

Abstract
We report on an efficient strategy for the asymmetric synthesis of trifluoromethyl-substituted cyclopropanes by means of myoglobin-catalyzed olefin cyclopropanation reactions in the presence of 2-diazo-1,1,1-trifluoroethane (CF3CHN2) as the carbene donor. These transformations were realized using a two-compartment setup in which ex situ generated gaseous CF3CHN2 is processed by engineered myoglobin catalysts expressed in bacterial cells. This approach was successfully applied to afford a variety of trans-1-trifluoromethyl-2-arylcyclopropanes in high yields (61–99%) and excellent diastereo- and enantioselectivity (97–99.9% de and ee). Furthermore, mirror-image forms of these products could be obtained using myoglobin variants featuring stereodivergent selectivity. These reactions provide a convenient and effective biocatalytic route to the stereoselective synthesis of key fluorinated building blocks of high value for medicinal chemistry and drug discovery. This work expands the range of carbene-mediated transformations accessible via metalloprotein catalysts and introduces a potentially very general strategy for exploiting gaseous and/or hard-to-handle carbene donor reagents in biocatalytic carbene transfer reactions.
Trifluoromethyl-substituted cyclopropanes constitute attractive synthons in medicinal chemistry as they combine the conformational rigidity of three-membered rings with the unique and often highly beneficial features of fluorinated substituents.1 Reflecting this notion, these structural motifs have represented key building blocks for the design and development of several bioactive molecules and investigational drugs.1a Various methods have been investigated to afford these structures, most of which involve starting materials that already incorporate the trifluoromethyl group.2 A convenient and most direct strategy to access this class of compounds is through the addition of trifluoromethylcarbene to an olefin. Early studies in this area have entailed the use of trifluoromethylcarbene generated by photolytic decomposition of 2-diazo-1,1,1-trifluorethane (CF3CHN2), resulting in a mixture of products.3 More synthetically useful strategies for olefin trifluoromethylcyclopropanation with this reagent were made available only recently using transition metal catalysis.4 To date, however, only a few studies have addressed the problem of developing enantioselective variants of these transformations.4a,5 In a first report, Simmoneaux and coworkers described the use of chiral metalloporphyrins for the asymmetric cyclopropanation of styrenes in the presence of 2-diazo-1,1,1-trifluoroethane, but only moderate enantioselectivity (30–79% ee) was observed using this system.4a Higher selectivity was more recently achieved in similar reactions using Co(III)-salen complexes by Carreira and coworkers.5 Despite these advances, the development of highly stereoselective strategies for olefin cyclopropanation with 2-diazo-1,1,1-trifluoroethane as the carbene donor remains an outstanding challenge. Here, we report a simple and efficient biocatalytic approach for promoting these transformations that hinges upon the use of engineered myoglobin variants in combination with a compartmentalized reaction setup. This strategy could be applied to the conversion of a broad range of vinylarene substrates, providing access to trifluoromethyl-substituted cyclopropanes with high diastereo- and enantioselectivity as well as stereocomplementary selectivity.
Our group has recently reported the development of engineered myoglobin catalysts for promoting a variety of carbene transfer reactions,6 including olefin cyclopropanation.7 These transformations are believed to be mediated by an electrophilic heme-bound carbenoid species generated upon reaction of a diazo compound with the heme cofactor embedded in the protein.7a This species can react with a number of nucleophiles (olefins, amines, thiols, phosphines) in order to form new C-C and C-heteroatom bonds, with the protein active site providing an asymmetric environment to affect the stereoselectivity of the carbene transfer process.6–7 In addition to myoglobin (Mb), other biocatalysts8 have been investigated for promoting carbene transfer reactions, but the scope of biocatalytic carbene transfer reactions has so far been restricted to α-diazoacetates as carbene donor reagents. Given the high reactivity of myoglobin variants toward the acceptor-only carbene donor reagent ethyl diazoacetate (EDA, 1), we envisioned that 2-diazo-1,1,1-trifluoroethane (DTE, 2) could provide a potentially viable carbene precursor for myoglobin-catalyzed cyclopropanations. A well-known challenge associated with the use of DTE in carbene transfer reactions, however, is the difficulty in handling this reagent due to its high toxicity and volatility. In situ generation of DTE via diazotization of 2,2,2-trifluoroethylamine has provided an attractive approach for utilizing this reagent in various reaction manifolds,4c,9 but these conditions are too harsh for protein-based catalysts. To overcome this problem, we envisioned the possibility to utilize a two-compartment reaction setup, in which ex situ produced DTE from a ‘reagent generation chamber’ is carried through a solution containing the myoglobin catalyst by inert gas (Figure 1). This system would effectively segregate the two incompatible reactions, thereby preserving the stability of the biocatalyst while eliminating the need for isolating and handling DTE.
Figure 1.
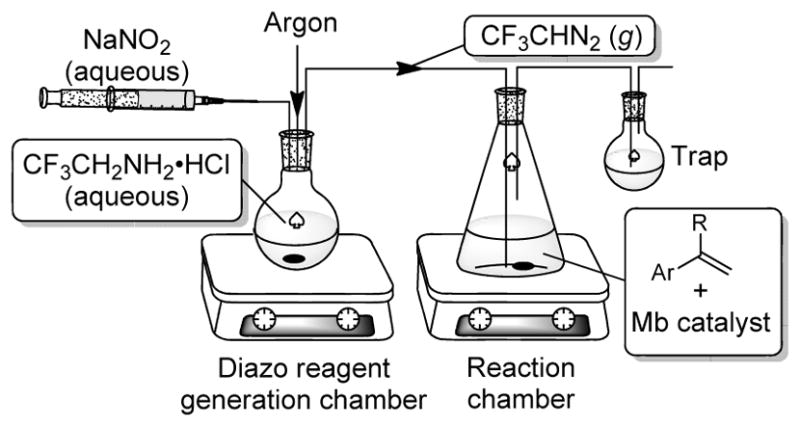
Compartmentalized reaction setup for coupling myoglobin-catalyzed cyclopropanation with ex situ generation of 2-diazo-1,1,1-trifluoroethane (DTE).
To implement this approach, we initially tested the possibility to carry out the myoglobin-catalyzed cyclopropanation of p-chloro-styrene (3) in the presence of ex situ generated EDA. The latter was produced by diazotization of glycine ethyl ester (4) in the presence of sodium nitrite and acid (Table 1).10 For these experiments, the Mb(H64V,V64A) variant was chosen as the catalyst based on its high activity and selectivity toward cyclopropanation of aryl-substituted olefins with EDA.7a Initial tests involving purified Mb(H64V,V64A) in the reaction chamber produced only small amounts of the cyclopropanation product 6 (Table 1, Entry 1), a result we attributed to protein inactivation by the gas stream flowing through the reaction mixture. Given our recent finding that Mb-catalyzed cyclopropanation reactions can be realized using whole cell systems,7b a suspension of E. coli cells expressing the Mb(H64V,V64A) variant was used as the biocatalytic system in the reaction chamber. Gratifyingly, this modified setup resulted in the accumulation of significantly larger amounts of 6 (Table 1, Entry 2). Further improvement of the reaction was possible by varying the cell density of the whole cell transformation and the molar ratio between the olefin substrate and the EDA precursor (Table 1, Entries 3–4). Under optimized conditions (i.e., OD600 = 40, corresponding to a cell density of ~3.5 g cdw L−1, 10 equivalents of glycine ethyl ester relative to the olefin), nearly quantitative conversion of the p-chloro-styrene substrate to 6 was observed within 12 hours (Table 1, Entry 5). Importantly, neither the reaction setup nor the whole cell setting was found to have a noticeable impact on the stereoselectivity of the Mb(H64V,V64A)-catalyzed reaction, leading to the formation of the trans-(1S,2S)-configured cyclopropanation product in excellent diastereo- and enantiomeric excess (98.5–99.9% de, 99.8–99.9% ee).
Table 1.
Mb(H64V,V68A)-catalyzed cyclopropanation of p-chlorostyrene in the presence of ex situ generated EDA and DTE.a
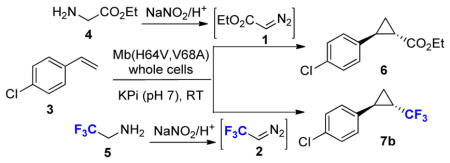
| |||||||
|---|---|---|---|---|---|---|---|
|
| |||||||
| Entry | Catal. | Prod. | Equiv 4 or 5b | Yieldc | TON | % de | % ee |
| 1 | protein | 6 | 2 | 4% | 180 | 99.9 | 99.8 |
| 2 | cellsd | 6 | 2 | 47% | 560 | 97.2 | 99.9 |
| 3 | cells | 6 | 5 | 80% | 365 | 99.9 | 99.8 |
| 4 | cells | 6 | 10 | 75% | 340 | 98.5 | 99.9 |
| 5 | cells | 6 | 10 | >99%e | 500 | 99.9 | 99.9 |
| 6 | protein | 7b | 5 | 22% | 1,110 | 98.5 | 99.9 |
| 7 | cells | 7b | 5 | 92% (67%) | 520 | 99.9 | 99.9 |
Reactions were carried out at a 20 mL-scale with 30 mM 4-chloro-styrene (3), purified Mb variant (20 μM) or Mb(H64V,V68A)-expressing E. coli (BL21(DE3)) cells (OD600 = 40) in KPi 50 mM (pH 7), RT, 5 hours. Diazo reagent was supplied using indicated equivalents of 4 or 5 (slow addition over 4 hours).
Relative to olefin.
As determined by GC using calibration curves generated with isolated product as reference. Isolated yields are reported in parentheses.
OD600 = 20.
Reaction time: 12 hours.
Based on these promising results, we next investigated the possibility to supply the Mb catalyst in the whole cell system with gaseous DTE as the carbene donor. In this case, 2,2,2-trifluoroethylamine (5) was subjected to the diazotization reaction in the reagent generation chamber (Table 1). To our delight, excellent conversion of p-chloro-styrene to the desired trifluoromethyl-substituted cyclopropane 7b (92%) was observed in less than five hours (Entry 7, Table 1) and using only 5 equivalents of the diazo reagent precursor 5. Because of the higher volatility of 7b compared to 6, product recovery in this case was improved by addition of a water trap downstream of the reaction vessel. No product was detected using cells not expressing the Mb variant, thus demonstrating the direct role of the hemoprotein in mediating the carbene transfer reaction. Under the applied conditions, the Mb variant was estimated to support about 520 catalytic turnovers (TON), as determined based the hemoprotein concentration in the reaction mixture measured via a CO-binding assay. This TON value is higher than that observed in the presence of ex situ generated EDA under identical operational conditions (Entry 7 vs. 3, Table 1). Higher yields (GC) and TON were also observed in the presence of DTE vs. EDA when purified Mb(H64V,V64A) was applied as the catalyst (Entry 6 vs. 1). These differences are likely to reflect the inherent reactivity of the diazo reagents as well as differences in the rate and efficiency with which the ex situ generated reagents are transferred from the generation chamber to the reaction vessel (Figure 1). Importantly, Mb(H64V,V68A)-catalyzed synthesis of 7b was found to occur with excellent diastereo- and enantioselectivity (99.9% de and ee).
To explore the scope of this reaction, a series of styrene derivatives and aryl-substituted olefins was investigated. As shown by the data in Table 2, all of these substrates could be efficiently converted to the corresponding trifluoromethyl-containing cyclopropanes (54–99%) using Mb(H64,V68A)-expressing cells in the two-compartment setup described in Figure 1. These included meta- and para-substituted styrene derivatives containing both electron-donating and electron-withdrawing groups. 4-Nitrostyrene displayed lower reactivity toward Mb-catalyzed cyclopropanation than electronrich styrene derivatives (e.g., products 9b, 11b, 12b), a result consistent with the electrophilic character of the putative heme-carbenoid intermediate expected to mediate these reactions.7a As demonstrated by the results with 13b and 14b, the Mb-catalyzed transformation could be readily extended to other aryl-substituted olefins such as 2-vinyl-naphthalene and 3-(propen-2-yl)thiophene, supporting the broad substrate scope of the Mb(H64,V64A) variant. In contrast to protocols involving chiral metalloporphyrins,4a the biocatalytic reactions proceed efficiently using the olefin as the limiting reagent, which increases their attractiveness for the transformation of valuable starting materials. Importantly, using Mb(H64V,V68A)-expressing cells, the cyclopropanation product was afforded in most cases with high diastereosteroselectivity (99.9% de) and enantioselectivity (92–99.9% ee). As an exception, moderate enantioselectivity (28–31% ee) was observed with the toluene derivatives 11a and 12a (Table 2, Entries 4–5). For these compounds, however, excellent diastereo- and enantioselectivity (97–99.9% de and ee) could be achieved using cells expressing Mb(H64V,V68G), a Mb variant previously identified as having similar selectivity properties to Mb(H64V,V68A) in cyclopropanation reactions with EDA.7b Altogether, the stereoselectivity offered by these Mb-based catalysts exceeds those provided by the most selective synthetic catalysts reported to date for the asymmetric synthesis of related CF3-containing cyclopropanes (84–91% ee).5 Compared to the latter, the Mb-catalyzed transformations also involve shorter reaction times (5 vs. 14 hours) and higher catalytic efficiency (e.g., 520 vs. ~10 TON for 7b). The use of whole cells bypasses the need for purification and isolation of the myoglobin catalyst, which further simplifies the overall procedure from a technical standpoint. From these reactions, the CF3-substituted cyclopropane products could be isolated in good yields (43–82%, Table 2). The synthetic utility of these transformations was further evidenced by a larger scale reaction with p-methoxy-styrene, from which 0.1 g of 9b was obtained in 76% isolated yield and excellent diastereo- and enantiomeric excess (99.9% de, 99.9% ee).
Table 2.
Substrate scope of myoglobin-catalyzed olefin cyclopropanation with ex situ generated DTE.a
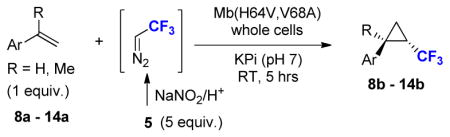
| |||||
|---|---|---|---|---|---|
|
| |||||
| Entry | Product | OD600 | Yield b | % de | % ee |
| 1 |
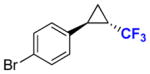 8b |
80 | 69% (68%) | 99.9 | 99.9 |
| 2 |
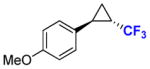 9b |
80 | 92% (76%) | 99.9 | 99.9 |
| 3 |
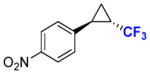 10b |
80 | 54% (43%) | 99.9 | 99.9 |
| 4 |
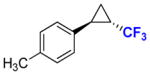 11b |
40 80 |
85% 88% (78%) |
96 96 c |
31 97c |
| 5 |
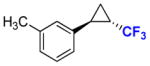 12b |
40 80 |
76% >99% (82%) |
99.8 99.9c |
28 99.9c |
| 6 |
 13b |
80 | 70% (58%) | 99.9 | 92 |
| 7 |
 14b |
40 | >99% (71%) | 99.9 | 99.9 |
Reactions were carried out at 20 mL-scale with Mb(H64V,V68A)-expressing E. coli at indicated cell density, 8–30 mM olefin in KPi 50 mM (pH 7), room temperature, 5 hours. DTE was supplied using 5 equiv. 5 (slow addition over 4 hours).
As determined by GC using calibration curves generated with isolated product as reference. Isolated yields are reported in parentheses.
Using cells expressing Mb(H64V,V68G).
To elucidate the stereoselectivity of the Mb(H64V,V68A) catalyst in these reactions, product 10b was crystallized and determined to have a trans-(1S,2S) configuration by X-ray diffraction analysis (Figure 2A). The same configuration was indirectly assigned to 7b and the other cyclopropanation products of Table 2 based on the similar order of elution for the corresponding enantiomers upon resolution via chiral GC or SFC (see Figure S2). The stereoselectivity of Mb(H64V,V68A) in the cyclopropanation reactions with DTE thus mirrors that observed in the reactions with EDA.7a Leveraging this insight and in consideration of the structural and electronic similarities between EDA and DTE, we propose a stereochemical scenario analogous to that proposed for Mb(H64V,V68A)-catalyzed cyclopropanation of aryl-substituted olefins with EDA7a. Specifically, the ‘cavity’ created by the Val68→Ala substitution (Figure S1) is expected to facilitate orientation of the bulkier –CF3 group (cp. to –H) in the heme-bound carbene intermediate in proximity to the N2 atom of the heme cofactor (Figure 2B). This active site configuration enforces high facial selectivity during end-on attack of the olefin to this reactive species, giving rise to the observed trans-(1S,2S)-configured product. Consistent with this scenario, the Mb(H64V,V68G) variant, which features a similar ‘large-to-small’ mutation at position 68 (Figure S1), is also trans-(1S,2S) selective (Table 2, Entries 4–5). An implication of the proposed model is that the aryl ring of the olefin projects toward the solvent-exposed side of the active site, a molecular arrangement that is consistent with Mb(H64V,V68A) ability to process structurally diverse vinylarene substrates with consistently high trans-(1S,2S) selectivity (Table 2).
Figure 2.
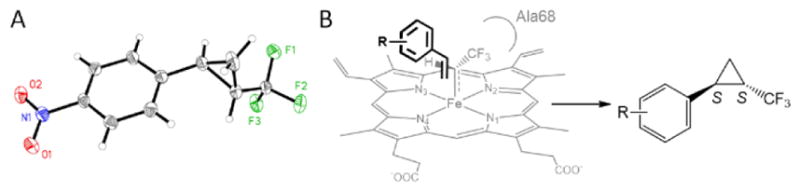
A) Crystal structure of (1S,2S)-2-(trifluoromethyl)cyclopropyl)-4-nitro-benzene product (10b). B) Plausible geometry for vinylarene attack to heme-bound trifluoromethylcarbene intermediate leading to the (1S,2S)-configured cyclopropane product.
Given the parallelism between the stereoselectivity exhibited by Mb(H64V,V68A) with EDA and DTE, we surmised that the stereoselectivity of Mb variants in cyclopropanation reactions with EDA could serve as a general predictor for their stereoselectivity in the cyclopropanation of related substrates with DTE. Supporting this notion, a very good correlation (R2 = 0.911) was found between the enantiomeric excess exhibited by a series of active site Mb variants in the cyclopropanation of p-methoxy-styrene with EDA vs. that of parallel reactions with DTE as the carbene donor (Figure 3A). In addition to Mb(H64V,V68A), this panel comprised other Mb variants with high trans-(1S,2S)-selectivity (Mb(H64V,V68G), Mb(H64V,V68S)) as well as a series of recently developed Mb catalysts with high trans-(1R,2R)-selectivity in olefin cyclopropanation with EDA (variants RR1 through RR5).7b Mb variants that span a range of enantiose-lectivity values from 13% ee (wild-type Mb) to 86% ee (Mb(V68A)) in the latter reaction were included as additional references (Table S1). As illustrated by the data of Figure 3A and Table S1, the diastereo- and enantioselectivity of the trifluoromethylcarbene transfer reaction was found to closely mirror the diastereo- and enantioselectivity of these Mb variants in the α-diazoester carbene transfer reaction (average IΔ(de)I = 3.3%; average IΔ(ee)I = 14.4%; Table S1). Based on these analyses, RR2 (= Mb(H64V,V68L,L29T)) was selected as a promising candidate for gaining access to the mirror-image forms of products 7b-14b. Gratifyingly, reactions with RR2-expressing E. coli cells produced the desired trans-(1R,2R) trifluoromethyl-substituted cyclopropanes 7c-14c with excellent diastereoselectivity (98% de for 9c, >99.5% de for the others) and high enantioselectivity (80–92% ee; Figure 3B; Figure S2). As an exception, 13c was obtained only in 21% ee. However, this compound could be synthesized in higher enantiomeric excess (58% ee) using RR4, as anticipated by the high trans-(1R,2R)-selectivity of this Mb variants in olefin cyclopropanation with EDA (Figure 3A). Altogether, these experiments demonstrate the versatility of the Mb scaffold toward providing access to both enantiomeric forms of the target trifluoromethyl-containing cyclopropanes. They also support the predictable reactivity of these biocatalysts in the presence of structurally and electronically related carbene donors.
Figure 3. Stereocomplementary selectivity.
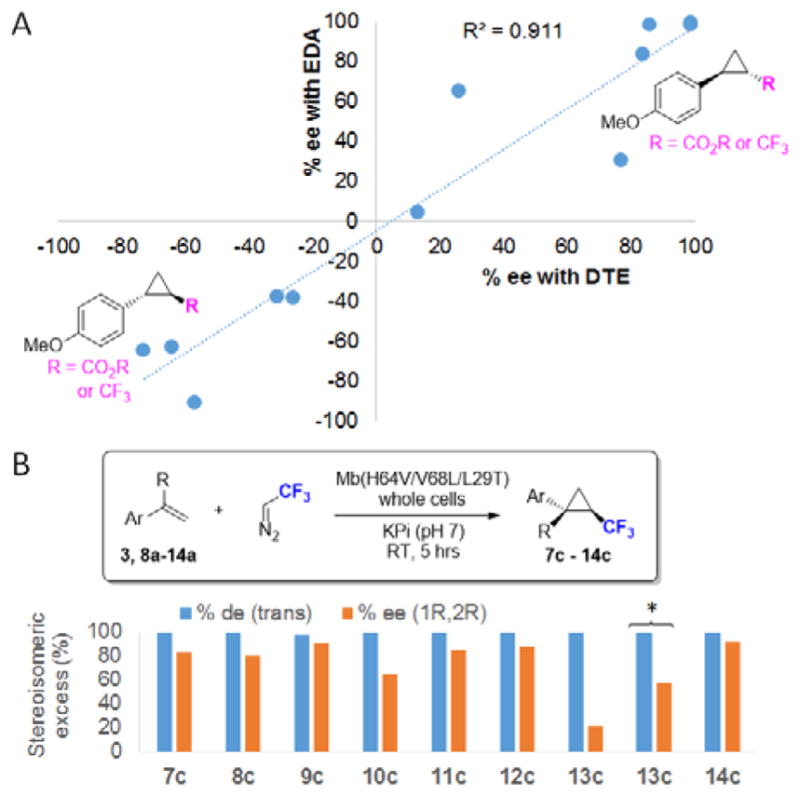
A) Correlation between enantioselectivity of Mb variants (n = 12) in cyclopropanation of p-methoxy-styrene with EDA vs. DTE. See Table S1 for additional data. B) Diastereo- and enantioselectivity of trans-(1R,2R)-selective Mb variant RR2 in cyclopropanation of aryl-substituted olefins in the presence of DTE. See Table S2 for details. * = with RR4-expressing cells.
In summary, we have developed a biocatalytic strategy for the asymmetric synthesis of trifluoromethyl-substituted cyclopro-panes via myoglobin-catalyzed addition of trifluoromethylcarbene to olefins. These transformations could be applied to a variety of vinylarene substrates, offering unprecedented levels of diastereo- and enantioselectivity. Furthermore, both enantiomers of the target CF3-containing products could be accessed using Mb catalysts with complementary stereoselectivity, whose choice was guided by their reactivity with EDA. The reactions presented here provide access to enantioenriched fluorinated building blocks of high value for medicinal chemistry. This study provides a first-time demonstration that carbene donor reagents other than α-diazoesters can be engaged in biocatalytic carbene transfer reactions. This finding, combined with the demonstrated feasibility of coupling these reactions with ex situ generated diazo compounds, is anticipated to enable extension of the present approach to a variety of other carbene precursors in order to expand the scope of carbene-mediated transformations accessible with myoglobins and other metalloprotein catalysts. Further studies in this direction are currently underway in our laboratory.
Supplementary Material
Acknowledgments
This work was supported in part by the U.S. National Institute of Health grant GM098628 and in part by the National Science Foundation grant CHE-1609550. The authors are grateful to Dr. William Brennessel (U. Rochester) and Dr. Eric Reinheimer (Rigaku Oxford Diffraction) for assistance with crystallographic analyses.
Footnotes
Supporting information includes Supplementary Tables and Figures, experimental procedures, compound characterization data andcrystallographic data.
References
- 1.(a) Grygorenko OO, Artamonov OS, Komarov IV, Mykhailiuk PK. Tetrahedron. 2011;67:803. [Google Scholar]; (b) Bos M, Poisson T, Pannecoucke X, Charette AB, Jubault P. Chem Eur J. 2017;23 doi: 10.1002/chem.201604564. in press. [DOI] [PubMed] [Google Scholar]
- 2.(a) Katagiri T, Yamaji S, Handa M, Irie M, Uneyama K. Chem Commun. 2001:2054. doi: 10.1039/b105602f. [DOI] [PubMed] [Google Scholar]; (b) Wang Y, Zhao XM, Li YH, Lu L. Tetrahedron Lett. 2004;45:7775. [Google Scholar]; (c) Risse J, Fernandez-Zumel MA, Cudre Y, Severin K. Org Lett. 2012;14:3060. doi: 10.1021/ol3011369. [DOI] [PubMed] [Google Scholar]; (d) Duncton MAJ, Singh R. Org Lett. 2013;15:4284. doi: 10.1021/ol401636d. [DOI] [PubMed] [Google Scholar]; (e) Zhu CL, Yang LJ, Li S, Zheng Y, Ma JA. Org Lett. 2015;17:3442. doi: 10.1021/acs.orglett.5b01450. [DOI] [PubMed] [Google Scholar]
- 3.Atherton JH, Fields R. J Chem Soc C. 1967:1450. [Google Scholar]
- 4.(a) Le Maux P, Juillard S, Simonneaux G. Synthesis. 2006:1701. [Google Scholar]; (b) Mykhailiuk PK, Afonin S, Ulrich AS, Komarov IV. Synthesis. 2008:1757.; (c) Morandi B, Carreira EM. Angew Chem Int Ed. 2010;49:4294. doi: 10.1002/anie.201000787. [DOI] [PubMed] [Google Scholar]
- 5.Morandi B, Mariampillai B, Carreira EM. Angew Chem Int Ed. 2011;50:1101. doi: 10.1002/anie.201004269. [DOI] [PubMed] [Google Scholar]
- 6.(a) Sreenilayam G, Fasan R. Chem Commun. 2015;51:1532. doi: 10.1039/c4cc08753d. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tyagi V, Bonn RB, Fasan R. Chem Sci. 2015;6:2488. doi: 10.1039/c5sc00080g. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Tyagi V, Fasan R. Angew Chem Int Ed. 2016;55:2512. doi: 10.1002/anie.201508817. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Tyagi V, Sreenilayam G, Bajaj P, Tinoco A, Fasan R. Angew Chem Int Ed. 2016;55:13562. doi: 10.1002/anie.201607278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Bordeaux M, Tyagi V, Fasan R. Angew Chem Int Ed. 2015;54:1744. doi: 10.1002/anie.201409928. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Bajaj P, Sreenilayam G, Tyagi V, Fasan R. Angew Chem Int Ed. 2016;55:16110. doi: 10.1002/anie.201608680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Coelho PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]; (b) Renata H, Wang ZJ, Kitto RZ, Arnold FH. Catal Sci Technol. 2014;4:3640. doi: 10.1039/C4CY00633J. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Wang ZJ, Renata H, Peck NE, Farwell CC, Coelho PS, Arnold FH. Angew Chem Int Ed. 2014;53:6810. doi: 10.1002/anie.201402809. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Weissenborn MJ, Low SA, Borlinghaus N, Kuhn M, Kummer S, Rami F, Plietker B, Hauer B. Chemcatchem. 2016;8:1636. [Google Scholar]; (e) Srivastava P, Yang H, Ellis-Guardiola K, Lewis JC. Nat Commun. 2015;6:7789. doi: 10.1038/ncomms8789. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Dydio P, Key HM, Nazarenko A, Rha JY, Seyedkazemi V, Clark DS, Hartwig JF. Science. 2016;354:102. doi: 10.1126/science.aah4427. [DOI] [PubMed] [Google Scholar]; (g) Key HM, Dydio P, Clark DS, Hartwig JF. Nature. 2016;534:534. doi: 10.1038/nature17968. [DOI] [PubMed] [Google Scholar]; (h) Rioz-Martinez A, Oelerich J, Segaud N, Roelfes G. Angew Chem Int Ed. 2016;55:14136. doi: 10.1002/anie.201608121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.(a) Morandi B, Carreira EM. Angew Chem Int Ed. 2010;49:938. doi: 10.1002/anie.200905573. [DOI] [PubMed] [Google Scholar]; (b) Morandi B, Carreira EM. Angew Chem Int Ed. 2011;50:9085. doi: 10.1002/anie.201103526. [DOI] [PubMed] [Google Scholar]; (c) Hyde S, Veliks J, Liegault B, Grassi D, Taillefer M, Gouverneur V. Angew Chem Int Ed. 2016;55:3785. doi: 10.1002/anie.201511954. [DOI] [PubMed] [Google Scholar]
- 10.(a) Barrett AGM, Braddock DC, Lenoir I, Tone H. J Org Chem. 2001;66:8260. doi: 10.1021/jo0108111. [DOI] [PubMed] [Google Scholar]; (b) Wurz RP, Charette AB. Org Lett. 2002;4:4531. doi: 10.1021/ol0270879. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.