

Abstract
The new strategy for chemical toxicity testing and modeling is to use in vitro human cell-based assays in conjunction with quantitative high-throughput screening (qHTS) technology, to identify molecular mechanisms and predict in vivo responses. Stem cells are more physiologically relevant than immortalized cell lines because of their unique proliferation and differentiation potentials. We established a robust two stem cells-two lineages assay system, encompassing human mesenchymal stem cells (hMSCs) along osteogenesis, and human induced pluripotent stem cells (hiPSCs) along hepatogenesis. We performed qHTS phenotypic screening of LOPAC®1280 and identified 38 preliminary hits for hMSCs. This was followed by validation of a selected number of hits and determination of their IC50 values, and mechanistic studies of idarubicin and cantharidin treatments using proteomics and bioinformatics. In general, hiPSCs were more sensitive than hMSCs to chemicals, and differentiated progenies were less sensitive than their progenitors. We showed that chemical toxicity depends on both stem cell types and their differentiation stages. Proteomics identified and quantified over 3,000 proteins for both stem cells. Bioinformatics identified apoptosis and G2/M as the top pathways conferring idarubicin toxicity. Our Omics-based assays of stem cells provide mechanistic insights into chemical toxicity, and may help prioritize chemicals for in-depth toxicological evaluations.
Keywords: Proteomics, LC-MS, qHTS, stem cells, hMSC, hiPSC, chemical toxicity, networks, LOPAC, idarubicin, cantharidin
Graphical Abstract
Introduction
There is a paradigm shift in toxicity testing and modeling 1–3. The new strategy is to use in vitro human cell-based assays (vs. animal-based) in conjunction with quantitative high-throughput screening (qHTS) technology, to identify molecular mechanisms and predict the in vivo responses. Stem cells (embryonic, induced pluripotent, and adult multipotent) hold great potential for regenerative medicine because they can self-renew and differentiate along different lineages both in vitro and in vivo 4, 5. However, stem cells may also undergo genotoxic (e.g., ionizing radiation) and non-genotoxic stresses (e.g., environmental chemicals), which may lead to transient cell-cycle arrest, apoptosis, mitotic catastrophe, cellular senescence, and malignant transformation. Therefore, stem cells are involved in cancer and aging 6, 7. Compared to immortalized cell lines, stem cells are more physiologically relevant for toxicity testing because of their unique proliferation and differentiation characteristics 8.
Both pluripotent stem cells, such as hiPSCs, and multipotent stem cells, such as hemopoietic stem cells (HSCs) and hMSCs have been utilized in drug screening 9 and chemical toxicity testing 10–12, respectively. However, the readouts were mostly conventional phenotypic (cytotoxicity) assays and genomics analyses. In order to understand the mechanisms underlying chemical toxicity, it is imperative to perform Omics-based assays that integrate cellular- and molecular-level information. For example, proteomic and metabolomic analysis of target cells will elucidate the cellular pathways and networks responsible for drug mechanisms of action 13. The information will help us devise optimal countermeasures for chemical toxicity. Furthermore, few studies have directly compared the dependency of chemical responses on stem cell types and their differentiation stages along various lineages. Mechanistic understanding of these dependencies will provide insights into organ-, tissue-, and cell type-specific chemical toxicity. For example, studies of hiPSC-derived hepatocytes could provide information on liver toxicity of a chemical. On the other hand, studies of hMSC-derived osteocytes could provide information on how bone cells and their progenitors respond to a chemical. Together, the information may help us understand the potential in vivo on-target and off-target effects of drug candidates, and facilitate rapid triage before expensive preclinical and clinical studies.
In this work, we demonstrated an integrated platform for screening chemical toxicity using stem cells. We first established an assay system of two stem cells-two lineages, encompassing hMSCs at different stages of osteogenic differentiation and hiPSCs at different stages of hepatogenesis. We identified potential chemical hits by performing qHTS phenotypic screening of The Library of Pharmacologically Active Compounds (LOPAC®1280), followed by validation of a selected number of hits. Finally, we gained mechanistic understanding of chemical toxicity on proliferation and various differentiation stages of these stem cells, through pathway and network analysis of their cellular proteomes in response to idarubicin and cantharidin treatments.
Materials and Methods
Cell culture of hMSCs and hiPSCs
Two hMSC lines (Cat. No. PT-2501) were purchased from Lonza (Walkersville, MD). They were isolated from the bone marrow of a 30-year-old male (Lot No. 0000429365) and a 25-year-old male (Lot No. 0000451491), respectively. hMSCs were cultured in the basal medium (Lonza, Cat. No. PT-3001) at 37°C/5% CO2, and expanded from 2 frozen stocks (≥750,000 cells per stock) at Passage 2 (P2) to 36 frozen stocks (5 × 105 hMSCs/stock) at P6, and stored in a liquid nitrogen tank. A cell stock was thawed, grown to ~90% confluency in a T75 flask, and seeded on the appropriate plates (i.e., 96-well plate) where experiments were conducted for hMSCs at P7. A retroviral reprogrammed hiPSC line (Cat. No. ACS-1007; Lot No. 0189) derived from the primary hepatic fibroblast of a 31-year-old male was obtained from ATCC (Manassas, VA). hiPSCs were expanded in mTeSR, a defined, feeder-free maintenance medium, and split with Dispase or Accutase from STEMCELL Technologies (Vancouver, BC, Canada). Detailed timeline of stem cell proliferation, differentiation, and chemical treatments is shown in Figure S1.
hMSCs osteogenic differentiation in vitro
The hMSCs osteogenesis was achieved by Osteogenic BulletKit (Cat. No.: PT-3002) from Lonza. After seeding and overnight culturing, basal culture medium was replaced with osteogenic medium which was changed once every three days. Cell morphology was monitored using an Olympus IX83 microscope. SIGMAFAST 5-bromo-4-chloro-3-indolyl phosphate (BCIP)/nitro blue tetrazolium (NBT) (Sigma, Cat. No. B5655) was used to stain the cells for alkaline phosphatase (ALP) activity, in order to confirm the osteogenic differentiation. SIGMAFAST p-Nitrophenyl phosphate (pNPP) Tablets (Cat. No. N1891) were used for quantitation of alkaline phosphatase activity using a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA) following the vendor’s instructions. We studied three timepoints (1 day, 3 days, and 6 days) after addition of the osteogenic media, including osteogenesis Day 1 (OD1), osteogenesis Day 3 (OD3), and osteogenesis Day 6 (OD6).
hiPSCs hepatogenic differentiation vitro
There are three phases in the hiPSC hepatic differentiation including Phase I, definitive endoderm induction; Phase II, hepatic specification; and Phase III, hepatocytes maturation 14–17. We optimized the differentiation protocol by combining commercial kits and off-the-shelf reagents. In our experiment, Phase I was achieved by STEMdiff™ Definitive Endoderm Kit (Catalog number: 05110) from STEMCELL Technologies. Phase II and III differentiation were achieved by following the protocols provided by Siller et al. 16. More specifically, Phase II consisted of 5 days of treatment with knockout DMEM media containing 20% knockout serum replacement, 2 mM GlutaMAX, 100 μM 2-mercaptoethanol, 1× MEM non-essential amino acids, and 1% DMSO; and Phase III consisted of 10 days of treatment with Leibovitz L-15 media containing 8.3% tryptose phosphate broth, 10 μM hydrocortisone 21-hemisuccinate, 50 μg/ml sodium-L-ascorbate, 100 nM dexamethasone, 0.58% insulin-transferrin-selenium (ITS), 2 mM GlutaMAX, 8.3% fetal bovine serum, and 100 nM dihexa. During phase I, cells were fed daily. During Phases II and III, cells were fed every 48 hrs. Knockout DMEM, knockout serum replacement, GlutaMAX supplement, 2-Mercaptoethanol, MEM non-essential amino acids solution (100×), insulin-transferrin-selenium (100×), and fetal bovine serum were obtained from Thermo Fisher. DMSO, L-15 medium, and tryptose phosphate broth solution were obtained from Sigma-Aldrich. Hydrocortisone 21-hemisuccinate sodium salt, (+)-sodium L-ascorbate, and dexamethasone were ordered from BioReagent through Sigma-Aldrich. Dihexa (Cat. No.: AP201016) was ordered from Activepeptide (Massachusetts, USA). For immunostaining, Oct4 and Sox2 antibodies (Cat. No. 09-0023 and 09-0024) were ordered from Stemgent, Sox17 antibody (Cat. No. AF1924) was ordered from R&D Systems, α-Fetoprotein (AFP) and albumin (ALB) antibodies (Cat. No. and A6684) were obtained from Sigma-Aldrich, and HDF4A antibody was obtained from Millipore. For functional assays on induced hepatocytes-like cells, we used Cardiogreen (Cat. No. I2633-100MG) and Periodic Acid-Schiff (PAS) Kit (Cat. NO. 395B-1KT) from Sigma-Aldrich. Primary human hepatocytes (hNHEPS™ Adherent Cells, Cat. No. CC-2591) from Lonza was used as a positive control for immunostaining and functional assay for our induced hepatocyte-like cells generated from hiPSCs.
qHTS assay in the 1536-well plate format
qHTS was performed at the National Center for Advancing Translational Sciences (NCATS) following the protocols previously described 18–20. The qHTS platform for chemical toxicity screening utilizes the CellTiter-Glo (CTG) assay which measures ATP content to indicate cell viability and the readout is chemiluminescence (Promega, Madison, WI). The higher sensitivity of the CTG assay (over the colorimetric Cell Counting Kit-8 (CCK-8) assay) was required for the small volume in a 1536-well format. The LOPAC®1280 was obtained from Sigma-Aldrich and tested on the basal hMSCs and osteogenic-day 1 cells. We screened 9 plates including 2 DMSO (control) and 7 LOPAC plates. The 7 concentrations for each compound ranged from 0.29 nM to 46 μM (0.0029, 0.0147, 0.0736, 0.368, 1.84, 9.2, 46 μM), with 1:5 serial dilutions starting from 46 μM. hMSCs were seeded at a density of 500 cells/well in 5 μL culture media. After incubation at 37°C/5% CO2 for 24 hours, 23 nL of each compound at desired concentration was added into each well on the assay plates. After 48-hour treatment, 5 μL of CTG reagent was added into each well. The assay plates were incubated under room temperature for 30 minutes and luminescence intensity was measured by a ViewLux microplate reader (PerkinElmer, Waltham, MA). The IC50 and efficacy of each compound were calculated based on the dose-response curves. The potential hits with significant toxicity to hMSCs were identified based on cutoff criteria of efficacy ≥ 50% and IC50 ≤ 20 μM.
In vitro assays for chemical cytotoxicity on 96-well plates
We conducted detailed studies of chemical toxicity on hiPSCs, hMSCs, and their differentiated progenies at various stages of differentiation (Figure S1). Cells were treated with different chemicals in 96-well plates and cell viability was measured with the CCK-8 assay (Sigma-Aldrich, Cat. No. 96992). For all chemical toxicity assays, 6,000 hMSCs in 100 μL medium were seeded per well in a 96-well plate, and treated for 48 hours at 37°C/5% CO2 before cell viability readout on a VersaMax microplate reader (Molecular Devices, Sunnyvale, CA). A total of 14 different concentrations ranged from 0.0029 to 223 μM were used for each chemical in triplicates. The four chemicals, AC-93253, idarubicin, NSC 95397, and cantharidin, were purchased from Sigma, and dissolved in 100% DMSO. For chemical toxicity test on hMSCs, the final DMSO concentration in the media was 0.5%. For hiPSCs and hepatic differentiation Phase I cells, the final DMSO concentration was 0.1%. For hepatic differentiation Phase II and Phase III chemical toxicity tests, the final DMSO concentration for NSC 95397 was 0.3%, while 0.1% for the other 3 chemicals. Each chemical toxicity tests were repeated at least three times. The cell viabilities were plotted versus concentrations for each chemical. GraphPad Prism 6 was used to fit the data to a 3-parameter dose-inhibition curve to obtain the IC50 values.
Proteomics studies on chemical cytotoxicity
We performed large-scale proteomics profiling of basal hiPSCs and hMSCs in response to idarubicin and cantharidin at various stage of osteogenesis differentiation. The cells were grown in T75 flasks and treated with the chemicals at their IC50 concentrations at basal and various differentiation stages. Cells were harvested 24 hours after the chemical treatment. hMSCs were washed three times with ice-cold 1× PBS buffer and then lysed directly on T75 flasks with a lysis buffer containing 25 mM Tris-HCl, 150 mM NaCl, 6 M guanidine hydrochloride, 0.1% ALS Progenta Zwitterionic acid labile surfactant I (Protea Biosciences), 1% phosphatase inhibitor cocktail (Thermo Fisher), at pH 7.6. The cell lysates were collected in a microcentrifuge tube, vortexed for 30 minutes, and centrifuged at 10,000 g for 15 minutes. The supernatant was transferred into a new tube and protein concentration was measured by a BCA assay kit (Pierce). hiPSCs were treated with accuses (Corning) to release the cells from Matrigel to minimize the contamination of background proteins. Cells were pelleted after centrifuge at 2,000 g for 5 minutes and lysed with the same lysis buffer. Prior to trypsin digestion, desired amounts (e.g., ~100 μg) of total proteins were reduced by Tris(2-carboxyethyl)phosphine hydrochloride (TCEP.HCl) and alkylated using iodoacetamide. Proteins were then digested with sequencing grade trypsin (Promega) in 50 mM ammonium bicarbonate with trypsin: protein, 1:100 (w/w) at 37°C for 4 hours, followed by a second addition of trypsin with trypsin: protein, 1:100 (w/w) at 37°C, and incubated overnight. Tryptic peptides were desalted on Sepak C18 cartridges (Waters Corp.), dried under a SpeedVac (Thermo Fisher), and stored in a −80°C freezer.
Tryptic peptides were re-solubilized with 20% acetonitrile/1% formic acid (FA) and further diluted 1:3 with mobile phase A containing 3% acetonitrile/0.2% formic acid in water. They were analyzed on a Q Exactive™ Plus (QE plus) Hybrid Quadrupole-Orbitrap™ Mass Spectrometer interfaced with an UltiMate 3000 nanoUPLC system (Thermo Fisher Scientific, USA). A Waters M-Class BEH C18 (75 μm × 150 mm, 1.7 μm) column was used for LC analysis and the trap column was a Waters Symmetry C18 column (150 μm × 150 mm, 3.5 μm). The mobile phase consisted of 3% acetonitrile/0.2% formic acid in water (A) and 97% acetonitrile/0.2% formic acid in water (B). The flow rates and the gradient steps for the 70 minutes LC-MS run were as follows: (1) 750 nL/min, 5%–15% B for 1 minute; (2) 250 nL/min, 15%–35% B for 56 minutes; (3) 250 nL/min, 35%–65% B for 2 minutes; (4) 750 nL/min, 65% B for 5 minutes; and (5) 750 nL/min, 5% B for 6 minutes. Data dependent MS acquisition was employed on QE plus with 70,000 of resolution, 3e6 of AGC target, 30 ms of maximum IT, 375 to 1,500 m/z range for full MS scan. MS2 spectra were acquired with 17,500 of resolution, 5e4 of AGC target, 45 ms of maximum IT, loop count of 15, and NCE 27. The samples were analyzed in triplicate LC-MS/MS runs and the run order was randomized for a batch of samples to minimize any bias due to the run sequence.
Data analysis
The MS raw data were loaded onto Proteome Discoverer 2.1 (Thermo Fisher, USA) and searched against SwissProt human sequence DBs using Sequest HT with 10 ppm of precursor mass tolerance, 0.02 Da of fragment mass tolerance. Protein modifications considered included carbamidomethylation of cysteine (fixed), N-terminal acetylation, N-terminal Gln to pyroGlu, oxidation of methionine, and phosphorylation of serine, threonine, and tyrosine. Percolator was employed to calculate the protein false discovery rate (FDR) by searching decoy database and the cutoff was FDR = 0.01 and p value < 0.05. The search results were exported as an Excel file. To calculate the fold change of each protein between two cellular conditions, the MS intensity was first normalized using the average intensity of all peptide precursor ions detected across all samples. Protein intensity was then calculated using the sum of top 3 peptides corresponding to the protein. OriginPro 2017 (Origin Lab, USA) and R were used for further statistical analysis. Student’s t-test (two-tailed) was used to calculate the p value for the difference between treatment groups for the same protein in triplicate runs.
Pathway analysis was done using Ingenuity Pathway Analysis (IPA) software from QIAGEN Bioinformatics (Redwood City, California, USA). Protein lists from proteomics studies were uploaded into IPA in Excel format. The files contained the protein accession number, experimental fold changes, and p values. Core analysis function of IPA was used to perform the pathway analysis. Cutoff of the experimental fold change of > 1.5 (upregulated) and < 0.67 (downregulated) with a p value < 0.05 was used in the analysis. The significance value for canonical pathways was calculated using Benjamini-Hochberg corrected Fisher’s extract test, by comparing the number of proteins that were in involved in a given function or pathway, relative to the total number of occurrences of these proteins in all functional/pathways annotations stored in the Ingenuity Pathway Knowledge Base.
Results and Discussion
Workflow for screening chemical toxicity using stem cells
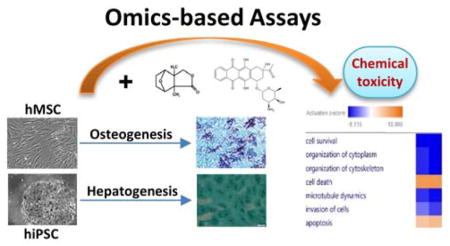
We established an integrated platform, with Omics-based assays at its core, for screening chemical toxicity using hiPSCs and hMSCs, by taking advantage of their distinct proliferation and differentiation potentials (Figure 1). We focused on osteogenic differentiation of hMSCs and hepatogenic differentiation of hiPSCs because they are relevant to chemical toxicity on bone and liver cells, respectively. The cell viability assay using these cells was screened against LOPAC®1280 from Sigma-Aldrich, which contains 1,280 marketed drugs and pharmaceutically relevant compound structures. This biologically annotated collection of inhibitors, receptor ligands, pharma-developed tools, and approved drugs impacts most signaling pathways and covers many drug target classes (Figure 1A).
Figure 1. Overall workflow for screening chemical toxicity using stem cells.

A. A two-stem cell system including hMSCs along osteogenesis and hiPSCs along hepatogenesis is used to screen LOPAC containing 1,280 chemicals.
B. Three key steps of our integrated platform including qHTS on 1536-well plates, validation of a selected number of hits on 96-well plates, and Omics studies on the confirmed hits.
C. Four kinds of chemical toxicity responses for stem cells and their progenies.
D. Potential impact of our integrated platform for screening chemical toxicity using stem cells.
The workflow included three major steps (Figure 1B). First, we used qHTS assays on 1536-well plates to identify potential hits. Second, we selected a few compounds from the hit list (4 chosen in this work) for validation on 96-well plates. Third, we performed detailed Omics studies on chemical toxicity of the validated hits (2 chosen in this work) to gain mechanistic understandings.
Given the diversity of chemicals and cell types, the chemical toxicity responses are diverse (Figure 1C). With our 2-stem cells, 2-lineage system, we classified chemical toxicity into four different classes: (1) stem cell-dependent, differentiation stage-dependent; (2) stem cell-dependent, differentiation stage-independent; (3) stem cell-independent, differentiation stage-dependent; and (4) stem cell-independent, differentiation stage-independent. This classification provided some mechanistic insights. For example, if the chemical toxicity is stem cell-independent but differentiation stage-dependent, it may suggest that this chemical affects pathways common to stem cells, i.e., stemness. As stem cells undergo differentiation, the unique pathways in the different progenies will be impacted differently. On the other hand, if the chemical toxicity is both stem-cell independent and differentiation stage-independent, it may suggest that the chemical targets the core pathways shared by most cell types. As a result, this chemical as a drug candidate may have more side effects due to its broad targeting.
Our generic platform can be expanded to any other stem cell-lineage systems and chemical libraries (Figure 1D). The goal is to streamline the in vitro toxicological evaluation of environmental chemicals, facilitate the prediction of their in vivo toxicity, thereby helping develop potential countermeasures for their mitigation.
Potential hits identified from LOPAC screening
We performed the CTG assay in a 1536-well format against LOPAC, containing 1,280 compounds with known pharmacological activity, on hMSCs at time points Basal-Day 1 and Osteogenesis-Day 1. For the qHTS assay in 1536-well plates, we only performed assays for basal hMSCs and OD1 cells, because it was unfeasible to change medium in 1536-well plates several times during osteogenesis. hMSCs were seeded directly in hMSCs basal or osteogenesis differentiation medium, and treated with seven concentrations of each chemical.
We validated the assay performance including specificity, sensitivity, coefficient of variation (CV) within and between assay plates, and quantitation (linearity and limit). The detailed procedures for curve fitting and data analysis had been described previously 18–23. The compound’s IC50 and efficacy were calculated based on the dose-response curves. We applied the cutoff criteria of efficacy ≥ 50% and IC50 ≤ 20 μM to identify compounds with significant toxicity to hMSCs. From the primary screening, we identified 38 hits for basal hMSCs and 23 hits for hMSCs after 1 day of osteogenic differentiation (Table S1). The majority of the hits for osteogenic hMSCs were also the hits for basal hMSCs, but not vice versa. For example, we observed much higher toxicity (i.e., lower IC50) for AC-93253 to basal hMSCs than osteogenic hMSCs. This might suggest that osteogenic differentiation reprogrammed the pathways and networks of hMSCs, and therefore the resistance to certain chemicals for osteogenic hMSCs increased.
To cover a wide range of IC50 while maintaining the diversity of putative drug targets, we selected four hits including AC-93253, a RARα agonist and SIRT2 inhibitor; cantharidin, a specific inhibitor of protein phosphatase 2A (PP2A); idarubicin hydrochloride, a topoisomerase II inhibitor that is used as an anthracycline antibiotic and anti-leukemia agent; and NSC 95397, an irreversible Cdc25 dual specificity phosphatase inhibitor; for further studies including: (1) validation of toxicity in a 96-well format for hMSCs at various stages of osteogenic differentiation, and for hiPSCs at various stage of hepatogenic differentiation; and (2) Omics-based assays for elucidating cellular pathways and networks involved in chemical toxicity responses.
Validation of chemical cytotoxicity on hMSCs at various stages of osteogenic differentiation
We implemented a protocol for osteogenic differentiation of hMSCs (Figure 2A). Basal hMSCs exhibited a fibroblast-like, spindle-shaped morphology when cultured in the basal culture medium. After cultured in the osteogenesis medium for 3 days (OD3), the cells started to show a branched shape. They exhibited a cuboidal morphology on day 6 (OD6). We confirmed the osteogenic differentiation using ALP activity, a marker of osteogenic differentiation. ALP staining (blue) was consistent with the osteogenic progression from OD1 to OD6.
Figure 2. The toxicity of four chemicals (AC-93253, idarubicin, NSC 95397, and cantharidin) on hMSCs at the different stages of osteogenic differentiation.

A. Representative phase contrast images showing the morphological changes (top) and ALP staining (bottom) for basal hMSCs, and hMSCs 1 day (OD1), 3 days (OD3), and 6 days (OD6) after osteogenic differentiation. Scale bar: 100 μm.
B. Representative phase contrast images of basal hMSCs and hMSCs 1 day and 3 days after osteogenic differentiation, and 24 hours after idarubicin treatment and DMSO control. The idarubicin concentration used in each group was the corresponding IC50 values for 48 hours treatment. Scale bar: 100 μm.
C. Cell survival curves for basal hMSCs, and hMSCs-OD1, OD3, and OD6 cells, in response to 48-hour treatments of AC-93253, idarubicin, NSC 95397, and cantharidin, respectively. Each chemical was measured in triplicates on 96-well plates. Data are represented as mean ± s.e.m. (s.e.m.: standard error of the mean).
D. IC50 values of the four chemicals on basal hMSCs, and hMSCs-OD1, OD3, and OD6 cells. For each group, the experiments were repeated 3 times (n=3). Data are represented as mean ± s.e.m. *p < 0.05; **p < 0.01; ***p < 0.001, by the unpaired two-tailed Student’s t-test.
Chemical treatment also changed the morphology of hMSCs (Figure 2B). For example, idarubicin caused both basal and osteogenic progenies to shrink significantly 48 hours after treatment. We validated chemical cytotoxicity and measured IC50 values using 96-well plates for AC-93253, idarubicin, NSC 95397, and cantharidin (Figure 2C and 2D, and Table 1). The qHTS platform for chemical toxicity screening utilizes the CTG assay which measures ATP content to indicate cell viability with luminescence readout. There are several strategies available for detecting cell viability. For example, the MTT assay measures mitochondrial dehydrogenase activity in living cells and has been used for determining the in vitro cytotoxicity of chemical toxins to MSCs 11, 12. Unlike the MTT assay that only measures the dehydrogenase activity from mitochondria, the CCK-8 assay utilizes WST-8 and the electron mediator, 1-methoxy PMS, to measure most of the dehydrogenase activity in a cell and is more sensitive than the MTT assay. Therefore, we chose the colorimetric CCK-8 assay, independent of CTG assay, to reduce the bias and achieve an independent validation of chemical toxicity.
Table 1.
IC50 values for hMSCs at different stages of osteogenic differentiation
| Chemical | IC50 (μM) | |||
|---|---|---|---|---|
|
| ||||
| hMSC | hMSC-OD1 | hMSC-OD3 | hMSC-OD6 | |
| AC-93253 | 3.94 ± 1.18 | 8.06 ± 0.88 | 5.65 ± 0.50 | 10.62 ± 1.67 |
| Idarubicin | 0.38 ± 0.074 | 2.96 ± 0.22 | 7.43 ± 4.56 | 10.76 ± 0.43 |
| NSC 95397 | 6.55 ± 1.76 | 5.33 ± 0.60 | 7.51 ± 2.33 | 9.73 ± 0.80 |
| Cantharidin | 1.54 ± 0.50 | 3.98 ± 0.69 | 4.95 ± 1.33 | 6.17 ± 2.55 |
OD1, OD3, OD6: osteogenic differentiation day 1, 3, 6.
Among these four chemicals, idarubicin demonstrated the highest toxicity on basal hMSCs (IC50 = 0.38 μM) and the most dramatic reduction in toxicity as hMSCs underwent osteogenic differentiation (28-fold decrease from basal to OD6, p<0.001). On the other hand, the toxicity of NSC 95397 were similar for basal hMSCs and hMSCs at OD1 to OD6, suggesting its effects were not differentiation-stage dependent. The toxicities of AC-93253 and cantharidin were in between, so were their differentiation-stage dependencies, with a reduction of 3~4 fold from basal hMSCs to their osteogenic progenies (OD1-OD6).
Compared to the qHTS using seven concentrations, our 96-well plate assays measured 14 concentrations of each compound. Furthermore, we could perform toxicity studies for osteogenic progenies beyond day 1 because differentiation media could be readily replaced every 3 days on 96-well plates. These allowed us to obtain more accurate IC50 values and study the dependency of chemical toxicity on osteogenic differentiation stages. We studied the acute cytotoxicity of chemicals (i.e., 48 hours treatment) on hMSCs at early stages of osteogenic differentiation (day 1 to day 6). Alternatively, we can treat hMSCs in osteogenic media on day 0, and monitor the cytotoxicity and the effects on osteogenesis using ALP activity over a longer time period, e.g., 3–4 weeks, to further elucidate the differentiation-stage dependency of chemical toxicity.
Validation of chemical toxicity on hiPSCs at various stages of hepatogenic differentiation
We implemented a robust protocol for hepatogenic differentiation of hiPSCs (Figure 3A). The protocol included three phases and hepatocyte maturation was achieved within 20 days. We confirmed the induction, specification, and maturation of hepatocytes by immunostaining using characteristic markers of hiPSCs and hepatocytes including Oct4 (stem cell marker), Sox17 (a definitive endoderm marker), AFP and HNF4A (hepatic progenitor markers), and Alb (hepatocyte marker) (Figure 3B). Finally, we validated the functionalities of our hiPSCs-derived hepatocytes by performing head-to-head comparison of morphology, PAS staining, and ICG uptake functional assay to primary human hepatocytes (Figure 3C).
Figure 3. The toxicity of four chemicals (AC-93253, idarubicin, NSC 95397, and cantharidin) on hiPSCs at the different stages of hepatogenic differentiation.

A. Flow chart showing three phases of hiPSCs hepatogenic differentiation. Phase I: 5 days of definitive endoderm (DE) induction; Phase II: 5 days of hepatic specification; and Phase III: 10 days of hepatocytes maturation. The key chemicals used and the molecular markers during each phase are specified. The bottom panel shows the representative cell morphology during the key timepoints of hepatogenic differentiation.
B. Immunofluorescence staining of the selected molecular markers on hiPSCs, and the cells at the end of hepatogenic differentiation Phase I, II, and III, respectively, in comparison to primary hepatocytes.
C. Functional assays including PAS staining and ICG update of the cells at the end of Phase III. hiPSCs were used as the negative control, and human primary hepatocytes were used as the positive control.
D. Cell survival curves for basal hiPSCs, and hiPSCs at the end of hepatogenic differentiation Phase I, II, and III, respectively, in response to 48-hour treatments of AC-93253, idarubicin, NSC 95397, and cantharidin, respectively. Each chemical was measured in triplicates on 96-well plates. Data are represented as mean ± s.e.m.
E. IC50 values of the four chemicals on basal hiPSCs, and hiPSCs at the end of hepatogenic differentiation Phase I, II, and III, respectively. For each group, the experiments were repeated 3 times (n=3). Data are represented as mean ± s.e.m. *p < 0.05; **p< 0.01; ***p < 0.001, by unpaired two-tailed Student’s t-test.
F. Comparison of IC50 values of hiPSCs, hepatic differentiation Phase I cells, and hMSCs in response to treatment of the four chemicals. For each group, the experiments were repeated 3 times (n=3). Data are represented as mean ± s.e.m. *p < 0.05; **p < 0.01; ***p < 0.001, by unpaired two-tailed Student’s t-test.
We performed toxicity tests on hiPSCs for the same 4 chemicals on hMSCs (Figure 3D–3F, and Table 2). For hiPSCs, we used 0.1% DMSO in the cell culture medium to minimize the cytotoxicity effects of DMSO. Compared to basal hMSCs, hiPSCs showed more than 30 times higher sensitivity to AC-93253 and idarubicin (IC50 = 0.090 vs. 3.94 μM, and 0.004 vs. 0.378 μM, respectively). In contrast, basal hMSCs and hiPSCs showed similar sensitivity to NSC 95397 and cantharidin. Interestingly, the effects of idarubicin were significantly dependent on the stages of hepatogenic differentiation of hiPSCs, similar to what we observed for osteogenic differentiation-stage dependency of hMSCs (Figure 3E and 2D). Surprisingly, the effects of cantharidin were not significantly dependent on the stages of hepatogenic differentiation of hiPSCs, in contrast to those of hMSCs. Therefore, the chemical toxicity responses depended on the stem cell types as well as their differentiation stages. We observed the most dramatic difference in sensitivity and differentiation stage-dependency between hMSCs and hiPSCs in response to idarubicin and cantharidin treatment. Although idarubicin is considered a TOP2A inhibitor, its exact functions in vivo have not been fully elucidated 24, 25. On the other hand, there is strong evidence to support that cantharidin is a specific inhibitor of PP2A 26, 27. However, how these two chemicals affect the proliferation and differentiation of hMSCs and hiPSCs, and the pathways and networks responsible for the chemical toxicity, need to be further investigated.
Table 2.
IC50 values for hiPSCs at different stages of hepatogenic differentiation
| Chemical | IC50 (μM) | |||
|---|---|---|---|---|
|
| ||||
| hiPSC | Hepa Diff Phase I | Hepa Diff Phase II | Hepa Diff Phase III | |
| AC-93253 | 0.090 ± 0.038 | 0.26 ± 0.01 | 2.16 ± 0.25 | 0.77 ± 0.39 |
| Idarubicin | 0.004 ± 0.001 | 0.07 ± 0.003 | 1.25 ± 0.23 | 4.99 ± 1.38 |
| NSC 95397 | 6.91 ± 3.01 | 11.30 ± 1.56 | 33.55 ± 7.43 | 83.03 ± 13.50 |
| Cantharidin | 4.36 ± 1.40 | 2.70 ± 0.49 | 2.58 ± 0.28 | 1.12± 0.34 |
Hepa Diff: hepatogenic differentiation
Proteomics studies of pathways and networks involved in toxicity responses to idarubicin and cantharidin
We established a comprehensive platform for proteomics profiling of stem cells and their progenies 7, 28–30. Our platform allowed us to identify and quantify over 3,000 proteins (including isoforms) starting from 100,000 – 200,000 cells. Proteins with a fold change > 1.5 or < 0.67 between two conditions, and with a p value < 0.05, were considered to be differentially-expressed. We performed pair-wise proteome comparisons between idarubicin or cantharidin-treated and DMSO controls, for basal hMSCs and hiPSCs, and hMSCs at different stages of osteogenic differentiation (OD1, OD3, and OD6).
Figure 4A and 4B show the representative volcano-plots for up- and down-regulated proteins in hiPSCs (4A) and hMSCs-OD6 (4B) after idarubicin treatment for 24 hours (Table S2 and S3, respectively). We detected higher number of proteins (n = 3,659 vs. 3,097) and observed more significant changes in the number of up- and down-regulation of cellular proteins (n = 202 vs. 73, and n = 568 vs. 155, respectively), for hiPSCs compared to hMSCs-OD6. This is consistent with the much higher sensitivity of hiPSCs to idarubicin (IC50 = 0.004 vs. 10.76 μM).
Figure 4. Proteomics studies of pathways and networks involved in toxicity responses to idarubicin.

A. Volcano-plot of differentially-expressed proteins in hiPSCs in response to 24-hour idarubicin treatment. Fold change > 1.5 or < 0.67, and p value < 0.05 were considered significant. Red dots: upregulated proteins after idarubicin treatment (n=202); blue dots: downregulated proteins after idarubicin treatment (n=568); grey dots: proteins with non-significant changes after idarubicin treatment (n=2,889). A total of 3,659 proteins were quantified in this proteomics assay.
B. Volcano-plot of differentially-expressed proteins in hMSCs-OD6 in response to 24-hour idarubicin treatment. Fold change > 1.5 or < 0.67, and p value < 0.05 were considered significant. Red dots: upregulated proteins after idarubicin treatment (n=73); blue dots: downregulated proteins after idarubicin treatment (n=155); grey dots: proteins with non-significant changes after idarubicin treatment (n=2,869). A total of 3,097 proteins were quantified in this proteomics assay.
C. The top canonical pathways enriched after 24-hour idarubicin treatment in different cells (hiPSCs, hMSCs, OD1 and OD6 cells). The activation Z-score indicates either activation (yellow) or inhibition (blue).
D. The top canonical pathways enriched due to the stemness effect. Comparisons were made between hiPSCs and hMSCs, and between basal hMSCs and OD1 and OD6 cells, respectively. The activation Z-score indicates either activation (yellow) or inhibition (blue).
To further understand the mechanism underlying chemical toxicity and its dependence on stem cell types and differentiation stages, we performed pathway and network analysis using IPA software. We clustered the top 10 canonical pathways affected by idarubicin treatment (Figure 4C) and those conferred stemness effects (Figure 4D) based on our proteomics data. In both cases, the elongation initiation factor 2 (EIF2) signaling pathway came out as the top pathway, although the activations were in the opposite direction. After idarubicin treatment, EIF2 signaling pathway was dramatically inhibited in hiPSCs, but to a less degree in basal hMSCs, and even less for OD1 and OD6. On the other hand, the EIF2 signaling was much more activated in pluripotent hiPSCs than hMSCs, OD1 and OD6 (Figure 4D and Figure S2). EIF2 signaling pathway plays an important role in regulating the initiation of protein synthesis; and a set of EIFs are required during the process that ribosomes recognize the mRNA to be translated and identify the translational start site 31–34. Our results suggested that translation elongation and protein synthesis in hiPSCs might be much faster than basal hMSCs, and the protein synthesis might gradually decrease during the osteogenic differentiation in hMSCs. This is consistent with the higher self-renewal capability of the pluripotent hiPSCs than those of multipotent hMSCs, and the inhibition of cell proliferation during differentiation. Nevertheless, our proteomics studies measured protein expression levels but not the reaction rates. Additional experiments are needed to confirm the increase in protein synthesis rates due to the activation of EIF2 signaling in hiPSCs.
We observed activation and inhibition of multiple pathways involved in actin polymerization and cytoskeleton reorganization, and cell motility (Figure 4C and 4D). These included Rho family GTPases, ephrin receptor signaling, integrin signaling, Rac signaling, actin cytoskeleton signaling, and integrin signaling etc. The actin cytoskeleton functions in the generation and maintenance of cell morphology, motility and cell division. Indeed, we observed significant changes in cell morphology after chemical treatments and cell differentiation (Figure 2 and 3). Two of the top pathways activated after idarubicin treatments were RhoGDI signaling and HIPPO signaling. RhoGDIs negatively regulate Rho-family GTPases to promote cytoskeletal and membrane changes associated with apoptosis 35, 36. HIPPO signaling pathway is involved in restraining cell proliferation and promoting apoptosis, and by finely tuning this pathway, mammals control organ sizes through the regulation of cell proliferation and apoptosis 37–39.
The pathway data suggested activation of apoptosis rather than inhibition of proliferation as the key toxicity response of hiPSCs to idarubicin treatment. Consistently, we identified the activation of chromatin condensation, DNA fragmentation, cell shrinkage, and inhibition of DNA repair, all leading to apoptosis, in response to idarubicin treatment (Figure 5A(i)). Our proteomics data supported the phenotypic observations that hiPSCs were more sensitive, and osteogenic progenies were less sensitive, to idarubicin treatment, than hMSCs. We observed significant activation of the G2/M DNA damage checkpoint pathway for hiPSCs but not for hMSCs-OD6 (Figure S3), presumably through idarubicin targeting of Top2A and its pathways. Additionally, the cell proliferation pathway is highly enriched in hiPSCs and much more than in hMSCs (Figure S4). These observations are consistent with the biological functions regulated by idarubicin, in which cell death and apoptosis were highly activated, while cell survival, cell cycle progression, and mitosis were significantly inhibited (Figure 5A(ii)). In contrast to the opposite responses of multiple pathways to idarubicin for hiPSCs and OD6, cantharidin conferred similar modulation of most of the top pathways for hiPSCs and OD6 (Figure 5B(i)–(ii) and Table S4). This might be due to the specific and potent inhibition of PP2A by cantharidin in both cell lines. On the other hand, idarubicin was considered to target multiple pathways besides Top2A.
Figure 5. Pathways and networks involved in toxicity responses to idarubicin and cantharidin for hiPSC and hMSCs at different stages of osteogenic differentiation.

A. (i) Apoptosis pathway was enriched and activated in hiPSCs after 24-hour idarubicin treatment. Protein increased in abundance are in red, and decreased are in green, not-significantly-changed are in grey. Proteins predicted by IPA to be activated are in orange; predicted to be inhibited are in blue. Only proteins with significant changes in abundance at p value < 0.05 between with or without idarubicin treatment were included in analysis. (ii) The predicted activated (yellow) or inhibited (blue) bio-functions by IPA on different cells (hiPSCs, hMSCs, OD1, and OD6), after 24-hour idarubicin treatment.
B. (i) The top canonical pathways enriched in hiPSCs and hMSC-OD6 cells, after 24-hour cantharidin treatment. (ii) Top bio-functions activated (yellow) or inhibited (blue) in hiPSCs and hMSC-OD6 cells, after 24-hour cantharidin treatment.
Our studies may have some limitations. For example, the pathways and networks might be biased towards the proteins involved in particular pathways, most of which are soluble cytoplasmic proteins that could be readily detected by LC-MS/MS using our proteomics platform. Future work will further increase the number of proteins detected by including enrichment and prefractionation steps for total cell lysates to increase the detectability of membrane and nuclear proteins. Our goal is to increase the total to 6,000–8,000 proteins in order to cover more comprehensively the global pathways and networks. On the other hand, the pathways and networks discovered by our phenotypic and mechanistic studies are mostly speculative at this point. Our main goal for the current work is to demonstrate the feasibility of an integrated Omics-based platform for screening chemical toxicity using stem cells. Future detailed biochemical work will validate our preliminary discoveries and provide more biological insights.
Conclusions
In this study, we established an integrated platform by combining phenotypic and Omics-based mechanistic studies, to evaluate the effects of chemical toxicants on proliferation and differentiation of stem cells, in particular, human mesenchymal stem cells at the different stages of osteogenesis, and human induced pluripotent stem cells at the different stages of hepatogenesis. Using qHTS, we identified the potential hits of chemical toxicity on hMSCs using LOPAC containing 1,280 compounds in a 1,536-well format. The chemical toxicity of AC-93253, cantharidin, idarubicin, and NSC 95397 identified from the primary screening was then confirmed in an orthogonal cell survival assay using a 96-well plate format. We identified four different types of chemical toxicity responses that are dependent on both the stem cell types and their differentiation stages. Proteomics and bioinformatics studies of the effects of idarubicin and cantharidin on hiPSCs and hMSCs as well as their progenies identified key pathways responsible for chemical-specific and stem cell-specific toxicity responses.
Our approach may be utilized in the future to test the toxicity of larger chemical libraries, for example, Tox21 10k library containing 10,000 compounds (~ 8,300 unique chemicals), or combinatory libraries for drug discovery that contain tens of thousands of compounds. Alternatively, we can utilize different stem cells such as hemopoietic stem cells and embryonic stem cells along different lineages.
In summary, our stem cell-based assays provide mechanistic insights into chemical toxicity, and may help prioritize chemicals for in-depth toxicological evaluation, and even help develop new intervention strategies for mitigating chemical toxicity, thereby contributing to public health.
Supplementary Material
Acknowledgments
The work was supported by the National Institutes of Health awards ES022360 and ES023529 (to Newomics Inc.). The authors also acknowledged supports from NIH awards AT008297, GM109682, AG046025, AI106100, and HHSN261201300033C. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. We thank current and former colleagues at Newomics Inc. including Drs. Henry Chang, Geuncheol Gil, and Pan Mao for technical assistance and helpful discussions.
Footnotes
Author Contributions
Y.H., J.Z., R.H. and M.X. designed and performed experiments and data analysis, and wrote the manuscript. D.W. conceived and directed the experiments and wrote the manuscript.
Conflict of Interest Disclosure
Y.H. and D. W. are employees of Newomics Inc., which may commercialize some of the assays described in this work.
Supporting Information: The following files are available free of charge at ACS website http://pubs.acs.org.
Supporting information: Figure S1–S4
Figure S1: Time courses of chemical toxicity tests on two stem cells and their progenies.
Figure S2: Enrichment of EIF2 signaling pathways in stem cells and their progenies.
Figure S3: G2/M DNA damage checkpoint pathway is activated and enriched in hiPSCs.
Figure S4: Enrichment of pathways for cell proliferation and protein synthesis in stem cells.
Table S1: LOPAC hits_50% and 20 μM
Table S2: hiPSC_Idarubicin vs DMSO
Table S3: OD6_Idarubicin vs DMSO
Table S4: hiPSC and hMSC_Cantharidin vs DMSO
References
- 1.Hartung T. Toxicology for the twenty-first century. Nature. 2009;460:208–12. doi: 10.1038/460208a. [DOI] [PubMed] [Google Scholar]
- 2.Sun H, Xia M, Austin CP, Huang R. Paradigm shift in toxicity testing and modeling. AAPS J. 2012;14:473–80. doi: 10.1208/s12248-012-9358-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Stephens ML, Andersen M, Becker RA, Betts K, Boekelheide K, Carney E, Chapin R, Devlin D, Fitzpatrick S, Fowle JR, Harlow P, Hartung T, Hoffmann S, Holsapple M, Jacobs A, Judson R, Naidenko O, Pastoor T, Patlewicz G, Rowan A, Scherer R, Shaikh R, Simon T, Wolf D, Zurlo J. Evidence-based toxicology for the 21st century: Opportunities and challenges. ALTEX. 2013;30:74–104. doi: 10.14573/altex.2013.1.074. [DOI] [PubMed] [Google Scholar]
- 4.Bianco P, Robey PG. Stem cells in tissue engineering. Nature. 2001;414:118–21. doi: 10.1038/35102181. [DOI] [PubMed] [Google Scholar]
- 5.Caplan AI, Bruder SP. Mesenchymal stem cells: building blocks for molecular medicine in the 21st century. Trends Mol Med. 2001;7:259–64. doi: 10.1016/s1471-4914(01)02016-0. [DOI] [PubMed] [Google Scholar]
- 6.Beausejour CM, Campisi J. Ageing: balancing regeneration and cancer. Nature. 2006;443:404–5. doi: 10.1038/nature05221. [DOI] [PubMed] [Google Scholar]
- 7.Wang D, Jang DJ. Protein kinase CK2 regulates cytoskeletal reorganization during ionizing radiation-induced senescence of human mesenchymal stem cells. Cancer Res. 2009;69:8200–7. doi: 10.1158/0008-5472.CAN-09-1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davila JC, Cezar GG, Thiede M, Strom S, Miki T, Trosko J. Use and application of stem cells in toxicology. Toxicol Sci. 2004;79:214–23. doi: 10.1093/toxsci/kfh100. [DOI] [PubMed] [Google Scholar]
- 9.Sharma A, Burridge PW, McKeithan WL, Serrano R, Shukla P, Sayed N, Churko JM, Kitani T, Wu H, Holmstrom A, Matsa E, Zhang Y, Kumar A, Fan AC, Del Alamo JC, Wu SM, Moslehi JJ, Mercola M, Wu JC. High-throughput screening of tyrosine kinase inhibitor cardiotoxicity with human induced pluripotent stem cells. Sci Transl Med. 2017:9. doi: 10.1126/scitranslmed.aaf2584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shuga J, Zhang J, Samson LD, Lodish HF, Griffith LG. In vitro erythropoiesis from bone marrow-derived progenitors provides a physiological assay for toxic and mutagenic compounds. Proc Natl Acad Sci U S A. 2007;104:8737–42. doi: 10.1073/pnas.0701829104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee HJ, Cha KE, Hwang SG, Kim JK, Kim GJ. In vitro screening system for hepatotoxicity: comparison of bone-marrow-derived mesenchymal stem cells and Placenta-derived stem cells. J Cell Biochem. 2011;112:49–58. doi: 10.1002/jcb.22728. [DOI] [PubMed] [Google Scholar]
- 12.Scanu M, Mancuso L, Cao G. Evaluation of the use of human Mesenchymal Stem Cells for acute toxicity tests. Toxicol In Vitro. 2011;25:1989–95. doi: 10.1016/j.tiv.2011.07.006. [DOI] [PubMed] [Google Scholar]
- 13.Norris JL, Farrow MA, Gutierrez DB, Palmer LD, Muszynski N, Sherrod SD, Pino JC, Allen JL, Spraggins JM, Lubbock AL, Jordan A, Burns W, Poland JC, Romer C, Manier ML, Nei YW, Prentice BM, Rose KL, Hill S, Van de Plas R, Tsui T, Braman NM, Keller MR, Rutherford SA, Lobdell N, Lopez CF, Lacy DB, McLean JA, Wikswo JP, Skaar EP, Caprioli RM. Integrated, High-Throughput, Multiomics Platform Enables Data-Driven Construction of Cellular Responses and Reveals Global Drug Mechanisms of Action. J Proteome Res. 2017;16:1364–1375. doi: 10.1021/acs.jproteome.6b01004. [DOI] [PubMed] [Google Scholar]
- 14.Hannan NR, Segeritz CP, Touboul T, Vallier L. Production of hepatocyte-like cells from human pluripotent stem cells. Nat Protoc. 2013;8:430–7. doi: 10.1038/nprot.2012.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Tasnim F, Phan D, Toh YC, Yu H. Cost-effective differentiation of hepatocyte-like cells from human pluripotent stem cells using small molecules. Biomaterials. 2015;70:115–25. doi: 10.1016/j.biomaterials.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 16.Siller R, Greenhough S, Naumovska E, Sullivan GJ. Small-molecule-driven hepatocyte differentiation of human pluripotent stem cells. Stem Cell Reports. 2015;4:939–52. doi: 10.1016/j.stemcr.2015.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaserman JE, Wilson AA. Protocol for Directed Differentiation of Human Induced Pluripotent Stem Cells (iPSCs) to a Hepatic Lineage. Methods Mol Biol. 2017;1639:151–160. doi: 10.1007/978-1-4939-7163-3_15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hsu CW, Zhao J, Huang R, Hsieh JH, Hamm J, Chang X, Houck K, Xia M. Quantitative high-throughput profiling of environmental chemicals and drugs that modulate farnesoid X receptor. Sci Rep. 2014;4:6437. doi: 10.1038/srep06437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Huang R, Sakamuru S, Martin MT, Reif DM, Judson RS, Houck KA, Casey W, Hsieh JH, Shockley KR, Ceger P, Fostel J, Witt KL, Tong W, Rotroff DM, Zhao T, Shinn P, Simeonov A, Dix DJ, Austin CP, Kavlock RJ, Tice RR, Xia M. Profiling of the Tox21 10K compound library for agonists and antagonists of the estrogen receptor alpha signaling pathway. Sci Rep. 2014;4:5664. doi: 10.1038/srep05664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Attene-Ramos MS, Huang R, Michael S, Witt KL, Richard A, Tice RR, Simeonov A, Austin CP, Xia M. Profiling of the Tox21 Chemical Collection for Mitochondrial Function to Identify Compounds that Acutely Decrease Mitochondrial Membrane Potential. Environ Health Perspect. 2015;123:49–56. doi: 10.1289/ehp.1408642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shukla SJ, Huang R, Simmons SO, Tice RR, Witt KL, Vanleer D, Ramabhadran R, Austin CP, Xia M. Profiling environmental chemicals for activity in the antioxidant response element signaling pathway using a high throughput screening approach. Environ Health Perspect. 2012;120:1150–6. doi: 10.1289/ehp.1104709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sakamuru S, Li X, Attene-Ramos MS, Huang R, Lu J, Shou L, Shen M, Tice RR, Austin CP, Xia M. Application of a homogenous membrane potential assay to assess mitochondrial function. Physiol Genomics. 2012;44:495–503. doi: 10.1152/physiolgenomics.00161.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Attene-Ramos MS, Huang R, Sakamuru S, Witt KL, Beeson GC, Shou L, Schnellmann RG, Beeson CC, Tice RR, Austin CP, Xia M. Systematic study of mitochondrial toxicity of environmental chemicals using quantitative high throughput screening. Chem Res Toxicol. 2013;26:1323–32. doi: 10.1021/tx4001754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mandelli F, Diverio D, Avvisati G, Luciano A, Barbui T, Bernasconi C, Broccia G, Cerri R, Falda M, Fioritoni G, Leoni F, Liso V, Petti MC, Rodeghiero F, Saglio G, Vegna ML, Visani G, Jehn U, Willemze R, Muus P, Pelicci PG, Biondi A, Lo Coco F. Molecular remission in PML/RAR alpha-positive acute promyelocytic leukemia by combined all-trans retinoic acid and idarubicin (AIDA) therapy. Gruppo Italiano-Malattie Ematologiche Maligne dell’Adulto and Associazione Italiana di Ematologia ed Oncologia Pediatrica Cooperative Groups. Blood. 1997;90:1014–21. [PubMed] [Google Scholar]
- 25.Chen T, Sun Y, Ji P, Kopetz S, Zhang W. Topoisomerase IIalpha in chromosome instability and personalized cancer therapy. Oncogene. 2015;34:4019–31. doi: 10.1038/onc.2014.332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li YM, Casida JE. Cantharidin-binding protein: identification as protein phosphatase 2A. Proc Natl Acad Sci U S A. 1992;89:11867–70. doi: 10.1073/pnas.89.24.11867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li W, Xie L, Chen Z, Zhu Y, Sun Y, Miao Y, Xu Z, Han X. Cantharidin, a potent and selective PP2A inhibitor, induces an oxidative stress-independent growth inhibition of pancreatic cancer cells through G2/M cell-cycle arrest and apoptosis. Cancer Sci. 2010;101:1226–33. doi: 10.1111/j.1349-7006.2010.01523.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang D, Gao L. Proteomic analysis of neural differentiation of mouse embryonic stem cells. Proteomics. 2005;5:4414–26. doi: 10.1002/pmic.200401304. [DOI] [PubMed] [Google Scholar]
- 29.Wang D, Park JS, Chu JS, Krakowski A, Luo K, Chen DJ, Li S. Proteomic profiling of bone marrow mesenchymal stem cells upon transforming growth factor b1 stimulation. J Biol Chem. 2004;279:43725–34. doi: 10.1074/jbc.M407368200. [DOI] [PubMed] [Google Scholar]
- 30.Gil G, Mao P, Avula B, Ali Z, Chittiboyina AG, Khan IA, Walker LA, Wang D. Proteoform-Specific Protein Binding of Small Molecules in Complex Matrices. ACS Chem Biol. 2017;12:389–397. doi: 10.1021/acschembio.6b01018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Petryshyn R, Rosa F, Fagard R, Levin D, London IM. Control of protein synthesis in human reticulocytes by heme-regulated and double-stranded RNA dependent eIF-2 alpha kinases. Biochem Biophys Res Commun. 1984;119:891–9. doi: 10.1016/0006-291x(84)90857-x. [DOI] [PubMed] [Google Scholar]
- 32.Kimball SR. Eukaryotic initiation factor eIF2. Int J Biochem Cell Biol. 1999;31:25–9. doi: 10.1016/s1357-2725(98)00128-9. [DOI] [PubMed] [Google Scholar]
- 33.Asano K, Clayton J, Shalev A, Hinnebusch AG. A multifactor complex of eukaryotic initiation factors, eIF1, eIF2, eIF3, eIF5, and initiator tRNA(Met) is an important translation initiation intermediate in vivo. Genes Dev. 2000;14:2534–46. doi: 10.1101/gad.831800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shrestha N, Bahnan W, Wiley DJ, Barber G, Fields KA, Schesser K. Eukaryotic initiation factor 2 (eIF2) signaling regulates proinflammatory cytokine expression and bacterial invasion. J Biol Chem. 2012;287:28738–44. doi: 10.1074/jbc.M112.375915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dovas A, Couchman JR. RhoGDI: multiple functions in the regulation of Rho family GTPase activities. Biochem J. 2005;390:1–9. doi: 10.1042/BJ20050104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Spiering D, Hodgson L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh Migr. 2011;5:170–80. doi: 10.4161/cam.5.2.14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huang J, Wu S, Barrera J, Matthews K, Pan D. The Hippo signaling pathway coordinately regulates cell proliferation and apoptosis by inactivating Yorkie, the Drosophila Homolog of YAP. Cell. 2005;122:421–34. doi: 10.1016/j.cell.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 38.Pan D. The hippo signaling pathway in development and cancer. Dev Cell. 2010;19:491–505. doi: 10.1016/j.devcel.2010.09.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Heallen T, Zhang M, Wang J, Bonilla-Claudio M, Klysik E, Johnson RL, Martin JF. Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science. 2011;332:458–61. doi: 10.1126/science.1199010. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.