar]
- Lindorff-Larsen K, Piana S, Palmo K, Maragakis P, Klepeis JL, Dror RO, et al. Improved side-chain torsion potentials for the Amber ff99SB protein force field. Proteins. 2010 doi: 10.1002/prot.22711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Chun E, Thompson AA, Chubukov P, Xu F, Katritch V, et al. Structural basis for allosteric regulation of GPCRs by sodium ions. Science. 2012;337:232–236. doi: 10.1126/science.1219218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luitz MP, Zacharias M. Protein-ligand docking using Hamiltonian replica exchange simulations with soft core potentials. J Chem Inf Model. 2014;54:1669–1675. doi: 10.1021/ci500296f. [DOI] [PubMed] [Google Scholar]
- Marchi M, Ballone P. Adiabatic bias molecular dynamics: a method to navigate the conformational space of complex molecular systems. J Chem Phys. 1999;110:3697–3702. doi: 10.1063/1.478259. [DOI] [Google Scholar]
- Mollica L, Decherchi S, Zia SR, Gaspari R, Cavalli A, Rocchia W. Kinetics of protein-ligand unbinding via smoothed potential molecular dynamics simulations. Sci Rep. 2015;5:11539. doi: 10.1038/srep11539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollica L, Theret I, Antoine M, Perron-Sierra F, Charton Y, Fourquez J-M, et al. Molecular dynamics simulations and kinetic measurements to estimate and predict protein-ligand residence times. J Med Chem. 2016;59:7167–7176. doi: 10.1021/acs.jmedchem.6b00632. [DOI] [PubMed] [Google Scholar]
- Nguyen ATN, Baltos J-A, Thomas T, Nguyen TD, Munoz LL, Gregory KJ, et al. Extracellular loop 2 of the adenosine A1 receptor has a key role in orthosteric ligand affinity and agonist efficacy. Mol Pharmacol. 2016;90:703–714. doi: 10.1124/mol.116.105007. [DOI] [PubMed] [Google Scholar]
- Pan AC, Borhani DW, Dror RO, Shaw DE. Molecular determinants of drug–receptor binding kinetics. Drug Discov Today. 2013;18:667–673. doi: 10.1016/j.drudis.2013.02.007. [DOI] [PubMed] [Google Scholar]
- Parrinello M, Rahman A. Polymorphic transitions in single crystals: a new molecular dynamics method. J Appl Phys. 1981;52:7182–7190. doi: 10.1063/1.328693. [DOI] [Google Scholar]
- Patel JS, Berteotti A, Ronsisvalle S, Rocchia W, Cavalli A. Steered molecular dynamics simulations for studying protein-ligand interaction in cyclin-dependent kinase 5. J Chem Inf Model. 2014;54:470–480. doi: 10.1021/ci4003574. [DOI] [PubMed] [Google Scholar]
- Pierce LCT, Salomon-Ferrer R, de Augusto Oliveira F, McCammon JA, Walker RC. Routine access to millisecond time scale events with accelerated molecular dynamics. J Chem Theory Comput. 2012;8:2997–3002. doi: 10.1021/ct300284c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polosa R, Blackburn MR. Adenosine receptors as targets for therapeutic intervention in asthma and chronic obstructive pulmonary disease. Trends Pharmacol Sci. 2009;30:528–535. doi: 10.1016/j.tips.2009.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Radić Z, Kirchhoff PD, Quinn DM, McCammon JA, Taylor P. Electrostatic influence on the kinetics of ligand binding to acetylcholinesterase distinctions between active center ligands and fasciculin. J Biol Chem. 1997;272:23265–23277. doi: 10.1074/jbc.272.37.23265. [DOI] [PubMed] [Google Scholar]
- Rich RL, Myszka DG. Grading the commercial optical biosensor literature—Class of 2008: “The Mighty Binders”. J Mol Recognit. 2009;23:1–64. doi: 10.1002/jmr.1004. [DOI] [PubMed] [Google Scholar]
- Rich RL, Errey J, Marshall F, Myszka DG. Biacore analysis with stabilized G-protein-coupled receptors. Anal Biochem. 2011;409:267–272. doi: 10.1016/j.ab.2010.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richardson PJ, Kase H, Jenner PG. Adenosine A2A receptor antagonists as new agents for the treatment of Parkinson’s disease. Trends Pharmacol Sci. 1997;18:338–344. doi: 10.1016/S0165-6147(97)01096-1. [DOI] [PubMed] [Google Scholar]
- Rivera-Oliver M, Díaz-Ríos M. Using caffeine and other adenosine receptor antagonists and agonists as therapeutic tools against neurodegenerative diseases: a review. Life Sci. 2014;101:1–9. doi: 10.1016/j.lfs.2014.01.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos H, Karlsson R, Nilshans H, Persson A. Thermodynamic analysis of protein interactions with biosensor technology. J Mol Recognit. 1998;11:204–210. doi: 10.1002/(SICI)1099-1352(199812)11:1/6<204::AID-JMR424>3.0.CO;2-T. [DOI] [PubMed] [Google Scholar]
- Sabbadin D, Moro S. Supervised molecular dynamics (SuMD) as a helpful tool to depict GPCR–ligand recognition pathway in a nanosecond time scale. J Chem Inf Model. 2014;54:372–376. doi: 10.1021/ci400766b. [DOI] [PubMed] [Google Scholar]
- Sahlan M, Zako T, Tai PT, Ohtaki A, Noguchi K, Maeda M, et al. Thermodynamic characterization of the Interaction between prefoldin and group II chaperonin. J Mol Biol. 2010;399:628–636. doi: 10.1016/j.jmb.2010.04.046. [DOI] [PubMed] [Google Scholar]
- Schmidtke P, Luque FJ, Murray JB, Barril X. Shielded hydrogen bonds as structural determinants of binding kinetics: application in drug design. J Am Chem Soc. 2011;133:18903–18910. doi: 10.1021/ja207494u. [DOI] [PubMed] [Google Scholar]
- Segala E, Guo D, Cheng RKY, Bortolato A, Deflorian F, Doré AS, et al. Controlling the dissociation of ligands from the adenosine A2A receptor through modulation of salt bridge strength. J Med Chem. 2016;59:6470–6479. doi: 10.1021/acs.jmedchem.6b00653. [DOI] [PubMed] [Google Scholar]
- Seibt BF, Schiedel AC, Thimm D, Hinz S, Sherbiny FF, Müller CE. The second extracellular loop of GPCRs determines subtype-selectivity and controls efficacy as evidenced by loop exchange study at A2 adenosine receptors. Biochem Pharmacol. 2013;85:1317–1329. doi: 10.1016/j.bcp.2013.03.005. [DOI] [PubMed] [Google Scholar]
- Shaw DE, Dror RO, Salmon JK, Grossman JP, Mackenzie KM, Bank JA et al (2009) Millisecond-scale molecular dynamics simulations on Anton. In: proc. conf. high perform. Comput Netw Storage Anal SC 09 no. c: 1–11
- Shepherd CA, Hopkins AL, Navratilova I. Fragment screening by SPR and advanced application to GPCRs. Prog Biophys Mol Biol. 2014;116:113–123. doi: 10.1016/j.pbiomolbio.2014.09.008. [DOI] [PubMed] [Google Scholar]
- Sinko W, Miao Y, de Oliveira CAF, McCammon JA. Population based reweighting of scaled molecular dynamics. J Phys Chem B. 2013;117:12759–12768. doi: 10.1021/jp401587e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley N, Pardo L, De Fabritiis G. The pathway of ligand entry from the membrane bilayer to a lipid G protein-coupled receptor. Sci Rep. 2016;6:22639. doi: 10.1038/srep22639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone TW, Ceruti S, Abbracchio MP. Adenosine receptors and neurological disease: neuroprotection and neurodegeneration. In: Wilson CN, Mustafa SJ, editors. Adenosine receptors in health and disease, vol 193. Berlin: Springer; 2009. pp. 535–587. [DOI] [PubMed] [Google Scholar]
- Torrie GM, Valleau JP. Nonphysical sampling distributions in Monte Carlo free-energy estimation: umbrella sampling. J Comput Phys. 1977;23:187–199. doi: 10.1016/0021-9991(77)90121-8. [DOI] [Google Scholar]
- Vauquelin G, Bostoen S, Vanderheyden P, Seeman P. Clozapine, atypical antipsychotics, and the benefits of fast-off D2 dopamine receptor antagonism. Naunyn Schmiedebergs Arch Pharmacol. 2012;385:337–372. doi: 10.1007/s00210-012-0734-2. [DOI] [PubMed] [Google Scholar]
- Wang J, Wolf RM, Caldwell JW, Kollman PA, Case DA. Development and testing of a general amber force field. J Comput Chem. 2004;25:1157–1174. doi: 10.1002/jcc.20035. [DOI] [PubMed] [Google Scholar]
- Wang K, Chodera JD, Yang Y, Shirts MR. Identifying ligand binding sites and poses using GPU-accelerated Hamiltonian replica exchange molecular dynamics. J Comput Aided Mol Des. 2013;27:989–1007. doi: 10.1007/s10822-013-9689-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf MG, Hoefling M, Aponte-Santamaría C, Grubmüller H, Groenhof G. g_membed: efficient insertion of a membrane protein into an equilibrated lipid bilayer with minimal perturbation. J Comput Chem. 2010;31:2169–2174. doi: 10.1002/jcc.21507. [DOI] [PubMed] [Google Scholar]
- Xu F, Wu H, Katritch V, Han GW, Jacobson KA, Gao Z-G, et al. Structure of an agonist-bound human A2A adenosine receptor. Science. 2011;332:322–327. doi: 10.1126/science.1202793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu R, Tabassum N, Jiang T. Investigation of α-conotoxin unbinding using umbrella sampling. Bioorg Med Chem Lett. 2016;26:1296–1300. doi: 10.1016/j.bmcl.2016.01.013. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.