

Abstract
Engineered hemoproteins have recently emerged as promising systems for promoting asymmetric cyclopropanations, but variants featuring predictable, complementary stereoselectivity in these reactions have remained elusive. In this study, a rationally driven strategy was implemented and applied to engineer myoglobin variants capable of providing access to 1-carboxy-2-aryl-cyclopropanes with high trans-(1R,2R) selectivity and catalytic activity. The stereoselectivity of these cyclopropanation biocatalysts complements that of trans-(1S,2S)-selective variants developed here and previously. In combination with whole-cell biotransformations, these stereocomplementary biocatalysts enabled the multigram synthesis of the chiral cyclopropane core of four drugs (Tranylcypromine, Tasimelteon, Ticagrelor, TRPV1 inhibitor 24) in high yield and with excellent diastereo- and enantioselectivity (98–99.9% de; 96–99.9% ee). These biocatalytic strategies outperform currently available methods to produce these drugs.
Chiral cyclopropanes à la carte
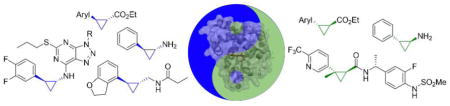
Myoglobin-based cyclopropanation catalysts featuring complementary stereoselectivity for the synthesis of 1-carboxy-2-aryl-cyclopropanes were developed. The engineered hemoproteins were applied in whole-cell reactions to afford cyclopropane-containing drugs and precursors thereof at the gram scale, in high yield and with excellent diastereo- and stereoselectivity.
Catalytic methods for the cyclopropanation of olefins cover a prominent role in organic and medicinal chemistry, owing to the recurrence of cyclopropane motifs among biologically active natural products and pharmaceuticals.[1] Significant progress has been made in the development of synthetic methods for asymmetric cyclopropanation, in particular through the transition metal-catalyzed insertion of carbenoid species into carbon-carbon double bonds.[2] More recently, the Arnold group and our own laboratory have shown that engineered cytochrome P450s[3] and myoglobins (Mb)[4], respectively, constitute promising catalysts for mediating the cyclopropanation of styrenes in the presence of α-diazoacetate reagents, thus providing a biocatalytic alternative to afford this valuable transformation. Variants of the bacterial cytochrome P450BM3 were found to favor cis-selectivity in the cyclopropanation of styrene in the presence of ethyl α-diazoacetate (EDA) (P450BM3-CIS-T438S: 86% de(cis), 97% ee(1S,2R)).[3a] By utilizing a different P450, opposite enantioselectivity was reported by Brustad and coworkers for the cis-cyclopropanation of this substrate, albeit with moderate diastereoselectivity (P450Biol-T238A: 42% de(cis), 95% ee(1R,2S)).[5] Unfortunately, varying cis : trans ratios and degrees of stereoselectivity were exhibited by these enzymes in the presence of other styrene derivatives.[3a, 5] We previously reported the development of an engineered myoglobin variant, Mb(H64V,V68A), capable of catalyzing the cyclopropanation of styrene with EDA with excellent trans diastereoselectivity and (1S,2S) enantioselectivity (99.9% de, 99.9% ee).[4] Promisingly, the high trans-(1S,2S) selectivity of this Mb variant extended to the cyclopropanation of a variety of styrene derivatives (97–99.9% de, 96–99.9% ee).[4]
The ability to access both enantiomeric forms of a target cyclopropane pharmacophore is critical in the context of the synthesis of bioactive molecules, as such stereoisomers often exhibit remarkably divergent pharmacological and/or toxicity profiles.[1] However, developing stereo- or enantiocomplementary variants of an enzyme is far from being a trivial task,[6] as mirror-image forms of these biomolecules are not readily available.[7] Reflecting this notion, cyclopropanation biocatalysts that can reliably offer complementary stereoselectivity have so far remained unavailable.[3, 5, 8] Here, we report the development and characterization of a panel of engineered Mb catalysts that give access to trans-(1R,2R) configured 1-carboxy-2-aryl-cyclopropanes with high selectivity and catalytic activity across a broad range of olefin substrates. We further demonstrate that these Mb-catalyzed reactions can be carried out and scaled up using whole cell systems. Using these stereocomplementary biocatalysts in combination with whole-cell transformations, it was possible to realize the asymmetric synthesis of the cyclopropane core of four different drugs, featuring both a trans-(1S,2S) and a trans-(1R,2R) configuration, at the multigram scale.
Previously, we found that mutations at the five amino acid positions defining the distal pocket in Mb (i.e., Leu29, Phe43, His64, Val68, and Ile107; Figure S1) significantly affected the activity and selectivity of this hemoprotein in carbene[4, 9] and nitrene[10] transfer reactions. In particular, mutation of the distal histidine residue (H64V) was determined to have a general activity enhancing effect in these reactions, possibly due to an increased accessibility of the heme pocket to the reactants. This mutation also slightly increases the trans-(1S,2S) selectivity of Mb for styrene cyclopropanation with EDA (93% de(trans), 10% ee(1S,2S) compared to 86% de(trans), 6% ee(1S,2S) for wild type Mb). This effect could be combined with that of a stronger trans-(1S,2S) selectivity inducing mutation, i.e., V68A (96% de(trans), 68% ee(1S,2S)), to yield the aforementioned, highly trans-(1S,2S) selective Mb(H64V,V68A) catalyst (Figure 1, grey path).[4] These results supported our hypothesis that additive effects can be leveraged to fine-tune the stereoselectivity of Mb in cyclopropanation reactions. At the same time, none of the active site Mb variants (>10) examined in these earlier studies showed any preference for formation of the trans-(1R,2R) product, indicating that achieving this type of stereoselectivity would require exploration of a wider active-site sequence space. On the basis of these considerations, we sought to implement a systematic active site mutagenesis approach combined with structure-reactivity guided design to achieve our goal of developing trans-(1R,2R)-selective Mb catalysts.
Figure 1.
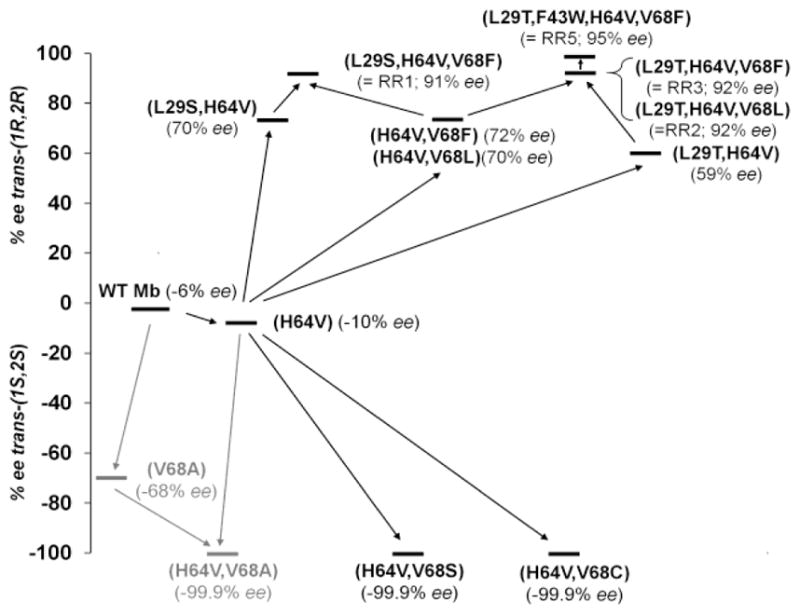
Structure-reactivity guided design of Mb cyclopropanation biocatalysts with complementary stereoselectivity. The path in grey is described in ref. [4].
Accordingly, starting from Mb(H64V) as the parent protein, each of the remaining four active site positions was systematically mutated to any of the other 19 amino acids by site-directed mutagenesis. This process resulted in a library of 76 Mb(H64V)-derived variants, which were tested individually for their activity (TON) and selectivity in the model cyclopropanation reaction with styrene and EDA (Table S2). Nearly half of these Mb variants (33/76; 43%) could be expressed in E. coli in correctly folded form as determined by the signature Soret band for their CO-bound complex (λmax ~ 420 nm). Moreover, the large majority of these proteins (75%) was catalytically competent (TON > 50) toward styrene cyclopropanation (Table S2), highlighting the robustness of Mb active site to mutagenesis. More importantly, screening of the library led to the identification of a number of Mb variants with significantly improved trans-(1R,2R) selectivity, as summarized in Table 1, compared to the parent protein, Mb(H64V), or wild-type Mb. Most effective in favouring such selectivity was the introduction of a leucine or phenylalanine residue in place of Val68 (i.e., 86→99% de, (−6)→70–72% ee(1R,2R)). Residue 68 lines along the side of the heme group and is expected to be in close proximity to the putative heme-bound carbene intermediate implicated in the cyclopropanation reaction.[4] Interestingly, substitution of the more remote Leu29 with either serine or threonine was also beneficial toward enhancing trans-(1R,2R) selectivity, as indicated by the 93–95% de and 59–70% ee values obtained with Mb(L29S,H64V) and Mb(L29T,H64V) (Table 1). Mutations of position Phe43 or Ile107 showed noticeable but comparatively more moderate effect on favouring trans-(1R,2R) selectivity (i.e., 16–55% ee; Table S2). Moreover, a number of highly trans-(1S,2S) selective variants were identified among the Val68 site-saturation sub-library, including Mb(H64V,V68S) and Mb(H64V,V68C), which produce 3b in >99% de and ee (Tables 1 and S2).
Table 1.
Activity and selectivity of representative myoglobin variants for styrene cyclopropanation with ethyl α-diazoacetate (EDA).[a] See also SI Tables S2 and S3.
| |||||
|---|---|---|---|---|---|
|
| |||||
| Catalyst | Conv. %[b] | TON | detrans [%] | eetrans [%][c] | eecis [%][c] |
| Mb | 36 | 180 | 86 | −6 | 0 |
| Mb(H64V) | 47 | 235 | 93 | −10 | 8 |
| Mb(L29S,H64V) | 60 | 300 | 93 | 70 | 18 |
| Mb(L29T,H64V) | 93 | 460 | 95 | 59 | 39 |
| Mb(H64V,V68F) | 57 | 285 | >99.9 | 72 | - |
| Mb(H64V,V68L) | 53 | 265 | >99.9 | 70 | - |
| Mb(H64V,V68S) | 82 | 410 | >99.9 | −99 | - |
| Mb(L29T,H64V,V68L) | 61 | 305 | >99.9 | 92 | - |
| Mb(L29T,H64V,V68F) | 58 | 290 | >99.9 | 92 | - |
| Mb(L29T,F43W,H64V,V68F) | 53 | 265 | 99 | 95 | - |
Reactions conditions: 20 μM Mb variant, 10 mM styrene, 20 mM EDA, 10 mM dithionite, 16 h.
% conversion based on GC analysis and relative to olefin.
trans = (1R,2R); cis = (1S,2R) as determined by chiral GC or SFC.
To obtain Mb catalysts with further improved trans-(1R,2R) selectivity, the beneficial mutations at position 68 (V68L, V68F) were combined with those at position 29 (L29T, L29S) or 107 (I107H) to yield a set of triple site variants (Table S3). Gratifyingly, this process resulted in the identification of two closely related variants, Mb(L29T,H64V,V68L) and Mb(L29T,H64V,V68F), with excellent trans diastereoselectivity (99% de) along with high (1R,2R) stereoselectivity (92% ee) (Table 1). Next, these Mb variants were further mutated at position Ile107 or Phe43, with the choice of the target substitutions being guided by the structure-reactivity relationship (SRR) data collected during screening of the initial library. Although none of the resulting quadruple site variants with substitutions at position 107 showed improved trans-(1R,2R) selectivity, the introduction of F43W mutation in the Mb(L29T,H64V,V68F) background led to the desired additivity effect. Indeed, the corresponding Mb(L29T,F43W,H64V,V68F) variant catalyzes the transformation of styrene and EDA into 3a with excellent diastereo- and stereoselectivity (>99% de; 95% ee; Table 1). Of note, refinement of the trans-(1R,2R) selectivity came with no loss in catalytic efficiency as indicated by the comparable TON supported by the trans-(1R,2R) selective Mb variants compared to the parent protein (265–460 vs. 235 TON). Figure 1 provides a graphical representation of the SRR-driven strategy implemented here for the development of stereocomplementary Mb cyclopropanation catalysts. As anticipated, achieving high trans-(1R,2R) selectivity involved a more extensive remodeling of the Mb active site (4 mutations) than required for obtaining high trans-(1S,2S) selectivity (2 mutations).
The substrate scope of the trans-(1R,2R) selective Mb catalysts (called ‘RR1’ through ‘RR5’, Table S3) was then probed against substituted styrene derivatives. As shown in Table 2, quantitative product conversion (95–99%) was obtained for different para, meta, ortho, and alpha functionalized styrenes (4b-6b, 8b-10b), with more moderate conversion (45%) being observed only with 4-trifluoromethyl-styrene (product 7b). Importantly, for all these substrates the desired trans-(1R,2R) configured cyclopropane product was obtained with good to excellent diastereoselectivity (92–99% de) and enantioselectivity (49–99% ee) with one or more of the Mb variants. Next, we extended these analyses to other aryl-substituted olefins, including pyridine, thiophene, N-methyl-imidazole, and benzothiazole derivatives. These molecules contain N- and S-containing heterocycles which are notoriously problematic in the context of cyclopropanation reactions with rhodium- or other metal-based complexes due to catalyst poisoning effects. For example, the reaction of 1-methyl-2-vinyl-imidazole with EDA and Rh2(OAc)4 as the catalyst failed to give any product (see SI for details). Notably, all of these olefins could be efficiently processed by the newly engineered Mb catalysts, resulting in product conversions up to 84% (11b-15b; Table 2). Furthermore, the corresponding trans-(1R,2R) cyclopropane product could be obtained in each case with good to very good selectivity (65–99% de, 65–98% ee). For all of these transformations, complementary stereoselectivity is provided by the trans-(1S,2S)-selective Mb(H64V,V68A) variant (Table S4).
Table 2.
Substrate scope for trans-(1R,2R)-selective Mb variants.[a]
| |||||
|---|---|---|---|---|---|
|
| |||||
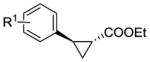
| Product | Catalyst[b] | Conv. [%] | TON | de [%] | ee [%] |
|
|||||
| R1 = 4-CH3 (4b) | RR5 | >99[c] | 165 | 96 | 63 |
| 4-MeO (5b) | RR5 | 95[c] | 160 | 97 | 74 |
| 4-Cl (6b) | RR2 | >99 | >500 | 97 | 49 |
| 4-CF3 (7b) | RR4 | 45[c] | 75 | 97 | 64 |
| 3-Me (8b) | RR4 | >99 | >500 | 92 | 84 |
| 2-Me (9b) | RR3 | >99 | >500 | >99 | >99 |
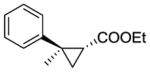 10b |
RR3 | >99[c] | 165 | 78 | 78 |
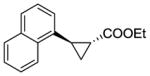 11b |
RR4 | 45[c] | 75 | 80 | 88 |
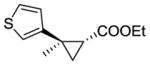 12b |
RR2 | 84 | 420 | 96 | 65 |
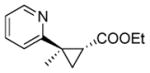 13b |
RR2 | 78 | 390 | 88 | 67 |
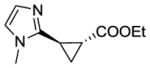 14b |
RR1 | 56[c] | 95 | 99 | 98 |
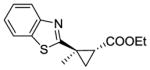 15b |
RR4 | 54[c] | 90 | 65 | 71 |
Whole-cell biotransformations constitute a convenient approach to enhance the scalability of biocatalytic processes supported by heme-dependent enzymes.[3b, 11] Importantly, we established that the Mb(H64V,V68A)-catalyzed cyclopropanation of styrene with EDA to give 3b can be readily carried out using E. coli cells (BL21DE3) expressing this Mb variant, resulting in high product conversion (68–100%) even under aerobic conditions (Table S5). Furthermore, the substrate loading in these whole-cell reactions could be increased up to 0.5 M styrene and 0.5 M EDA in a 1:1 ratio, while maintaining high conversion ratio (62%) along with excellent selectivity (99.9% de, 99.9% ee, Table S5).
Aryl-substituted trans-cyclopropanes are found in several marketed and investigational drugs (Scheme 1),[1] including the MAO inhibitor Tranylcypromine ((±)-16, former Parnate®),[12] the melatonin receptor agonist Tasimelteon (20),[13] the TRPV1 antagonist 24,[14] and the platelet aggregation inhibitor Ticagrelor (28, Brilinta®)[15]. Although tranylcypromine has been used in clinical settings in racemic form, the two enantiomers display different biological potency.[16] The other drugs are used in enantiomerically pure form. Each of these molecules feature a cyclopropane unit that can be retrosynthetically derived from either a trans-(1R,2R)- ((−)-16, 19, 28) or trans-(1S,2S)-configured 1-carboxy-2-aryl-cyclopropane ((+)-16, 24). As such, these drugs were chosen as relevant targets to assess the synthetic utility of the stereocomplementary cyclopropanation biocatalysts developed herein.
Scheme 1.

Total and formal synthesis of (a) levo- and dextrorotatory enantiomer of tranylcypromine, (b) Tasimelteon, (c) TRPV1 inhibitor 24, and (d) Ticagrelor, via myoglobin-catalyzed cyclopropanation in whole cells. See SI for details on synthetic steps. DPPA = diphenylphosphoryl azide, PTSA = para-toluenesulfonic acid, T3P = propane phosphonic acid anhydride, TFA = trifluoroacetic acid.
Accordingly, to obtain the dextrorotatory form of Tranylcypromine, a large scale reaction with E. coli cells expressing Mb(H64,V68A) was carried out in the presence of 2.9 g styrene and 3.1 g EDA (1 equiv.), resulting in the isolation of 3b as the only product (99.9% de, 99.9% ee) in 91% yield (4.7 g; Scheme 1a). Under similar reaction conditions but using cells expressing Mb(L29T,H64V,V68L), 1.2 g of the opposite enantiomer 3a were obtained in 80% yield and excellent selectivity (99.9% de, 95% ee. Both intermediates can be then converted to the desired (+)- and (−)-Tranylcypromine, respectively, via conversion to the corresponding acyl azides followed by a stereoretentive Curtius rearrangement[16] and hydrolysis (93% yield over three steps for (+)-16, see SI for details). By comparison, the same cyclopropanation reaction catalyzed by two prominent Cu(Box) catalysts was reported to proceed with high stereoselectivity (90–99% ee) but only moderate diastereoselectivity (46–50% de(trans)).[2d, 2e]
The asymmetric cyclopropanation of olefin 18 to give 19 represents a key step in the synthesis of Tasimelteon as reported by chemists at BMS.[17] Under best performing conditions, this transformation was realized in 91% de and 85% ee using Nishiyama’s Ru(iPr-PyBox) catalyst in the presence of excess EDA. After affording intermediate 18 according to published procedures,[17] this compound was subjected to Mb-catalyzed cyclopropanation using whole-cells expressing the trans-(1R,2R)-selective variant Mb(L29T,H64V,V68F,I107L) in presence of stoichiometric amounts of EDA (Scheme 1b). Notably, the desired (1R,2R)-configured cyclopropanation product 19 was obtained in 99.9% de and 96% ee and in 91% isolated yield (0.96 g), thus furnishing the key intermediate en route to Tasimelteon (20).[17]
The stereoselective cyclopropanation of 22 into 23 was denoted by Pfizer chemists as “the most challenging step” in the preparation of the advanced drug candidate 24,[14] due to the presence of a pyridine functionality and formation of a trisubstituted cyclopropane.[2k] Upon screening over 120 different catalytic conditions, the best results were obtained using Ru(iPr-PyBox) as the catalyst to give 23 in 40% de and 65% ee.[14] Using whole cells expressing Mb(H64V,V68A), 0.83 g of 23 could be obtained in high yield (75%) with much greater diastereo- and stereoselectivity (99.9% de, 99.9% ee) (Scheme 1c). The cyclopropanation product was further processed to afford 24 (see SI for details).
Lastly, we targeted the synthesis of (1R,2R)-ethyl 2-(3,4-difluorophenyl)-cyclopropanecarboxylate 26 (Scheme 1d) which can be readily hydrolysed to 27, a key synthetic intermediate for the preparation of Ticagrelor.[15] Of note, this intermediate is currently accessible through synthetic routes involving no less than four to five steps.[15, 18] Conveniently, the key cyclopropane building block 26 could be obtained in a single step in high yield (94%) and selectivity (98% de, 58% ee) via whole-cell cyclopropanation of commercially available 3,4-difluoro-styrene with cells expressing the (1R,2R)-selective Mb(L29T,H64V,V68L) variant. Taken together, these results demonstrate the promise and scalability of stereoselective myoglobin-catalyzed cyclopropanations in the context of relevant and challenging building blocks for drug synthesis.
In summary, we report the successful design and application of myoglobin-based cyclopropanation biocatalysts capable of offering high trans-selectivity along with complementary stereoselectivity across a broad panel of aryl-substituted olefins. In addition, we demonstrate that these myoglobin-mediated transformations can be performed in the context of whole-cell systems, which further simplifies their use for synthetic applications. The biocatalytic systems developed here have enabled the stereoselective synthesis of multiple cyclopropane-containing drugs at the preparative scale, offering superior performance over currently available methods for asymmetric cyclopropanation (i.e., (+)- and (−)-16, 20, and 24) or granting a more concise route to their preparation (28). Along with the growing number of abiotic reactions accessible using engineered myoglobins,[4, 9–10, 19] these results support the promise of these metalloproteins for the asymmetric synthesis of chiral drugs and synthons at a practical scale.
Supplementary Material
Acknowledgments
This work was supported by the U.S. National Institute of Health grant GM098628. LC-MS instrumentation was supported by the U.S. NSF grant CHE-0946653.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- 1.Talele TT. J Med Chem. 2016;59(19):8712–8756. doi: 10.1021/acs.jmedchem.6b00472. [DOI] [PubMed] [Google Scholar]
- 2.Doyle MP, Forbes DC. Chem Rev. 1998;98:911–936. doi: 10.1021/cr940066a.Lebel H, Marcoux JF, Molinaro C, Charette AB. Chem Rev. 2003;103:977–1050. doi: 10.1021/cr010007e.Pellissier H. Tetrahedron. 2008;64:7041–7095.Lowenthal RE, Abiko A, Masamune S. Tetrahedron Lett. 1990;31:6005–6008.For representative examples with acceptor-only diazo reagents: Evans DA, Woerpel KA, Hinman MM, Faul MM. J Am Chem Soc. 1991;113:726–728.Doyle MP, Winchester WR, Hoorn JAA, Lynch V, Simonsen SH, Ghosh R. J Am Chem Soc. 1993;115:9968–9978.Nishiyama H, Itoh Y, Matsumoto H, Park SB, Itoh K. J Am Chem Soc. 1994;116:2223–2224.Uchida T, Irie R, Katsuki T. Synlett. 1999:1163–1165.Che CM, Huang JS, Lee FW, Li Y, Lai TS, Kwong HL, Teng PF, Lee WS, Lo WC, Peng SM, Zhou ZY. J Am Chem Soc. 2001;123:4119–4129. doi: 10.1021/ja001416f.Huang LY, Chen Y, Gao GY, Zhang XP. J Org Chem. 2003;68:8179–8184. doi: 10.1021/jo035088o.Fantauzzi S, Gallo E, Rose E, Raoul N, Caselli A, Issa S, Ragaini F, Cenini S. Organometallics. 2008;27:6143–6151.Carminati DM, Intrieri D, Caselli A, Le Gac S, Boitrel B, Toma L, Legnani L, Gallo E. Chemistry. 2016;22:13599–13612. doi: 10.1002/chem.201602289.
- 3.a) Coelho PS, Brustad EM, Kannan A, Arnold FH. Science. 2013;339:307–310. doi: 10.1126/science.1231434. [DOI] [PubMed] [Google Scholar]; b) Coelho PS, Wang ZJ, Ener ME, Baril SA, Kannan A, Arnold FH, Brustad EM. Nat Chem Biol. 2013;9:485–487. doi: 10.1038/nchembio.1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bordeaux M, Tyagi V, Fasan R. Angew Chem Int Ed. 2015;54:1744–1748. doi: 10.1002/anie.201409928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gober JG, Rydeen AE, Gibson-O’Grady EJ, Leuthaeuser JB, Fetrow JS, Brustad EM. Chembiochem. 2016;17:394–397. doi: 10.1002/cbic.201500624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.a) Wu Q, Soni P, Reetz MT. J Am Chem Soc. 2013;135:1872–1881. doi: 10.1021/ja310455t. [DOI] [PubMed] [Google Scholar]; b) Walton AZ, Sullivan B, Patterson-Orazem AC, Stewart JD. ACS Catal. 2014;4:2307–2318. doi: 10.1021/cs500429k. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Scheller PN, Fademrecht S, Hofelzer S, Pleiss J, Leipold F, Turner NJ, Nestl BM, Hauer B. Chembiochem. 2014;15:2201–2204. doi: 10.1002/cbic.201402213. [DOI] [PubMed] [Google Scholar]; d) van der Meer JY, Poddar H, Baas BJ, Miao YF, Rahimi M, Kunzendorf A, van Merkerk R, Tepper PG, Geertsema EM, Thunnissen AMWH, Quax WJ, Poelarends GJ. Nat Commun. 2016:7. doi: 10.1038/ncomms10911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mugford PF, Wagner UG, Jiang Y, Faber K, Kazlauskas RJ. Angew Chem Int Ed. 2008;47:8782–8793. doi: 10.1002/anie.200705159. [DOI] [PubMed] [Google Scholar]
- 8.a) Wang ZJ, Renata H, Peck NE, Farwell CC, Coelho PS, Arnold FH. Angew Chem Int Ed. 2014;53:6810–6813. doi: 10.1002/anie.201402809. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Renata H, Wang ZJ, Kitto RZ, Arnold FH. Catal Sci Technol. 2014;4:3640–3643. doi: 10.1039/C4CY00633J. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Srivastava P, Yang H, Ellis-Guardiola K, Lewis JC. Nat Commun. 2015;6:7789. doi: 10.1038/ncomms8789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.a) Sreenilayam G, Fasan R. Chem Commun. 2015;51:1532–1534. doi: 10.1039/c4cc08753d. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tyagi V, Bonn RB, Fasan R. Chem Sci. 2015;6:2488–2494. doi: 10.1039/c5sc00080g. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tyagi V, Fasan R. Angew Chem Int Ed. 2016;55:2512–2516. doi: 10.1002/anie.201508817. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Giovani S, Singh R, Fasan R. Chem Sci. 2016;7:234–239. doi: 10.1039/c5sc02857d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bordeaux M, Singh R, Fasan R. Bioorg Med Chem. 2014;22:5697–5704. doi: 10.1016/j.bmc.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.a) Lundemo MT, Woodley JM. Appl Microbiol Biotechnol. 2015;99:2465–2483. doi: 10.1007/s00253-015-6403-x. [DOI] [PubMed] [Google Scholar]; b) Sowden RJ, Yasmin S, Rees NH, Bell SG, Wong LL. Org Biomol Chem. 2005;3:57–64. doi: 10.1039/b413068e. [DOI] [PubMed] [Google Scholar]; c) Fasan R, Chen MM, Crook NC, Arnold FH. Angew Chem Int Ed. 2007;46:8414–8418. doi: 10.1002/anie.200702616. [DOI] [PubMed] [Google Scholar]; d) Fasan R, Crook NC, Peters MW, Meinhold P, Buelter T, Landwehr M, Cirino PC, Arnold FH. Biotechnol Bioeng. 2011;108:500–510. doi: 10.1002/bit.22984. [DOI] [PubMed] [Google Scholar]; e) Ringle M, Khatri Y, Zapp J, Hannemann F, Bernhardt R. Appl Microbiol Biotechnol. 2013;97:7741–7754. doi: 10.1007/s00253-012-4612-0. [DOI] [PubMed] [Google Scholar]; f) Muller CA, Dennig A, Welters T, Winkler T, Ruff AJ, Hummel W, Groger H, Schwaneberg U. J Biotechnol. 2014;191:196–204. doi: 10.1016/j.jbiotec.2014.06.001. [DOI] [PubMed] [Google Scholar]; g) Tieves F, Erenburg IN, Mahmoud O, Urlacher VB. Biotechnol Bioeng. 2016;113:1845–1852. doi: 10.1002/bit.25953. [DOI] [PubMed] [Google Scholar]
- 12.Burger A, Yost WL. J Am Chem Soc. 1948;70:2198–2201. [Google Scholar]
- 13.Catt J, Johnson G, Keavy D, Mattson R, Parker M, Takaki K, Yevich J. 5,981,571. Squibb B-M, editor. US Pat. 1999
- 14.Butcher KJ, Denton SM, Field SE, Gillmore AT, Harbottle GW, Howard RM, Laity DA, Ngono CJ, Pibworth BA. Org Process Res Dev. 2011;15:1192–1200. [Google Scholar]
- 15.Springthorpe B, Bailey A, Barton P, Birkinshaw TN, Bonnert RV, Brown RC, Chapman D, Dixon J, Guile SD, Humphries RG, Hunt SF, Ince F, et al. Bioorg Med Chem Lett. 2007;17:6013–6018. doi: 10.1016/j.bmcl.2007.07.057. [DOI] [PubMed] [Google Scholar]
- 16.Riley TN, Brier CG. J Med Chem. 1972;15:1187–1188. doi: 10.1021/jm00281a028. [DOI] [PubMed] [Google Scholar]
- 17.Simpson JH, Godfrey J, Fox R, Kotnis A, Kacsur D, Hamm J, Totelben M, Rosso V, Mueller R, Delaney E, Deshpande RP. Tetrahedron-Asymmetr. 2003;14:3569–3574. [Google Scholar]
- 18.a) Zhang H, Liu J, Zhang L, Kong L, Yao H, Sun H. Bioorg Med Chem Lett. 2012;22:3598–3602. doi: 10.1016/j.bmcl.2012.04.050. [DOI] [PubMed] [Google Scholar]; b) Guile S, Hardern D, Springthorpe B, Willis P. 2000/034283 WO. 2000; c) Clark A, Jones E, Larsson U, Minidis A. 2001/092200 WO. 2001; d) Hugentobler KG, Sharif H, Rasparini M, Heath RS, Turner NJ. Org Biomol Chem. 2016;14:8064–8067. doi: 10.1039/c6ob01382a. [DOI] [PubMed] [Google Scholar]
- 19.Key HM, Dydio P, Clark DS, Hartwig JF. Nature. 2016;534:534–537. doi: 10.1038/nature17968. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.