

Abstract
Rationale
Cardiac lipotoxicity, characterized by increased uptake, oxidation and accumulation of lipid intermediates, contributes to cardiac dysfunction in obesity and diabetes. However, mechanisms linking lipid overload and mitochondrial dysfunction are incompletely understood.
Objective
To elucidate the mechanisms for mitochondrial adaptations to lipid overload in postnatal hearts in vivo.
Methods and Results
Using a transgenic mouse model of cardiac lipotoxicity overexpressing long-chain acyl-CoA synthetase 1 in cardiomyocytes, we show that modestly increased myocardial fatty acid uptake leads to mitochondrial structural remodeling with significant reduction in minimum diameter. This is associated with increased palmitoyl-carnitine oxidation and increased reactive oxygen species (ROS) generation in isolated mitochondria. Mitochondrial morphological changes and elevated ROS generation are also observed in palmitate-treated neonatal rat ventricular cardiomyocytes (NRVCs). Palmitate exposure to NRVCs initially activates mitochondrial respiration, coupled with increased mitochondrial membrane potential and adenosine triphosphate (ATP) synthesis. However, long-term exposure to palmitate (> 8h) enhances ROS generation, which is accompanied by loss of the mitochondrial reticulum and a pattern suggesting increased mitochondrial fission. Mechanistically, lipid-induced changes in mitochondrial redox status increased mitochondrial fission by increased ubiquitination of A-kinase anchor protein (AKAP121) leading to reduced phosphorylation of DRP1 at Ser637 and altered proteolytic processing of OPA1. Scavenging mitochondrial ROS restored mitochondrial morphology in vivo and in vitro.
Conclusions
Our results reveal a molecular mechanism by which lipid overload-induced mitochondrial ROS generation causes mitochondrial dysfunction by inducing post-translational modifications of mitochondrial proteins that regulate mitochondrial dynamics. These findings provide a novel mechanism for mitochondrial dysfunction in lipotoxic cardiomyopathy.
Keywords: Cardiac lipotoxicity, mitochondrial dynamics, oxidative stress, heart failure, diabetic cardiomyopathy, metabolism, reactive oxygen species
Introduction
To maintain high energy requirements for contractile function, the heart requires an uninterrupted delivery of oxygen and substrates that are oxidized to promote adenosine triphosphate (ATP) synthesis. ATP consumption primarily fuels sarcomere contraction and ionic pumps that maintain membrane potential 1. In the healthy well-oxygenated heart, mitochondria are central to cardiomyocyte energy metabolism 1. Mitochondrial oxidation of fatty acids (FAs), glucose, ketone bodies and lactate accounts for the majority of ATP generation in cardiomyocytes. FAs derived from circulating triglyceride-rich lipoproteins and albumin bound non-esterified FAs are oxidized in the mitochondrial matrix via β-oxidation, whereas pyruvate derived from glucose and lactate is oxidized by the pyruvate-dehydrogenase complex, localized within the inner mitochondrial membrane. Acetyl-CoA formed from FAs and pyruvate is oxidized within the mitochondrial matrix by the citric acid cycle. Healthy adult cardiomyocytes preferentially utilize fatty acids as an energy substrate 2-4. However, energy substrate usage is flexible in normal hearts and varies to adapt to physiological or pathological stresses 5.
Diabetes and insulin resistance are the most common metabolic disorders in the world. Cardiovascular complications are the main cause of death from diabetes mellitus, given the high prevalence of comorbid risk factors, such as coronary artery disease. In addition, diabetes mellitus increases the risk for heart failure by mechanisms that may be independent of underlying myocardial ischemia or hypertension 6. This increased vulnerability termed “diabetic cardiomyopathy”, has a complex pathophysiology that includes altered myocardial substrate utilization 7, 8. An important characteristic of the metabolic abnormality in diabetic hearts is “metabolic inflexibility”, characterized by reduced glucose utilization and increased FA utilization. An imbalance between substrate uptake and utilization may lead to the accumulation of toxic metabolic intermediates 9. Studies by us and others reveal that, in addition to altered substrate utilization, diabetes or myocardial insulin resistance impairs mitochondrial bioenergetics characterized by decreased oxidative capacity, increased ROS generation and mitochondrial uncoupling 10-12. It is likely that mitochondrial dysfunction is multifactorial, although the relative contributions of increased lipid uptake versus changes in insulin signaling are incompletely understood.
To understand the mechanisms by which increased cardiac lipid uptake could alter mitochondrial function, we investigated mice with cardiomyocyte-restricted low-level overexpression of long chain acyl-CoA synthetase 1 (ACSL1) in which myocardial palmitate biodistribution was moderately increased (2-fold) 13. In this murine model, the modest increase in FA uptake dramatically remodeled the mitochondrial network, leading to the accumulation of elongated mitochondria with reduced diameter in cardiomyocytes. Similar changes, appearing on 2-D microscopy as numerous “fragmented” mitochondria, were recapitulated in rat neonatal cardiomyocytes (NRVCs) and L6 myotubes following short-term exposure to the long chain FA palmitate. Mechanistically, these changes in mitochondrial morphology reflected changes in mitochondrial dynamics that were mediated via ROS-induced post-translational modification of mitochondrial proteins, AKAP121, DRP1 and OPA1. Thus, lipid overload leads to dysregulation of mitochondrial dynamics, which may impair mitochondrial bioenergetics in diabetic cardiomyopathy.
Methods
The authors declare that all supporting data are available within the article [and its online supplementary files].
Detailed methods can be found in the online supplement.
Results
Increased FA uptake elevates myocardial ceramide and diacylglycerol content and induces cardiac hypertrophy and modest systolic dysfunction
To investigate the consequences of persistent lipid overload in the heart, we examined mice with low-level over-expression of long chain acyl-CoA synthetase 1 (ACSL1) driven by the cardiomyocyte-specific α-MHC promoter (ACStg mice). Because our goal was to examine mitochondrial adaptations prior to the onset of lipotoxic cardiomyopathy, we studied the line with the lowest level of ACSL1 overexpression, which does not develop overt heart failure until animals are 4-months old 13. Transgene expression was examined through development and ACSL1 protein was undetectable by Western blot in both wild-type (WT) and ACStg mice at birth (P0) despite ACSL1 mRNA expression being 13-fold greater in ACStg neonates compared to WT neonates (Fig.1A,1B). Increased ACSL1 protein in ACStg hearts was observed after P7 (Fig.1A). Palmitate biodistribution measured by in vivo positron emission tomography (PET) at 12 weeks of age was increased 2-fold, indicating increased cardiac FA uptake in ACStg hearts (Fig.1C). At this time point, there was no increase of myocardial triglyceride content in ACStg hearts under random fed conditions (Fig.1D). However, myocardial ceramide and diacylglycerol (DAG) content were significantly increased in ACStg hearts (Fig.1E,1F). Consistent with the previous report, this moderate increase of lipid uptake induced a modest increase in ventricular weight to body weight ratio and ventricular wall thickness (IVS) on the basis of cardiomyocyte hypertrophy in ACStg hearts (Fig.1G-I, Online Fig. I) 13. Although there was no statistically discernable difference in LV end-diastolic volume (LVEDV) and LV ejection fraction (LVEF) measured by echocardiography (Fig.1J, K), cardiac catheterization revealed a significant reduction in arterial blood pressure in ACStg mice accompanied by decreased max dP/dt and min dP/dt by the age of 24 weeks (Fig.1L-N). Furthermore, BNP and alpha skeletal muscle actin (Acta1) expression were modestly increased in ACStg hearts by 2-fold (BNP) and 1.3-fold (Acta1) at the age of 12 weeks (Fig.1O). Taken together, a 2-fold increase of lipid uptake induces modest cardiac hypertrophy with preserved systolic function at the age of 12 weeks, but impairs systolic function at the age of 24 weeks.
Figure 1. ACStg mice develop mild LVH with modest systolic dysfunction.
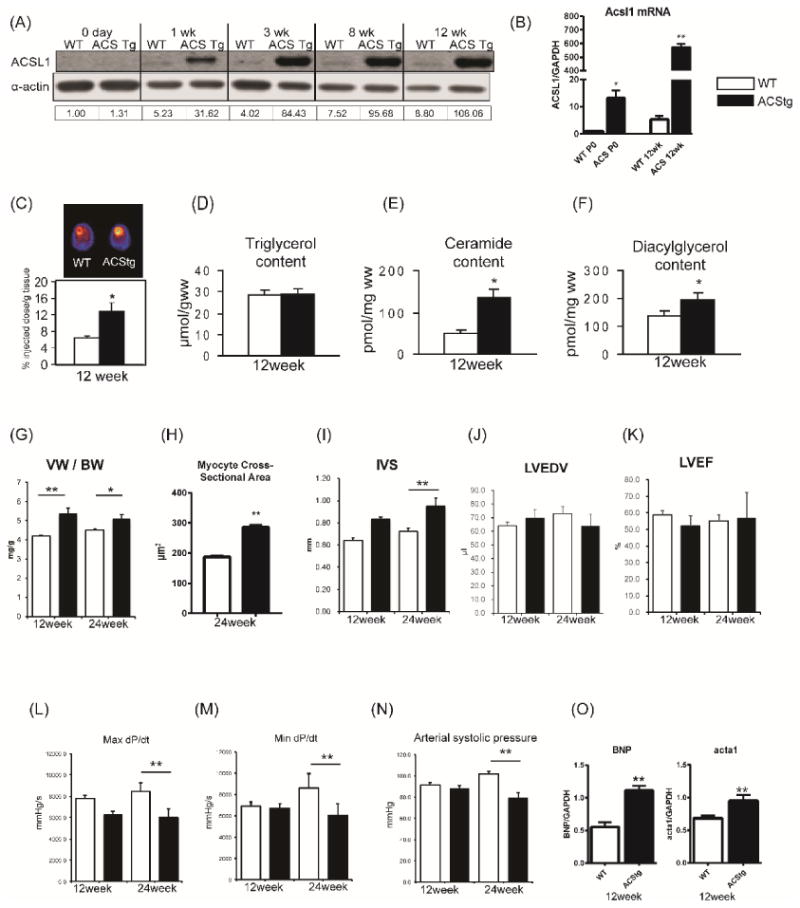
(A) Representative western blot of Acsl1 protein expression in wild-type (WT) and Acsl1 transgenic mice (ACStg) from birth to 12-weeks of age. Numbers beneath each lane represents ACSL1 densitometry. The antibody recognizes both endogenous ACSL1 and the transgene. (B) mRNA expression of Acsl1 was quantified by RT-PCR at the age of P0 and 12-weeks. * P<0.05, ** P<0.01 vs. WT. (C) Representative PET image and quantification of 1-11C palmitate biodistribution in 12-wk-old WT and ACStg hearts; n =10-12. * P <0.05 (D-F) Cardiac triacylglycerol (D), ceramide (E), or diacylglycerol (F) content in 12-week-old WT and ACStg hearts; n =5. * P<0.05 vs. WT. (G) Quantification of ventricular weight (VW) vs body weight (BW) ratio. n =4 in each group. (H) Myocyte cross-sectional areas estimated from WGA-stained cross sections obtained from 24-wk-old ACStg mice and age-matched controls (n=2 hearts per genotype). See Online Fig.I for representative image. (I-K) Echocardiographic analysis. Interventricular septal thickness (I), Left ventricle end-diastolic volume (LVEDV) (J), LV ejection fraction (LVEF) (K), at 12 and 24-weeks of age. n= 5 at 12-weeks of age and n = 4 at 24-weeks of age. (L-M) Cardiac catheterization. Max dP/dt (L) and Min dP/dt (M) and arterial blood pressure (N) in ACStg hearts at 12 and 24-weeks of age. n =5 at 12-weeks of age and n =4 at 24-weeks of age. (O) BNP and Acta1 mRNA expression were quantified by RT-PCR at 12-weeks of age. n =4, ** P < 0.01. All data are mean±sem.
Increased respiration and ROS production in isolated mitochondria from ACStg hearts
In ACStg mice, oxygen consumption rates and ATP synthesis rates in isolated mitochondria incubated with palmitoyl-carnitine as a substrate, were increased at 12-weeks of age, but were unchanged at 24-weeks of age relative to WT controls (Fig.2A,2B). In contrast, oxygen consumption rates were not impaired with pyruvate or glutamate as substrates at 12- and 24-weeks of age respectively (Fig.2C,2D). In addition, in-gel activities (in blue-native PAGE) of oxidative phosphorylation (OXPHOS) complexes I and IV were unchanged but complex V activity was increased at 12-weeks of age in ACStg hearts (Fig.2E-G). Mitochondrial superoxide production (measured as H2O2 release from mitochondria) was increased 2-fold in ACStg heart mitochondria exposed to succinate or palmitoyl-carnitine as a substrate, and was completely inhibited by addition of rotenone (Fig.2H). Increased ROS was not observed when glutamate was used as a substrate (Online Fig.IIA), suggesting that ROS production derives from reducing equivalents that are oxidized by complex II. Oxidation of 2′,7′-dichlorofluorescein-diacetate (DCFDA) was increased in whole cell extracts of ACStg hearts (Fig.2I). Moreover, increased mitochondrial 4-hydroxy-2-nonenal (4HNE) adducts in ACStg mitochondrial proteins, a highly toxic aldehyde byproduct of lipid peroxidation caused by ROS production, also supports increased mitochondrial ROS production (Fig.2J). However, activity of mitochondrial aconitase was not reduced, suggesting that there was no increase in oxidative stress in the mitochondrial matrix of ACStg hearts, potentially the result of increased SOD2 content (Fig.2K, Online Fig.IIB). Thus, short-term low-level lipid overload does not initially impair mitochondrial oxidative capacity in the murine heart despite increasing ROS generation.
Figure 2. Mitochondrial respiratory function and ROS production in isolated mitochondria from ACStg hearts.
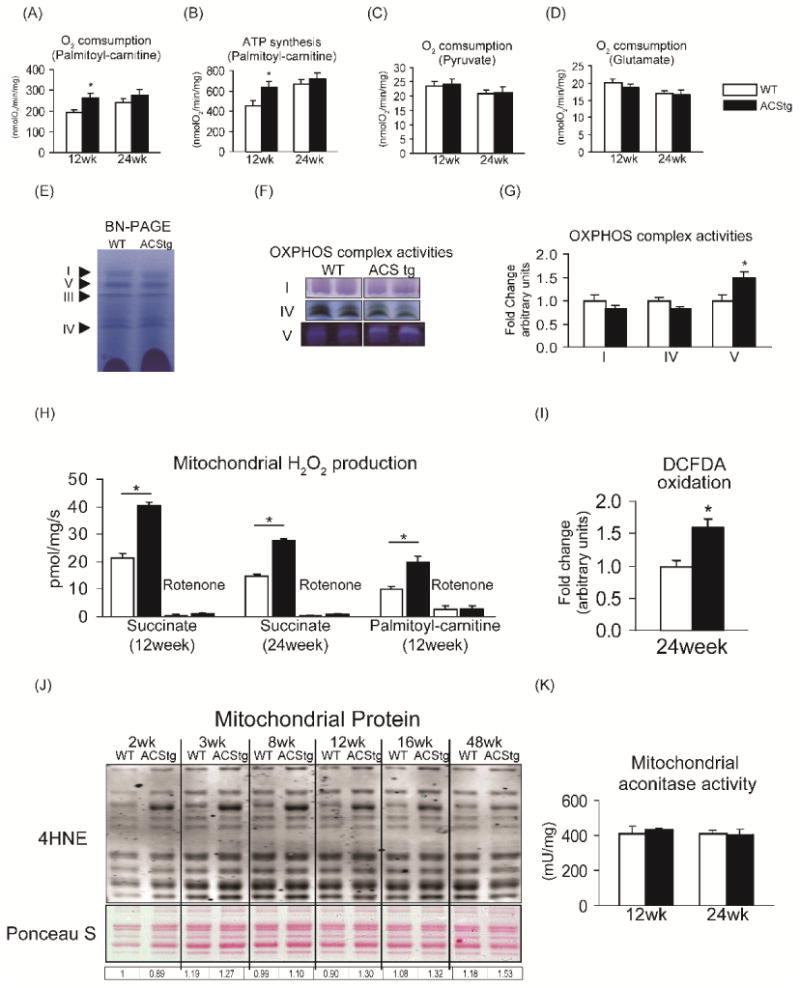
(A,B) Maximal ADP-stimulated mitochondrial oxygen consumption (A) and ATP synthesis rates (B) in mitochondria isolated from 12- and 24-week-old WT and ACStg hearts using palmitoyl-carnitine as a substrate; n =4-5. * P<0.05 vs. WT. (C,D) Maximal ADP-stimulated mitochondrial oxygen consumption in saponin-permeabilized cardiac fibers of 12- and 24-week-old WT and ACStg hearts using pyruvate (C) or glutamate (D) as a substrate; n =6. (E-G) Electrophoretic separation of OXPHOS complexes by blue-native PAGE (E), representative images of in-gel activities of complexes I, IV, and V (F), and quantification of OXPHOS complex activities (G), measured in 12-week-old WT and ACStg hearts; n =4. *P<0.05 vs. WT. (H) H2O2 production with succinate as a substrate in mitochondria isolated from 12- or 24- week-old WT and ACStg hearts, or with palmitoyl-carnitine as a substrate in mitochondria from 12-week-old WT and ACStg hearts, in the absence or presence of rotenone (Rot); n =3 at 12wks, n =6 at 24wks. *P<0.05 vs. WT. (I) Oxidation of DCFDA in whole tissue extracts of 24-week-old WT and ACStg hearts; n =5-6. *P<0.05 vs. WT. (J) Western blot for 4HNE protein adducts in mitochondrial protein isolated from WT and ACStg hearts. Numbers beneath each lane represent densitometry of 4HNE immunoreactivity of all bands in that lane. (K) Aconitase activity measured in mitochondria isolated from 12- or 24-week-old WT and ACStg hearts; n =3 at 12weeks, n =6 at 24weeks. All data are mean ± sem.
Postnatal increase in mitochondrial cross-sectional area is attenuated in lipid-overloaded cardiomyocytes
We next investigated mitochondrial morphology. During the early postnatal period, cardiomyocytes dynamically change their structure and function to adapt to the increased workload and oxygen consumption that parallels the switch of substrate utilization from glycolysis to mitochondrial fatty acid oxidation 3. Mitochondrial morphology also changes and volume density increases as they become larger and more ovoid in shape 14. 2-D Electron microscopy revealed that mitochondrial dimension in WT hearts increased 2-fold within the first 3 weeks after birth before plateauing (Fig3A, Online Fig. IIIA). This postnatal mitochondrial enlargement was absent in ACStg mice (Fig.3A, Online Fig. IIIA). In contrast, mitochondrial volume density was significantly increased in ACStg hearts (Fig.3A,3B, Online Fig.III B, III C), based upon the appearance of accumulated small mitochondria. To evaluate mitochondrial morphology more precisely, we performed 3D-transmission electron microscopic tomography at the age of 8 weeks. 3D mitochondrial images revealed the presence of narrow but elongated mitochondria whose tortuous shapes crossed individual sectioning planes more than once (Fig.3B,3C). This is associated with an increase in number of cytosol-mitochondrial membrane transitions on any projection between the M-lines of neighboring myofilament bundles (Fig.3D) The short axis diameter of mitochondria was significantly reduced in ACStg hearts by 60% (Fig.3E). Taken together, perinatal lipid overload significantly alters age-dependent structural mitochondrial remodeling, characterized by a reduction in minimum diameter and morphological changes that are consistent with fragmentation of the mitochondrial network.
Figure 3. Mitochondrial fragmentation in ACStg hearts.
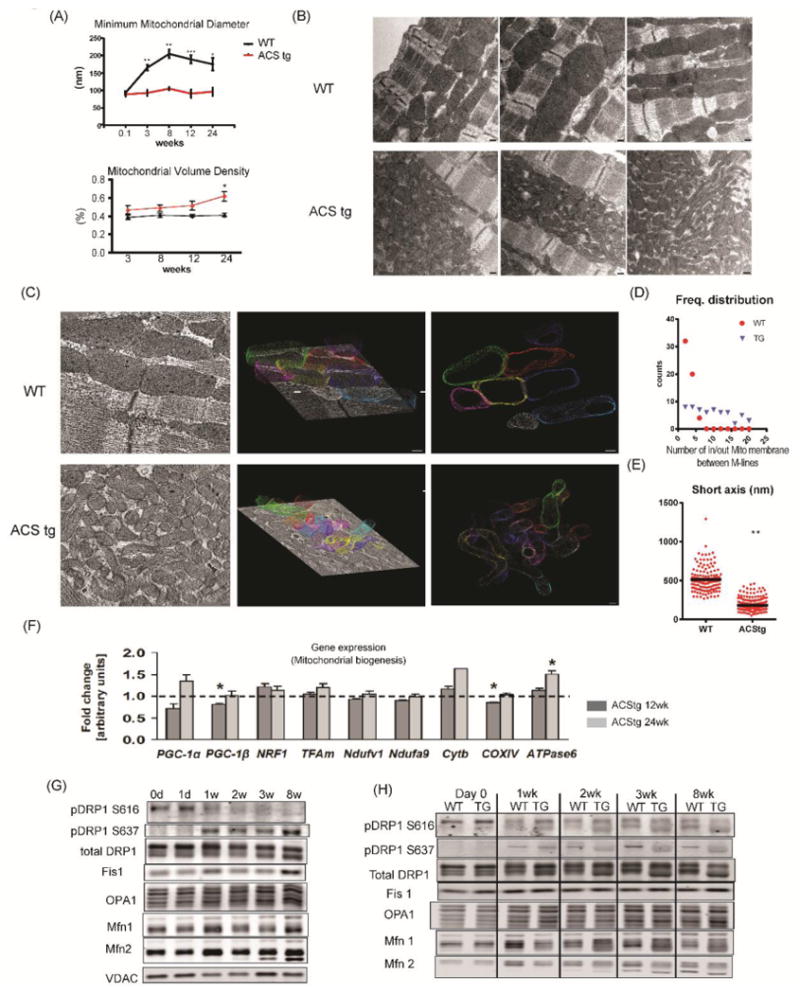
(A) Postnatal mitochondrial enlargement was attenuated in ACStg hearts. Stereologic quantification of mitochondrial minimum diameter and volume density was performed at the ages as indicated in 2-D electron micrographs (EM) presented in Online Fig.III A. ; n =3-4. * P<0.05, ** P<0.01, *** P<0.001. (B) Representative electron micrographs of longitudinal sections of WT and ACStg cardiomyocytes at the age of 8weeks, 3 hearts per genotype. Scale bars indicate 200nM. (C-E) Representative electron tomographic micrographs and corresponding 3D models of the mitochondrial network. Scale bars indicate 100nM (C). Number of mitochondrial outer membranes transitioned on a straight line trajectory between M-lines of two neighbouring myofilament bundles, counted in 22 (WT) and 26 (ACStg) electron tomograms (3 hearts per genotype) (D). Short axis diameter was measured in 132 (WT) and 290 (ACStg) mitochondria of 22 (WT) and 26 (ACStg) electron tomograms from 3 hearts per genotype; ** P <0.01(E). (F) Myocardial gene expression in 12- and 24-week-old WT and ACStg mice normalized to 16S RNA transcript levels. Values represent fold change in mRNA transcript levels relative to WT, which was assigned as 1 (dashed line), n=8 *; P<0.05 vs WT. (G) Western blot of whole heart lysates probed with antibodies as indicated at postnatal ages as shown. d=day, w=week. See also Online Fig.IV A. Each sample at 0d, 1w and 3w was isolated more than 3 times and immunoblotted separately, and each sample at day1, 2w and 8w was repeated twice. A representative western blot is shown. (H) Western blot of whole heart lysates from WT and ACStg mice probed with antibodies as indicated and harvested at the ages shown. See also Online Fig.IV B. All data are mean ± sem. Each sample at day0, 1wk and 3wk was isolated more than 3times and immunoblotted separately, and each sample at 2wk and 8wk was repeated twice. A representative western blot is shown.
These morphological changes could also reflect increased biogenesis or altered dynamics of the mitochondrial network. To address this, we examined mRNA expression levels of peroxisome proliferator-activated receptor γ coactivators 1 α/β (PGC-1α/β) and their targets, but found no evidence for increased mitochondrial biogenesis signaling (Fig.3F). Thus, we hypothesized that lipid overload may alter signaling pathways that regulate mitochondrial dynamics.
Mitochondrial dynamics is regulated by the activity of the dynamin superfamily of large GTPases, specifically mitofusin 1 and 2 (Mfn1 and Mfn2), optic atrophy 1 (Opa1), and dynamin related protein 1 (DRP1) 15, 16. Mitochondrial fusion is mediated by OPA1 and Mfn1/2 which reside on mitochondrial inner and outer membranes respectively. Fission is controlled by the cytosolic protein DRP1 that translocates from the cytosol to mitochondrial membranes following post-translational modifications to interact with the outer mitochondrial membrane protein Fis1. We determined the protein levels of these mitochondrial fusion and fission proteins and DRP1 phosphorylation during postnatal cardiac growth. Western blot analysis of whole heart lysates from wild-type mice revealed that DRP1 phosphorylation dramatically changed during the first 3 weeks of life (Fig.3G, Online Fig.IV A). Consistent with increased postnatal mitochondrial dimensions (fusion), DRP1 phosphorylation at S616, which activates DRP1-mediated mitochondrial fission, declined with age. In contrast, DRP1 phosphorylation at S637, which induces mitochondrial fusion by inhibiting DRP1-mediated mitochondrial fission, increased over time (Fig.3G, Online Fig.IV A). This developmental increase in DRP1 S637 phosphorylation was not observed in ACStg mice (Fig3H, Online Fig.IV B). These data suggest that cardiac lipid overload impairs postnatal mitochondrial remodeling by modulating DRP1 phosphorylation.
Lipid overload alters ubiquitin-proteasome mediated AKAP121 degradation and OPA1 proteolysis
Protein kinase A (PKA) mediated DRP1 phosphorylation at S637 inhibits DRP1 localization to mitochondria and subsequent mitochondrial fission 17. The mitochondrial localization of PKA and its ability to phosphorylate DRP1 is mediated by ubiquitin-proteasome mediated degradation of A kinase anchoring protein 121 (AKAP121), which are mitochondrial outer membrane scaffold proteins 18-20. We therefore examined whether AKAP121 protein content might be decreased in ACStg hearts. In 12-week-old ACStg mouse hearts, western blotting revealed that DRP1 phosphorylation at S637 was significantly reduced and phosphorylation at S616 increased (Fig.4A-C). In addition, AKAP121 expression was also siginificantly decreased in ACStg hearts relative to WT (Fig.4A,4D), which was not related to any difference in AKAP121 mRNA expression (Fig.4E). Therefore we tested whether posttranslational regulation by the ubiquitin-proteasome system (UPS) might increase AKAP121 degradation in the face of lipid overload. WT or ACStg mice were injected intraperitoneally with MG132 or DMSO and whole heart homogenates were subjected to western blot analysis. The proteasome inhibitor MG132 blocked the reduction of AKAP121 protein content in ACStg mice, suggesting that AKAP121 is degraded by the UPS pathway in response to lipid overload (Fig.4F). AKAP121 is a target of the ubiquitin ligase Siah2 in the context of hypoxia 20. However, Siah2 mRNA expression was not increased in ACStg hearts (Fig.4G). Taken together, lipid overload altered DRP1 phosphorylation by enhancing AKAP121 degradation which may be independent of the hypoxia-stimulated pathway.
Figure 4. Differential post-translational modifications of DRP1 and OPA1 in ACStg mice.
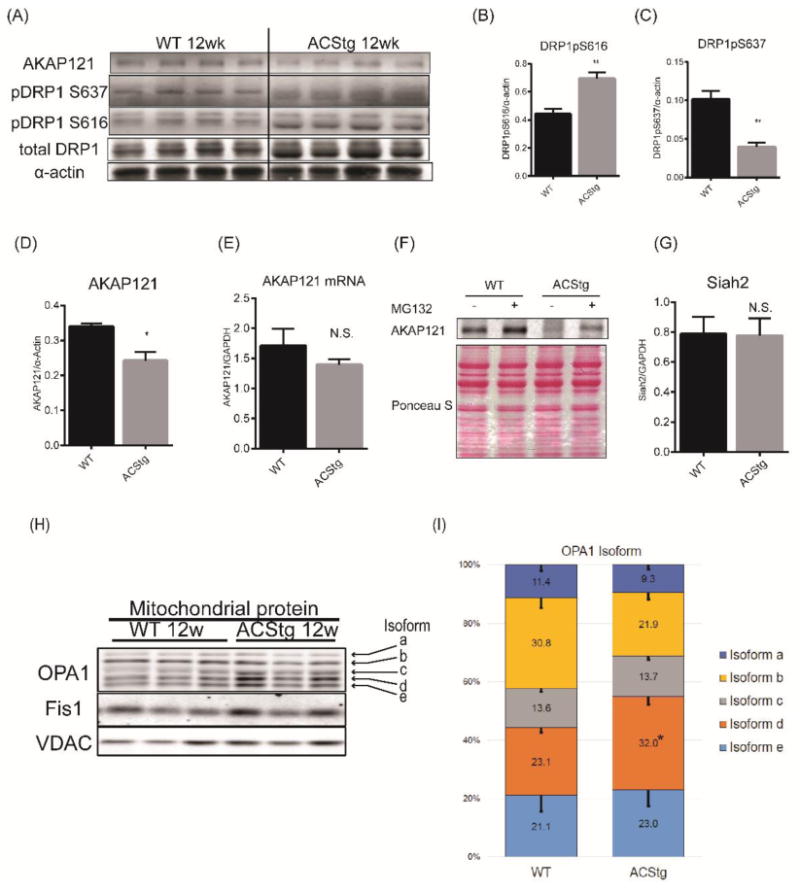
(A-D) Decreased protein content of AKAP121, and altered phosphorylation profile of DRP1. Whole heart lysates were subjected to western blot analysis (A) and densitometric quantification of DRP1pS616 (B), DRP1pS637 (C) and AKAP121 (D), n =4. ** P <0.01, * P <0.05. (E) AKAP121 mRNA expression of 12-week-old WT and ACStg mice was determined by quantitative RT-PCR (n=4) (F) AKAP121 degradation is mediated by the Ubiquitin-Proteasome pathway in ACStg hearts. WT or ACStg mice were injected intraperitoneally twice with DMSO or MG132 (18hrs and 6hrs before euthanasia). Whole-heart lysates were subjected to western blot analysis. (G) Siah2 gene expression in 12-week-old WT and ACStg mouse hearts was determined by realtime-PCR. Values are normalized to GAPDH expression. n=4. (H and I) Increased OPA1 cleavage in ACStg hearts. Mitochondrial fractions were prepared from 12-week-old WT or ACStg hearts and subjected to western blot analysis. Each lane shows the sample from different animals. The top 2 bands correspond to non-cleaved isoforms of OPA1 (long-form) and the bottom 3 bands correspond to cleaved isoforms of OPA1 (short-form) (H), quantitative densitometric analysis of OPA1 isoforms (I), n =4. * P<0.05 vs. WT. All data are mean ± sem.
We also examined whether the mitochondrial fusion protein, OPA1, could be affected by lipid overload. There are at least 8 mRNA variants of OPA1 as a result of alternative splicing 21. Variants 1 and 7 are dominant variants in mammalian cells. In response to decreased mitochondrial membrane potential, OPA1 also undergoes proteolytic cleavage which impairs its ability to induce mitochondrial fusion 22. Western blot analysis of heart mitochondrial protein showed 5 bands of OPA1; 2 fusion-competent long-isoforms and the 3 fusion-incompetent short-isoforms generated by the proteolytic processing of the 2 long-isoforms (Fig.4H). Consistent with enhanced mitochondrial network fragmentation in ACStg hearts, the ratio of short-form to long-form was increased by 1.7-fold in ACStg hearts (Fig.4H,4I). Thus, these data support the hypothesis that mitochondrial dynamics shifts towards enhanced mitochondrial fission by at least two distinct mechanisms.
Effect of long chain FA, palmitate and oleate on mitochondrial respiration and ROS production in rat neonatal cardiomyocytes
To more directly determine the mechanistic basis for the mitochondrial adaptation to lipid overload, we treated cultured rat neonatal cardiomyocytes (NRVCs) with free fatty acids (FFAs). Because saturated fatty acids are the main mediators of lipotoxicity 23, 24, we treated NRVCs with bovine serum albumin (BSA)-conjugated palmitic acid (C16:0) to mimic lipid overload in vivo, and compared them with cells treated with the non-saturated fatty acid, oleic acid (C18:1). Oxygen consumption of lipid-treated NRVCs was determined with the Seahorse XF24 flux analyzer. Although short exposure to palmitate (< 4h) increased mitochondrial oxygen consumption rates (OCR) in NRVCs, continued exposure (> 8h) reduced OCR (Fig.5A,5B, Online Fig.VA, VB). In contrast, oleate treatment did not alter mitochondrial OCR (Fig.5A). To confirm that this increase of OCR is coupled to mitochondrial oxidative phosphorylation and ATP synthesis, we examined time course changes of mitochondrial membrane potential and ATP content after palmitate treatment. Consistent with OCR, palmitate treatment initially increased mitochondrial membrane potential and ATP content, but these parameters declined after continued incubation for more than 12h (Fig.5C, 5D). We also examined ROS production of cardiomyocytes after palmitate or oleate treatment. As expected, western blotting of 4HNE revealed that ROS production was increased after palmitate treatment (Fig.5E). CellROX Green (ROS probe) also accumulated in the nucleus after 12hrs of palmitate treatment (Fig 5F,5G). In contrast, oleate treatment did not increase 4HNE accumulation (Fig.5E). Taken together, palmitate supplementation initially accelerates mitochondrial respiration that is coupled to ATP synthesis. However, continued exposure to palmitate induces mitochondrial respiratory failure, potentially on the basis of ROS-induced injury.
Figure 5. Mitochondrial metabolism and ROS production in rat neonatal cardiomyocytes following increasing duration of free fatty acids exposure.
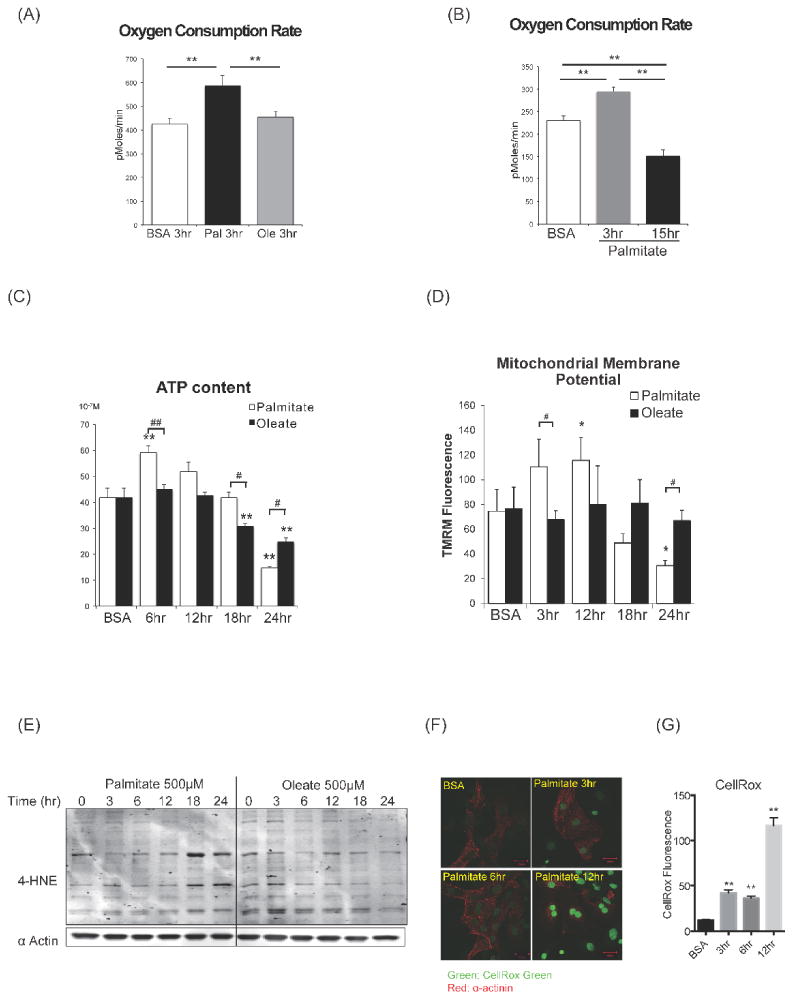
(A) Basal oxygen consumption rate (OCR) (assayed by the Seahorse XF24 system) in NRVCs incubated with BSA alone, palmitate-BSA (500μM) or oleate-BSA (500μM). ** P <0.01. (B) Time course analysis of OCR of NRVCs treated with palmitate-BSA. Increased basal OCR after short-term exposure to palmitate (3hr) and reduced basal OCR after long-term exposure (15hr) of palmitate. **P <0.01. See also Online Fig.V. (C) Time course analysis of ATP content in NRVCs after palmitate or oleate treatment. **P <0.01 vs BSA # P <0.05, ## P<0.01. (D) Time course analysis of mitochondrial membrane potential after palmitate or oleate treatment. Cardiomyocytes were stained with TMRM and Hoechst at indicated times and fluorescence intensity was assayed with a plate reader. The ratio of TMRM/Hoechst fluorescence are shown. n = 4, * P <0.05 vs BSA, # P <0.05. (E) Western blot for 4HNE protein adducts in NRVCs after palmitate or oleate treatment. (F and G) CellROX green staining in NRVCs after palmitate treatment. CellROX Green is a DNA dye, and upon oxidation, it binds to DNA. Representative confocal image (F) and fluorescence intensity was quantified (G). ** P <0.01 vs BSA. All data are mean±sem. Scale bars indicate 20μm.
Altered mitochondrial dynamics in response to lipid overload is mediated in part by post-translational modification of DRP1
We next determined if mitochondrial dynamics is modulated by fatty acids in NRVCs. To visualize the mitochondrial network, cardiomyocytes were stained with mitotracker in live cells or immunostained with an anti-Tom20 antibody in fixed cells. The normal mitochondrial network in NRVCs appeared as tubular mitochondria (Fig.6A, Online Fig.VI A). Palmitate supplementation changed mitochondrial morphology from a tubular to small rounded shape (Fig.6A,6B, Online Fig.VI A). Interestingly, these morphological changes were not observed in oleate-treated cardiomyocytes (Fig.6A,6B). We examined DRP1 phosphorylation profiles in NRVCs after palmitate treatment. Similar to P0 hearts (Fig.3G,3H), we detected no phosphorylation at S637 in isolated NRVCs throughout the time course. Instead, palmitate treatment increased DRP1 phosphorylation at S616 in NRVCs (Fig.6C). Immunohistochemistry revealed DRP1 translocation from the cytosol to mitochondria in response to palmitate treatment (Fig.6D). To validate that DRP1 mediates mitochondrial network fragmentation in lipid overload, we examined the effect of DRP1K38E, a dominant negative mutant of DRP1 25. DRP1K38E overexpression prevented palmitate-induced mitochondrial network fragmentation in NRVCs (Fig.6E, Online Fig.VII A). Furthermore, the reduced mitochondrial cross-section area observed was partially reversed in ACStg mice that lacked one DRP1 allele in cardiomyocytes (ACStg DRP1+/-) (Fig.6F, Online Fig.VII B-E). Taken together, palmitate exposure shifts mitochondrial dynamics towards fission and away from fusion, in part via a DRP1-mediated mechanism.
Figure 6. DRP1 mediates mitochondrial fission after lipid overload.
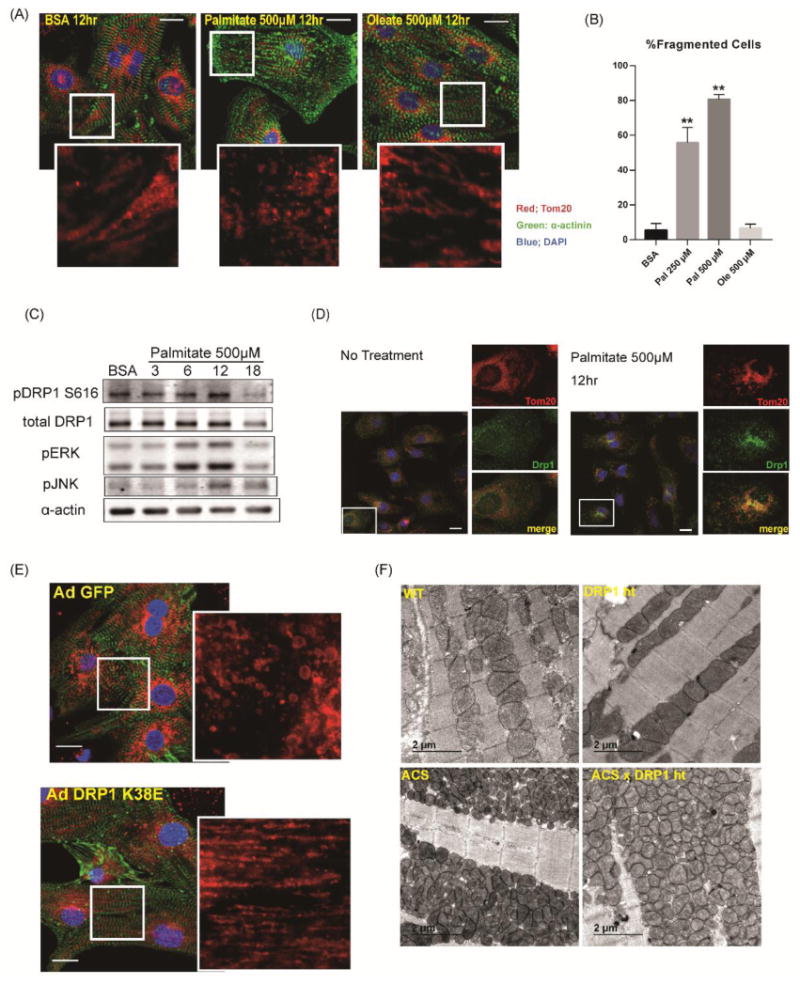
(A) Rat neonatal cardiomyocytes were stimulated with growth medium with or without 500μM palmitate or oleate. 12hrs after stimulation, cells were fixed with 4% paraformaldehyde and immunostained with α-actinin (green), Tom20 (red) and DAPI (blue). Scale bars indicate 20μm. (B) Quantification of mitochondrial fragmentation presented in Figure 6A. More than 100 cells were counted to determine the percentage (%) of cells with fragmented mitochondria. n=3, **: P<0.01. (C) Increased phosphorylation of DRP1 at Ser616 after palmitate treatment. NRVCs were treated in growth medium with 500μM palmitate and cell lysates were harvested at the indicated times in hr. (D) NRVCs were treated in growth medium with or without 500μM palmitate and subjected to immunohistochemistry for DRP1 (green) and Tom20 (red). Note that DRP1 is co-localized with Tom20 after palmitate treatment. Scale bars indicate 20μm. (E) NRVCs were infected with AdGFP or Ad DRP1K38E and were treated in growth medium with 500μM palmitate. NRVCs were subjected to immunohistochemistry for α-actinin (green) and Tom20 (red). See also Online Fig.VII A. Scale bars indicate 20μm. (F) Representative electron micrographs of longitudinal heart sections obtained from WT, DRP1+/-, ACStg and ACStg × DRP1+/-. DRP1 knockdown partially rescued mitochondrial morphology in ACStg hearts. See also Online Fig.VII B-E.
Mitochondrial morphology reflects the balance of mitochondrial fusion and fission. To directly evaluate if palmitate exposure inhibits mitochondrial fusion leading to mitochondrial network fragmentation, we co-cultured L6 myoblasts expressing either mitoGFP or mitoRFP and induced cell fusion with polyethylene glycol (PEG). L6 myoblasts displayed mitochondrial network fragmentation which was evident after 3hrs of palmitate supplementation (Fig.7A,7B). Mitochondrial network fragmentation was further augmented when expression of ACSL1 was increased following retroviral transduction (Online Fig.VI B). After the induction of cell fusion with PEG, fusion of GFP and RFP-labeled mitochondria was evident in cells treated with control media when imaged 8hr following PEG-induced cell fusion (Fig.7C top). In contrast, mitochondrial fusion was not observed in the cells incubated with palmitate-supplemented medium (Fig.7C middle). To determine if palmitate pre-exposure permanently impacted mitochondrial fusion in L6 myoblasts, cells were pretreated with palmitate supplemented medium for 3hr that disrupted the mitochondrial network and then the medium was replaced with normal growth medium. Mitochondria regained their tubular structure following removal of palmitate, and fusion of GFP and RFP mitochondrial populations was observed, suggesting that palmitate-induced defects in mitochondrial fusion are reversible (Fig.7C bottom).
Figure 7. Mitochondrial fusion in L6 myoblasts was impaired by palmitate exposure.
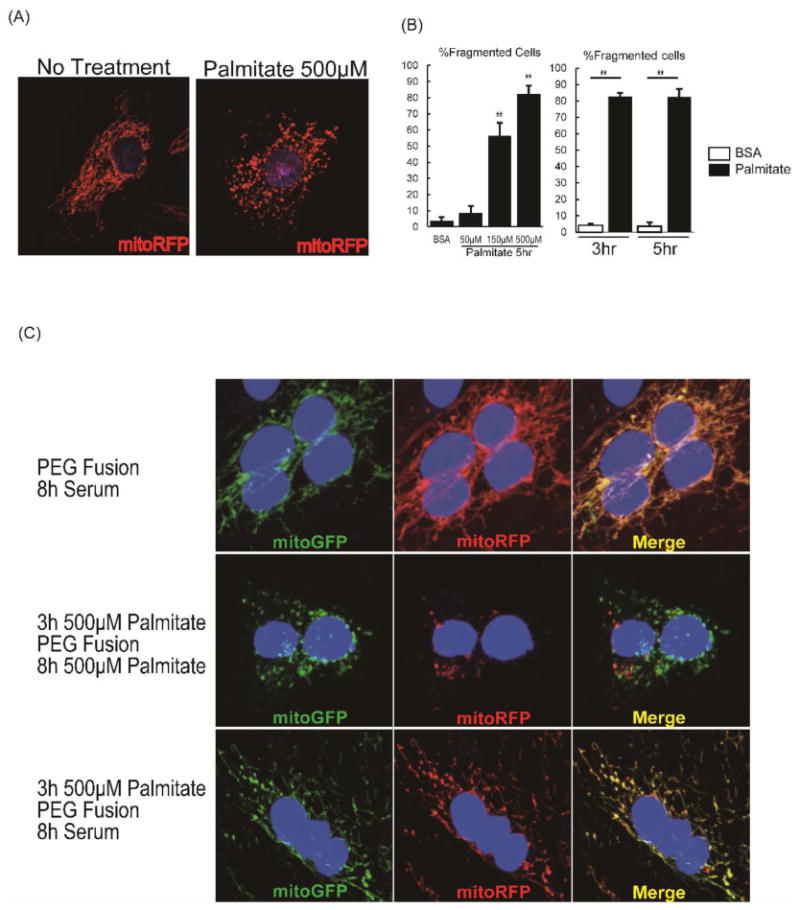
(A) L6 myoblasts transfected with mitoRFP were incubated in growth medium with or without 500μM palmitate for 5h, and representative images are shown. (B) Cells were evaluated to quantify tubular or fragmented mitochondrial networks, and cells with fragmented mitochondria were expressed as percentage of all viewed cells, n=4 and 80 cells counted per group. ** P < 0.01 vs BSA. All data are mean±sem. (C) L6 myoblasts were transfected either with mitoRFP or mitoGFP, co-plated (25,000 cells each) on cover slips for 24h, and then cell fusion was induced with polyethylene glycol (PEG). Cells were fused for 8h in regular growth medium with no palmitate (top row), 8h in growth medium with 500μM palmitate, following 3h pre-incubation with 500μM palmitate prior to PEG fusion (middle row), or 8h in the regular growth medium with no palmitate, following 3h pre-incubation with 500μM palmitate prior to PEG fusion (bottom row).
Contribution of mitochondrial ROS generation to FA-mediated mitochondrial fragmentation
In light of our observations of increased lipid-mediated ROS production in vivo and in vitro that correlated with mitochondrial network fragmentation, we tested the hypothesis that elevated mitochondrial ROS production may initiate downstream signaling events to alter mitochondrial dynamics. Because mitochondrial superoxide dismutase 2 (SOD2) mediates superoxide detoxification at a diffusion limiting-rate in the mitochondrial matrix 26, we examined whether SOD2 overexpression could rescue altered mitochondrial morphology in ACStg hearts. We generated a cardiac-specific double-transgenic mouse harboring SOD2 and ACSL1 transgenes (ACS × SOD2 mice). As expected, enhanced superoxide production and increased 4HNE adducts observed in ACStg mitochondria were almost completely rescued by the overexpression of SOD2 (Fig.8A,8B, Online Fig.VIII A). Mitochondrial cross-sectional area and apparent number were normalized in ACStg hearts by SOD2 overexpression (Fig.8C, Online Fig.VIII B). Surprisingly, SOD2 overexpressing cardiomyocytes with reduced ROS production, demonstrated a 4-fold increase in mitochondrial cross-sectional area estimated from 2-D images, suggesting that mitochondrial ROS levels are inversely correlated with mitochondrial dimensions (Fig.8A,8C).
Figure 8. Mitochondrial superoxide dismutase (SOD2) overexpression partially rescued abnormal mitochondrial dynamics in ACStg mice.
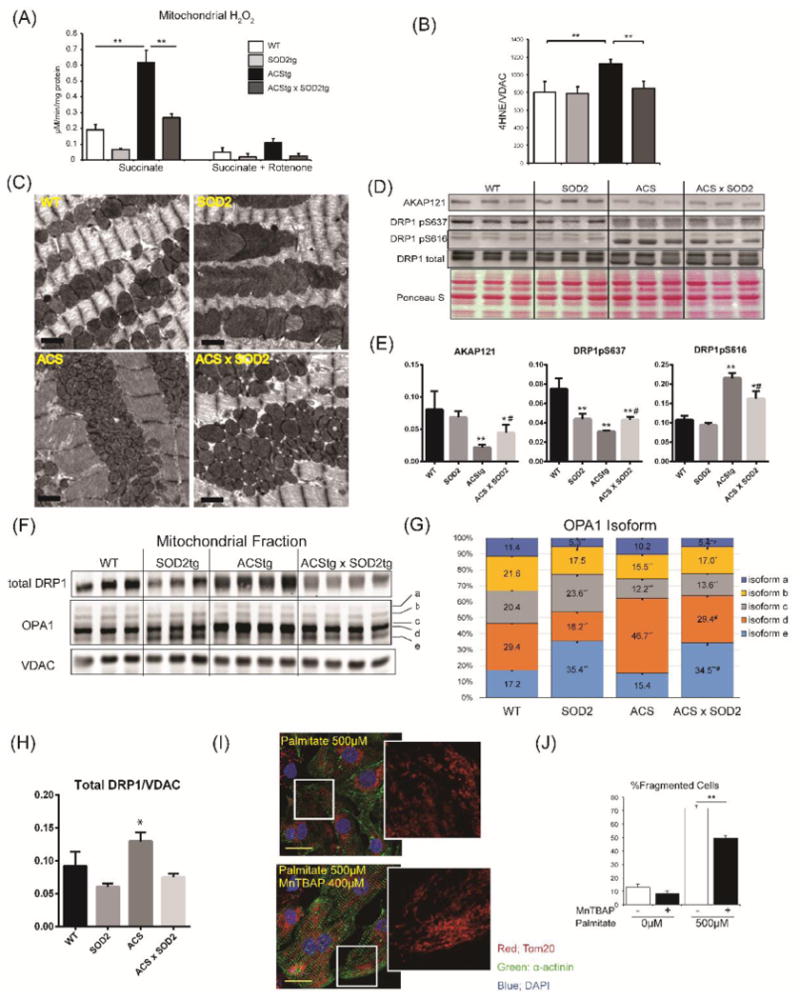
(A,B) Enhanced ROS production in ACStg hearts was rescued by SOD2 overexpression. H2O2 production was determined in isolated mitochondria from 12-week-old WT, SOD2tg, ACStg and SOD2 × ACS double tg hearts in the absence or presence of rotenone (A). Mitochondrial fractions were prepared from 12-week-old WT, SOD2tg, ACStg and SOD2 × ACS double transgenic hearts and subjected to western blot for 4HNE and quantified by densitometry (B), n =3-4 in each group. ** P <0.01. See also Online Fig.VIII A (C) SOD2 overexpression partially rescued fragmented mitochondria in ACStg hearts. Representative electron micrographs from WT, SOD2tg, ACStg and SOD2 × ACS double tg mice. Bar indicates 1μm. See also Online Fig.VIII B. (D-H) SOD2 overexpression reversed altered phosphorylation pattern of Drp1 and processing of OPA in ACStg hearts. Mitochondrial fractions were prepared from 12-week-old WT, SOD2tg, ACStg and SOD2 × ACS double tg hearts and subjected to western blot analysis for AKAP121 and total and phosphorylated Drp1at Ser637 and Ser616 and the distribution of OPA1 isoforms were quantified (F-H). n =3-4 in each group. * P <0.05, ** P <0.01 vs WT, # P <0.05 vs ACStg. (I,J) MnTBAP prevented palmitate-induced mitochondrial fragmentation in NRVCs. NRVCs were treated with or without 400μM MnTBAP, a SOD mimetic and peroxynitrite scavenger, and then stimulated with or without growth medium containing 500μM Palmitate. After 12h of stimulation, cells were fixed and stained with Tom20 antibody. Representative image from each group. (I) and quantification of the ratio of fragmented cells (J), n =3 in each group. ** P <0.01. Scale bars indicate 20μm.
Finally, we determined if altered DRP1 phosphorylation or changes in OPA1 processing in ACStg mice were prevented by SOD2 overexpression. Western blot analysis showed that protein expression of AKAP121 and DRP1 phosphorylation at S637 was increased and DRP1 phosphorylation at S616 was reduced by SOD2 overexpression (Fig.8D,8E). In addition, total levels of DRP1 normalized to VDAC, which were increased in ACStg hearts, were normalized by SOD2 overexpression (Fig.8F,8H). Overexpression of ACSL1 enhanced OPA1 proteolysis to specifically increase short isoform-d formation (Fig.8F,8G). Relative to ACStg, in SOD2/ACS double transgenic mice there was a reduction in isoform-d and an increase in isoform-e (Fig.8F,8G). In vitro, MnTBAP, the SOD mimetic and peroxynitrite scavenger, partially normalized palmitate-induced mitochondrial network fragmentation in NRVCs (Fig.8I,8J). Taken together, these data indicate that mitochondrial redox signaling regulates mitochondrial dynamics by post-translational modulation of mitochondrial fusion and fission proteins.
Discussion
Recent studies have suggested that impaired mitochondrial energetics may contribute to the increased risk of heart failure in type-2 diabetes 8. The pathophysiology of mitochondrial dysfunction in diabetes is complex and may include altered insulin signaling, glucotoxicity and lipotoxicity. Prior work from our group suggested that impaired myocardial insulin signaling could precipitate mitochondrial dysfunction in the heart 11. Given the likelihood that lipotoxicity could have distinct effects on mitochondria, the present study was designed to model increased myocardial lipid accumulation to a level that was similar in magnitude to that reported in animal models of obesity and diabetes 27. We therefore utilized mice with cardiomyocyte -specific low-level overexpression of ACSL1, which gradually developed myocardial dysfunction and recapitulated key observations in neonatal cardiomyocytes that were incubated with palmitate. First, we observed that lipid overload initially enhances mitochondrial respiration coupled to ATP synthesis. However, prolonged lipid overload enhanced mitochondrial ROS generation, which is followed by reduced mitochondrial respiration and ATP synthesis. Second, we observed differential metabolic fates of palmitate and oleate in NRVCs. Our data indicates that increased mitochondrial respiration after palmitate supplementation results in accumulation of mitochondrial ROS and impaired mitochondrial energetics. Conversely, oleate supplementation did not increase mitochondrial respiration or ROS production and did not precipitate mitochondrial impairment. Third, mitochondrial redox status might influence mitochondrial morphology by modulating the post-translational modifications of proteins that regulate mitochondrial dynamics. Lipid overload increased AKAP121 ubiquitination, modulated DRP1 phosphorylation and altered OPA1 processing. Future studies in models with inducible increases in myocardial lipid uptake will be needed to determine if these mechanisms also alter mitochondrial dynamics and energetics in adult (postnatal) murine hearts. Prior studies from our laboratory showed altered mitochondrial morphology in db/db diabetic mice or type-1 diabetic Akita mice, which was accompanied by enhanced mitochondrial ROS production 10, 28. A recent study also revealed that right atrial cardiomyocytes isolated from type-2 diabetic patients exhibit increased mitochondrial ROS production and impaired mitochondrial bioenergetics 29. Taken together, these observations suggest that the mechanism observed in the present study could be universally applicable to the pathophysiology of mitochondrial dysfunction in lipotoxic cardiomyopathy associated with obesity or diabetes mellitus.
Following uptake of FAs in cardiomyocytes, ACSLs immediately catalyze FAs to Acyl-CoA esters, which enter a variety of lipid metabolic pathways such as FA oxidation, TG synthesis/storage, phospholipid metabolism, and de novo ceramide synthesis. Cardiac ACSL1 over expression in vivo increases mitochondrial ROS production and the content of reactive lipid metabolites. We used a genetic approach to modulate mitochondrial ROS production by overexpressing ACSL1, SOD2, or both and demonstrated that mitochondrial redox status correlates with mitochondrial morphology. In addition, mitochondrial redox status is also associated with processing of mitochondrial proteins, such as OPA1 proteolysis, ubiquitin-proteasome mediated degradation of AKAP121 and DRP1 phosphorylation at S637. DRP1 phosphorylation at S616, a target site for MAP kinases and CDK1 30-32, also correlated with mitochondrial redox status and was recapitulated by in vitro palmitate treatment of NRVCs. These data are also supported by a recent publication showing that the ROS modulator 1 (ROMO1), a mitochondrial protein, is a redox-sensitive factor that regulates mitochondrial morphology by affecting OPA1 cleavage and oligomerization 33.
A recently described mechanism that may also induce mitochondrial fission is hypoxia. Siah2, a RING finger ubiquitin ligase induced by hypoxia, mediates ubiquitin-proteasome degradation of AKAP121. Subsequent reduction of PKA-dependent inhibitory phosphorylation of DRP1 is an important regulatory pathway for hypoxia induced mitochondrial fission 20. In ACStg hearts, we also observed substantial proteasome mediated degradation of AKAP121 and mitochondrial redox status was correlated with AKAP121 protein levels and DRP1 phosphorylation at S637. However, in contrast to hypoxia, Siah2 mRNA induction was not observed in ACStg hearts (Fig.4G), suggesting that an alternative mechanism may link the AKAP121 and DRP1 pathways in cardiac lipid overload.
Our data also reveals that substrate availability and mitochondrial dynamics plays an important role in the developmental maturation of mitochondria in the perinatal period. In fetal cardiomyocytes, energy substrate utilization mainly depends on lactate and glucose supplied from the maternal circulation. After birth, energy substrate utilization shifts rapidly from lactate oxidation and glycolysis to fatty acid oxidation in mitochondria, and cardiomyocytes preferentially utilize fatty acid as an energy source throughout their life 2, 34. In this process, mitochondria undergo profound remodeling in terms of shape and volume, which is accompanied by an increase in the activities of enzymes required for lipid oxidation, such as LCAD, MCAD, CPT1 and ACSL1 4, 35. The progressive post-natal increase in mitochondrial dimensions was absent in ACStg heart, which showed a multitude of apparently small mitochondria occupying a larger fractional cell volume than WT. 3-D EM tomographic reconstructions revealed a narrow, elongated and tortuous morphology of mitochondria that is principally different from WT and which would be sectioned multiple times in a single plane. Our novel imaging methodology underscores the challenges in estimating mitochondrial size and number from 2-D images and adds new insight into the structural adaptations of mitochondria in the heart when signaling pathways that regulate mitochondrial dynamics are perturbed.
The substantial increase of fatty acid availability in the perinatal circulation activates PPARα that drives fatty acid oxidation in mitochondria 36, 37. We show here that DRP1 phosphorylation is rapidly modulated during post-natal cardiac development. Furthermore, forced lipid uptake in the early post-natal period alters the developmental changes in DRP1 phosphorylation and prevented physiological and maturation-dependent mitochondrial fusion. An indispensable role of DRP1 in perinatal cardiac development has been recently reported 38-40. Taken together, post translational modifications of DRP1 after birth, plays an important role in normal perinatal mitochondrial maturation.
Along with the alteration of mitochondrial membrane dynamics, mitochondrial membrane lipid content was also affected in our cardiac lipid overload model. Although the detailed molecular mechanisms involved are incompletely understood, the lipid composition of cristae membrane, specifically cardiolipin and phospholipids, plays an important role in mitochondrial membrane fluidity, cristae structure and oxidative phosphorylation.41 Consistent with an elevation of cardiac ceramide and DAG levels (Fig.1E,1F), phospholipid composition and the side chain pattern of cardiolipin were significantly altered in ACStg mitochondrial membranes (Online Fig.IX A, IX B). Interestingly, there was a significant reduction in tetra-linoleic (18:2)4 cardiolipin (L4CL) (Online Fig.IX A). It is widely accepted that L4CL is the fully functional CL species and the reduction of L4CL has been associated with diabetes, heart failure, ischemia reperfusion injury, all of which are associated with impaired mitochondrial function.42, 43 These changes are due in part to an important role for cardiolipin in mitochondrial supercomplex formation and cytochrome oxidase activity.44 Moreover, L4CL is thought to promote more efficient proton flow and as such the reduction observed in ACStg mice could account in part for the increased ROS generation.
We also addressed the contribution of increased ceramide content in altering mitochondrial morphology in our lipid overload model in vivo and vitro. Using myriocin, an inhibitor for serine-palmitoyl transferase, which catalyzes the first step of de novo ceramide synthesis, we determined that the inhibition of de novo ceramide synthesis exacerbated ROS production in palmitate treated NRVCs (Online Fig.X A-E). Furthermore, in vivo inhibition of ceramide synthesis by heterozygous deletion of Des1, an enzyme that catalyzes de novo ceramide synthesis, failed to reverse the altered mitochondrial network (Online Fig. X F). These data indicate that increased ROS resulting from mitochondrial lipid oxidation likely represents a primary mechanism responsible for augmenting mitochondrial fission in lipid overload, and that ceramide accumulation does not contribute to this process.
In summary, we demonstrate here that increased myocardial lipid uptake impairs mitochondrial dynamics by increasing mitochondrial ROS generation, which modulates post-translational modification of OPA1 and DRP1 (Online Fig. XI). We also provide evidence that mitochondrial dynamics is involved in perinatal mitochondrial maturation and forced lipid uptake interferes with that process. These findings add a new dimension to understanding the regulation of mitochondrial morphology and physiology in the heart and its dysregulation by lipid excess.
Supplementary Material
Novelty and Significance.
What Is Known?
Obesity and diabetes induce myocardial lipotoxicity that may reduce cardiac function.
Cardiac lipotoxicity is associated with mitochondrial dysfunction including decreased oxidative phosphorylation via incompletely understood mechanisms.
What New Information Does This Article Contribute?
Myocardial lipid overload by saturated fatty acids (FA) transiently increases mitochondrial respiration, membrane potential and ROS production.
FA-induced mitochondrial ROS overproduction induces post-translational modifications of mitochondrial fusion/fission regulatory proteins that precipitate increased mitochondrial fission.
3D-electron tomography revealed that lipid overload in post-natal hearts induces ROS-mediated fragmentation of the mitochondrial network, characterized by narrow tortuous and tubular morphology.
Obesity, insulin resistance and diabetes are associated with increased fatty acids (FAs) utilization and reduced glucose utilization. One proposed mechanism for cardiac dysfunction called “lipotoxicity” is excess intracellular lipid accumulation secondary to a mismatch between lipid supply and utilization. In this study, using a mouse model with increased myocardial lipid uptake, we demonstrate that myocardial lipid overload induces mitochondrial ROS production, which alters the activity of DRP1 and OPA1, leading to fragmentation of the mitochondrial network characterized by a narrow tubular morphology. Thus, dysregulation of mitochondrial dynamics following lipid overload may represent an important mechanism contributing to mitochondrial and contractile dysfunction in lipotoxic cardiomyopathies.
Acknowledgments
We thank Dr. Elizabeth A. Amiott and Dr. Janet M. Shaw for providing critical reagents and experimental guidance in the L6 mitochondrial fusion assays; Heather Theobald for technical help, and Dr. Eric T. Weatherford and Mr. Paul Casella for the editing of the manuscript.
Sources of Funding: Japan Heart Foundation/Bayer Yakuhin Research Grant Abroad (to K.T.)
American Heart Association Post-doctoral fellowship (12POST12030309) (to K.T.)
Deutsche Forschungsgemeinschaft (DFG) (to H.B.)
National Institute of Health (RO1 DK064989, P20 HL113444) (to J.E.S)
National Institute of Health (UO1 HL087947, RO1HL108379) (to E.D.A.)
Juvenile Diabetes Research Foundation (to E.D.A.)
European Research Council Advanced Grant CardioNECT (20120314) (to P.K.).
Nonstandard Abbreviations and Acronyms
- 4HNE
4-hydroxy-2-nonenal
- ACSL1
Long-chain acyl-CoA synthetase 1
- ACTA1
alpha skeletal muscle actin
- AKAP121
A-kinase anchor protein 121
- ATP
Adenosine triphosphate
- BNP
B-type natriuretic peptide
- BSA
Bovine serum albumin
- CDK1
Cyclin-dependent kinase 1
- CPT1
Carnitine palmitoyl transferase I
- DAG
Diacylglycerol
- DCFDA
2′,7′-dichlorofluorescein-diacetate
- DES1
Dihydroceramide desaturase 1
- DRP1
Dynamin related protein 1
- FA
Fatty acid
- IVS
Interventricular septum
- L4CL
Tetra-linoleic cardiolipin
- LCAD
Long-chain acyl-CoA dehydrogenase
- LVEDV
Left-ventricular end-diastolic volume
- LVEF
Left-ventricular ejection fraction
- MCAD
Medium-chain acyl-CoA dehydrogenase
- Mfn
Mitofusin
- NRVC
Neonatal rat ventricular cardiomyocyte
- OCR
Oxygen consumption rate
- OPA1
Optic atrophy 1
- OXPHOS
Oxidative phosphorylation
- PAGE
Polyacrylamide gel electrophoresis
- PPAR
Peroxisome proliferator-activated receptor
- ROS
Reactive oxygen species
- SOD2
Superoxide dismutase 2
- UPS
ubiquitin-proteasome system
- VDAC
Voltage-dependent anion channel
Footnotes
Author Contributions: K.T., H.B., A.R.W., J.S., G.A.J., A.R.T., R.M., Y.Z., R.S., X.X.H, C.L.S, R.O.P., V.A.L., K.W.S, T.L.S., K.I.S G.C.S., P.K., E.R-Z and O.K performed experiments and analyzed the data; J.E.S. provided critical reagents and experimental guidance. K.T., H.B. and E.D.A. designed the study and prepared a draft of the manuscript. E.D.A. coordinated the project and the writing of the paper, to which all authors contributed.
Disclosures: The authors have declared no conflict of interest exists.
References
- 1.Doenst T, Nguyen TD, Abel ED. Cardiac metabolism in heart failure: implications beyond ATP production. Circ Res. 2013;113:709–24. doi: 10.1161/CIRCRESAHA.113.300376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.de Jong H, Neal AC, Coleman RA, Lewin TM. Ontogeny of mRNA expression and activity of long-chain acyl-CoA synthetase (ACSL) isoforms in Mus musculus heart. Biochim Biophys Acta. 2007;1771:75–82. doi: 10.1016/j.bbalip.2006.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lopaschuk GD, Collins-Nakai RL, Itoi T. Developmental changes in energy substrate use by the heart. Cardiovasc Res. 1992;26:1172–80. doi: 10.1093/cvr/26.12.1172. [DOI] [PubMed] [Google Scholar]
- 4.Lopaschuk GD, Witters LA, Itoi T, Barr R, Barr A. Acetyl-CoA carboxylase involvement in the rapid maturation of fatty acid oxidation in the newborn rabbit heart. J Biol Chem. 1994;269:25871–8. [PubMed] [Google Scholar]
- 5.Kolwicz SC, Jr, Purohit S, Tian R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ Res. 2013;113:603–16. doi: 10.1161/CIRCRESAHA.113.302095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Duncan JG. Mitochondrial dysfunction in diabetic cardiomyopathy. Biochim Biophys Acta. 2011;1813:1351–9. doi: 10.1016/j.bbamcr.2011.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Boudina S, Abel ED. Diabetic cardiomyopathy, causes and effects. Rev Endocr Metab Disord. 2010;11:31–9. doi: 10.1007/s11154-010-9131-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bugger H, Abel ED. Mitochondria in the diabetic heart. Cardiovasc Res. 2010;88:229–40. doi: 10.1093/cvr/cvq239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zorzano A, Liesa M, Palacin M. Role of mitochondrial dynamics proteins in the pathophysiology of obesity and type 2 diabetes. Int J Biochem Cell Biol. 2009;41:1846–54. doi: 10.1016/j.biocel.2009.02.004. [DOI] [PubMed] [Google Scholar]
- 10.Boudina S, Sena S, Theobald H, Sheng X, Wright JJ, Hu XX, Aziz S, Johnson JI, Bugger H, Zaha VG, Abel ED. Mitochondrial energetics in the heart in obesity-related diabetes: direct evidence for increased uncoupled respiration and activation of uncoupling proteins. Diabetes. 2007;56:2457–66. doi: 10.2337/db07-0481. [DOI] [PubMed] [Google Scholar]
- 11.Boudina S, Bugger H, Sena S, O'Neill BT, Zaha VG, Ilkun O, Wright JJ, Mazumder PK, Palfreyman E, Tidwell TJ, Theobald H, Khalimonchuk O, Wayment B, Sheng X, Rodnick KJ, Centini R, Chen D, Litwin SE, Weimer BE, Abel ED. Contribution of impaired myocardial insulin signaling to mitochondrial dysfunction and oxidative stress in the heart. Circulation. 2009;119:1272–83. doi: 10.1161/CIRCULATIONAHA.108.792101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Belke DD, Betuing S, Tuttle MJ, Graveleau C, Young ME, Pham M, Zhang D, Cooksey RC, McClain DA, Litwin SE, Taegtmeyer H, Severson D, Kahn CR, Abel ED. Insulin signaling coordinately regulates cardiac size, metabolism, and contractile protein isoform expression. J Clin Invest. 2002;109:629–39. doi: 10.1172/JCI13946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chiu HC, Kovacs A, Ford DA, Hsu FF, Garcia R, Herrero P, Saffitz JE, Schaffer JE. A novel mouse model of lipotoxic cardiomyopathy. J Clin Invest. 2001;107:813–22. doi: 10.1172/JCI10947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Piquereau J, Novotova M, Fortin D, Garnier A, Ventura-Clapier R, Veksler V, Joubert F. Postnatal development of mouse heart: formation of energetic microdomains. The Journal of physiology. 2010;588:2443–54. doi: 10.1113/jphysiol.2010.189670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kanamaru Y, Sekine S, Ichijo H, Takeda K. The phosphorylation-dependent regulation of mitochondrial proteins in stress responses. Journal of signal transduction. 2012;2012:931215. doi: 10.1155/2012/931215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta. 2013;1833:1256–68. doi: 10.1016/j.bbamcr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 17.Chang CR, Blackstone C. Cyclic AMP-dependent protein kinase phosphorylation of Drp1 regulates its GTPase activity and mitochondrial morphology. J Biol Chem. 2007;282:21583–7. doi: 10.1074/jbc.C700083200. [DOI] [PubMed] [Google Scholar]
- 18.Merrill RA, Strack S. Mitochondria: a kinase anchoring protein 1, a signaling platform for mitochondrial form and function. Int J Biochem Cell Biol. 2014;48:92–6. doi: 10.1016/j.biocel.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Carlucci A, Adornetto A, Scorziello A, Viggiano D, Foca M, Cuomo O, Annunziato L, Gottesman M, Feliciello A. Proteolysis of AKAP121 regulates mitochondrial activity during cellular hypoxia and brain ischaemia. EMBO J. 2008;27:1073–84. doi: 10.1038/emboj.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kim H, Scimia MC, Wilkinson D, Trelles RD, Wood MR, Bowtell D, Dillin A, Mercola M, Ronai ZA. Fine-tuning of Drp1/Fis1 availability by AKAP121/Siah2 regulates mitochondrial adaptation to hypoxia. Mol Cell. 2011;44:532–44. doi: 10.1016/j.molcel.2011.08.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Delettre C, Griffoin JM, Kaplan J, Dollfus H, Lorenz B, Faivre L, Lenaers G, Belenguer P, Hamel CP. Mutation spectrum and splicing variants in the OPA1 gene. Hum Genet. 2001;109:584–91. doi: 10.1007/s00439-001-0633-y. [DOI] [PubMed] [Google Scholar]
- 22.Ishihara N, Fujita Y, Oka T, Mihara K. Regulation of mitochondrial morphology through proteolytic cleavage of OPA1. EMBO J. 2006;25:2966–77. doi: 10.1038/sj.emboj.7601184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wende AR, Abel ED. Lipotoxicity in the heart. Biochim Biophys Acta. 2010;1801:311–9. doi: 10.1016/j.bbalip.2009.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Russo SB, Baicu CF, Van Laer A, Geng T, Kasiganesan H, Zile MR, Cowart LA. Ceramide synthase 5 mediates lipid-induced autophagy and hypertrophy in cardiomyocytes. J Clin Invest. 2012;122:3919–30. doi: 10.1172/JCI63888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Germain M, Mathai JP, McBride HM, Shore GC. Endoplasmic reticulum BIK initiates DRP1-regulated remodelling of mitochondrial cristae during apoptosis. EMBO J. 2005;24:1546–56. doi: 10.1038/sj.emboj.7600592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J. 2009;417:1–13. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mazumder PK, O'Neill BT, Roberts MW, Buchanan J, Yun UJ, Cooksey RC, Boudina S, Abel ED. Impaired cardiac efficiency and increased fatty acid oxidation in insulin-resistant ob/ob mouse hearts. Diabetes. 2004;53:2366–74. doi: 10.2337/diabetes.53.9.2366. [DOI] [PubMed] [Google Scholar]
- 28.Bugger H, Boudina S, Hu XX, Tuinei J, Zaha VG, Theobald HA, Yun UJ, McQueen AP, Wayment B, Litwin SE, Abel ED. Type 1 diabetic akita mouse hearts are insulin sensitive but manifest structurally abnormal mitochondria that remain coupled despite increased uncoupling protein 3. Diabetes. 2008;57:2924–32. doi: 10.2337/db08-0079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Montaigne D, Marechal X, Coisne A, Debry N, Modine T, Fayad G, Potelle C, El Arid JM, Mouton S, Sebti Y, Duez H, Preau S, Remy-Jouet I, Zerimech F, Koussa M, Richard V, Neviere R, Edme JL, Lefebvre P, Staels B. Myocardial contractile dysfunction is associated with impaired mitochondrial function and dynamics in type 2 diabetic but not in obese patients. Circulation. 2014;130:554–64. doi: 10.1161/CIRCULATIONAHA.113.008476. [DOI] [PubMed] [Google Scholar]
- 30.Kashatus JA, Nascimento A, Myers LJ, Sher A, Byrne FL, Hoehn KL, Counter CM, Kashatus DF. Erk2 phosphorylation of Drp1 promotes mitochondrial fission and MAPK-driven tumor growth. Mol Cell. 2015;57:537–51. doi: 10.1016/j.molcel.2015.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Strack S, Wilson TJ, Cribbs JT. Cyclin-dependent kinases regulate splice-specific targeting of dynamin-related protein 1 to microtubules. J Cell Biol. 2013;201:1037–51. doi: 10.1083/jcb.201210045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taguchi N, Ishihara N, Jofuku A, Oka T, Mihara K. Mitotic phosphorylation of dynamin-related GTPase Drp1 participates in mitochondrial fission. J Biol Chem. 2007;282:11521–9. doi: 10.1074/jbc.M607279200. [DOI] [PubMed] [Google Scholar]
- 33.Norton M, Ng AC, Baird S, Dumoulin A, Shutt T, Mah N, Andrade-Navarro MA, McBride HM, Screaton RA. ROMO1 is an essential redox-dependent regulator of mitochondrial dynamics. Science signaling. 2014;7:ra10. doi: 10.1126/scisignal.2004374. [DOI] [PubMed] [Google Scholar]
- 34.Makinde AO, Kantor PF, Lopaschuk GD. Maturation of fatty acid and carbohydrate metabolism in the newborn heart. Mol Cell Biochem. 1998;188:49–56. [PubMed] [Google Scholar]
- 35.Nagao M, Parimoo B, Tanaka K. Developmental, nutritional, and hormonal regulation of tissue-specific expression of the genes encoding various acyl-CoA dehydrogenases and alpha-subunit of electron transfer flavoprotein in rat. J Biol Chem. 1993;268:24114–24. [PubMed] [Google Scholar]
- 36.Barger PM, Kelly DP. PPAR signaling in the control of cardiac energy metabolism. Trends Cardiovasc Med. 2000;10:238–45. doi: 10.1016/s1050-1738(00)00077-3. [DOI] [PubMed] [Google Scholar]
- 37.Lehman JJ, Barger PM, Kovacs A, Saffitz JE, Medeiros DM, Kelly DP. Peroxisome proliferator-activated receptor gamma coactivator-1 promotes cardiac mitochondrial biogenesis. J Clin Invest. 2000;106:847–56. doi: 10.1172/JCI10268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ishihara T, Ban-Ishihara R, Maeda M, Matsunaga Y, Ichimura A, Kyogoku S, Aoki H, Katada S, Nakada K, Nomura M, Mizushima N, Mihara K, Ishihara N. Dynamics of mitochondrial DNA nucleoids regulated by mitochondrial fission is essential for maintenance of homogeneously active mitochondria during neonatal heart development. Molecular and cellular biology. 2015;35:211–23. doi: 10.1128/MCB.01054-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res. 2015;116:264–78. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 40.Kageyama Y, Hoshijima M, Seo K, Bedja D, Sysa-Shah P, Andrabi SA, Chen W, Hoke A, Dawson VL, Dawson TM, Gabrielson K, Kass DA, Iijima M, Sesaki H. Parkin-independent mitophagy requires Drp1 and maintains the integrity of mammalian heart and brain. The EMBO journal. 2014;33:2798–813. doi: 10.15252/embj.201488658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shen Z, Ye C, McCain K, Greenberg ML. The Role of Cardiolipin in Cardiovascular Health. Biomed Res Int. 2015;2015:891707. doi: 10.1155/2015/891707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sparagna GC, Chicco AJ, Murphy RC, Bristow MR, Johnson CA, Rees ML, Maxey ML, McCune SA, Moore RL. Loss of cardiac tetralinoleoyl cardiolipin in human and experimental heart failure. J Lipid Res. 2007;48:1559–70. doi: 10.1194/jlr.M600551-JLR200. [DOI] [PubMed] [Google Scholar]
- 43.Sparagna GC, Johnson CA, McCune SA, Moore RL, Murphy RC. Quantitation of cardiolipin molecular species in spontaneously hypertensive heart failure rats using electrospray ionization mass spectrometry. J Lipid Res. 2005;46:1196–204. doi: 10.1194/jlr.M500031-JLR200. [DOI] [PubMed] [Google Scholar]
- 44.Shinzawa-Itoh K, Aoyama H, Muramoto K, Terada H, Kurauchi T, Tadehara Y, Yamasaki A, Sugimura T, Kurono S, Tsujimoto K, Mizushima T, Yamashita E, Tsukihara T, Yoshikawa S. Structures and physiological roles of 13 integral lipids of bovine heart cytochrome c oxidase. EMBO J. 2007;26:1713–25. doi: 10.1038/sj.emboj.7601618. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.