

Abstract
DDX3 is a DEAD box RNA helicase with oncogenic properties. RK-33 is developed as a small molecule inhibitor of DDX3 and showed potent radiosensitizing activity in preclinical tumor models. This study aimed to assess DDX3 as a target in breast cancer and to elucidate how RK-33 exerts its anti-neoplastic effects. High DDX3 expression was present in 35% of breast cancer patient samples and correlated with markers of aggressiveness and shorter survival. With a quantitative proteomics approach, we identified proteins involved in the mitochondrial translation and respiratory electron transport pathways to be significantly downregulated after RK-33 or DDX3 knockdown. DDX3 localized to the mitochondria and DDX3 inhibition with RK-33 reduced mitochondrial translation. As a consequence, oxygen consumption rates and intracellular ATP concentrations decreased and reactive oxygen species (ROS) increased. RK-33 antagonized the increase in oxygen consumption and ATP production observed after exposure to ionizing radiation and reduced DNA repair. Overall, we conclude that DDX3 inhibition with RK-33 causes radiosensitization in breast cancer through inhibition of mitochondrial translation, which results in reduced oxidative phosphorylation capacity and increased ROS levels, culminating in a bioenergetic catastrophe.
Keywords: DDX3, breast cancer, mitochondrial translation, oxidative phosphorylation, radiosensitizer
Introduction
DEAD box RNA helicases are a family of proteins with ATPase-dependent helicase activity, which allows for the restructuring of complex RNA structures and unwinding of double-stranded RNA1. DDX3, also known as DDX3X, is an RNA helicase that has been associated with several cytosolic steps of mRNA processing2. Recent functional studies have demonstrated that DDX3 plays an oncogenic role in the development of breast3 and several other types of cancer4–7. DDX3 was found to have anti-apoptotic properties8, 9 and to play a role in cell cycle progression4,5, migration3, 10 and invasion11, 12. However, the oncogenic role of DDX3 in breast cancer remains to be validated in patient samples.
To target DDX3 for cancer treatment, a small molecule inhibitor, RK-33, was recently developed13. RK-33 is designed to fit into the ATP-binding pocket of DDX3 and thereby inactivate it. It was shown to selectively bind DDX3 over other DEAD box RNA helicases and to potently inhibit RNA helicase activity4. Furthermore, RK-33 was found to have selective anti-cancer activity in mouse models, both as a monotherapy6 and as a radiosensitizer4, 7. However, the exact working mechanisms behind the action of RK-33 in cancer remain to be elucidated.
Using a quantitative proteomics approach, we here identified mitochondrial translation as a potential target of RK-33. Mitochondria have their own ribosomal machinery, responsible for translating the thirteen genes that are located on the mitochondrial genome, which all play a role in facilitating oxidative phosphorylation (OXPHOS). Increasing evidence indicates that cancer cells are dependent on upregulation of OXPHOS when encountering cellular stressors, like chemotherapy14, 15, or during metastasis16. Irradiated cells also increase oxygen consumption and mitochondrial ATP production17 allowing for more efficient repair of radiation induced DNA damage18. Since we have previously shown that RK-33 has radiosensitizing abilities, we hypothesized that this might be due to RK-33 inhibiting mitochondrial translation and thereby limiting the cellular capacity to upregulate OXPHOS. In this work, we evaluate DDX3 as a target in breast cancer and investigated the effect of DDX3 inhibition on the bioenergetic profile of breast cancer cells. We show that RK-33 radiosensitizes breast cancer cells through inhibition of mitochondrial translation, resulting in reduced OXPHOS and increased production of reactive oxygen species (ROS).
Results
DDX3 in human breast cancer samples
To evaluate DDX3 as a target in breast cancer, we assessed DDX3 expression by immunohistochemistry in 366 breast cancer patient samples (Figure 1A). As shown in Table 1 high cytoplasmic DDX3 expression was present in 127 cases (35%) and correlated with slightly higher age at diagnosis (p = 0.042), higher mitotic index (MAI; p = 0.002), ductal histological type (p = 0.003), higher grade (p = 0.002) and negative estrogen (p = 0.005), progesterone (p = 0.007) and positive HER2 receptor (p = 0.001). In addition, tumors with high DDX3 expression more frequently had a basal like (p = 0.032) or HER2 driven (p = 0.002) molecular subtype. High DDX3 expression was also associated with worse outcome in breast cancer patients. The five-year survival rate was 78.9% in patients with high DDX3 expressing tumors, as compared to 87.4% in those with low DDX3 expression (Hazard ratio (HR) 2.01, p = 0.042; Figure 1B). In a Cox regression model including tumor size, lymph node status, grade, mitotic activity index (MAI) and DDX3 expression, only lymph node status was an independent prognostic factor (Supplementary Table 1). Analysis of multiplicative interaction terms indicated that both MAI and lymph node status acted as effect modifiers and the relation between DDX3 and survival was most strong in tumors with low MAI and positive lymph node status (Supplementary Figure 1A & B). Together, these findings indicate that DDX3 associates with markers of aggressive disease in breast cancer patient samples, making it a suitable target for further evaluation.
Figure 1. DDX3 is a therapeutic target in breast cancer.
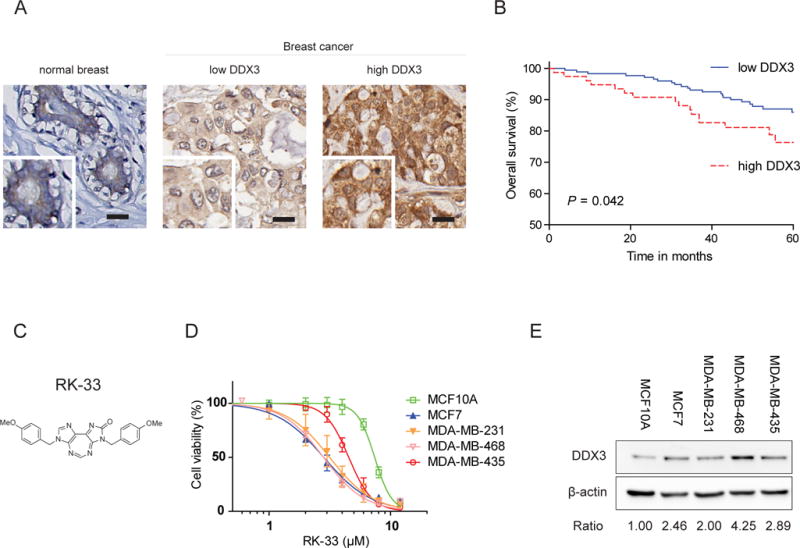
A. Example of low DDX3 cytoplasmic expression in normal breast cells and low and high cytoplasmic DDX3 expression assessed by immunohistochemistry in breast cancer patient samples. Scale bar indicates 20 μm. B. Kaplan-Meier curve plot showing worse overall survival for breast cancer patients with high DDX3 expression. N = 250. P-value calculated by Breslow test. C. Molecular structure of the small molecule inhibitor of DDX3, RK-33 D. MTS assay showing RK-33 cytotoxicity in a normal breast cell line (MCF10A) and four cancer cell lines (MCF7, MDA-MB-231, MDA-MB-468 & MDA-MB-435). Graphs represent mean ± SD of 3 biological replicates. E. Immunoblot showing the relative DDX3 expression in cell lines.
Table 1.
Clinicopathological correlations of cytoplasmic DDX3 expression in invasive breast cancer
| Cytoplasmic DDX3 | |||
|---|---|---|---|
| Low | High | P-value | |
| N | 239 | 127 | |
| Mean age (range) | 59.7 (28–88) | 62.4 (34–87) | 0.042a |
| Mean tumor size (range) | 2.4 (0.6–8.0) | 2.6 (0.2–10.0) | 0.233a |
| Mitotic index (mitosis/2mm2) | 15.0 (0–151) | 22.6 (0–131) | 0.002a |
| Histological type | |||
| Ductal | 78.2% (187) | 89.7% (113) | 0.003c |
| Lobular | 12.1% (29) | 2.4% (3) | |
| Other | 9.6% (23) | 7.9% (10) | |
| Grade | |||
| 1 | 23.5% (55) | 10.2% (13) | 0.002b |
| 2 | 37.6% (88) | 34.6% (44) | |
| 3 | 38.9% (91) | 55.1% (70) | |
| Estrogen receptor | |||
| Positive | 82.4% (197) | 69.3% (88) | 0.005b |
| Negative | 17.6% (42) | 30.7% (39) | |
| Progesterone receptor | |||
| Positive | 64.3% (153) | 49.6% (63) | 0.007b |
| Negative | 35.7% (85) | 50.4% (64) | |
| HER2 | |||
| Positive | 7.1% (17) | 18.9% (24) | 0.001b |
| Negative | 92.9% (222) | 81.1% (103) | |
| Lymph node status | |||
| Positive | 51.8% (118) | 49.2% (60) | 0.646b |
| Negative | 48.2% (110) | 50.8% (62) | |
| Molecular subtype | |||
| Luminal | 84.5% (202) | 70.1% (80) | |
| Basal like | 13.0% (31) | 20.5% (26) | 0.032d |
| HER2-driven | 2.5% (6) | 9.4% (12) | 0.002d |
Abbreviation: HER2, human epidermal growth factor receptor 2.
t-test.
X2-test.
Fisher’s exact test.
X2-test as compared to luminal.
The efficacy of the DDX3 inhibitor RK-33 in breast cancer cell lines
The efficacy of the DDX3 inhibitor RK-33 was evaluated in several cancer cell lines and the normal breast cell line MCF10A by an MTS assay (Figure 1C–D). The sensitivity to RK-33 was higher in cancer cell lines (IC50 2.8–4.5 μM) when compared to MCF10A (IC50 7.4 μM), with the latter also expressing lower amounts of DDX3 (Figure 1E). To ensure that this difference in potency reflected viable cell numbers and not just alterations in metabolism, we confirmed this finding in a calcein green based cytotoxicity assay (Supplementary Figure 1C).
DDX3 inhibition causes reduced expression of proteins involved in mitochondrial translation and OXPHOS
To further evaluate the effect of DDX3 inhibition, changes in protein expression levels were assessed after RK-33 or shDDX3 treatment (Figure 2A) in the metastatic cancer cell line MDA-MB-435 with quantitative proteomics. 666 identified proteins were significantly altered after RK-33 and 770 after shDDX3 (Supplementary Figure 2A and Supplementary Table 2). 186 proteins had altered protein expression after both treatments, which was 1.7 times more than could be expected based on chance (p <0.001; Supplementary Figure 1B). Gene set enrichment analysis in the Reactome database19 identified the “mitochondrial translation” (p < 0.001) and “respiratory electron transport” (p <0.001) pathways among the most enriched after RK-33 treatment. The proteins significantly altered by shDDX3 were also enriched for these pathways (Figure 2B). In addition, network analysis using the STRING database20 revealed tight networks of mitochondrial ribosome proteins and proteins that were part of the electron transport chain (ETC) complexes to be downregulated after both RK-33 and shDDX3 (Figure 2C). We identified two out of the thirteen proteins that the mitochondrial genome encodes for. Of which the complex IV protein MT-COII was among the significantly altered proteins (downregulated by −1.285 times, q = 0.001) and MT-NDI had a borderline significant fold change (−1.149, q = 0.009). Interestingly, the remainder of the ETC complex proteins was encoded on the nuclear genome, but belonged to OXPHOS complex I and IV, which are the ETC complexes that contain most mitochondrially encoded proteins. Since mutation or downregulation of a single subunit in an OXPHOS complex can cause downregulation of the entire supercomplex it belongs21, it is not surprising that we observed downregulation of nuclear encoded OXPHOS proteins as well.
Figure 2. DDX3 inhibition results in reduced expression of proteins involved in mitochondrial translation.
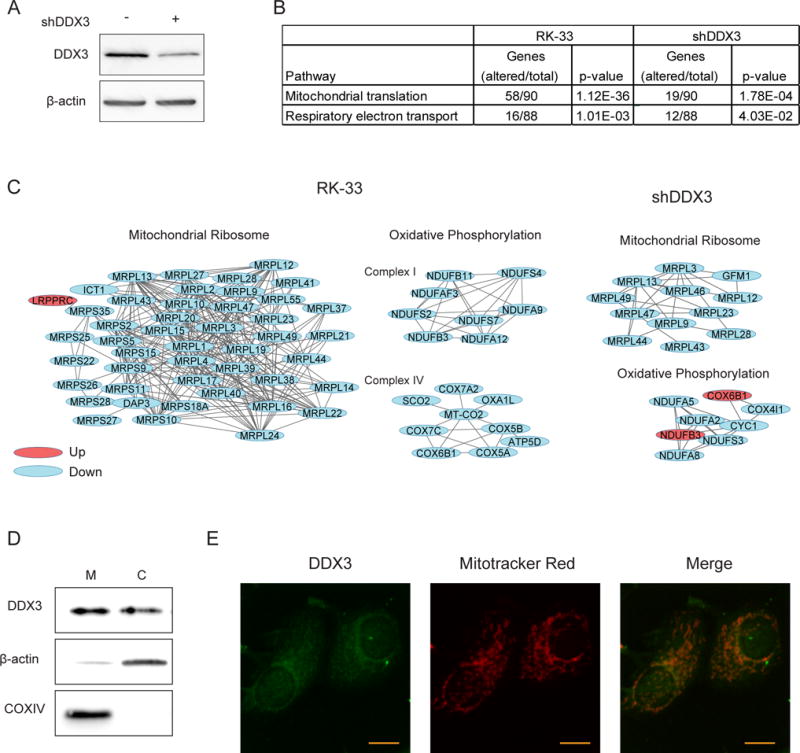
A. Immunoblot showing the DDX3 expression before and after transduction with shDDX3 in MDA-MB-435 cells. B. Table showing two overrepresented Reactome pathways identified by gene set enrichment analysis of significantly altered proteins in MDA-MB-435 cells after 24 hours exposure to 4.5 μM RK-33 or after shDDX3 transduction. C. Protein networks identified by string network analysis in the group of significantly altered proteins explained under B. D. Immunoblot showing the DDX3 expression in mitochondrial (M) and cytoplasmic (C) fractions of MCF7. E. 2-foton microscopy image of MCF7 immunofluorescently stained for DDX3 and labeled with mitotracker red. Scale bar indicates 5 μm. All experiments have been replicated a minimum of two independent times.
DDX3 localizes to the mitochondria
The potential involvement of DDX3 in mitochondrial translation made us evaluate whether DDX3 localizes to the mitochondria. DDX3 was identified in a mitochondrial extract, and also to a lesser extent in the free cytosolic fraction of MCF7 (Figure 2D). Even higher levels of mitochondrial DDX3 were observed in MDA-MB-231 (Supplementary Figure 2C). In addition, we found DDX3 to colocalize with mitotracker-red labeled mitochondria by immunofluorescence (Figure 2E).
RK-33 causes mitochondrial translation inhibition
To assess whether treatment with RK-33 resulted in inhibition of mitochondrial translation, we performed an S35-methionine pulse-labeling experiment in the presence of the cytoplasmic translation inhibitor emetine. As shown in Figure 3A, two hours of RK-33 treatment resulted in potent inhibition of S35-methionine incorporation, indicating a block of mitochondrial translation. A reduction of nascent (newly synthesized) mitochondrial proteins was observed to a lesser extent, in the more resistant MCF10A normal breast cell line. Equal loading of protein was insured by Coomassie blue staining of the gel (Supplementary Figure 3A). In addition, immunoblotting showed that low mitochondrial translation rates also significantly reduced total expression levels of the OXPHOS complexes in breast cancer cell lines after RK-33 treatment (Figure 3B–C) and shDDX3 (supplementary Figure 3B). Specifically, a decrease in COX II expression, which is mitochondrially encoded and translated, was observed in MCF7 (fold change 0.67; p = 0.017) and MDA-MB-231 (fold change 0.52; p = 0.012). It is not surprising that multiple OXPHOS complexes are downregulated as a result of reduced translation of mitochondrially encoded components, since the stability of these complexes is highly dependent on single components and mutations in one gene affects expression of the whole complex it belongs to and even other OXPHOS complexes22.
Figure 3. RK-33 reduces oxidative phosphorylation by blocking mitochondrial translation.
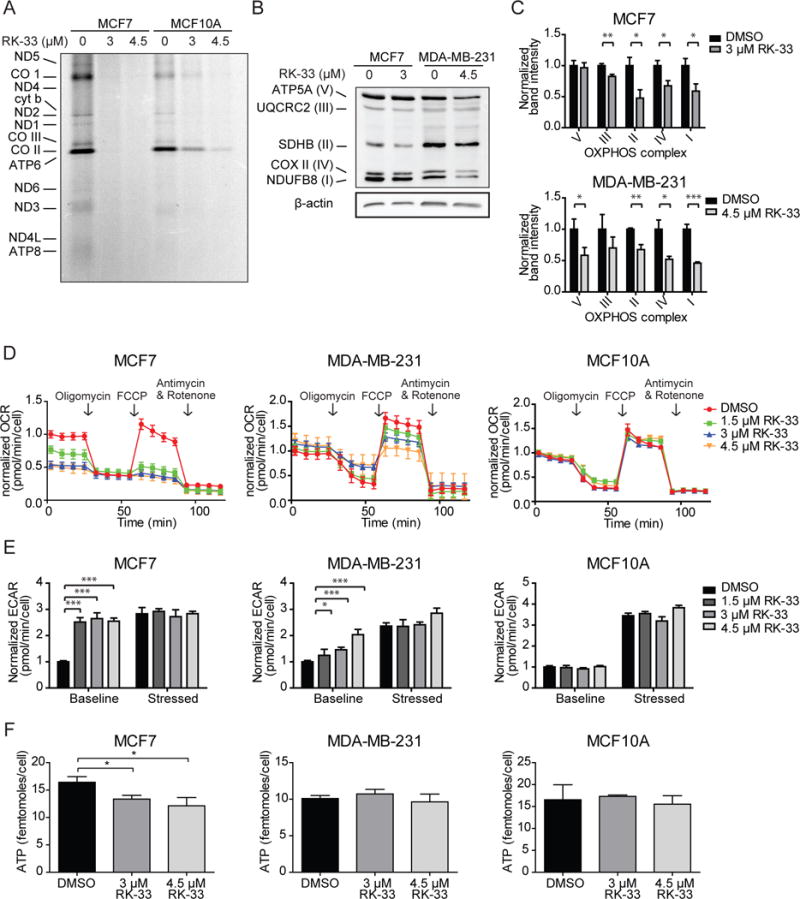
A. Autoradiograph showing the effect of two hours RK-33 exposure on mitochondrial translation, measured by a S35-methionine pulse labeling experiment. B. Immunoblot showing the expression of OXPHOS complexes after twelve hours of RK-33 exposure. C. Bar graphs showing the OXPHOS complex band intensities normalized for β-actin after twelve hours exposure to RK-33. Graphs represent mean ± SD of 3 biological replicates. D. Normalized oxygen consumption rates (OCR) per cell measured by a Seahorse assay after twelve hours exposure to RK-33. Graphs represent mean ± SD of 6 biological replicates. E. Extracellular acidification rates (ECAR) as measured by a Seahorse assay after twelve hours exposure to RK-33 at baseline and stressed (after oligomycin and FCCP) conditions. Graphs represent mean ± SD of 6 biological replicates. F. Intracellular ATP concentrations after twelve hours exposure to RK-33. Graphs represent mean ± SD of 2 biological replicates. *p < 0.05, **p < 0.01, ***p < 0.001. P-values were calculated by a Student’s t-test. All experiments have been replicated a minimum of two independent times.
RK-33 causes a bioenergetic shortage by reducing oxidative phosphorylation
Next we evaluated whether expression of OXPHOS complexes resulted in decreased oxygen consumption rates (OCR), as a measure of oxidative phosphorylation enzymatic activity. As shown in Figure 3D, basal respiration (fold change range 0.50–0.70; p <0.001) and maximum respiration rates (fold change range 0.32–0.44; p <0.001) were markedly reduced in MCF7 after 12 hours of RK-33 exposure. This result was confirmed by measuring OCR with a Clark’s electrode in both MCF-7 (fold change 0.61; p = 0.077, Supplementary Figure 3B) and MDA-MB-435 (fold change 0.78; p = 0.008). In MDA-MB-231 no reduction in basal OCR was observed, but incomplete decline of OCR after oligomycin indicated an increased proton leak (fold change range 2.28–3.26; p = 0.002) and reduced ATP production (fold change range 0.90–0.64; p = 0.001). In addition, maximum respiration rates after Carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) were also reduced (fold change range 0.79–0.48; p < 0.001). No changes in OCR were observed after RK-33 treatment in MCF10A cells. At this time point less than ten percent of cells were found to be apoptotic by Annexin-PI flow cytometry (Supplementary Figure 3C). As was previously reported4, RK-33 does not cause a reduction in OCR immediately after addition (Supplementary Figure 3D). As seen in Figure 2E, a compensatory increase in the extracellular acidification rate (ECAR), indicative of increased glycolysis, could be observed in MCF7 (fold change 2.55, p <0.001) and MDA-MB-231 12 hours after RK-33 treatment (fold change range 1.25–2.04; p = 0.031). In MCF10A, ECAR levels were unaffected. A significant drop in intracellular ATP levels was observed after exposure to RK-33 in MCF7 (fold change range 0.74–0.81; p = 0.015; Figure 3F). Increased glycolysis adequately compensated ATP levels in MDA-MB-231 and no change was observed in MCF10A. MCF7 were more sensitive to DDX3 inhibition by RK-33, when cells were pushed to derive more of their energy from oxidative phosphorylation, by substitution of glucose with pyruvate (IC50 1.9 μM vs 3.2 μM; Supplementary Figure 3E). No difference in sensitivity was observed in MDA-MB-231.
RK-33 causes a collapse in mitochondrial membrane potential
A reduction in mitochondrial translation can both reduce mitochondrial biogenesis and cause defective ETC complexes resulting in a reduced H+ membrane potential. To evaluate both the amount of mitochondria and their respective membrane potential, mitotracker-labeled MCF7 and MDA-MB-231 cells, were analyzed with flow cytometry, following RK-33 treatment (Figure 4A, Supplementary Figure 4A). DDX3 inhibition with RK-33 did not affect the total amount of mitochondria as measured by mitotracker green, but a significant reduction in mitotracker red staining was observed in MCF7 (fold change range 0.39–0.45, p = 0.012) and MDA-MB-231 (fold change range 0.39–0.58, p = 0.051), indicative of a collapsed membrane potential.
Figure 4. RK-33 treatment causes reduced mitochondrial potential and increased production of reactive oxygen species.
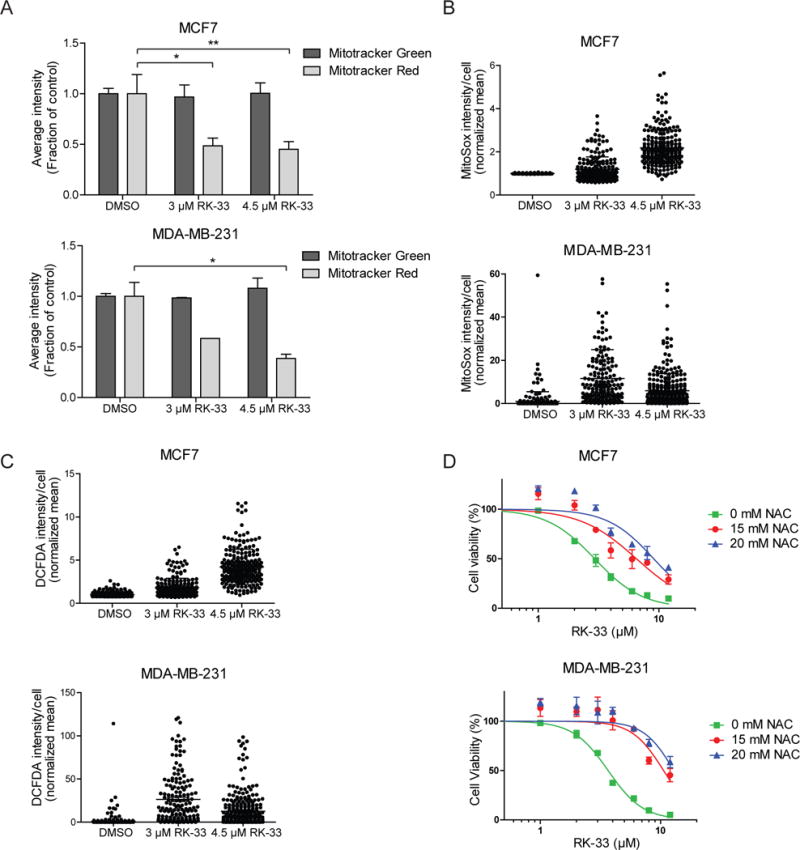
A. Bar graphs showing the mean mitochondrial mass (mitotracker green) and membrane potential (mitotracker red) per cell after twelve hours exposure to RK-33 as measured by flow cytometry. Graphs represent mean ± SD of two biological controls. B. Dot plots showing the amount of mitochondrial superoxide production after twelve hours exposure to RK-33 as measured by the MitoSox intensity per cell. Graphs represent mean ± SD. C. Dot plots showing the amount of ROS production per cell as measured by the DCFDA intensity per cell. Graphs represent mean ± SD. D. MTS assays showing the cytotoxicity of RK-33 in the absence and presence of 15–20 mM n-acetylcysteine (NAC). Graphs represent mean ± SD of two biological replicates. *p < 0.05, **p < 0.01. P-values were calculated by a Student’s t-test. All experiments have been replicated a minimum of two independent times.
RK-33 acts through generation of reactive oxygen species
Malfunctioning of the ETC complexes can elevate the extent to which electrons leak out of the chain prematurely and are being accepted by oxygen, resulting in elevated superoxide formation. RK-33 treatment resulted in increased mitochondrial superoxide and cytosolic reactive oxygen species (ROS) levels in MCF7 and MDA-MB-231 (Figure 4B & C; Supplementary Figure 4B). No change in ROS levels was observed in MCF10A (Supplementary Figure 4C). To evaluate whether ROS formation played a causative role in RK-33 induced cytotoxicity, cells were treated with the antioxidant n-acetylcysteine (NAC) and RK-33 simultaneously. Addition of 20 mM NAC significantly reduced the sensitivity to RK-33 in both MCF7 (IC50 9.05 μM vs. 2.93 μM) and MDA-MB-231 (IC50 12.91 μM vs. 3.66 μM; Figure 4D).
RK-33 induces apoptosis and autophagosome formation
Damaged mitochondria can ultimately trigger apoptosis and cause cell death. Immunoblotting for cleaved PARP and caspase 3 as shown in figure 5A indicated apoptosis occurred 24–48 hours after RK-33 exposure. Consistent with Annexin-PI flow cytometry (Supplementary Figure 3C), no apoptosis was observed at 12 hours at which time the changes in mitochondrial respiratory function were observed. Interestingly, acidic vesicular organelles were observed from 12 hours onward (Supplementary Figure 5),. Transmission electron microscopy was used to confirm that the vesicles were autophagosomes (Figure 5B). Immunoblotting for LC3-II revealed an induction after RK-33 treatment of 9.4 fold (p < 0.001) and 4.6 fold (p = 0.023) in MCF7 and MDA-MB-231 respectively, confirming an increase in the number of autophagosomes. However, inhibition of the breakdown of autophagosomes by the lysosomal inhibitor chloroquine, resulted only in limited further elevation of LC3-II levels in MCF7 (to 11.4 fold; p = 0.371) and MDA-MB-231 (to 7.0 fold; p = 0.101). Addition of 25 μM chloroquine did not affect the RK-33 sensitivity in MCF7 or MDA-MB-231 (Supplementary Figure 5B), indicating that autophagy is not the primary pathway in RK-33 mediated cell death.
Figure 5. RK-33 induces autophagosome formation.
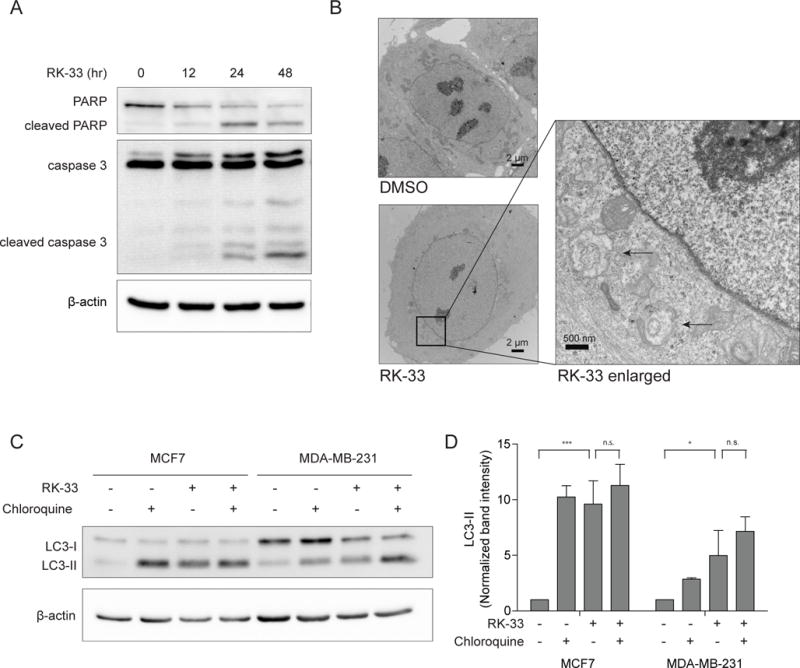
A. Immunoblot showing PARP and caspase 3 cleavage in RK-33 treated MDA-MB-231, indicating induction of apoptosis after 24 hours of exposure. B. Transmission electron microscopy images showing the presence of autophagosomes in MCF7 cells, as indicated by arrows, after 12 hours treatment with 3 μM RK-33. C. Immunoblot showing accumulation of autophagosome specific LC3-II after twelve hours exposure to 3 μM RK-33 or one hour exposure to 50 μM Chloroquine (CQ). D. Bar graph showing the LC3-II band intensities after twelve hours RK-33 exposure normalized for β-actin. Graphs represent mean ± SD of 2 (MDA-MB-231) or 3 (MCF7) biological replicates. *p < 0.05, **p < 0.01, ***P < 0.001. P-values were calculated by a Student’s t-test. All experiments have been replicated a minimum of two independent times.
Combination of RK-33 and radiation therapy results in a bioenergetic catastrophe
As RK-33 was previously demonstrated to have radiosensitizing abilities4, we explored whether these were attributable to reduced mitochondrial functions. In MCF7 basal respiration levels increased after radiation (fold change 1.21, p = 0.002; Figure 6A). Interestingly, RK-33 antagonized the radiation-induced increase in OCR (fold change 0.40; p < 0.001). In MDA-MB-231 a small reduction was observed after radiation (fold change 0.89, p = 0.022). Addition of RK-33 resulted in further reduction of respiration rates (fold change 0.58, p <0.001). Intracellular ATP levels increased after exposure to radiation in MCF7 (fold change 1.28, p = 0.036) and MDA-MB-231 radiation (fold change 1.37, p = 0.016), and this increase was blocked by RK-33 addition in both cell lines (Figure 6B). Evaluation of γH2AX foci showed that RK-33 significantly slows down DNA double strand break repair at 6 (fold change range 1.7–2.9; p = 0.003) and 24 hours (fold change range 3.2–5.1; p < 0.001) after radiation (Figure 6C–D). Furthermore, we showed that treatment with 3 μM (p = 0.009) and 4.5 μM RK-33 (p = 0.006) synergized with radiation therapy in these cells and this effect can be reversed by addition of the antioxidant NAC (Figure 6E).
Figure 6. RK-33 radiosensitizes through reduced OXPHOS and increased ROS production.
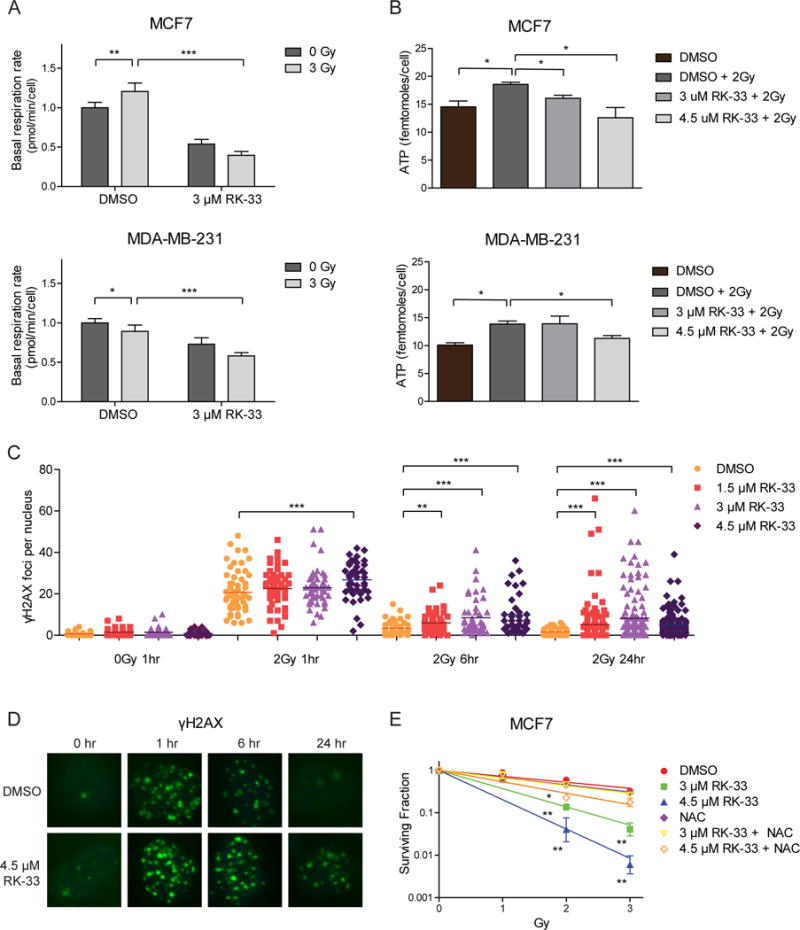
A. Bar graphs showing basal respiration rates (OCR) as measured by the Seahorse XF96 Extracellular Flux Analyzer 15 hours after exposure to 3 μM RK-33 and twelve hours after exposure to 3 Gy ionizing radiation. Graphs represent mean ± SD of 6 biological replicates. P-values were calculated by a student’s t-test. B. Intracellular ATP levels 15 hours after exposure to RK-33 and twelve hours after 3 Gy ionizing radiation. Graphs represent mean ± SD of 2 biological replicates. P-values were calculated by a student’s t-test. C. Dot plot showing the number of γH2AX foci after exposure to 2 Gy radiation, preceded by three hours exposure to RK-33. P-values calculated by a Mann-Whitney U test. D. Immunofluorescent image of γH2AX foci (green) as a measure of DNA double strand breaks at different timepoints after exposure to 2 Gy ionizing radiation preceded by three hours exposure to RK-33. Nuclei are labeled with DAPI. E. Colony forming assay showing the surviving fraction after ionizing radiation preceded by three hours exposure to RK-33. Values are normalized to the response to RK-33 alone. Graphs represent mean ± SD of 2 biological replicates. P-values calculated with a Student’s t-test. *p < 0.05; ** p < 0.01 ***p < 0.001. All experiments have been replicated a minimum of two independent times.
Discussion
This study aimed to elucidate the working mechanism of the DDX3 inhibitor RK-33 by studying its effect on the metabolic profile of breast cancer cells. High DDX3 expression is present in 35% of breast cancers and is associated with an aggressive phenotype and worse overall survival, making it an attractive target in breast cancer patients. We showed that RK-33 functions as a potent inhibitor of mitochondrial translation and thereby reduced the mitochondrial OXPHOS capacity and increased ROS production in cancers cells. Our results explain the selective anti-cancer activity observed after RK-33 treatment, especially in combination with radiation (Figure 7). Since normal cells have a relatively low baseline ATP demand as compared to cancer cells, the effect of RK-33 on non-transformed cells is limited. However, in cancer cells, energy use is higher and a larger OXPHOS reserve capacity is required to deal with sudden increases in ATP demand as a result of exposure to cellular stressors like ionizing radiation14–17, especially in tumor areas that are glucose deprived23. The cytotoxic effect of RK-33 in these cells is therefore much larger. In addition, both ionizing radiation and RK-33 increase the intracellular ROS levels. Together these treatments result in metabolic catastrophe.
Figure 7. DDX3 inhibition with RK-33 causes metabolic synthetic lethality.
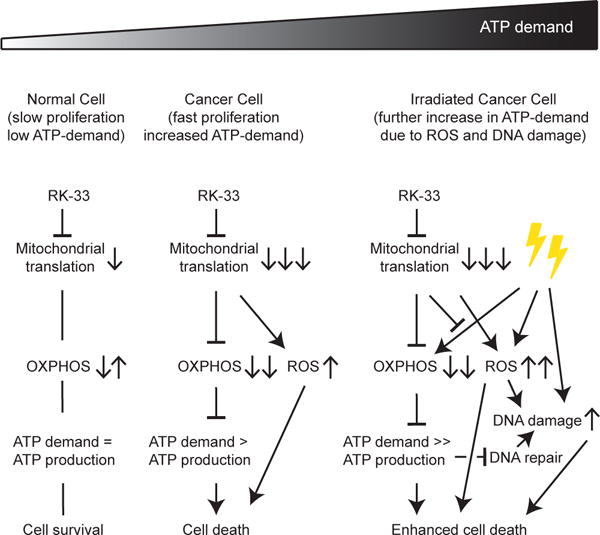
Schematic overview of the mechanism behind RK-33 cytotoxicity in breast cancer cells. ATP = adenosine triphosphate, OXPHOS = oxidative phosphorylation, ROS = reactive oxygen species.
We found that the effect of RK-33 treatment on mitochondrial respiration was greater in MCF7, when compared to MDA-MB-231. However, ROS levels were increased to a similar extent in both cell lines. Addition of the antioxidant NAC was especially protective in MDA-MB-231, indicating that the effect of RK-33 might be more ROS-mediated in this cell line. This finding is also in line with MCF7 being relatively more reliant on OXPHOS for ATP production24. Promotion of mitochondrial ROS production has been recognized as an effective strategy to induce cancer cell death and increase chemosensitivity25.
Mitochondrial damage often induces the formation of autophagosomes26. We indeed observed an accumulation of autophagosomes after RK-33. Only a minor further increase was observed after addition of the autophagy inhibitor chloroquine, implying that the increased number of autophagosomes after RK-33 is the result of both increased production and decreased breakdown of autophagosomes. Although, autophagy has also been reported to have a chemoprotective role27, we did not find that inhibiting autophagy with chloroquine altered the response to RK-33. This indicates that accumulation of autophagosomes following RK-33 exposure may not be a major determinant of RK-33 mediated cell death.
Although concurrent chemoradiation strategies are not mainstay treatments in breast cancer, certain breast cancer subtypes, like triple negative breast cancer, are relatively radioresistant and have higher local recurrence rates corresponding with worse overall survival28. The development of radiosensitizers in breast cancer could benefit this group of patients that we found to have particularly high DDX3 expression levels. In addition, radiosensitizers could allow for reduced radiation dose and consequently reduce normal tissue toxicity, commonly occurring after axillary and internal mammary nodal radiation for breast cancer treatment. We previously showed that the DDX3 inhibitor RK-33 is a potent radiosensitizer in cancer types where local control is particularly challenging, like lung4 and prostate cancer7. Although our study focuses on breast cancer cells, it is likely that inhibition of mitochondrial translation is also part of the working mechanism of RK-33 radiosensitization in these cancers. One study in Ewing sarcoma showed RK-33 can be used as a monotherapy as well6. More research focusing on the use of DDX3 inhibitors as a single agent or in combination with chemotherapeutics is warranted.
Importantly, normal cell toxicity can be a concern with treatments targeting mitochondrial function. However, no toxicity was observed after RK-33 treatment in extensive toxicology studies performed in mice4. This is in line with the fact that normal cells have lower energy demands in general, encounter less stressors causing sudden increases in ATP demand (eg. DNA damage), have lower ROS levels and express lower amounts of DDX3.
This is the first study suggesting that DDX3 is involved in mitochondrial translation and could therefore be of paramount importance for maintenance of the bioenergetics machinery of oxidative phosphorylation. Although, we do not proof a direct role for DDX3 in mitochondrial translation, the fact that we and others29 found DDX3 to localize to the mitochondria intuitively supports the idea that RK-33 mediates its effect on mitochondrial translation, directly through DDX3 inhibition. In addition, another DEAD/H box RNA helicase family members, DDX28 and DHX30, were recently found to be responsible for the assembly of mitochondrial ribosomes30, 31. Interestingly, DDX3 was identified by mass spectrometry analysis of the mitochondrial DDX28 and DHX30 interactome31, implying that DDX3 might also be involved in mitochondrial ribosome assembly. This potentially explains why we observed a reduction in mitoribosomal proteins that are encoded on the nuclear genome as well, since disassembly of the mitoribosome could reduce its stability and therefore protein expression levels. Geissler, et al. previously showed that DDX3 has a role in the assembly of functional 80S ribosomes2 The role of DDX3 in cap-dependent cytosolic translation is disputed in the literature2, 32, 33, with most studies concluding that DDX3 inhibition does not result in major changes in general protein synthesis34–37, but could play a role in translation of mRNAs with complex features in their 5′UTR37, 38,35.39 It is possible that DDX3 functions in both cytoplasmic and mitochondrial translation. However, the timeline of events with a profound decrease in mitochondrial translation as early as two hours after treatment onset and apoptosis occurring only after 24 hours does suggest that the effect of RK-33 on mitochondrial translation is direct. Our finding that DDX3 inhibition results in increased ROS production is also in line with a recent study in Leishmania, that indicated a central role for DDX3 in regulating the mitochondrial stress response of this parasitic protozoan40. In addition, that DDX3 inhibition ultimately reduces DNA damage repair, was also supported by a study by Chen. et al., which showed that DDX3 knockout resulted in higher levels of genomic damage murine embryonic cells41.
Decades after Otto Warburg’s initial observation that tumor cells upregulate glycolysis in the presence of oxygen, so-called aerobic glycolysis, cancer metabolism is an area of renewed attention15, 42. Increased aerobic glycolysis is often erroneously interpreted as a sign of reduced and damaged oxidative phosphorylation in cancer cells. In fact, accumulating evidence now indicates that cancer cells are reliant on the mitochondria for their bioenergetic machinery and macromolecule synthesis function15, 43. Consequently, mitochondrial respiration is increasingly recognized as a viable target for anti-cancer therapy43 and (triple negative) breast cancer treatment in particular44,45. OXPHOS was found to be upregulated in cancer stem cells46 and during metastases16. In addition, chemo- and radioresistant cells exhibit increased respiration rates17, 47. A recent study showed that irradiated cells increase OXPHOS to favor DNA repair and cell survival48 and inhibitors of electron transport complexes can enhance radiosensitivity49, 50. In addition, mitochondrial translation has previously been identified as a therapeutic target in the treatment of acute myeloid leukemia51.
We conclude that DDX3 is involved in mitochondrial translation and could therefore be of paramount importance for maintenance of the bioenergetics machinery of oxidative phosphorylation. The DDX3 inhibitor RK-33 causes radiosensitization in breast cancer cells through direct inhibition of mitochondrial translation, which results in reduced OXPHOS capacity and increased intracellular ROS levels, culminating in a bioenergetic catastrophe and eventual apoptosis.
Material and methods
Patient samples
Tissue microarrays (TMAs) with 540 breast cancers archived in the UMC Utrecht between 1993 and 2009 were used52. The patient group consisted out of 422 consecutive breast cancer patients, supplemented with a collection of 95 invasive lobular carcinomas and 23 distant metastases. Cases were subclassified in molecular subtypes as was described before53, 54. We used anonymous archival leftover pathology material. Therefore, no ethical approval or informed consent is required according to Dutch legislation55 as use of leftover material is part of the standard agreement with patients in our hospital. The UMC Utrecht Medical Research Ethics Committee confirmed that official approval of this study is not required by law (reference number WAG/mb/16/021628).
Immunohistochemistry
Immunohistochemical staining for DDX3 has been described in detail previously56. Briefly, sections were labeled for 1 hour with anti-DDX3 (1:1000, pAb r647)57. Cases with absent to moderate DDX3 expression were classified as having low DDX3 expression and evaluated against cases with high expression, as before5.
Statistics
Discrete variables were compared by χ2 or Fisher’s exact test. Student’s t-test and Mann Whitney U-tests were calculated for normal and non-normal distributed variables respectively. Survival was compared by Kaplan-Meier curves and Breslow tests. Multivariate analysis was performed by Cox regression. Effect modifiers were identified by including multiplicative interaction terms into the model. Statistical analyses were performed with SPSS 20.0 (IBM Inc, Armonk, NY, USA) or R version 3.2.0 regarding p-values smaller than 0.05 as significant.
Cell culture
MCF10A, MCF7, MDA-MB-231 and MDA-MB-435 cells were originally purchased from the American Type Culture Collection (ATCC, Manassas, VA, USA) and regularly STR-profiled and mycoplasma tested. DDX3 knockdown in MDA-MB-435 cells was achieved by lentiviral transduction with an shDDX3 construct or empty vector control, as before3. As there is an ongoing debate over the MDA-MB-435 cell line58, we decided to use MDA-MB-231 as a triple negative cell line for all our functional experiments. In terms of receptor expression, MDA-MB-231 is similar to MDA-MB-435.
Cell viability assays
For cell viability assays 1 × 103 − 3 × 103 cells were plated per well in a 96-well plate and allowed to attach overnight. The number of viable cells was estimated after 72 hours of drug exposure with an MTS assay (CellTiter 96 Aqueous One Solution, Promega, Madison, WI, USA). RK-33 cytotoxicity was assessed in the presence of: 5.5 mM glucose or sodium pyruvate, 15–20 mM n-acetyl-L-cysteine (Sigma-Aldrich, St Louis, MO, USA), 25 μM chloroquine (Sigma-Aldrich).
Immunoblotting
Whole cellular protein extracts were lysed in SDS-extraction buffer. Mitochondrial and cytoplasmic extracts were prepared using a mitochondria isolation kit (ThermoFisher Scientific, Waltham, MA, USA) and Dounce homogenizer. The following primary antibodies were used: DDX3 (1:1000, mAb AO19657), β-actin (1:10000, A5441, Sigma-Aldrich), OXPHOS complexes (1:1000, ab110411, Abcam, Cambridge UK), COX IV (1:5000, #4850, Cell Signaling Technology, Danvers, MA, USA), PARP (1:1000, #9542, Cell Signaling Technology), Caspase 3 (1:500, #9665, Cell Signaling Technology), and LC3 (1:1000, #1775S, Cell Signaling Technology).
Proteomics
MDA-MB-435 cells were exposed for 24 hours to 4.5 μM RK-33 or were harvested 72 hours after shDDX3 transduction in extraction buffer. More information about protein extraction and digestion, TMT-labeling, mass spectrometry, the Mascot search strategy and bioinformatics processing is available in the supplementary methods.
Bioinformatics
Protein abundances were compared between three RK-33 treated vs DMSO treated samples, and between two shDDX3 and control samples. Moderated t-test statistics and multiple comparison corrected q-values were calculated59, 60. Proteins with a q < 0.05 and a fold change larger than 1.15 were considered significantly altered. Gene set expression analysis was performed by searching the Reactome database using the Enrichr web tool (http://amp.pharm.mssm.edu/Enrichr/)61. In this analysis enriched pathways are identified by comparing the number of significantly altered proteins in each pathway of the reactome database to the number that could be expected based the size of each pathway and the total amounts of proteins identified. In addition, protein interactions within the group of significantly altered proteins were surveyed by searching the STRING database (http://string-db.org/)20 version 9.1, using a confidence level of 0.9.
Immunofluorescence
For mitochondrial colocalization experiments, cells were labeled with 100 nM Mitotracker red (CMXRos #9082, Cell Signaling Technology) and fixed in methanol. For DNA damage experiments, cells were treated with RK-33 two hours before exposure to 2 Gy ionizing radiation (CIDX, XStrahl, Camberley, United Kingdom), followed by fixation in formalin and permeabilization with 0.2% Triton-X. The following primary antibodies were used: DDX3 (1:50; AO19657), γH2AX (1:1600, DAM1782241, EMD Millipore, Billirica, MA, USA). Anti-mouse Alexa488 (1:200; 1 hour, Life Technologies, Carlsbad, CA, USA) was used as a secondary antibody. Photographs were taken with Nikon Eclipse 80i fluorescence microscope for the DNA damage experiments and with an Olympus FV10MP-LACDS/BX61W1 multiphoton microscope for colocalization analysis (Olympus, Center Valley, PA, USA).
Mitochondrial translation assay
Pulse labeling was performed as described by Leary, et al.62. Briefly, 4 × 105 cells were plated in 60 mm dishes and allowed to attach overnight. After 2 hour exposure to RK-33, cells were pulse labeled for 60 minutes with [S35]-methionine (150 μCi/ml, Easytag, PerkinElmer, Waltham, MA, USA) and 100 μg/ml Emetine. Cells were chased for 10 minutes and subsequently harvested. 50 μg whole cellular protein extract was run over a 15–20% gradient SDS-PAGE gel. After fixation and Coomassie blue staining the gel was dried and imaged by autoradiography.
Measurements of oxygen consumption
Real time oxygen consumption (OCR) and extracellular acidification (ECAR) rates were measured using a Seahorse XF96 Extracellular Flux Analyzer (Agilent Technologies, Santa Clara, CA, USA). If applicable, cells were irradiated three hours after the onset of RK-33 treatment (CIDX, XStrahl). Complex inhibitors were used in the following final concentrations in Seahorse media: oligomycin (1 μM), FCCP (0.35 μM), antimycin (4 μM) and rotenone (4 μM). Respiratory measurements were normalized by cell numbers as estimated by a DNA assay. For this the wells were lysed in TE lysis buffer containing 0.2% Triton-X and proteinase K for 10 minutes on ice and stained with PicoGreen (ThermoFisher scientific; 1:200). Fluorescent intensity was measured using a Victor3V plate reader (PerkinElmer). The range of fold changes and highest p-value as calculated by student’s t-test is reported in the text.
ATP quantification
7.5–17.5 × 104 cells were plated in a 12 well plate and allowed to attach overnight. Cells were treated with RK-33 and three hours later with ionizing radiation (CIDX, XStrahl) if applicable. Intracellular ATP concentrations of 2 × 104 cells were measured 12 hours after RK-33 addition by CellTiter-Glo (Promega) luminometry.
Mitotracker flow cytometry
For detection of mitochondrial mass and membrane potential cells were treated with RK-33 and subsequently labeled with 100 nM Mitotracker green FM (#9074 Cell Signaling Technology) and 100 nM Mitotracker red (CMXRos #9082, Cell signaling Technology) respectively, after which cells were harvested by trypsinization. Fluorescent intensity of cells was detected by flow cytometry on a FACSCalibur instrument (BD Biosciences, San Jose, CA, USA). Data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR, USA).
Measurement of reactive oxygen species
1.4−6 × 105 cells were plated on collagen coated 35 mm dishes and after 12 hour of RK-33 treatment labeled with 5 μM DCFDA and 2.5 μM MitoSox for 30 minutes. After a recovery time of 30 minutes cells were imaged with Olympus FV10MP-LACDS/BX61W1 multiphoton microscope. Fluorescent intensity per cell was measured with ImageJ63.
Electron microscopy
Fixation took place in 2% paraformaldehyde, 2.5% glutaraldehyde, 0.1 M cacodylate buffer. Cells were post-fixed in 0.1 M cacodylate buffer containing 1% osmium tetroxide and 0.8% ferrocyanide, subsequently stained with 2% aqueous uranyl acetate and dehydrated in increasing ethanol concentrations, followed by overnight infiltration with equal parts EPON 8/2 and ethanol. After five hours in 100% EPON 1.5% DMP-30, catalyst was added and the resin was allowed to polymerize overnight at 60 °C and cure for three days at 37 °C. Blocks were sectioned at a 65 nm thickness with a diamond knife and imaged with a Hitachi H7600 transmission electron microscope at 25.000 × magnification.
Colony forming assay
200 MCF7 cells were plated per well in a 6-well plate and allowed to attach overnight. Cells were exposed to RK-33 three hours before irradiation (CIDX, XStrahl). 24 hours after radiation treatment the media was refreshed. After 7 days, colonies were stained with 0.5% crystal violet in methanol and counted.
Supplementary Material
Acknowledgments
We would like to thank Bob Cole, Tatiana Boronina and Bob O’ Meally of the Johns Hopkins Mass Spectrometry and Proteomics core facility for their help with the proteomics experiments, Tri Nguyen for his help with interpretation of the electron microscopy images, the Dawson laboratory at the Johns Hopkins School of Medicine for their help with the mitochondrial translation assay and Beth Rodgers, who kindly provided us with S35-methionine.
Funding: This work was financially supported by Utrecht University Alexandre Suerman Stipend (MHVV), the Dutch Cancer Foundation (UU2013–5851; MHVV), NIH (R01CA166348 to PT and R01-CA193895 to AL), FAMRI (VR) and Safeway (VR).
Footnotes
Conflict of Interest
Venu Raman and Phuoc Tran have received a patent for the use of RK-33 as a radiosensitizer (US8,518,901). Venu, Raman Guus Bol, and Paul van Diest have received a patent for the use of DDX3 as a cancer biomarker (US9,322,831). Paul van Diest, Phuoc Tran and Venu Raman are on the advisory board of Natsar Pharmaceuticals.
Supplementary information accompanies the paper on the Oncogene website (http://www.nature.com/onc)
References
- 1.Tanner NK, Linder P. DExD/H box RNA helicases: from generic motors to specific dissociation functions. Molecular cell. 2001;8:251–262. doi: 10.1016/s1097-2765(01)00329-x. [DOI] [PubMed] [Google Scholar]
- 2.Geissler R, Golbik RP, Behrens SE. The DEAD-box helicase DDX3 supports the assembly of functional 80S ribosomes. Nucleic acids research. 2012;40:4998–5011. doi: 10.1093/nar/gks070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Botlagunta M, Vesuna F, Mironchik Y, Raman A, Lisok A, Winnard P, Jr, et al. Oncogenic role of DDX3 in breast cancer biogenesis. Oncogene. 2008;27:3912–3922. doi: 10.1038/onc.2008.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bol GM, Vesuna F, Xie M, Zeng J, Aziz K, Gandhi N, et al. Targeting DDX3 with a small molecule inhibitor for lung cancer therapy. EMBO molecular medicine. 2015;7:648–669. doi: 10.15252/emmm.201404368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Heerma van Voss MR, Vesuna F, Trumpi K, Brilliant J, Berlinicke C, de Leng W, et al. Identification of the DEAD box RNA helicase DDX3 as a therapeutic target in colorectal cancer. Oncotarget. 2015;6:28312–28326. doi: 10.18632/oncotarget.4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wilky BA, Kim C, McCarty G, Montgomery EA, Kammers K, DeVine LR, et al. RNA helicase DDX3: a novel therapeutic target in Ewing sarcoma. Oncogene. 2016;35:2574–2583. doi: 10.1038/onc.2015.336. [DOI] [PubMed] [Google Scholar]
- 7.Xie M, Vesuna F, Tantravedi S, Bol GM, Heerma van Voss MR, Nugent K, et al. RK-33 radiosensitizes prostate cancer cells by blocking the RNA helicase DDX3. Cancer research. 2016 doi: 10.1158/0008-5472.CAN-1116-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Li Y, Wang H, Wang Z, Makhija S, Buchsbaum D, LoBuglio A, et al. Inducible resistance of tumor cells to tumor necrosis factor-related apoptosis-inducing ligand receptor 2-mediated apoptosis by generation of a blockade at the death domain function. Cancer research. 2006;66:8520–8528. doi: 10.1158/0008-5472.CAN-05-4364. [DOI] [PubMed] [Google Scholar]
- 9.Shih JW, Wang WT, Tsai TY, Kuo CY, Li HK, Wu Lee YH. Critical roles of RNA helicase DDX3 and its interactions with eIF4E/PABP1 in stress granule assembly and stress response. The Biochemical journal. 2012;441:119–129. doi: 10.1042/BJ20110739. [DOI] [PubMed] [Google Scholar]
- 10.Sun M, Song L, Zhou T, Gillespie GY, Jope RS. The role of DDX3 in regulating Snail. Biochimica et biophysica acta. 2011;1813:438–447. doi: 10.1016/j.bbamcr.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hagerstrand D, Tong A, Schumacher SE, Ilic N, Shen RR, Cheung HW, et al. Systematic interrogation of 3q26 identifies TLOC1 and SKIL as cancer drivers. Cancer discovery. 2013;3:1044–1057. doi: 10.1158/2159-8290.CD-12-0592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen HH, Yu HI, Cho WC, Tarn WY. DDX3 modulates cell adhesion and motility and cancer cell metastasis via Rac1-mediated signaling pathway. Oncogene. 2015;34:2790–2800. doi: 10.1038/onc.2014.190. [DOI] [PubMed] [Google Scholar]
- 13.Kondaskar A, Kondaskar S, Kumar R, Fishbein JC, Muvarak N, Lapidus RG, et al. Novel, Broad Spectrum Anti-Cancer Agents Containing the Tricyclic 5:7:5-Fused Diimidazodiazepine Ring System. ACS medicinal chemistry letters. 2010;2:252–256. doi: 10.1021/ml100281b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Vellinga TT, Borovski T, de Boer VC, Fatrai S, van Schelven S, Trumpi K, et al. SIRT1/PGC1alpha-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clinical cancer research : an official journal of the American Association for Cancer Research. 2015;21:2870–2879. doi: 10.1158/1078-0432.CCR-14-2290. [DOI] [PubMed] [Google Scholar]
- 15.Viale A, Corti D, Draetta GF. Tumors and mitochondrial respiration: a neglected connection. Cancer research. 2015;75:3685–3686. doi: 10.1158/0008-5472.CAN-15-0491. [DOI] [PubMed] [Google Scholar]
- 16.LeBleu VS, O’Connell JT, Gonzalez Herrera KN, Wikman H, Pantel K, Haigis MC, et al. PGC-1alpha mediates mitochondrial biogenesis and oxidative phosphorylation in cancer cells to promote metastasis. Nature cell biology. 2014;16:992–1003. 1001–1015. doi: 10.1038/ncb3039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lu CL, Qin L, Liu HC, Candas D, Fan M, Li JJ. Tumor cells switch to mitochondrial oxidative phosphorylation under radiation via mTOR-mediated hexokinase II inhibition–a Warburg-reversing effect. PloS one. 2015;10:e0121046. doi: 10.1371/journal.pone.0121046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Qin L, Fan M, Candas D, Jiang G, Papadopoulos S, Tian L, et al. CDK1 Enhances Mitochondrial Bioenergetics for Radiation-Induced DNA Repair. Cell reports. 2015;13:2056–2063. doi: 10.1016/j.celrep.2015.11.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Croft D, Mundo AF, Haw R, Milacic M, Weiser J, Wu G, et al. The Reactome pathway knowledgebase. Nucleic acids research. 2014;42:D472–477. doi: 10.1093/nar/gkt1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Franceschini A, Szklarczyk D, Frankild S, Kuhn M, Simonovic M, Roth A, et al. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic acids research. 2013;41:D808–815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Y, D’Aurelio M, Deng JH, Park JS, Manfredi G, Hu P, et al. An assembled complex IV maintains the stability and activity of complex I in mammalian mitochondria. The Journal of biological chemistry. 2007;282:17557–17562. doi: 10.1074/jbc.M701056200. [DOI] [PubMed] [Google Scholar]
- 22.Acin-Perez R, Bayona-Bafaluy MP, Fernandez-Silva P, Moreno-Loshuertos R, Perez-Martos A, Bruno C, et al. Respiratory complex III is required to maintain complex I in mammalian mitochondria. Molecular cell. 2004;13:805–815. doi: 10.1016/s1097-2765(04)00124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhang X, Fryknas M, Hernlund E, Fayad W, De Milito A, Olofsson MH, et al. Induction of mitochondrial dysfunction as a strategy for targeting tumour cells in metabolically compromised microenvironments. Nature communications. 2014;5:3295. doi: 10.1038/ncomms4295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pelicano H, Zhang W, Liu J, Hammoudi N, Dai J, Xu RH, et al. Mitochondrial dysfunction in some triple-negative breast cancer cell lines: role of mTOR pathway and therapeutic potential. Breast cancer research : BCR. 2014;16:434. doi: 10.1186/s13058-014-0434-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen Y, McMillan-Ward E, Kong J, Israels SJ, Gibson SB. Oxidative stress induces autophagic cell death independent of apoptosis in transformed and cancer cells. Cell death and differentiation. 2008;15:171–182. doi: 10.1038/sj.cdd.4402233. [DOI] [PubMed] [Google Scholar]
- 26.Youle RJ, Narendra DP. Mechanisms of mitophagy. Nature reviews Molecular cell biology. 2011;12:9–14. doi: 10.1038/nrm3028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huang Z, Zhou L, Chen Z, Nice EC, Huang C. Stress management by autophagy: Implications for chemoresistance. International journal of cancer Journal international du cancer. 2016;139:23–32. doi: 10.1002/ijc.29990. [DOI] [PubMed] [Google Scholar]
- 28.Eiermann W, Vallis KA. Locoregional treatments for triple-negative breast cancer. Annals of oncology : official journal of the European Society for Medical Oncology / ESMO. 2012;23(Suppl 6):vi30–34. doi: 10.1093/annonc/mds192. [DOI] [PubMed] [Google Scholar]
- 29.Oshiumi H, Sakai K, Matsumoto M, Seya T. DEAD/H BOX 3 (DDX3) helicase binds the RIG-I adaptor IPS-1 to up-regulate IFN-beta-inducing potential. European journal of immunology. 2010;40:940–948. doi: 10.1002/eji.200940203. [DOI] [PubMed] [Google Scholar]
- 30.Antonicka H, Shoubridge EA. Mitochondrial RNA Granules Are Centers for Posttranscriptional RNA Processing and Ribosome Biogenesis. Cell reports. 2015 doi: 10.1016/j.celrep.2015.01.030. [DOI] [PubMed] [Google Scholar]
- 31.Tu YT, Barrientos A. The Human Mitochondrial DEAD-Box Protein DDX28 Resides in RNA Granules and Functions in Mitoribosome Assembly. Cell reports. 2015 doi: 10.1016/j.celrep.2015.01.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shih JW, Tsai TY, Chao CH, Wu Lee YH. Candidate tumor suppressor DDX3 RNA helicase specifically represses cap-dependent translation by acting as an eIF4E inhibitory protein. Oncogene. 2008;27:700–714. doi: 10.1038/sj.onc.1210687. [DOI] [PubMed] [Google Scholar]
- 33.Lee CS, Dias AP, Jedrychowski M, Patel AH, Hsu JL, Reed R. Human DDX3 functions in translation and interacts with the translation initiation factor eIF3. Nucleic acids research. 2008;36:4708–4718. doi: 10.1093/nar/gkn454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abaeva IS, Marintchev A, Pisareva VP, Hellen CU, Pestova TV. Bypassing of stems versus linear base-by-base inspection of mammalian mRNAs during ribosomal scanning. The EMBO journal. 2011;30:115–129. doi: 10.1038/emboj.2010.302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Soto-Rifo R, Rubilar PS, Limousin T, de Breyne S, Decimo D, Ohlmann T. DEAD-box protein DDX3 associates with eIF4F to promote translation of selected mRNAs. The EMBO journal. 2012;31:3745–3756. doi: 10.1038/emboj.2012.220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fukumura J, Noguchi E, Sekiguchi T, Nishimoto T. A temperature-sensitive mutant of the mammalian RNA helicase, DEAD-BOX × isoform, DBX, defective in the transition from G1 to S phase. Journal of biochemistry. 2003;134:71–82. doi: 10.1093/jb/mvg126. [DOI] [PubMed] [Google Scholar]
- 37.Lai MC, Lee YH, Tarn WY. The DEAD-box RNA helicase DDX3 associates with export messenger ribonucleoproteins as well as tip-associated protein and participates in translational control. Molecular biology of the cell. 2008;19:3847–3858. doi: 10.1091/mbc.E07-12-1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lai MC, Chang WC, Shieh SY, Tarn WY. DDX3 regulates cell growth through translational control of cyclin E1. Molecular and cellular biology. 2010;30:5444–5453. doi: 10.1128/MCB.00560-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Tarn WY, Chang TH. The current understanding of Ded1p/DDX3 homologs from yeast to human. RNA biology. 2009;6:17–20. doi: 10.4161/rna.6.1.7440. [DOI] [PubMed] [Google Scholar]
- 40.Padmanabhan PK, Zghidi-Abouzid O, Samant M, Dumas C, Aguiar BG, Estaquier J, et al. DDX3 DEAD-box RNA helicase plays a central role in mitochondrial protein quality control in Leishmania. Cell death & disease. 2016;7:e2406. doi: 10.1038/cddis.2016.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Chen CY, Chan CH, Chen CM, Tsai YS, Tsai TY, Wu Lee YH, et al. Targeted inactivation of murine Ddx3x: essential roles of Ddx3x in placentation and embryogenesis. Human molecular genetics. 2016;25:2905–2922. doi: 10.1093/hmg/ddw143. [DOI] [PubMed] [Google Scholar]
- 42.Koppenol WH, Bounds PL, Dang CV. Otto Warburg’s contributions to current concepts of cancer metabolism. Nature reviews Cancer. 2011;11:325–337. doi: 10.1038/nrc3038. [DOI] [PubMed] [Google Scholar]
- 43.Weinberg SE, Chandel NS. Targeting mitochondria metabolism for cancer therapy. Nature chemical biology. 2015;11:9–15. doi: 10.1038/nchembio.1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sansone P, Ceccarelli C, Berishaj M, Chang Q, Rajasekhar VK, Perna F, et al. Self-renewal of CD133(hi) cells by IL6/Notch3 signalling regulates endocrine resistance in metastatic breast cancer. Nature communications. 2016;7:10442. doi: 10.1038/ncomms10442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Jones RA, Robinson TJ, Liu JC, Shrestha M, Voisin V, Ju Y, et al. RB1 deficiency in triple-negative breast cancer induces mitochondrial protein translation. The Journal of clinical investigation. 2016 doi: 10.1172/JCI81568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Farnie G, Sotgia F, Lisanti MP. High mitochondrial mass identifies a sub-population of stem-like cancer cells that are chemo-resistant. Oncotarget. 2015;6:30472–30486. doi: 10.18632/oncotarget.5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Matassa DS, Amoroso MR, Lu H, Avolio R, Arzeni D, Procaccini C, et al. Oxidative metabolism drives inflammation-induced platinum resistance in human ovarian cancer. Cell death and differentiation. 2016 doi: 10.1038/cdd.2016.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Qin B, Minter-Dykhouse K, Yu J, Zhang J, Liu T, Zhang H, et al. DBC1 functions as a tumor suppressor by regulating p53 stability. Cell reports. 2015;10:1324–1334. doi: 10.1016/j.celrep.2015.01.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wardman P, Anderson RF, Hodgkiss RJ, Parrick J, Smithen CE, Wallace RG, et al. Radiosensitization by non-nitro compounds. International journal of radiation oncology, biology, physics. 1982;8:399–401. doi: 10.1016/0360-3016(82)90647-2. [DOI] [PubMed] [Google Scholar]
- 50.Zhang X, Zhou X, Chen R, Zhang H. Radiosensitization by inhibiting complex I activity in human hepatoma HepG2 cells to X-ray radiation. Journal of radiation research. 2012;53:257–263. doi: 10.1269/jrr.11124. [DOI] [PubMed] [Google Scholar]
- 51.Skrtic M, Sriskanthadevan S, Jhas B, Gebbia M, Wang X, Wang Z, et al. Inhibition of mitochondrial translation as a therapeutic strategy for human acute myeloid leukemia. Cancer cell. 2011;20:674–688. doi: 10.1016/j.ccr.2011.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moelans CB, de Weger RA, van Blokland MT, Ezendam C, Elshof S, Tilanus MG, et al. HER-2/neu amplification testing in breast cancer by multiplex ligation-dependent probe amplification in comparison with immunohistochemistry and in situ hybridization. Cellular oncology : the official journal of the International Society for Cellular Oncology. 2009;31:1–10. doi: 10.3233/CLO-2009-0461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Carey LA, Perou CM, Livasy CA, Dressler LG, Cowan D, Conway K, et al. Race, breast cancer subtypes, and survival in the Carolina Breast Cancer Study. JAMA : the journal of the American Medical Association. 2006;295:2492–2502. doi: 10.1001/jama.295.21.2492. [DOI] [PubMed] [Google Scholar]
- 54.Vermeulen JF, van de Ven RA, Ercan C, van der Groep P, van der Wall E, Bult P, et al. Nuclear Kaiso expression is associated with high grade and triple-negative invasive breast cancer. PloS one. 2012;7:e37864. doi: 10.1371/journal.pone.0037864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.The Medical Research Involving Human Subjects Act [In Dutch: Wet medisch-wetenschappelijk onderzoek met mensen, WMO] Burgerlijk Wetboek. 1998 [Google Scholar]
- 56.Bol GM, Raman V, van der Groep P, Vermeulen JF, Patel AH, van der Wall E, et al. Expression of the RNA helicase DDX3 and the hypoxia response in breast cancer. PloS one. 2013;8:e63548. doi: 10.1371/journal.pone.0063548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Angus AG, Dalrymple D, Boulant S, McGivern DR, Clayton RF, Scott MJ, et al. Requirement of cellular DDX3 for hepatitis C virus replication is unrelated to its interaction with the viral core protein. The Journal of general virology. 2010;91:122–132. doi: 10.1099/vir.0.015909-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chambers AF. MDA-MB-435 and M14 cell lines: identical but not M14 melanoma? Cancer research. 2009;69:5292–5293. doi: 10.1158/0008-5472.CAN-09-1528. [DOI] [PubMed] [Google Scholar]
- 59.Kammers K, Cole RN, Tiengwe C, Ruczinski I. Detecting Significant Changes in Protein Abundance. EuPA open proteomics. 2015;7:11–19. doi: 10.1016/j.euprot.2015.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smyth GK. Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Statistical applications in genetics and molecular biology. 2004;3 doi: 10.2202/1544-6115.1027. Article3. [DOI] [PubMed] [Google Scholar]
- 61.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC bioinformatics. 2013;14:128. doi: 10.1186/1471-2105-14-128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Leary SC, Sasarman F. Oxidative phosphorylation: synthesis of mitochondrially encoded proteins and assembly of individual structural subunits into functional holoenzyme complexes. Methods Mol Biol. 2009;554:143–162. doi: 10.1007/978-1-59745-521-3_10. [DOI] [PubMed] [Google Scholar]
- 63.Rasband WS. ImageJ. Vol. 2015. U. S. National Institutes of Health; Bethesda, Maryland, USA: [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.