

SUMMARY
Alternative mRNA processing is a critical mechanism for proteome expansion and gene regulation in higher eukaryotes. The SR family proteins play important roles in splicing regulation. Intriguingly, mammalian genomes encode many poorly characterized SR-like proteins, including subunits of the mRNA 3′ processing factor CFIm, CFIm68 and CFIm59. Here we demonstrate that CFIm functions as an enhancer-dependent activator of mRNA 3′ processing. CFIm regulates global alternative polyadenylation (APA) by specifically binding and activating enhancer-containing poly(A) sites (PAS). Importantly, the CFIm activator functions are mediated by the arginine-serine repeat (RS) domains of CFIm68/59, which bind specifically to an RS-like region in the CPSF subunit Fip1, and this interaction is inhibited by CFIm68/59 hyper-phosphorylation. The remarkable functional similarities between CFIm and SR proteins suggest that interactions between RS-like domains in regulatory and core factors may provide a common activation mechanism for mRNA 3′ processing, splicing, and potentially other steps in RNA metabolism.
Keywords: mRNA 3′ processing, alternative polyadenylation, SR proteins
eTOC blurb
Zhu et al show that CFIm is an enhancer-dependent activator that promotes mRNA 3′ processing complex assembly. CFIm activator function requires the RS-like domains of CFIm68/59 and involves a mechanism similar to SR protein-mediated splicing regulation, suggesting a unified activation mechanism for mRNA 3′ processing and splicing.
INTRODUCTION
The transcripts of most human genes undergo alternative splicing and/or polyadenylation to produce multiple mRNA isoforms that encode distinct proteins or have different regulatory properties (Braunschweig et al., 2013; Nilsen and Graveley, 2010; Tian and Manley, 2017). Alternative mRNA processing is regulated in a tissue- and/or developmental stage-specific manner and aberrant mRNA processing has been linked to a wide range of human diseases. It is therefore important to understand how mRNA processing is regulated. Splicing regulation requires both cis elements and trans acting factors. Many regulatory sequences, such as enhancers and silencers, have been identified and they recruit regulatory proteins, including SR proteins and hnRNPs, to modulate splicing (Nilsen and Graveley, 2010; Wang and Burge, 2008). The SR family proteins contain one or two N-terminal RNA recognition motif (RRM) domains and a C-terminal RS domain that is rich in arginine-serine dipeptide repeats (Graveley, 2000; Tacke and Manley, 1999; Zhong et al., 2009). SR proteins function as both essential splicing factors and important alternative splicing regulators. In the latter function, SR proteins often bind to exonic enhancer sequences and promote the recruitment of core splicing factors, including U1-70K and U2AF35, to nearby splice sites through RS domain-mediated interactions (Graveley, 2000). Interestingly, core splicing factors bind to SR proteins via their own RS or RS-like domains. For example, an arginine-aspartate/glutamate (RE/D)-rich region in U1-70K is necessary and sufficient for SR protein binding (Cao and Garcia-Blanco, 1998). SR proteins are extensively phosphorylated in vivo and both hyper- and hypo-phosphorylated SR proteins are inactive in splicing (Kanopka et al., 1998; Prasad et al., 1999; Sanford and Bruzik, 1999).
The regulatory mechanisms for APA remain poorly understood. Although some cis elements have been shown to promote efficient processing of certain viral or cellular PASs, their mechanisms and impact on the transcriptome are unclear (Zhao et al., 1999). Recent studies have identified a number of global APA regulators (Tian and Manley, 2017). Among them, the essential mRNA 3′ processing factor CFIm seems particularly important as CFIm-mediated APA regulation has been linked to tumor suppression and neurological disorders (Gennarino et al., 2015; Masamha et al., 2014). CFIm consists of a small subunit CFIm25 and two alternative large subunits, CFIm68 and CFIm59, both of which are members of the SR superfamily proteins (Ruegsegger et al., 1998). CFIm25 binds specifically to a UGUA motif (Brown and Gilmartin, 2003; Yang et al., 2010b). CFIm25 forms a dimer and CFIm68/59 binds to the CFIm25 dimer via their RRM domains to form a tetrameric CFIm complex (Yang et al., 2010a). CFIm, CPSF and CstF bind to PAS RNA cooperatively to assemble the core mRNA 3′ processing complex, but the exact functions of CFIm in mRNA 3′ processing remain poorly understood (Chan et al., 2011; Shi and Manley, 2015). Intriguingly, depletion of CFIm25 or CFIm68, but not CFIm59, results in widespread shift to proximal PASs and 3′ UTR shortening (Gruber et al., 2012; Hwang et al., 2016; Martin et al., 2012). At least two models have been proposed for CFIm-mediated APA regulation. First, CFIm has been suggested to suppress proximal PASs, possibly by binding to sub-optimal target sites and blocking CPSF recruitment (Martin et al., 2012; Masamha et al., 2014). Alternatively, it was proposed that the CFIm25 dimer could simultaneously bind to two copies of UGUA, each located upstream of an alternative PAS, such that the proximal PAS is looped out and thus inhibited (Yang et al., 2011). However, these models have not been experimentally tested.
Here we demonstrate that CFIm is an enhancer-dependent activator of mRNA 3′ processing that regulates APA by binding and activating enhancer-containing PASs. Importantly, our results revealed that the RS domains of CFIm68/59 play a central role in activating mRNA 3′ processing, at least in part, by binding to the RE/D domain in the CPSF subunit Fip1. Our results suggest that SR superfamily proteins may activate mRNA 3′ processing and splicing through a common mechanism.
RESULTS
UGUA is not an essential cis-element, but an enhancer for mammalian mRNA 3′ processing
To characterize CFIm functions, we first examined the role of its cognate sequence UGUA in mammalian mRNA 3′ processing. By comparing the frequency of UGUA in annotated human PASs (from −100 nucleotides (nt) to +100 nt relative to the cleavage site) and that in randomly selected genomic sequences, we calculated its enrichment score: log2 (frequency in PAS/frequency in random sequence). As shown in Fig. 1A, UGUA is modestly enriched at around −50 nt, but depleted near the cleavage site. By contrast, the poly(A) signal A(A/U)UAAA was more enriched than UGUA and displayed a distinct peak at −19 nt. Additionally, at least half of human and mouse PASs do not harbor a UGUA motif within the 100 nt region upstream of the cleavage sites while nearly 70% of PASs have at least one A(A/U)UAAA in the same region. Therefore UGUA is only found in a subset of human PASs.
Figure 1. UGUA is not an essential cis-element, but an enhancer for mRNA 3′ rocessing.

(A) Enrichment score (log2(frequency in PAS/frequency in random sequence)) for UGUA and A(A/U)UAAA. (B) RNA substrates used in this study: L3 and p14/Robld3 PAS. (C) Compare L3 wild type (WT), Δ1, Δ1-2, Δ1-3UGUA mutant PASs using in vitro mRNA 3′ processing assays. Top panel: coupled cleavage/polyadenylation assay. Bottom panel: cleavage assay. Quantification results are shown below of the gel: % processed= (5′cleavage product)/(pre-mRNA). (D) Compare p14 WT, 1x and 2xUGUA mutant PASs using in vitro mRNA 3′ processing assays. Results are shown similar to (C).
To characterize the functional role of UGUA motifs, we used two natural PASs (Fig. 1B): 1) L3, a commonly used adenovirus PAS that contains three copies of UGUA, located at −50, −39, and +3 nt respectively; 2) the human p14/Robld3 PAS, which lacks UGUA. For L3, we generated the wild type (WT) and several mutant RNAs, in which the first one (Δ1), two (Δ1-2) or all three (Δ1-3) UGUAs were mutated (see Table S1 for sequence information). Conversely we introduced one or two copies of UGUAs into the p14 PAS at −57 and −46 nt respectively. We then tested these RNA substrates using in vitro 3′ processing assays with HeLa nuclear extract (NE). The WT L3 was efficiently processed in both coupled cleavage/polyadenylation (Fig. 1C, top panel) and cleavage assays (lower panel). The L3-Δ1 showed similar processing efficiencies as the WT. However, the processing efficiency of L3-Δ1-2 decreased by ~50% compared to the WT, and Δ1-3 showed little additional decrease. Further studies of these UGUAs individually and in combinations suggest that both upstream UGUAs stimulated mRNA 3′ processing (Fig. S1). The second UGUA had higher activities, but adding the first UGUA further enhanced activity. Conversely, the p14 WT showed very low activity in mRNA 3′ processing assays (Fig. 1D). When one or two copies of UGUA were inserted in p14 PAS, its mRNA 3′ processing efficiency progressively increased (Fig. 1D). p14 PAS with the addition of UGUAs was still weaker than L3, indicating that additional sequences are involved in determining PAS strength. Together, our computational and experimental results strongly suggest that the UGUA motif is not an essential cis-element, but an enhancer for mammalian mRNA 3′ processing. As similar changes were observed for both coupled cleavage/polyadenylation and cleavage assays (Fig. 1C and D), we concluded that UGUA activates mRNA 3′ processing primarily at the cleavage step.
The enhancer activity of UGUA is position-dependent
Next we tested if the UGUA position can affect its enhancer activity. To this end, we used L3Δ1-3 as a template and inserted UGUAs at different positions (see Table S1 for sequence information). As CFIm forms a dimer and is capable of binding two copies of UGUA simultaneously (Yang et al., 2010b), we initially inserted two tandem copies of UGUA in these constructs (Fig. 2A). When we performed in vitro 3′ processing assays using these RNAs, we observed the highest 3′ processing activity with L3-2xUGUA-50 and L3-2xUGUA-60, and the activity decreased when UGUAs were inserted further upstream or downstream (Fig. 2B–C). We next tested the activities of a single UGUA inserted at different positions (Fig. 2D). The results showed that a single copy of UGUA had the highest activities at −39 nt and then −50 nt (Fig. 2E–F). A comparison with the results on two UGUAs suggests a combinatorial effect between UGUA enhancers.
Figure 2. The enhancer activity of UGUA is position dependent.

(A and D) Diagrams to show the design of PAS RNAs. Two tandem copies of UGUA (A) or one copy (D) were inserted at different positions in tL3-Δ1-3. (B and E) In vitro cleavage/polyadenylation assay using L3 PAS with 2x or 1xUGUA inserted at different positions. (C and F) Quantification of the results shown in (B) and (F): mean ± s.e.m (n=3). (G) Design of the pPASPORT reporter. CMV: promoter; Rluc: renilla luciferase; PAS: poly(A) site to be tested; IRES: internal ribosomal entry site; Fluc: firefly luciferase. (H) PAS activity (Rluc/Fluc): mean ± s.e.m (n=3).
To complement our in vitro results, we also tested the positional effect of two UGUAs on mRNA 3′ processing in living cells using the dual luciferase reporter pPASPORT (Fig. 2G) (Lackford et al., 2014; Yao et al., 2012). In this construct, both Renilla (Rluc) and Firefly luciferase (Fluc) genes are expressed in a bicistronic mRNA and both luciferases can be translated (Fluc translation is driven by an internal ribosomal entry site (IRES)). A PAS to be tested is inserted between the two luciferase genes. With strong PASs, cleavage/polyadenylation occurs efficiently so that only Rluc is expressed. For weak PASs, inefficient 3′ processing leads to more transcription read-through and the expression of both Rluc and Fluc. Therefore, the Rluc/Fluc ratio provides a quantitative measurement of PAS strength. To test the effect of UGUA positions, we cloned all PASs shown in Fig. 2A into pPASPORT and carried out reporter assays. Consistent with the in vitro results, our reporter assays detected the highest PAS activity when the two UGUAs were located at −50 nt (Fig. 2H). Together our in vitro and in vivo reporter assays consistently demonstrated that the enhancer activities of UGUA are position-dependent.
Enhancer-bound CFIm promotes the assembly of mRNA 3′ processing complex
We next tested how the UGUA enhancer affected the CFIm-PAS interaction and mRNA 3′ processing complex assembly. First, we incubated L3 and p14 PASs and their derivatives with HeLa NE under 3′ processing conditions and then performed gel mobility shift assays to monitor the assembly of mRNA 3′ processing complexes (P complex). Our results showed that the P complex assembled efficiently on WT L3 while no P complex was observed on a mutant L3 in which the poly(A) signal AAUAAA was mutated to AAGAAA (Fig. 3A, lanes 1 and 3), consistent with the essential role of AAUAAA in mRNA 3′ processing. P complex assembled on L3-Δ1-3, but less efficiently when compared to WT (Fig. 3A, compare lanes 1 and 2). On the other hand, the insertion of two copies of UGUA into p14 PAS enhanced the P complex assembly (Fig. 3A, compare lanes 4 and 5). These results strongly suggest that the UGUA enhancer promotes P complex assembly. Secondly, we purified the P complexes assembled on PAS RNAs using a previously described RNA affinity approach (Fig. 3B) (Shi et al., 2009), and monitored their protein compositions by western blotting analyses. The results showed that the P complex assembled on the WT L3 contained all known CPSF, CstF, and CFIm subunits (Fig. 3C, lane 1). In the pulldown sample with the AAGAAA mutant, none of these factors were detected (Fig. 3C, lane 3). These results were consistent with previous studies and our gel mobility shift assay results (Fig. 3A). Strikingly, the P complex assembled on L3-Δ1-3 essentially lacked all three CFIm subunits (Fig. 3C, lane 2). Although CPSF and CstF subunits were present, their levels were reduced (Fig. 3C, compare lanes 1 and 2). Conversely, adding UGUAs to p14 PAS significantly promoted the recruitment of CFIm (Fig. 3C, compare lane 5 and 6). Together, these data suggest that CFIm binds to PAS in an UGUA-dependent manner and the enhancer-bound CFIm promotes the assembly of the mRNA 3′ processing complex.
Figure 3. Enhancer-bound CFIm promotes the assembly of mRNA 3′ processing complex.

(A) Gel mobility shift assays with L3 or p14-derived PASs. P: mRNA 3′ processing complex; H: heterogenous complex. (B) A diagram showing the RNA affinity purification procedure. MBP-MS2: a fusion protein between maltose binding protein and MS2. (C) The complexes assembled on the 3MS2-tagged L3 or p14-derived PASs were purified and analyzed by western blotting. The red arrows mark the CFIm subunits. (D) mRNA 3′ processing complex assembly on L3-derived PASs as shown in Fig. 2(A). (E) The mRNA 3′ processing complexes assembled on the 3MS2-tagged L3 derivatives as shown in Fig. 2(A) were purified and analyzed by western blotting. (F) Quantification of western blot signals in (E) using ImageJ.
As the enhancer activity of UGUA is position-dependent (Fig. 2), we next tested how UGUA position affected CFIm recruitment and the P complex assembly using the series of L3-derived PASs in which two UGUAs were inserted at different positions (Fig. 2A). Gel mobility shift assays showed that optimal P complex formation was achieved on L3-2xUGUA-50, and that the P complex levels decreased when UGUAs were placed further upstream or downstream (Fig. 3D), which mirrored our in vitro processing assay results (Fig. 2B). We next purified the P complexes assembled on these RNAs and examined the levels of mRNA 3′ processing factors. Interestingly, CFIm subunits were present at the highest levels in the L3-2xUGUA-50 complex and decreased precipitously when the UGUA motifs were located further upstream or downstream (Fig. 3E and quantification in Fig. 3F). CPSF and CstF subunits were detected in most samples, but their highest levels were observed in the L3-2xUGUA-50 complex (Fig. 3E and quantification in Fig. 3F). It was also noted that CstF recruitment seemed to be affected more than CPSF. These data suggest that CFIm is optimally recruited to PASs in vitro only when the UGUA enhancers are located at a specific location and the enhancer-bound CFIm promotes the recruitment of CPSF and CstF.
CFIm activates mRNA 3′ processing via its RS-like domains
To determine which CFIm subunit is responsible for activating mRNA 3′ processing, we tethered individual CFIm subunits to a PAS using the λN-Box B system (Baron-Benhamou et al., 2004), and tested their effects on mRNA 3′ processing efficiency by using a reporter assay. To create the RNA substrate, we modified L3-2xUGUA-50 (Fig. 2A) by replacing its two UGUAs with Box B hairpins. The resultant PAS, called L3-2xBoxB, was cloned into the pPASPORT reporter (Fig. 4A). We then co-expressed the L3-2xBoxB reporter and individual CFIm subunits with or without an N-terminal λN tag (Fig. S2), and measured the PAS activities. Interestingly, λN-tagged CFIm25, CFIm59, and CFIm68 all caused significant activation of mRNA 3′ processing compared to the untagged proteins (Fig. 4B, p value<0.001 t-test; Fig. S2B). CFIm25 had the most significant effect, causing a ~6-fold increase in PAS activity compared to control (Fig. 4B). As CFIm25 binds to CFIm68 and CFIm59, the tethered CFIm25 may activate mRNA 3′ processing directly by itself or indirectly by recruiting CFIm68 or CFIm59. To distinguish between these possibilities, we sought to specifically disrupt CFIm25-CFIm68/59 interaction while maintaining the integrity of CFIm25. As CFIm68/59 binds to the CFIm25 dimer interface (Yang et al., 2010a), we introduced a mutation L218R into this region to specifically disrupt the hydrophobic interactions (Fig. S3A). CFIm25-L218R was stable in cells but its interactions with CFIm59 and CFIm68 were significantly compromised (Fig. S3B). When tethered to a PAS, CFIm25-L218R displayed significantly reduced activity compared to the WT (Fig. 4B, p value<0.001 t-test). These data suggest that CFIm25 activates mRNA 3′ processing primarily by recruiting CFIm68 and/or CFIm59.
Figure 4. The RS-like domain of CFIm68/59 is necessary and sufficient for activating mRNA 3′ processing.

(A) A diagram of the tethering assay. RRM: RNA recognition motif; PRR: proline-rich region; RS: arginine-serine repeat region. (B–E) Tethering assay results obtained by co-expressing the L3-2xBoxB reporter and the proteins as labeled. The CFIm25 mutant L218R was labeled vertically. The results were plotted as mean ± s.e.m (n=3). PAS activities for tagged and untagged proteins were compared. L218R was compared to the wild type CFIm25. *** indicates that the p-values<0.001 (t-test). All samples were compared with the vector and *** indicates that the p-values<0.001 (t-test).
In the tethering assay, both CFIm68 and CFIm59 activated mRNA 3′ processing and CFIm68 displayed greater activity (Fig. 4B). Both proteins have an N-terminal RRM, a central proline-rich region (PRR), and a C-terminal RS-like domain (Fig. 4A). To map which domain(s) are required, we created several deletion mutants: ΔRRM, ΔPRR, and ΔRS. A nuclear localization signal was attached to the C-terminus of each truncated protein to ensure proper localization. When tested in the tethering assays, ΔRRM and ΔPRR displayed similar or modestly reduced activities compared to the full-length (FL) proteins (Fig. 4C and Fig. S2A). Strikingly, however, the activation was abolished in both ΔRS mutants (Fig. 4C). These data demonstrated that the RS domains of CFIm68 and CFIm59 are necessary for activating mRNA 3′ processing.
We next wanted to test if the CFIm68/59 RS domains were sufficient to activate mRNA 3′ processing using the tethering assay. However, the RS domains alone did not express well in cells (data not shown). To overcome this limitation, we expressed GST-RS fusion proteins and tested them in our tethering assays. Interestingly, both GST-RS(CFIm68) and GST-RS(CFIm59) activated mRNA 3′ processing to comparable levels as the FL proteins while tethering GST alone had no effect (Fig. 4D and Fig. S2A). Based on these results, we concluded that the RS domains of CFIm68 and CFIm59 are both necessary and, when tethered to a PAS, sufficient for activating mRNA 3′ processing.
As CFIm68 had higher activities than CFIm59 in tethering assays (Fig. 4B–C), we tested the contribution of all protein domains to their functional difference. To this end, we generated a series of chimeric proteins between CFIm68 and CFIm59 (Fig. 4E), and tested them in our tethering system. Chimeras 1 and 2, which contained the CFIm68 RS domain, showed similar activities as CFIm68 itself, whereas those containing CFIm59 RS domain (chimeras 3 and 4) showed similar activity as CFIm59 (Fig. 4E). These data strongly suggest that the RS-like domains are the primary determinant of CFIm68/59 activities.
CFIm68/59 RS domains directly bind to the RE/D domain of the CPSF subunit Fip1
Our in vitro assay results suggest that the enhancer-bound CFIm activates mRNA 3′ processing by stimulating the recruitment of CPSF and CstF (Fig. 3), and that the RS domains of CFIm68/59 are necessary and sufficient for activation (Fig. 4). As the RS domains of SR proteins are extensively phosphorylated in vivo, we determined if the CFIm68/59 RS domains were also phosphorylated. To this end, we treated HeLa NE with alkaline phosphatase (CIP), and compared the gel mobility of CFIm68 and CFIm59 by SDS-PAGE followed by western blotting analyses. Using this assay, we failed to detect any significant change in the gel mobilities of either protein (Fig. S4A). As relatively large changes in phosphorylation are needed to cause visible gel mobility shift on regular SDS-PAGE, we analyzed the same samples on Phos-tag gels (Kinoshita et al., 2009). The Phos-tag reagent in acrylamide gels binds specifically to phosphorylated amino acids and causes slower migration of phosphorylated proteins. Using Phos-tag gels, we observed that CIP treatment significantly increased the mobilities of CFIm68 (Fig. 5A, top panel). A similar, but less pronounced, mobility shift was also detected for CIP-treated CFIm59 (Fig. 5A, lower panel), suggesting that both CFIm68 and CFIm59 are phosphorylated in vivo. The same analyses suggested that recombinant CFIm25-68 and CFIm25-59 complexes or GST-RS(CFIm68/59) fusion proteins purified from baculovirus-infected Sf9 insect cells were also phosphorylated at near physiological levels (Fig. 5A and Fig. S4).
Figure 5. The CFIm68/59 RS-like domain binds to Fip1.

(A) HeLa nuclear extract (NE) or recombinant CFIm25-68 or CFIm25-59 complexes purified from baculovirus-infected Sf9 insect cells with or without alkaline phosphatase (CIP) treatment, were resolved by Phos-tag gel and analyzed by western blotting. The red arrows point to the phosphorylated proteins and the green arrows dephosphorylated proteins. (B) CFIm59 and CFIm68 RS domain sequences. (C) GST pulldown assay with GST, GST-RS(CFIm59) or (CFIm68) (purified from Sf9 cells) and in vitro translated 35S-labeled individual CPSF and CstF subunits. GST pulldown samples were resolved on SDS-PAGE and visualized by phosphorimaging (top panel). The same pulldown assay was performed with 6xHis-Fip1 expressed in Sf9 cells and pulldown samples were resolved on SDS-PAGE and analyzed by western blotting (lower panel). (D) A diagram of the Fip1 domain/regions. The Fip1-N and -C fragments were marked. Pulldown assays were similar to (C) with in vitro translated and 35S-labeled Fip1-N and Fip1-C. (E) Top panel: the sequences of the Fip1-RD and -RA peptides. Lower panel: Fip1-RD and –RA pulldown with purified 6xHis-CFIm25 (E. coli), 6xHis-CFIm25-59 (Sf9), and 6xHis-CFIm25-68 complexes (Sf9) and the bound proteins were resolved on SDS-PAGE and analyzed by western blotting. Negative control: streptavidin beads (beads). (F) Top panel: GST-RS(CFIm59/68) purified from E. coli were mock treated (−) or treated (+) with SRPK1 and then used in pulldown assays with 6xHis-Fip1. The pulldown samples were analyzed by western blotting. Lower panel: GST-RS(CFIm59/68) purified from Sf9 cells were mock untreated (−) or treated (+) with CIP, and then used in pulldown assays with purified 6xHis-Fip1. (G) Nuclear extracts from control, CFIm59-KO, or CFIm68-KO HEK293T cell lines were used for IP with anti-CFIm25 antibody and the IP samples were analyzed by western blotting. The red arrows mark the CFIm59 or CFIm68 that are absent in KO cell lines.
We hypothesized that the RS domains of CFIm68/59 might directly bind to one or more subunits of CPSF and CstF. To test this hypothesis, we used GST-RS (CFIm68/59) purified from Sf9 cells (Fig. 5B and Fig. S4B and F) and performed GST pulldown assays with in vitro translated individual CPSF or CstF subunits. Interestingly, both GST-RS(CFIm68) and GST-RS(CFIm59) specifically pulled down Fip1, but not other CPSF or CstF subunits tested (Fig. 5C). Additionally, we noted that slightly higher amounts of Fip1 seemed to be precipitated by GST-RS(CFIm68) (Fig. 5C, top panel). To further characterize this interaction, we used recombinant 6xHis-Fip1 expressed in Sf9 cells (Fig. S4C) and repeated the GST pulldown assays. Again, GST-RS(CFIm68) pulled down significantly more Fip1 compared to GST-RS(CFIm59) (Fig. 5C, bottom panel). These results suggest that the RS domains of CFIm68 and CFIm59 can directly interact with Fip1.
Next we wanted to map the Fip1 region/domains involved in this interaction. Fip1 is a largely disordered protein with several distinct regions, including an N-terminal acidic region, a conserved central domain, and a RE/D-rich C-terminal region (Fig. 5D, top panel). We expressed a C-terminal fragment of Fip1 that contained the RE/D-rich region (Fip1-C) and another fragment that covered the rest of the protein (Fip1-N, Fig. 5D) by in vitro translation and performed GST pulldown assays. We found that the CFIm68/59 RS domains specifically pulled down Fip1-C, but not Fip1-N (Fig. 5D, lower panel), suggesting that the Fip1 C-terminal region mediates direct interactions with CFIm RS domains.
As shown in Fig. 5E (top panel), the Fip1 RE/D region contains a 34-amino acid fragment that consists largely of RD/E dipeptide repeats. We next determined if the Fip1 RE/D region interacts with CFIm. To this end, we chemically synthesized this 34-amino acid peptide with an N-terminal biotin tag (Fip1-RD). To determine if the alternating charges on the RE/D peptide are important, we also synthesized another peptide in which all aspartate or glutamate residues in Fip1-RD were mutated to alanines (Fip1-RA). We then immobilized the Fip1-RD or -RA peptides on streptavidin beads and carried out pulldown assays with purified 6xHis-CFIm25 protein or 6xHis-CFIm25-59 and 6xHis-CFIm25-68 complexes (Fig. S4D). Fip1-RD peptide specifically pulled down CFIm25-68 complex (Fig. 5E, middle panels) and, to a lesser degree, CFIm25-59 complex (lower panel), but not CFIm25 alone (top panel). Additionally, the Fip1-RA peptide pulled down significantly less CFIm25-68 complex compared to the Fip1-RD peptide (Fig. 5E, middle panel), but both peptides pulled down similar amounts of CFIm25-59 complex (Fig. 5E, bottom panel). These results suggest that the Fip1 RE/D region is sufficient to interact with CFIm68/59.
It is well known that SR protein-mediated interactions are modulated by phosphorylation of their RS domains (Graveley, 2000; Tacke and Manley, 1999). Therefore we tested if and how the phosphorylation levels of the CFIm68/59 RS domains may affect their interactions with Fip1. To this end, we compared GST-RS(CFIm68/59) expressed and purified from Sf9 cells (Fig. S4B), which were phosphorylated at near physiological levels, and those from E. coli (Fig. S4E), thus unphosphorylated. First, after incubating GST-RS(CFIm68/59) from E. coli with the SR protein kinase SRPK1 in the presence of ATP, we observed dramatic mobility shifts on Phos-tag gels (Fig. S4F, compare lanes 2 and 4, 6 and 8), suggesting that CFIm68/59 RS domains were phosphorylated by SRPK1. When these proteins were used for GST pulldown assays, the SRPK1-treated GST-RS pulled down significantly less Fip1 (Fig. 5F, top panel), indicating that phosphorylation of CFIm68/59-RS inhibited their interactions with Fip1. On the other hand, CIP treatment of the GST-RS(CFIm68/59) protein purified from Sf9 led to partial dephosphorylation (Fig. S4F, compare lanes 1 and 3, 5 and 7), but only modest changes in their pulldown efficiency of Fip1 proteins (Fig. 5F, lower panel), arguing against a significant role for phosphorylation. To understand this inconsistency, we compared the SRPK1-treated GST-RS(CFIm68/59) and those purified from Sf9 cells and found that the former had lower mobility on Phos-tag gels (Fig. S4F, compare lanes 1 and 2, 5 and 6), suggesting that SRPK1 hyper-phosphorylated GST-RS(CFIm68/59). Based on these results, we concluded that CFIm68/59 RS-like domains are phosphorylated in vivo, but such phosphorylation is not required for its interaction with Fip1 under normal physiological conditions. However, hyper-phosphorylation of CFIm68/59 RS domains by SRPK1 could inhibit their interactions with Fip1.
Our in vitro binding assays provided evidence that Fip1 may have higher affinity for CFIm68 than CFIm59. To test this in vivo, we have generated CFIm59 knockout (KO) HEK293T cell lines using the CRISPR/Cas9 system and obtained several previously reported CFIm68 KO HEK293T cell lines (Sowd et al., 2016). As demonstrated by western blotting (Fig. 5G), CFIm68 and CFIm59 were specifically depleted in these cell lines without significant effect on the protein levels of other CFIm subunits. Using these cells, we immunoprecipitated endogenous CFIm complexes using an antibody against CFIm25 and examined the levels of CPSF and CstF subunits that were co-precipitated (Fig. 5G). Similar levels of CFIm complexes were recovered from wild type and the KO cell lines as indicated by the similar amounts of CFIm25 as well as CFIm68 and CFIm59. All CPSF and CstF subunits tested were co-precipitated in CFIm59 KO cells at comparable levels as the wild type cells (Fig. 5G, compare lanes 5 and 6). Interestingly, however, significantly lower amounts of CPSF and CstF subunits were co-precipitated with CFIm in CFIm68 KO cells, (Fig. 5G, compare lanes 5–6 and 7). Together these data suggest that CFIm68 plays a more important role than CFIm59 in mediating interactions between CFIm and CPSF.
Mechanisms for CFIm-mediated APA regulation
Having established that CFIm is a UGUA enhancer-dependent activator of mRNA 3′ processing, we wanted to determine if this function is involved in CFIm-mediated APA regulation. First, we analyzed the global APA profiles of wild type HEK293T and our CFIm68 KO and CFIm59 KO cell lines by mRNA 3′ end mapping. To ensure reproducibility, we have used two independent KO cell lines for both CFIm59 and CFIm68. By comparing the APA profiles of the control and KO cell lines, we found that CFIm68 KO led to significant APA changes in 422 genes while CFIm59 KO only affected 9 genes (APA change>=15%, FDR<0.05; see Methods for details) (Fig. 6A). Among CFIm68 target genes, the vast majority (96%) showed significant shift to proximal PASs, leading to 3′ UTR shortening (Fig. 6A, red dots: distal-to-proximal). Two representative examples of CFIm68 KO-induced APA change were shown in Fig. 6B. Our data is highly consistent with previously published datasets of CFIm25, 59, and 68 knockdown in human and mouse cells (Gruber et al., 2012; Li et al., 2015; Martin et al., 2012; Masamha et al., 2014). A direct comparison of our KO cell line data and previous knockdown data in HEK293 showed that 42% and 37% of genes with distal-to-proximal shifts in CFIm68 KO cells also displayed similar APA changes in CFIm68 and CFIm25 knockdown cells (Fig. S5A), suggesting that CFIm68 and CFIm25 depletion induced similar APA changes in an overlapping set of genes.
Figure 6. Mechanism for CFIm-mediated APA regulation.

(A) Scatter plots to show an APA comparison between control HEK293T cells (y axis) and CFIm68 or CFIm59 KO cells (x axis). Genes with significant APA changes (FDR<0.05 and at least 15% change) were highlighted: red dots represent genes with distal to proximal (DtoP) APA changes while blue dots proximal to distal (PtoD). (B) Poly(A) site sequencing (PAS-seq) and PAR-CLIP data for Vma21 and Ddx3x genes. The proximal and distal PASs were marked by dotted boxes and labeled on the top. (C) UGUA distribution at the proximal (dotted lines) and distal (solid lines) of CFIm25 or CFIm68 target (red) and non-target (green) genes. The UGUA distribution curves at proximal and distal PASs were compared. ***: p value<0.001; n.s.: not significant (K-S test). (D) CFIm68 PAR-CLIP signals at the proximal (dotted lines) and distal (solid lines) of CFIm25 or CFIm68 targets (red) and non-target (green) genes. (E) Gel mobility shift assays to characterize interactions between CFIm25-68 complex and the specificied PASs. Free RNAs and RNA-protein complexes are marked. (F) Vma21 APA profiles were measured by RT-qPCR with one primer set for the common (comm) region and another for the extended (ext) 3′ UTR region. The over-expressed proteins are marked on the x-axis.
To determine the role of the UGUA enhancer in CFIm-mediated APA regulation, we first compared the distribution of UGUA in the proximal and distal PASs of CFIm25 or CFIm68 target mRNAs that displayed distal-to-proximal APA shifts. Interestingly, UGUA was highly enriched in the distal PASs compared to the proximal sites for both CFIm25 and CFIm68 target mRNAs (Fig. 6C) (p=1.6×10−29 and 1.6×10−13 respectively, K-S test). By contrast, when we compared the UGUA distribution in the proximal and distal PASs of non-target mRNAs, we found similar distribution patterns (Fig. 6C), suggesting that the distribution of the UGUA enhancer at alternative PASs within a transcript may determine whether its APA profile is regulated by CFIm levels. Additionally, the peak of UGUA was located near −55 nt for all samples (Fig. 6C), similar to the optimal position for UGUA to function as an enhancer as demonstrated earlier (Fig. 2).
We next examined the CFIm-RNA interactions using a previously published CFIm PAR-CLIP dataset (Martin et al., 2012). As the CFIm25 CLIP signals were much lower and more variable, we focused on CFIm68 and CFIm59 CLIP data. Interestingly we detected significantly more concentrated CFIm68 CLIP signals at the distal PASs of CFIm25 and CFIm68 target mRNAs compared to the proximal PASs (Fig. 6D, p value=4.9×10−7 and 1.3×10−8 respectively, K-S test). CFIm59 CLIP signals showed a similar pattern (Fig. S5B). This trend was also evident in both example genes (Fig. 6B). By contrast, the distribution of CFIm68 and CFIm59 CLIP signals were very similar for the proximal and distal PASs in non-target mRNAs (Fig. 6D and S5B). Therefore the CFIm-PAS interaction patterns are highly consistent with the UGUA enhancer distribution (Fig. 6C and D) and suggest that CFIm preferentially binds to distal PASs in the target mRNAs.
We next validated the CFIm-PAS binding patters for a few representative CFIm APA targets, including Vma21 and Ddx3x (Fig. 6B). To validate the CLIP results, we synthesized the proximal and distal PASs of these genes and performed gel mobility shift assays with purified 6xHis-CFIm25-68 complexes. Our results showed that CFIm25-68 had higher affinity for distal PASs than the proximal sites for all genes tested (Fig. 6E and Fig. S6A). These results confirmed that CFIm preferentially bound to the distal PASs in target mRNAs.
To test the distinct roles of CFIm68 and CFIm59 in regulating the APA of endogenous mRNAs, we selected Vma21 mRNA as a model system (Fig. 6B), and validated its APA changes in CFIm68 KO cells by RT-qPCR (Fig. 6F). We then asked whether over-expression of CFIm68 or CFIm59 can restore the Vma21 APA profile in CFIm68 KO cells. Interestingly, over-expression of CFIm68 and, to a lesser degree, CFIm59, reverted the Vma21 APA change in CFIm68 KO cells (Fig. 6F and Fig. S6B). This is consistent with our data suggesting that CFIm59 is a weaker activator than CFIm68. Finally, we tested the role of the individual domains of CFIm68 and CFIm59 in APA regulation. To this end, in CFIm68 KO cells, we over-expressed CFIm68, CFIm59, or the series of chimeric proteins as described earlier (Fig. 4E), and measured their effect on Vma21 APA by RT-qPCR. Interestingly, chimeras 1 and 2, which contained the RS domain of CFIm68, showed higher activities in restoring Vma21 APA than chimeras 3 and 4, which harbored the CFIm59 RS domain (Fig. 6F). These data suggest that the RS domains of CFIm68 and CFIm59 play an important role in CFIm-mediated APA regulation and that CFIm68 RS domain has more potent activity, consistent with its higher activity as an activator of mRNA 3′ processing. We conclude that CFIm is a UGUA enhancer-dependent activator of mRNA 3′ processing and this activity contributes to its role in regulating global APA.
DISCUSSION
Based on the data presented here, we propose the following model for CFIm-mediated APA regulation (Fig. 7A): CFIm is an UGUA enhancer-dependent activator of mRNA 3′ processing. In a subset of mRNAs, the enrichment of UGUA enhancers at the distal PASs leads to higher CFIm recruitment and, in turn, specific activation of these sites. CFIm depletion will cause decreased activities of the distal PASs in these mRNAs while the proximal sites are less affected, thus resulting in a net shift to proximal PASs. For mRNAs in which UGUA enhancers are distributed similarly at alternative PASs, changes in CFIm levels would affect these sites to a similar degree, thus their overall profiles are unaffected. Finally, as CFIm59 is a weaker activator than CFIm68, CFIm59 depletion has less impact on APA. Our model provides a mechanistic explanation not only for the 3′ UTR shortening phenotype in CFIm25- and CFIm68-depleted cells, but also for the target specificity and the different impact of CFIm59 and CFIm68 on CFIm-mediated APA regulation. Additionally, although mRNA 3′ processing takes place co-transcriptionally, our model argues that commitment to an upstream PAS could still occur after the downstream PAS has been transcribed. Recent studies demonstrated that RNAP II pauses within several kilobases after PASs (Nojima et al., 2015). If there are multiple upstream PASs, these sites could compete for mRNA 3′ processing factors. This is consistent with the current model that the usage of the proximal PAS is determined by the distance between the proximal and distal PASs, the RNAP II elongation rate, and the efficiency of PAS recognition at both proximal and distal sites (Li et al., 2015; Shi, 2012; Weng et al., 2016).
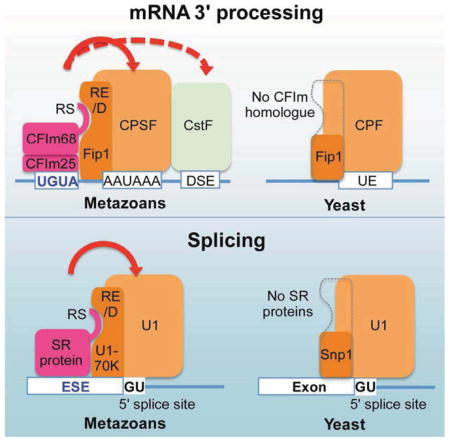
Figure 7. A unified activation mechanism for mRNA 3′ processing and splicing.

The solid red line with arrow indicates that CFIm helps to recruits CPSF through direct interactions and the dotted red line with arrow indicates that CFIm promotes CstF recruitment indirectly (A–B). The dotted grey lines indicate the lack of RE/D regions in the yeast Fip1 and Snp1 (C–D). UE: U-rich elements. CFIm25-68 is a dimer, but shown as a monomer due to space limitation (A). The blue arrows represent cleavage and the widths of the arrows represent the frequencies of PAS usage.
Fip1 mediates, at least in part, the interactions between CFIm and CPSF (Fig. 5). However, CFIm depletion induces primarily 3′ UTR shortening while Fip1 knockdown causes 3′ UTR lengthening (Lackford et al., 2014; Li et al., 2015). These seemingly contradictory observations can be explained by two aspects of Fip1 functions. First, Fip1 is an essential component of CPSF complex and is required for mRNA 3′ processing (Zhao et al., 1999). In Fip1-depleted cells, the intact CPSF complexes become limiting so that proximal PASs, which are generally weaker, cannot be efficiently recognized. The resultant read-through leads to transcription of the stronger distal PASs, which will outcompete the proximal sites in recruiting the limited amounts of CPSF (Lackford et al., 2014). Secondly, in Fip1-depleted cells, the limited CPSF complexes become more dependent on activators such as CFIm for recruitment to PASs. As CFIm preferentially bind to distal PASs in its targets, CPSF is preferentially recruited to these sites. These mechanisms, perhaps working in concert, may explain why distal PASs are favored in Fip1-depleted cells.
Our results suggest that CFIm68 and CFIm59 are functionally similar to SR proteins in many important aspects: 1) both CFIm68/CFIm59 and SR proteins can bind to enhancer sequences to regulate mRNA processing; 2) the enhancer-bound CFIm68/CFIm59 and SR proteins stimulate mRNA processing by promoting the recruitment of core processing machineries; 3) the activator functions of CFIm68/CFIm59 and SR proteins require their RS or RS-like domains; 4) both CFIm68/CFIm59 and SR proteins bind to RS-like domains of core processing factors: CFIm68/59 binds to the RE/D region of the CPSF subunit Fip1. SR proteins bind to the RE/D or RS-like regions in U1-70K and U2AF35 (Fig. 7B) (Kohtz et al., 1994; Wu and Maniatis, 1993); 5) CFIm68/CFIm59 and SR proteins have dual functions, both as essential processing factors and as regulators (Graveley, 2000). Although previous studies identified CFIm as an essential mRNA 3′ processing factor (Ruegsegger et al., 1996), our study revealed that it is also a sequence-dependent activator. CFIm68 and CFIm59 may have redundant functions in constitutive cleavage/polyadenylation as neither one is essential for cell viability (Fig. 5E and Sowd et al., 2016), but they clearly have distinct activities in APA regulation (Fig. 6A). Similarly, SR proteins function both as essential splicing factors and as critical splicing regulators (Graveley, 2000; Tacke and Manley, 1999; Zhong et al., 2009). The role of SR proteins in constitutive splicing seems redundant, but each SR protein has specific functions in regulating alternative splicing. The same interactions between CFIm68/59 and SR proteins with the core processing factors may be responsible for recruiting SR proteins or CFIm in constitutive as well as alternative splicing and mRNA 3′ processing. Together, our results revealed that, despite the fact that splicing and mRNA 3′ processing require distinct machineries, the activation of both processes involve a very similar mechanism. Finally, CFIm68/59 seem to share similar evolutionary paths as SR family proteins. Budding yeast does not have homologues of CFIm or SR proteins. Interestingly, although Fip1 is conserved in yeast, the yeast Fip1 homologue lacks the RE/D region (Fig. 7C and Fig. S7A). Similarly the yeast U1-70K homologue Snp1 does not contain a RE/D region (Fig. 7D and Fig. S7B). These results suggest that the activators for mRNA 3′ processing (CFIm68/59) and splicing (SR proteins) have co-evolved with their respective target proteins in the core processing machinery, allowing for more elaborate regulation in higher eukaryotes.
Our study revealed an interesting difference in the role of phosphorylation in the function of SR proteins and CFIm68/59. Unphosphorylated SR proteins bind weakly to U1-70K and this interaction is stimulated by SR protein phosphorylation (Xiao and Manley, 1997). By contrast, unphosphorylated CFIm68 or CFIm59 RS-like domains bind to Fip1 efficiently (Fig. 5F). This difference could be due to the sequences of their RS domains: the RS domains of the canonical SR proteins consist largely of RS dipeptide repeats, but the RS-like domains of CFIm68/59 contain not only RS, but also RE/D dipeptides (Fig. 5B). As RE/D may mimic phosphorylated RS, this may explain why CFIm68/59 interactions with Fip1 may be less dependent on phosphorylation than canonical SR proteins. Nonetheless, hyper-phosphorylation seems to inhibit the functions of both SR proteins and CFIm68/59.
Finally this common activation mechanism may be flexible enough to allow cross regulation. Indeed, CFIm subunits have been detected in purified human spliceosomes (Rappsilber et al., 2002; Zhou et al., 2002), indicating that CFIm may be involved in splicing regulation. CPSF has recently been shown to bind to U1-70K to regulate global alternative splicing (Misra et al., 2015). U2AF65 has been shown to interact with CFIm59 to stimulate mRNA 3′ processing (Millevoi et al., 2006). Additionally SR proteins have been shown to regulate mRNA 3′ processing and APA (Hudson and McNally, 2011; Lou et al., 1998; Muller-McNicoll et al., 2016). Together these studies provided evidence that the RS and RE/D domains provide a common binding platform to allow cross regulation and coordination of multiple steps of RNA metabolism.
STAR * METHODS
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CPSF160 | Bethyl | A301-580A; RRID:AB_1078859 |
| CPSF100 | Bethyl | A301-581A; RRID:AB_1078861 |
| Fip1 | Bethyl | A301-091A; RRID:AB_2084528 |
| CstF64 | Bethyl | A301-092A; RRID:AB_873014 |
| CFIm68 | Bethyl | A301-358A; RRID:AB_937785 |
| CFIm59 | Bethyl | A301-360A; RRID:AB_937864 |
| CFIm25 | Santa Cruz | sc-81109; RRID:AB_2153989 |
| hnRNP A1 | Santa Cruz | sc-56700; RRID:AB_629651 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DMEM (high glucose) | Thermo Fisher | 11995-073 |
| Dynabeads Protein A | Thermo Fisher | 10002D |
| Dynabeads Streptavidin | Thermo Fisher | 658.01D |
| Glutathione Sepharose High Performance beads | GE Healthcare Life Sciences | 17527901 |
| Shrimp Alkaline Phosphatase (SAP) | Thermo Fisher | EF0511 |
| 6xHis-CFIm25/68 | This study | N/A |
| 6xHis-CFIm25/59 | This study | N/A |
| GST-CFIm59 RS | This study | N/A |
| GST-CFIm68 RS | This study | N/A |
| Fip1-RD peptide | GenScript | Custom synthesis: SC1208/U2711BI160_1 |
| Fip1-RA peptide | GenScript | Custom synthesis: SC1208/U2711BI160_4 |
| Critical Commercial Assays | ||
| TnT® Quick Coupled Transcription/Translation System Kit | Promega | L1170 |
| Dual-Luciferase Reporter Assay Kit | Promega | E1910 |
| FuGENE® HD Transfection Reagent | Promega | E2311 |
| Phos-tag™ | Wako | 304-93521 |
| Deposited Data | ||
| PAS-seq | This study | GSE101871 |
| Raw experimental data | This study | http://dx.doi.org/10.17632/r23kcs7s8n.1 |
| Experimental Models: Cell Lines | ||
| Human: CFIm68 KO cells | Dr. Alan Engelman | Sowd et al., 2016 |
| Human: CFIm59 KO cells | This study | N/A |
| Sf9 Insect cells | This study | N/A |
| Oligonucleotides | ||
| Primers for cloning and qPCR | This study | See Table S1 |
| Recombinant DNA | ||
| Plasmids for transfections and in vitro assays | This study | See Table S2 |
| Software and Algorithms | ||
| deepTools | Ramirez et al., 2016 | http://deeptools.readthedocs.io/en/latest/ |
| diffSpliceDGE and topSpliceDGE | Robinson et al., 2010 | http://bioconductor.org |
| BEDTools | Quinlan and Hall, 2010 | http://bedtools.readthedocs.io/en/latest/ |
| SAMtools | Li et al., 2009 | http://samtools.sourceforge.net/ |
Contact for reagent and resource sharing
For CFIm68 knockout cell lines: Dr. Alan Engelman (Alan_Engelman@dfci.harvard.edu)
For all other reagent and resources: Dr. Yongsheng Shi (yongshes@uci.edu)
Experimental Model and Subject Details
Cell lines and cell culture conditions
HEK293T cell lines were maintained in Dulbecco’s modified Eagle medium (DMEM) with 10% fetal bovine serum (FBS). Sf9 cells were maintained in SFM 900 III media. Baculovirus for making His-CFIm25/59 and His-CFIm25/568 were generated by using the Baculovirus Expression System (Fisher/Life Technologies).
Method details
In vitro cleavage/polyadenylation assay
All PASs were cloned into pBluescript vector, and the RNA substrates were synthesized by in vitro transcription with T7 polymerase in the presence of [α-32P]-UTP. In vitro coupled cleavage/polyadenylation reactions typically contain 20 cps radiolabled RNA per 10μl reaction, 40% NE, 8.8 mM HEPES (pH 7.9), 44 mM KCl, 0.4 mM DTT, 0.7 mM MgCl2, 1 mM ATP, and 20 mM creatine phosphate. In cleavage reactions, ATP was omitted and 0.2 mM 3′ dATP (Sigma), 2.5% PVA, and 40 mM creatine phosphate were added.
Gel shift assay
[α-32P]-UTP-labeled RNA was incubated with 1mM ATP, 20mM creatine phosphate, 100 ng/μl yeast tRNA and 44% HeLa nuclear extract in 10μl reaction at 30 °C for 20 min. The reactions were cooled on ice and heparin was added to 0.4μg/μl. 6μl of the reaction was resolved on 4% native PAGE in 1x Tris-Glycine running buffer at 100V for 210 min in cold room and visualized by phosphorimaging.
Reporter assay
The PAS sequences to be tested were cloned into the multiple cloning sites in pPASPORT. Reporter constructs were transfected into HEK293T cells using Lipofectamine 2000 (Fisher/Life Technologies). Cells were harvested 2 days post-transfection and the Rluc/Fluc ratio was determined using the Dual Luciferase Assay Kit (Promega). For λN Tethering assay, the λN-CFIm and 2xBoxB-L3-pPASPORT were co-transfected in 293T cells and the luciferase activities were measured by using the same method. A Myc nuclear localization signal sequence (PAAKRVKLD) was added to the C-termini of all truncated proteins to ensure proper nuclear entry.
3xMS2-based RNA affinity purification
10pmol 3MS2-PAS RNA was incubated with 500pmol of MBP-MS2 fusion protein on ice for 30 mins, and then add 1mM ATP, 20mM creatine phosphate, 100 ng/μl yeast tRNA and 200 μl HeLa nuclear extract (total reaction volume: 500 μl) and the reaction mix was incubated at 30 °C for 20 min. The reactions were chilled on ice and heparin was added to 0.4μg/μl. 30 μl pre-washed amylose resin was incubated with the reaction for 1 hour (h) at 4 °C. Beads were washed in Wash Buffer (20mM Hepes-KOH [pH7.9], 100mM KCl, 1mM MgCl2, 1% Triton X-100 and 0.5mM DTT) for 3×10 min, and then the complexes were eluted with 120μl wash buffer plus 12mM maltose at 4 °C for 2×20 min. Eluted proteins were precipitated with acetone at −20 °C overnight. Spin down at 12,000 rpm at 4 °C for 15 min to collect proteins and performed SDS-PAGE and western blotting with Enhanced Chemical Luminescence.
Generation of CFIm59 knockout (KO) cell line
Two pairs of CFIm59 sgRNA (Table S2) were designed using an online tool (http://crispr.mit.edu) and inserted into the px330 vector following the protocol listed online. We transfected 0.5μg px330-sgRNA plasmid in a 24-well plate of 293T cells, and re-seeded the cells in 15cm plates at ~20 cells/plate. When colonies are formed, they were picked and screened by western blotting to identify KO cell lines.
Protein purification
In E. coli: To make GST-RS(CFIm59) and GST-RS(CFIm68) in E. coli, the RS domains from the two proteins were cloned into the multiple cloning sites in pGEX4T-3 and purified using glutathione sepharose per manufacturer’s instructions (GE Healthcare). pET-SRPK1 (a kind gift from Dr. Joseph Adams) was used for producing 6xHis-SPRK1 in E. coli and the protein was purified using Cobalt beads per manufacturer’s instructions (Fisher).
In insect cells (Sf9): Fip1 cDNA was cloned into pFastBac, CFIm25 and CFIm59 or CFIm68 cDNAs were cloned into Multi-Bac vectors. Both Fip1 and CFIm25 had an N-terminal 6xHis tag. The pFastBac and MultiBac constructs were used to produce Bacmids and recombinant baculoviruses using standard procedures. Baculoviruses were used to infect Sf9 cells and these cells were harvested 2 days post-infection. Recombinant proteins were purified with Cobalt beads per manufacturer’s instructions. To make GST-RS(CFIm59) and GST-RS(CFIm68) in Sf9 cells, the whole GST-RS cDNAs were amplified by PCR from pGEX constructs and cloned into pFastBac, which were used to produce these proteins in Sf9 cells as described above.
Kinase and phosphatase treatment
2μg GST-CFIm59 RS and GST-CFIm68 RS purified from E. coli were phosphorylated with 6xhis-SRPK1 in presence of 1mM ATP, 50mM MgCl2 at 37 °C for 30min. GST-CFIm59 RS and GST-CFIm68 RS purified from sf9 cells were treated with 2 units Alkaline Phosphatase, Calf Intestinal (CIP) at 37 °C for 30min. After the treatment of SRPK1 and CIP, the GST proteins were purified by incubating with glutathione beads at 4 °C for 30min and then washed with buffer D300 (20mM HEPES-KOH [pH7.9], 300mM KCl, 1mM MgCl2, 0.2% NP40, proteinase inhibitor cocktail) for 3 times and buffer D100 (the same as D300 except that 100mM KCl was used) once.
Protein-protein interaction assay
For Fip1-RD peptide pull-down assay, 0.5μg 6xHis-CFIm25-59 or 6xHis-CFIm25-68 was incubated with 200ng Fip1-RD or RA peptides (synthesized by Genscript) immobilized on Streptavidin beads in D100 buffer (20mM HEPES [pH 7.9], 100mM NaCl, 1mM MgCl2, 0.2mM EDTA and 100x proteinase inhibitor) at 4 °C for 2h. The beads were washed with buffer D300 (0.2% NP40, 100x proteinase inhibitor) for 3 times and buffer D100 once.1xSDS loading buffer was added to the beads and boiled. For GST pulldown assays, 2μg GST- RS(CFIm59) or GST-RS(CFIm68) protein was pulled-down with purified His-Fip1 protein from Sf9 cells, E. coli or in vitro translated Fip1 protein. Binding reaction was made in D100 buffer. 2μg GST-CFIm59 RS and GST-CFIm68 RS purified from E. coli were phosphorylated with His-SRPK in presence of 1mM ATP, 50mM MgCl2 at 37 °C for 30min. GST-CFIm59 RS and GST-CFIm68 RS purified from sf9 cells were treated with 2 units Alkaline Phosphatase, Calf Intestinal (CIP) at 37°C for 30min. After the treatment of SRPK and CIP, removed those protein from the reaction containing phosphorylated GST-CFIm59 RS and GST-CFIm68 RS by incubating with GST beads at 4 °C for 30min and then washed with buffer D300 (0.2% NP40, 100x proteinase inhibitor) for 3 times and with buffer D100 once.
PAS-seq
Total RNA was extracted with Trizol as per manual (Life technologies), 10 μg total RNA was fragmented with fragmentation reagent (Ambion) at 70 °C for 10 minutes followed by precipitation with ethanol. After centrifugation, RNA was dissolved and Reverse transcription was performed with PASSEQ7-2 RT oligo:[phos]NNNNAGATCGGAAGAGCGTCGTGTTCGGATCCATTAGGATCCGAGACGTGTGCTCTTCCGATCTTTTTTTTTTTTTTTTTTTT[V-Q] and Superscript III. cDNA was recovered by ethanol precipitation and centrifugation. 120–200 nucleotides of cDNA was gel-purified and eluted from 8% Urea-PAGE. Recovered cDNA was circularized with Circligase™ II (Epicentre) at 60 °C overnight. Buffer E (Promega) was added in cDNA and heated at 95 °C for 2 minutes, and then cool to 37 °C slowly. Circularized cDNA was linearized by BamH I (Promega). cDNA was collected by centrifugation after ethanol precipitation. PCR was carried out with primers PE1.0 and PE2.0 containing index. Around 200 bp of PCR products was gel-purified and submitted for sequencing (single read 100 nucleotides).
Quantification and statistical analysis
PAS-Seq Data Analysis
From the raw PAS-seq reads, first those with no polyA tail (less than 15 consecutive “A”s) were filtered out. The rest were trimmed and mapped to hg19 genome using TopHat (v2.1.0) with -g 1 and strand specificity parameters (Kim et al., 2013). If 6 consecutive “A”s or more than 7 “A”s were observed in the 10 nucleotides downstream of poly(A) (PAS) for a reported alignment, it was marked as a possible internal priming event and that read was removed. The bigwig files were then generated for the remaining reads using deepTools (v2.4) with “normalizeUsingRPKM” and “ignoreDuplicates” parameters (Ramirez et al., 2016).
Next, the locations of 3′ ends of the aligned reads were extracted and those in 40nt of each other were merged into one to provide a list of potential poly(A) sites for human. This list was then annotated based on the canonical transcripts for known genes. The final count table was created using the reads with their 3′ ends in −40nt to 40nt of these potential PASs.
PASs with significant changes in different experimental conditions were identified using diffSpliceDGE and topSpliceDGE from edgeR package(v3.8.5) (Robinson et al., 2010). This pipeline first models the PAS read counts, then compared the log fold change of each PAS to the log fold change of the entire gene. This way, these functions, primarily used to find differential exon usage, generated a list of sites with significant difference between our PAS-seq samples. From this list, those with a FDR value less than 0.05 and more than 15% difference in the ratio of PAS read counts to gene read counts between samples were kept, and finally for each gene the top two were chosen based on P-value and marked distal or proximal based on their relative location on gene.
For the genes with significantly different APA profiles (target genes), the log2 of ratio of read counts in distal site to the read counts in proximal site was calculated and illustrated as a heatmap in Fig. 6A with heatmap.2 in R (v3.1.0). The heatmap was hierarchically clustered using Pearson correlation of the genes profiles in different experiments.
The sequence around distal and proximal PASs were extracted using BEDTools (v2.25.0) (Quinlan and Hall, 2010) for alternatively polyadenylated sites and the same number of sites with no significant changes between control and experiment chosen randomly. UGUA distributions were extracted from these sequences in the format of a histogram with 20 bps bin size. The smooth underlying function of the normalized histogram was then generated using interp1d class in SciPy library (http://www.scipy.org/) and then visualized as seen in Fig. 6C. Distribution of UGUA and A(A/U)UAAA used in Fig. 1A were generated following the same process on random regions besides the sequences around poly(A) sites.
PAR-CLIP Data Acquisition and Analysis
We normalized the CFI68 or CFI59 PAR-CLIP signals from GSE37401 (Martin et al. 2012) at proximal and distal PASs for target and non-target genes to count the binding frequency per transcript. For proximal PAS, CLIP read counts was divided by the PAS-seq read counts of that PAS plus all downstream PASs, and for distal PAS the CLIP read counts was divided by the PAS-seq read counts of the distal PAS. Wig files were converted to bigwigs, and the CLIP signals on −100nt to 100nt region around GSE37401d poly(A)s were extracted by deepTools (v2.4) (Ramírez et al. 2016) using those bigwig files, separately for each strand. Signals were combined, normalized, and averaged in Python. The averaged curve for each set of 201nt intervals, were then scaled by their own total coverage for comparison of distributions. The final plots are illustrated in Fig. 6D and Supplemental Figure S9.
General Analysis
The computational analyses and visualization if not specified otherwise, were done in Python 2.7. Where necessary, conversion between BAM and BED files were done using BEDTools (v2.25.0) (Quinlan and Hall, 2010) and BAM files were sorted or indexed via SAMtools (v1.1) (Li et al., 2009). Kolmogorov–Smirnov (K-S) test, implemented in Scipy library, was used in multiple cases (Fig. 6C, 6D, and S9) to determine if two samples are from the same distribution. The generated p-value quantifies the significance of the observations coming from different distributions.
Data and Software Availability
PAS -seq data have been deposited to the GEO database (accession number: GSE101871).
Raw image data have been deposited to Mendeley Data (http://dx.doi.org/10.17632/r23kcs7s8n.1)
Supplementary Material
Table S1. Sequences and plasmids used in this study, Related to Figure 1–6, Figure S1 and S6.
Table S2. Oligoes and plasmids used in this study, Related to Figure 1–6, Figure S1 and S2.
Highlights.
UGUA is a position-dependent enhancer for mRNA 3′ processing
CFIm is an enhancer-dependent activator of mRNA 3′ processing
The activator function of CFIm is mediated by its RS-like domains
mRNA 3′ processing and splicing share a common activation mechanism
Acknowledgments
We would like to thank Drs. Serena Chan and Joe Adams for providing reagents, Dr. Jin-Kwang Kim for help with graphics, and UCI GHTF for sequencing. This study was supported by the following grants: NIH GM090056, CA17488 and American Cancer Society RSG-12-186 to Y.S.; NIH AI052014 to A.N.E..
Footnotes
AUTHOR CONTRIBUTIONS
Y.Z., X.W., and Y.S. conceived and designed the experiments. Y.Z. and X.W performed the majority of the experiments. E. F., X.X., F.Q., and K. H. contributed to data analyses. J.J. contributed to protein expression. G.S., and A.E. provided reagents and technical assistance. Y.S. wrote the paper with input from all authors.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baron-Benhamou J, Gehring NH, Kulozik AE, Hentze MW. Using the lambdaN peptide to tether proteins to RNAs. Methods Mol Biol. 2004;257:135–154. doi: 10.1385/1-59259-750-5:135. [DOI] [PubMed] [Google Scholar]
- Braunschweig U, Gueroussov S, Plocik AM, Graveley BR, Blencowe BJ. Dynamic integration of splicing within gene regulatory pathways. Cell. 2013;152:1252–1269. doi: 10.1016/j.cell.2013.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown KM, Gilmartin GM. A mechanism for the regulation of pre-mRNA 3′ processing by human cleavage factor Im. Mol Cell. 2003;12:1467–1476. doi: 10.1016/s1097-2765(03)00453-2. [DOI] [PubMed] [Google Scholar]
- Cao W, Garcia-Blanco MA. A serine/arginine-rich domain in the human U1 70k protein is necessary and sufficient for ASF/SF2 binding. J Biol Chem. 1998;273:20629–20635. doi: 10.1074/jbc.273.32.20629. [DOI] [PubMed] [Google Scholar]
- Chan S, Choi EA, Shi Y. Pre-mRNA 3′-end processing complex assembly and function. Wiley Interdiscip Rev RNA. 2011;2:321–335. doi: 10.1002/wrna.54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarino VA, Alcott CE, Chen CA, Chaudhury A, Gillentine MA, Rosenfeld JA, Parikh S, Wheless JW, Roeder ER, Horovitz DD, et al. NUDT21-spanning CNVs lead to neuropsychiatric disease and altered MeCP2 abundance via alternative polyadenylation. Elife. 2015;4 doi: 10.7554/eLife.10782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graveley BR. Sorting out the complexity of SR protein functions. RNA. 2000;6:1197–1211. doi: 10.1017/s1355838200000960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber AR, Martin G, Keller W, Zavolan M. Cleavage factor Im is a key regulator of 3′ UTR length. RNA Biol. 2012;9:1405–1412. doi: 10.4161/rna.22570. [DOI] [PubMed] [Google Scholar]
- Hudson SW, McNally MT. Juxtaposition of two distant, serine-arginine-rich protein-binding elements is required for optimal polyadenylation in Rous sarcoma virus. J Virol. 2011;85:11351–11360. doi: 10.1128/JVI.00721-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang HW, Park CY, Goodarzi H, Fak JJ, Mele A, Moore MJ, Saito Y, Darnell RB. PAPERCLIP Identifies MicroRNA Targets and a Role of CstF64/64tau in Promoting Non-canonical poly(A) Site Usage. Cell Rep. 2016;15:423–435. doi: 10.1016/j.celrep.2016.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanopka A, Muhlemann O, Petersen-Mahrt S, Estmer C, Ohrmalm C, Akusjarvi G. Regulation of adenovirus alternative RNA splicing by dephosphorylation of SR proteins. Nature. 1998;393:185–187. doi: 10.1038/30277. [DOI] [PubMed] [Google Scholar]
- Kinoshita E, Kinoshita-Kikuta E, Koike T. Separation and detection of large phosphoproteins using Phos-tag SDS-PAGE. Nat Protoc. 2009;4:1513–1521. doi: 10.1038/nprot.2009.154. [DOI] [PubMed] [Google Scholar]
- Kohtz JD, Jamison SF, Will CL, Zuo P, Luhrmann R, Garcia-Blanco MA, Manley JL. Protein-protein interactions and 5′-splice-site recognition in mammalian mRNA precursors. Nature. 1994;368:119–124. doi: 10.1038/368119a0. [DOI] [PubMed] [Google Scholar]
- Lackford B, Yao C, Charles GM, Weng L, Zheng X, Choi EA, Xie X, Wan J, Xing Y, Freudenberg JM, et al. Fip1 regulates mRNA alternative polyadenylation to promote stem cell self-renewal. EMBO J. 2014 doi: 10.1002/embj.201386537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, You B, Hoque M, Zheng D, Luo W, Ji Z, Park JY, Gunderson SI, Kalsotra A, Manley JL, Tian B. Systematic profiling of poly(A)+ transcripts modulated by core 3′ end processing and splicing factors reveals regulatory rules of alternative cleavage and polyadenylation. PLoS Genet. 2015;11:e1005166. doi: 10.1371/journal.pgen.1005166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou H, Neugebauer KM, Gagel RF, Berget SM. Regulation of alternative polyadenylation by U1 snRNPs and SRp20. Mol Cell Biol. 1998;18:4977–4985. doi: 10.1128/mcb.18.9.4977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin G, Gruber AR, Keller W, Zavolan M. Genome-wide Analysis of Pre-mRNA 3′ End Processing Reveals a Decisive Role of Human Cleavage Factor I in the Regulation of 3′ UTR Length. Cell Rep. 2012;1:753–763. doi: 10.1016/j.celrep.2012.05.003. [DOI] [PubMed] [Google Scholar]
- Masamha CP, Xia Z, Yang J, Albrecht TR, Li M, Shyu AB, Li W, Wagner EJ. CFIm25 links alternative polyadenylation to glioblastoma tumour suppression. Nature. 2014;510:412–416. doi: 10.1038/nature13261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millevoi S, Loulergue C, Dettwiler S, Karaa SZ, Keller W, Antoniou M, Vagner S. An interaction between U2AF 65 and CF I(m) links the splicing and 3′ end processing machineries. Embo J. 2006;25:4854–4864. doi: 10.1038/sj.emboj.7601331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misra A, Ou J, Zhu LJ, Green MR. Global Promotion of Alternative Internal Exon Usage by mRNA 3′ End Formation Factors. Mol Cell. 2015;58:819–831. doi: 10.1016/j.molcel.2015.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller-McNicoll M, Botti V, de Jesus Domingues AM, Brandl H, Schwich OD, Steiner MC, Curk T, Poser I, Zarnack K, Neugebauer KM. SR proteins are NXF1 adaptors that link alternative RNA processing to mRNA export. Genes Dev. 2016;30:553–566. doi: 10.1101/gad.276477.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nilsen TW, Graveley BR. Expansion of the eukaryotic proteome by alternative splicing. Nature. 2010;463:457–463. doi: 10.1038/nature08909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nojima T, Gomes T, Grosso ARF, Kimura H, Dye MJ, Dhir S, Carmo-Fonseca M, Proudfoot NJ. Mammalian NET-Seq Reveals Genome-wide Nascent Transcription Coupled to RNA Processing. Cell. 2015;161:526–540. doi: 10.1016/j.cell.2015.03.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad J, Colwill K, Pawson T, Manley JL. The protein kinase Clk/Sty directly modulates SR protein activity: both hyper-and hypophosphorylation inhibit splicing. Mol Cell Biol. 1999;19:6991–7000. doi: 10.1128/mcb.19.10.6991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rappsilber J, Ryder U, Lamond AI, Mann M. Large-scale proteomic analysis of the human spliceosome. Genome Res. 2002;12:1231–1245. doi: 10.1101/gr.473902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruegsegger U, Beyer K, Keller W. Purification and characterization of human cleavage factor Im involved in the 3′ end processing of messenger RNA precursors. J Biol Chem. 1996;271:6107–6113. doi: 10.1074/jbc.271.11.6107. [DOI] [PubMed] [Google Scholar]
- Ruegsegger U, Blank D, Keller W. Human pre-mRNA cleavage factor Im is related to spliceosomal SR proteins and can be reconstituted in vitro from recombinant subunits. Mol Cell. 1998;1:243–253. doi: 10.1016/s1097-2765(00)80025-8. [DOI] [PubMed] [Google Scholar]
- Sanford JR, Bruzik JP. Developmental regulation of SR protein phosphorylation and activity. Genes Dev. 1999;13:1513–1518. doi: 10.1101/gad.13.12.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y. Alternative polyadenylation: new insights from global analyses. RNA. 2012;18:2105–2117. doi: 10.1261/rna.035899.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Di Giammartino DC, Taylor D, Sarkeshik A, Rice WJ, Yates JR, 3rd, Frank J, Manley JL. Molecular architecture of the human pre-mRNA 3′ processing complex. Molecular cell. 2009;33:365–376. doi: 10.1016/j.molcel.2008.12.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi Y, Manley JL. The end of the message: multiple protein-RNA interactions define the mRNA polyadenylation site. Genes & development. 2015;29:889–897. doi: 10.1101/gad.261974.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sowd GA, Serrao E, Wang H, Wang W, Fadel HJ, Poeschla EM, Engelman AN. A critical role for alternative polyadenylation factor CPSF6 in targeting HIV-1 integration to transcriptionally active chromatin. Proc Natl Acad Sci U S A. 2016;113:E1054–1063. doi: 10.1073/pnas.1524213113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tacke R, Manley JL. Determinants of SR protein specificity. Curr Opin Cell Biol. 1999;11:358–362. doi: 10.1016/S0955-0674(99)80050-7. [DOI] [PubMed] [Google Scholar]
- Tian B, Manley JL. Alternative polyadenylation of mRNA precursors. Nat Rev Mol Cell Biol. 2017;18:18–30. doi: 10.1038/nrm.2016.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z, Burge CB. Splicing regulation: from a parts list of regulatory elements to an integrated splicing code. RNA. 2008;14:802–813. doi: 10.1261/rna.876308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng L, Li Y, Xie X, Shi Y. Poly(A) code analyses reveal key determinants for tissue-specific mRNA alternative polyadenylation. RNA. 2016;22:813–821. doi: 10.1261/rna.055681.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu JY, Maniatis T. Specific interactions between proteins implicated in splice site selection and regulated alternative splicing. Cell. 1993;75:1061–1070. doi: 10.1016/0092-8674(93)90316-i. [DOI] [PubMed] [Google Scholar]
- Xiao SH, Manley JL. Phosphorylation of the ASF/SF2 RS domain affects both protein-protein and protein-RNA interactions and is necessary for splicing. Genes Dev. 1997;11:334–344. doi: 10.1101/gad.11.3.334. [DOI] [PubMed] [Google Scholar]
- Yang Q, Coseno M, Gilmartin GM, Doublie S. Crystal structure of a human cleavage factor CFI(m)25/CFI(m)68/RNA complex provides an insight into poly(A) site recognition and RNA looping. Structure. 2010a;19:368–377. doi: 10.1016/j.str.2010.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Gilmartin GM, Doublie S. Structural basis of UGUA recognition by the Nudix protein CFI(m)25 and implications for a regulatory role in mRNA 3′ processing. Proc Natl Acad Sci U S A. 2010b;107:10062–10067. doi: 10.1073/pnas.1000848107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q, Gilmartin GM, Doublie S. The structure of human cleavage factor I(m) hints at functions beyond UGUA-specific RNA binding: a role in alternative polyadenylation and a potential link to 5′ capping and splicing. RNA Biol. 2011;8:748–753. doi: 10.4161/rna.8.5.16040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao C, Biesinger J, Wan J, Weng L, Xing Y, Xie X, Shi Y. Transcriptome-wide analyses of CstF64-RNA interactions in global regulation of mRNA alternative polyadenylation. Proc Natl Acad Sci U S A. 2012;109:18773–18778. doi: 10.1073/pnas.1211101109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao J, Hyman L, Moore C. Formation of mRNA 3′ ends in eukaryotes: mechanism, regulation, and interrelationships with other steps in mRNA synthesis. Microbiol Mol Biol Rev. 1999;63:405–445. doi: 10.1128/mmbr.63.2.405-445.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong XY, Wang P, Han J, Rosenfeld MG, Fu XD. SR proteins in vertical integration of gene expression from transcription to RNA processing to translation. Mol Cell. 2009;35:1–10. doi: 10.1016/j.molcel.2009.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Licklider LJ, Gygi SP, Reed R. Comprehensive proteomic analysis of the human spliceosome. Nature. 2002;419:182–185. doi: 10.1038/nature01031. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Sequences and plasmids used in this study, Related to Figure 1–6, Figure S1 and S6.
Table S2. Oligoes and plasmids used in this study, Related to Figure 1–6, Figure S1 and S2.