

Abstract
Background
NRF2 is the key regulator of oxidative stress in normal cells and aberrant expression of the NRF2 pathway due to genetic alterations in the KEAP1 (Kelch-like ECH-associated protein 1)-NRF2 (nuclear factor erythroid 2 like 2)-CUL3 (cullin 3) axis leads to tumorigenesis and drug resistance in many cancers including head and neck squamous cell cancer (HNSCC). The main goal of this study was to identify specific genes regulated by the KEAP1-NRF2-CUL3 axis in HNSCC patients, to assess the prognostic value of this gene signature in different cohorts, and to reveal potential biomarkers.
Methods
RNA-Seq V2 level 3 data from 279 tumor samples along with 37 adjacent normal samples from patients enrolled in the The Cancer Genome Atlas (TCGA)-HNSCC study were used to identify upregulated genes using two methods (altered KEAP1-NRF2-CUL3 versus normal, and altered KEAP1-NRF2-CUL3 versus wild-type). We then used a new approach to identify the combined gene signature by integrating both datasets and subsequently tested this signature in 4 independent HNSCC datasets to assess its prognostic value. In addition, functional annotation using the DAVID v6.8 database and protein-protein interaction (PPI) analysis using the STRING v10 database were performed on the signature.
Results
A signature composed of a subset of 17 genes regulated by the KEAP1-NRF2-CUL3 axis was identified by overlapping both the upregulated genes of altered versus normal (251 genes) and altered versus wild-type (25 genes) datasets. We showed that increased expression was significantly associated with poor survival in 4 independent HNSCC datasets, including the TCGA-HNSCC dataset. Furthermore, Gene Ontology, Kyoto Encyclopedia of Genes and Genomes, and PPI analysis revealed that most of the genes in this signature are associated with drug metabolism and glutathione metabolic pathways.
Conclusions
Altogether, our study emphasizes the discovery of a gene signature regulated by the KEAP1-NRF2-CUL3 axis which is strongly associated with tumorigenesis and drug resistance in HNSCC. This 17-gene signature provides potential biomarkers and therapeutic targets for HNSCC cases in which the NRF2 pathway is activated.
Electronic supplementary material
The online version of this article (10.1186/s12885-017-3907-z) contains supplementary material, which is available to authorized users.
Keywords: Head and neck squamous cell cancer, KEAP1-NRF2-CUL3 mutations, Overall survival, Gene-expression signature
Background
Head and neck squamous cell cancer (HNSCC) is the sixth most prevalent form of cancer. It has a high incidence worldwide, and 90% of cases are histologically identified as squamous cell carcinomas [1, 2]. HNSCC is a broad category of cancers that predominantly arise in the oral cavity, oropharynx, hypopharynx, larynx, soft tissues of the neck, salivary glands, skin, and mucosal membranes [3, 4]. The most common causes are the consumption of tobacco and alcohol, and human papillomavirus infection [5].
NRF2 is the master transcription factor that regulates the genes involved in antioxidant and detoxification pathways. Under normal conditions, Kelch like-ECH-associated protein 1 (KEAP1) negatively regulates the NRF2 expression by cullin-3 (CUL3)-mediated ubiquitination and proteasomal degradation [6]. Under oxidative stress, NRF2 is liberated from the tight control of the KEAP1/CUL3 complex, is relocated to the nucleus where it forms heterodimers with small Maf proteins, and transactivates its downstream genes through binding with antioxidant responsive elements (AREs) [7]. Genetic alterations such as mutations (gain of function mutations of NRF2 and loss of function mutations in KEAP1 and CUL3), and copy-number changes (amplification of NRF2 and deletion of KEAP1 and CUL3) leads to oncogenesis and drug- and radio-resistance in different types of cancers including HNSCC [8, 9]. Due to the dysregulated NRF2 activity in different cancers, it is emerging as a promising therapeutic target in drug discovery [10, 11].
Stacy et al. [12] first reported the increased expression of NRF2 in HNSCC patients and suggested that NRF2 might be a biomarker. Another report from Huang et al. [13] found the increased expression of KEAP1 and NRF2 in oral squamous cell carcinoma. However, in their report, overall survival analysis of patients with increased expression of KEAP1 and NRF2 did not reveal significant differences. Recently, The Cancer Genome Atlas (TCGA) has provided a wealth of information about KEAP1-NRF2-CUL3 changes in HNSCC patients [14]. Therefore, examining the molecular mechanisms involved in these alterations by using publicly available data may contribute to the development and design of therapeutic targets for personalized/precision medicine in subsets of patients. Several emerging studies including our recent study on lung cancer have identified an NRF2-regulated gene signature and potential biomarkers for patient survival and NRF2 activity [15–18].
Given the importance of KEAP1-NRF2-CUL3 changes in HNSCC, it is important to identify the biomarkers that determine patient survival and NRF2 activity. A recent analysis on TCGA-HNSCC data revealed that patients with disruption of the KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex have significantly poorer survival than non-disrupted counterparts [19]. However, their study specifically focused on the data from patients with a disrupted KEAP1/CUL3/RBX1 complex, but not the data from samples in which NRF2 was altered. In addition they utilized 302 patients data which contains provisional information in their study and overall survival analysis was limited to one cohort. In our study, we restricted the patients samples number (n = 279) which were reported in the TCGA publication [14] and excluded provisional data. Moreover, we analyzed the TCGA-HNSCC [14] RNA-Seq data and identified a 17-gene signature that was highly expressed in samples with altered KEAP1-NRF2-CUL3 compared with both normal and wild-type counterparts. Further, we showed that genomic changes in KEAP1-NRF2-CUL3 were key effectors of the overexpression of genes dependent on the NRF2 pathway. Furthermore, we identified known NRF2-regulated genes involved in drug and glutathione metabolism, along with 4 putative KEAP1-NRF2-CUL3-regulated genes. Finally, we found that higher expression of this gene signature was significantly associated with poorer survival in 4 HNSCC cohorts.
Methods
Samples and transcriptomic profile datasets
We obtained RNA-Seq gene expression version2 (RNA-SeqV2) level 3 data (Illumina Hiseq platform) from HNSCC patients along with adjacent normal tissues from the Broad GDAC Firehose website (http://gdac.broadinstitute.org/). We carried out the analysis of RNA-Seq data of 279 tumor samples and 37 adjacent normal samples listed in the TCGA network study [14]. All the alteration data for KEAP1-NRF2-CUL3 (KEAP1-mutation/deletion, NRF2-mutation/amplification, and CUL3-muatation/deletion) used in the present study was obtained from cBioportal [20, 21]. In addition to the TCGA-HNSCC RNA-Seq data, three independent HNSCC cohorts microarray data– Saintigny et al. (GSE26549) [22], Jung et al. (E-MTAB-1328) [23], and Cohen et al. (GSE10300) [24] – were also used for overall survival analysis. Our study meets the publication guidelines listed by the TCGA network.
RNA-Seq data analysis
The conventional method of differentially-expressed gene (DEG) analysis involves the comparison of tumor transcriptomic data with normal cell data. However, in recent studies, due to the availability of large sets of tumor samples and fewer adjacent normal datasets, researchers have performed DEG analysis of TCGA data by applying a new method in which the DEGs are identified by comparing altered or mutated tumor samples (including a particular gene/set of genes) with wild-type tumors (caused by factors other than alterations or mutations) [15, 25, 26].
Despite the fact that these two methods have been used separately for DEG analysis, in this study, we applied a combinatorial approach to obtain DEGs from HNSCC patients by using both conventional and new methods. We then integrated the resulting upregulated genes from both datasets to obtain overlapping genes. This approach led to the robust identification of more markedly upregulated genes specific to the samples with altered KEAP1-NRF2-CUL3 than in both normal and wild-type samples. Moreover, our method not only identified specific genes targeted by the KEAP1-NRF2-CUL3 axis but also minimized false-positive results.
We segregated the 279 HNSCC tumor samples into two groups: 54 altered KEAP1-NRF2-CUL3 samples (referred to below as ‘altered’) and 225 wild-type samples. Before performing transcriptomic data analysis, the TCGA barcodes of patient data were cross-checked to avoid technical errors. First, we carried out DEG analysis in the 54 altered versus 37 normal samples followed by 54 altered versus 225 wild-type samples using the R/Bioconductor package [27] – edgeR [28]. To crosscheck how our combinatorial approach effectively found specific genes targeted by the KEAP1-NRF2-CUL3 axis, we also subjected the 225 wild-type and 37 normal samples to DEG analysis. Briefly, the raw counts of RNA-SeqV2 level 3 data were filtered by removing the genes containing zero values. We then considered the genes with >100 counts per million in at least two samples for normalization using the trimmed mean of M-values method, followed by the estimation of dispersions using generalized linear models. Up- and down-regulated genes for altered versus normal and altered versus wild-type samples were identified separately by applying a Benjamini-Hochberg (BH) false-discovery rate (FDR) p < 0.01 with a log-fold change (logFC) > 1.5 and <−1.5. Finally, we used the overlapping upregulated genes obtained from both datasets using ‘Venny 2.1’ (http://bioinfogp.cnb.csic.es/tools/venny/index.html) for further analysis. Hierarchical clustering of overlapping upregulated genes was performed using the ‘Heatmapper’ web tool [29]. Box plots of the overlapping upregulated genes that represent the log (counts per million) expression values were generated using R-package ‘ggplot2’ [30]. The overall workflow of the study design is presented in Fig. 1.
Fig. 1.
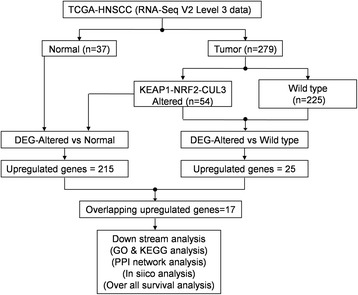
Overview of transcriptomic analysis of TCGA-HNSCC RNA-Seq data. DEG, differentially-expressed genes
Functional annotation and protein-protein interaction (PPI) network analysis
Functional annotation (Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis) of overlapping upregulated genes was performed using the updated version of the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8 web tool [31]. PPI network analysis was performed using the STRING v10 database [32].
Identification of NRF2-binding sites by in silico analysis
To identify the NRF2 binding sites within the promoter regions of the putative KEAP1-NRF2-CUL3-regulated genes, we used the transcription factor-binding site finding tool LASAGNA-Search 2.0 [33] with cutoff p-values ≤ 0.001. The search was limited to the -5 kb upstream promoter region relative to the transcription start site.
Survival analysis
Cox proportional hazard regression was performed using the online survival analysis and biomarker validation tool SurvExpress [34]. We considered the data from a total of 502 patients in 4 independent HNSCC cohorts available in the SurvExpress database: the TCGA-HNSCC cohort (n = 283) with other three HNSCC cohorts – Saintigny et al. (GSE26549) (n = 86) [22], Jung et al. (E-MTAB-1328) (n = 89) [23], and Cohen et al. (GSE10300) (n = 44) [24] – for survival analysis. In the case of microarray-based survival data, we considered the average values for genes whose expression was associated with multiple probe sets such as duplicates or alternatives. SurvExpress separated the patient samples into two groups, high - and low-risk, based on average expression of the 17 genes signature values, and performed statistical analysis of survival probability of the two groups using the log-rank method. SurvExpress used the log-rank test to generate Kaplan-Meir plots based on the ‘Survival’ package of the R platform, which is integrated into its website. Log-rank test p-values < 0.05 were considered to be statistically significant.
Results
Overview of genetic alterations in the KEAP1-NRF2-CUL3axis
In HNSCC, changes in the KEAP1-NRF2-CUL3 axis occurred in ~20% of patients; of these, KEAP1 alterations accounted for 4.6%, NRF2 for 11.8%, and CUL3 for 5.7%. However, few samples overlapped (Fig. 2a). In order to better understand the KEAP1-NRF2-CUL3 mutational landscape in HNSCC, we used the cBioportal cancer genomics website [20, 21] to examine the types of mutation and their positions in the domain structure of proteins. All 13 KEAP1 and 18 NRF2 mutations were missense mutations, while 70% of the CUL3 mutations (7/10) were missense, 20% (2/10) were nonsense, and 10% (1/10) were splice mutations (Fig. 2b).
Fig. 2.
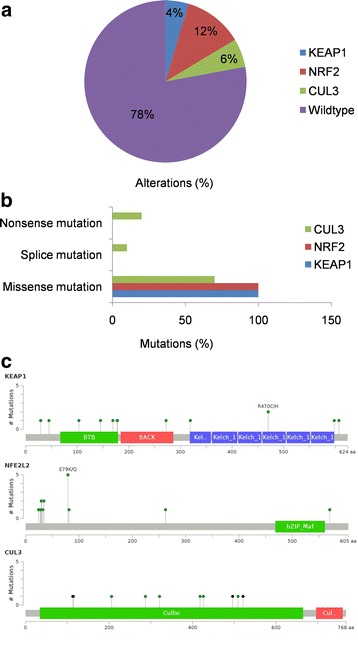
Overview of genetic changes in KEAP1-NRF2-CUL3 in TCGA-HNSCC patients. a Pie chart showing individual percentages of genetic alterations in the KEAP1-NRF2-CUL3 complex. b Bar chart showing the types and percentages of mutations of the KEAP1-NRF2-CUL3 complex. c cBioportal-predicted mutation maps (lollipop plots) showing the positions of mutations on the functional domains of KEAP1, NRF2, and CUL3 proteins. The colored lollipops show the positions of the mutations as identified by whole-exon sequencing
KEAP1 consists of 605 amino-acids with 3 domains in which 6 mutations were reported in the BTB (broad-complex, tramtrack, and bric-a-brac) domain, 1 in the IVR (intervening region), 1 in the C-terminal, 1 in the N-terminal region, and another 4 were in the Kelch domain, which is essential for the binding of NRF2. In the case of NRF2 structure, the majority of mutations (16) occurred in the crucial KEAP1-binding domain Neh2, and another 2 were found in each of the Neh7 and Neh3 domains. CUL3 contained 4 mutations in the N-terminal domain, 5 in the C-terminal domain, and 1 in the cullin repeat 3 domain (Fig. 2c). Overall, two samples contained both KEAP1 and NRF2 mutations, while one sample contained both NRF2 and CUL3 mutations. KEAP1 and CUL3 mutations were mutually exclusive.
Identification of genes regulated by the KEAP1-NRF2-CUL3 axis in HNSCC
In order to identify the genes regulated by the KEAP1-NRF2-CUL3 axis in HNSCC, we focused on the identification of differentially expressed genes by analyzing the RNA-Seq expression profiles in 54 altered versus 37 normal, and 54 altered versus 225 wild-type samples. A total of 215 upregulated genes and 9 downregulated genes were found in the altered versus normal analysis (Additional file 1: Table S1), and 25 upregulated genes and 13 downregulated genes in the altered versus wild-type analysis (Additional file 2: Table S2) with logFC >1.5 (p < 0.01 with BH-FDR adjustment). Since the ultimate effect of KEAP1-NRF2-CUL3 axis gene alterations leads to overexpression of NRF2 and its downstream genes, we focused on the upregulated genes for further analysis. By integrating both datasets using Venny web tool (http://bioinfogp.cnb.csic.es/tools/venny/index.html), we obtained 17 overlapping upregulated genes (Fig. 3a). We carried out literature survey to verify whether the downregulated genes obtained from both methods contains previously reported NRF2 regulated genes or not. Notably, we didn’t observe any previously reported NRF2 target genes among all downregulated genes.
Fig. 3.
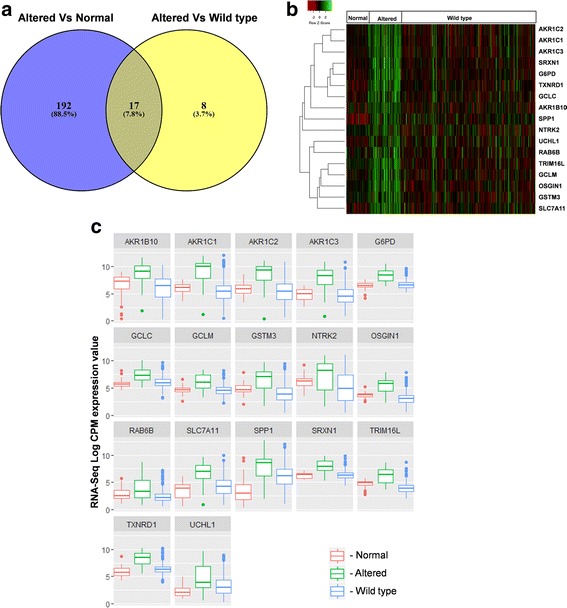
Identification of expression signature of genes regulated by KEAP1-NRF2-CUL3 axis in TCGA-HNSCC. a Venn diagram of overlapping genes from both altered versus normal and altered versus wild-type upregulated gene analysis in HNSCC. b Hierarchical clustering of normal, altered, and wild-type cases showing the specific expression pattern of the 17-gene signature. Green, relatively high expression; red, relatively low expression. c Box plots of 17-gene signature illustrating significant differences of expression in normal, altered, and wild-type cases. X-axis, RNA-Seq V2 log CPM (counts per million) values
We also carried out DEG analysis in 225 wild-type versus 37 normal samples to assess the specificity of the 17 genes regulated by the KEAP1-NRF2-CUL3 axis. Strikingly, none of the 17 genes were found in the list of upregulated genes in the wild-type versus normal samples with logFC > 1.5 (p < 0.01 with BH-FDR adjustment; Additional file 3: Table S3). Thus, our analysis clearly showed that these 17 genes were significantly overexpressed in altered KEAP1-NRF2-CUL3 samples compared with their normal and wild-type counterparts (Fig. 3b, c). We then designated these 17 genes as the signature of gene expression regulated by the KEAP1-NRF2-CUL3 axis based on their specificity and higher expression (Table 1). Among these 17 genes, 13 – AKR1B10, AKR1C1, AKR1C2, AKR1C3, G6PD, GCLC, GCLM, GSTM3, OSGIN1, SRXN1, TXNRD1, SLC7A11 [11, 35, 36], and SPP1 [37]– are well-known NRF2-regulated genes, listed and reviewed in a wide variety of studies.
Table 1.
List of 17 upregulated KEAP1-NRF2-CUL3 axis genes identified in HNSCC
| Gene symbol | Description |
|---|---|
| AKR1B10 | Aldo-keto reductase family 1 member B10 |
| AKR1C1 | Aldo-keto reductase family 1 member C1 |
| AKR1C2 | Aldo-keto reductase family 1 member C2 |
| AKR1C3 | Aldo-keto reductase family 1 member C3 |
| G6PD | Glucose-6-phosphate dehydrogenase |
| GCLC | Glutamate-cysteine ligase catalytic subunit |
| GCLM | Glutamate-cysteine ligase modifier subunit |
| GSTM3 | Glutathione S-transferase mu 3 |
| NTRK2 | Neurotrophic receptor tyrosine kinase 2 |
| OSGIN1 | Oxidative stress induced growth inhibitor 1 |
| RAB6B | RAB6B, member RAS oncogene family |
| SLC7A11 | Solute carrier family 7 member 11 |
| SPP1 | Secreted phosphoprotein 1 |
| SRXN1 | Sulfiredoxin 1 |
| TRIM16L | Tripartite motif containing 16-like |
| TXNRD1 | Thioredoxin reductase 1 |
| UCHL1 | Ubiquitin C-terminal hydrolase L1 |
NRF2 binds with the ARE sequences of 3 putative genes identified in the 17-gene signature
Since the ultimate effect of KEAP1-NRF2-CUL3 gene alterations results in the overexpression of NRF2 and its target genes, it was not surprising that the majority of genes in our results were well-characterized NRF2-regulated genes. In addition, we found 4 putative KEAP1-NRF2-CUL3-regulated genes, NTRK2 (neurotrophic receptor tyrosine kinase 2), RAB6B, TRIM16L, and UCHL1 and investigated whether they were also regulated by NRF2. Interestingly, further in silico analysis using the ‘LASAGNA-Search 2.0’ [33] bioinformatics tool identified NRF2-ARE sequences within the -5 kb upstream promoter regions of the human RAB6B, UCHL1 and TRIM16L genes (Fig. 4a,b,c; Additional file 4: Table S4). However, we did not find an ARE sequence in the promoter region of the NTRK2 gene. Together, our results suggest that NRF2 directly binds with the promoter regions of 16 of the genes in the signature and triggers their overexpression; NTRK2 is the exception.
Fig. 4.
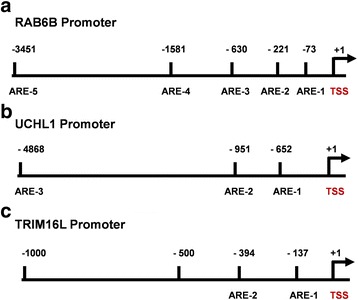
In silico analysis of NRF2 binding sites. Schematic representation shows positions of in silico predicted NRF2 binding sites (AREs) in the promoter regions of human (a), RAB6B, (b), UCHL1, (c), TRIM16L genes
Functional annotation of the gene expression signature regulated by the KEAP1-NRF2-CUL3 axis
Functional annotation analysis from GO and KEGG pathway predictions using both DAVID and STRING v10 revealed that the 17 genes were significantly enriched (p < 0.001) in the biological processes daunorubicin metabolic process, doxorubicin metabolic process, oxidation-reduction process, cellular response to jasmonic acid stimulus, progesterone metabolic process, response to oxidative stress, and steroid metabolic process. In KEGG pathway analysis, we found significant enrichment (p < 0.005) in the three pathways glutathione metabolism, steroid hormone biosynthesis, and metabolism of xenobiotics by cytochrome P450 (Table 2).
Table 2.
GO and KEGG pathway analysis of 17 KEAP1-NRF2-CUL3 axis regulated genes in HNSCC
| Term | p-value | Genes |
|---|---|---|
| GO_Biological Proceess (GO_BP) | ||
| GO:0044597~daunorubicin metabolic process | 3.22E-08 | AKR1C3, AKR1C2, AKR1B10, AKR1C1 |
| GO:0044598~doxorubicin metabolic process | 3.22E-08 | AKR1C3, AKR1C2, AKR1B10, AKR1C1 |
| GO:0055114~oxidation-reduction process | 3.29E-07 | AKR1C3, AKR1C2, G6PD, AKR1B10, OSGIN1, TXNRD1, AKR1C1, SRXN1 |
| GO:0071395~cellular response to jasmonic acid stimulus | 4.46E-06 | AKR1C3, AKR1C2, AKR1C1 |
| GO:0042448~progesterone metabolic process | 2.67E-05 | AKR1C3, AKR1C2, AKR1C1 |
| GO:0006979~response to oxidative stress | 1.18E-04 | GCLC, GCLM, SRXN1, SLC7A11 |
| GO:0008202~steroid metabolic process | 6.58E-04 | AKR1C3, AKR1C2, AKR1B10 |
| KEGG Pathway | ||
| hsa00480:Glutathione metabolism | 5.9956E-05 | GSTM3, G6PD, GCLC, GCLM |
| hsa00140:Steroid hormone biosynthesis | 0.00362777 | AKR1C3, AKR1C2, AKR1C1 |
| hsa00980:Metabolism of xenobiotics by cytochrome P450 | 0.00584594 | AKR1C2, GSTM3, AKR1C1 |
The 17-gene signature is significantly associated with poor survival in TCGA-HNSCC patients
To evaluate the prognostic value of the 17-gene signature in patient survival, we first analyzed overall survival in the TCGA-HNSCC cohort available in the SurvExpress web tool. A total of 283 patient samples were divided into high-risk (n = 141) and low-risk groups (n = 142) based on their expression pattern (Fig. 5a). The survival probability estimates in the two risk groups were visualized as Kaplan-Meier plots. Strikingly, overall survival analysis revealed that the patients in the high-risk group had poorer survival (HR = 2.28; CI = 1.56–3.32; p = 1.221e-05) than the low-risk group (Fig. 5b). Thus, our analysis strongly suggests that genes regulated by the KEAP1-NRF2-CUL3 axis are powerful predictors of a poor prognosis in HNSCC patients. In addition, we also carried out the multivariate analysis with the limited variables present in Survexpress database. Consistent with the above results, patients with high-risk scores for clinical variables such as tumor grades G2 and G3, pathological stages T1 and T2, and pathological disease stages II and III were significantly associated with poor survival whereas the results were insignificant in other variables (Additional file 5: Table S5). Kaplan-Meier survival plots with log-rank test results for the significant clinical variables are shown in Additional file 6: Figure S1.
Fig. 5.
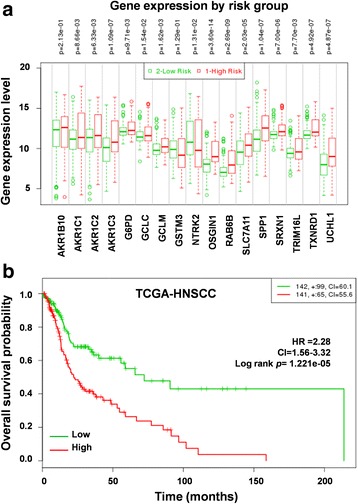
Correlation of 17-gene signature with poor survival in TCGA-HNSCC patients. a Box plots of the expression differences of the 17-gene signature in low (green) and high (red) risk groups of TCGA-HNSCC patients. X-axis, gene expression value of each gene; above the box plot, p-values of the expression difference between risk groups. b Kaplan-Meier survival plots showing that high expression of the 17-gene signature is associated with poor survival in TCGA-HNSCC patients. Red, high-risk group; green, low-risk group; top right corner inset, numbers of high- and low-risk samples, numbers of censored samples marked with + and concordance index (CI) of each risk group; X-axis, time (months); Y-axis, overall survival probability; HR, hazard ratio; CI, confidence interval
Association of 17-gene signature with disease-free survival (DFS), metastasis-free survival (MFS), and recurrence in HNSCC patients
After analyzing the prognostic value of the 17-gene signature in the TCGA cohort, we evaluated its prognostic value in another 3 HNSCC cohorts containing DFS, MFS, and recurrence data. Among these, Saintigny et al. (GSE26549) [22] contains DFS data, while Jung et al. (E-MTAB-1328) [23] contains MFS data. The third cohort, Cohen et al. (GSE10300) [24], contains recurrence data. Interestingly, our DFS analysis using the Saintigny et al. (GSE26549) [22] cohort showed that patients in the high-risk group with increased expression of the 17-gene signature had poorer survival (HR = 2.28; CI = 1.56–3.32; p = 1.221e-05) than the low-risk group (Fig. 6a). Likewise, we found a markedly shorter MFS (HR = 2.83, CI = 1.47–5.48; p = 0.001) in the high-risk group of the Jung et al. (E-MTAB-1328) [23] cohort (Fig. 6b). In the Cohen et al. (GSE10300) [24] cohort, we found lower recurrence-free survival (HR = 4.15; CI = 1.14–15.05; p < 0.01) in the high-risk group with the17-gene signature than in the low-risk group (Fig. 6c). Thus, log-rank analysis revealed that the 17-gene signature was associated with a significantly increased risk of recurrence in HNSCC. The multivariate analysis results for the above cohorts were listed in Additional file 5: Table S5.
Fig. 6.
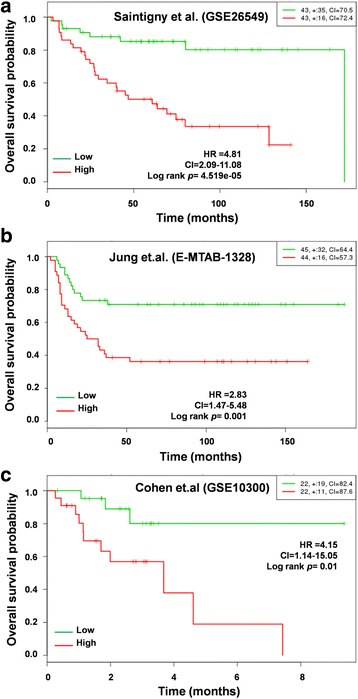
17-gene signature predicts poor survival in three independent cohorts. Kaplan-Meier survival plots showing that high expression of the 17-gene signature is associated with poor survival in 3 independent HNSCC cohorts: a Saintigny et al. (GSE26549). b Jung et al. (E-MTAB-1328). c Cohen et al. (GSE10300). Red, high-risk group; green, low-risk group
Discussion
The TCGA network provides valuable information about genetic changes in key genes involved in the oxidative-stress pathway, such as KEAP1, NRF2, and CUL3, in HNSCC patients. These particular data permit researchers to identify potential biomarkers, druggable mutations, and therapeutic targets for personalized medicine. In this study, using a new approach that consisted of two RNA-Seq DEG analysis methods, we identified a common set of 17 genes regulated by the KEAP1-NRF2-CUL3 axis that constitute an expression signature in TCGA-HNSCC patients. We further tested this signature in 4 independent clinical cohorts including the TCGA-HNSCC cohort. Kaplan-Meier survival plots generated for all 4 cohorts showed that higher expression of this gene signature is significantly correlated with poor survival outcomes.
The DFS data of Saintigny et al. (GSE26549) [22] suggested that patients with an increased 17-gene signature had poor benefit from chemotherapy because of aggressive expression of genes downstream of NRF2 that are involved in chemoresistance. Our GO and KEGG analysis of the 17-gene signature strongly supported the above conclusion. The top two enriched GO biological process terms were ‘daunorubicin metabolic process’ and ‘doxorubicin metabolic process’, clearly indicating that the genes involved in these processes, such as AKR (aldo-keto reductase) 1C3, AKR1C2, AKR1B10, and AKR1C1, are crucial drug-metabolizing enzymes whose overexpression is strongly associated with drug resistance in many cancers [38, 39] (Table 2). Aldo-keto reductases are well-characterized NRF2-regulated genes which contain consensus ARE sequences in their promoter regions for the binding and transactivation of NRF2 [39–41]. A recent lung cancer study emphasized that a panel of aldo-keto reductase family genes are markedly upregulated in patients harboring somatic alterations in the NRF2 pathway and considered to be biomarkers of NRF2 hyperactivation in lung cancer [17]. Consistent with their study, we showed that aldo-keto reductases were not only highly expressed in lung cancer but also in HNSCC patients with a dysregulated NRF2 pathway and could be used as biomarkers.
More interestingly, the top hit in the KEGG pathway analysis of the 17-gene signature identified an important pathway involved in oxido-reductase activity known as ‘glutathione metabolism’(Table 2). The genes listed in this pathway, such as GSTM3, G6PD, GCLC, and GCLM, play major roles in redox balance in normal cells. The redox imbalance in cancer cells because of the overexpression of these genes mainly leads to tumor growth and drug resistance [42]. Thus, our study revealed that NRF2 drives the expression of genes involved in glutathione metabolism, so the development of NRF2 inhibitors could be a means of altering tumor growth and drug resistance in HNSCC. A very interesting recent study on the inhibition of NRF2, glutathione (GSH), and thioredoxin (Trx) in head and neck cancer (HNC) strongly supports our prediction that combined inhibition of the GSH, Trx, and NRF2 pathways could be an effective strategy to overcome therapeutic resistance in HNC [43].
In addition to the GO and KEGG analyses, we used the STRING v10 database to construct a PPI network of the 17-gene signature along with the KEAP1, NRF2, and CUL3 genes to reveal the complex associations between these genes. The enrichment results based on functional association between these genes revealed that the majority were closely associated with each other through a coordinated interactive network (Fig. 7). Thus, PPI network analysis suggested that the cross-talk of KEAP1, NRF2, and CUL3 with the 17-gene signature coordinately drives tumor progression and therapeutic resistance in HNSCC.
Fig. 7.
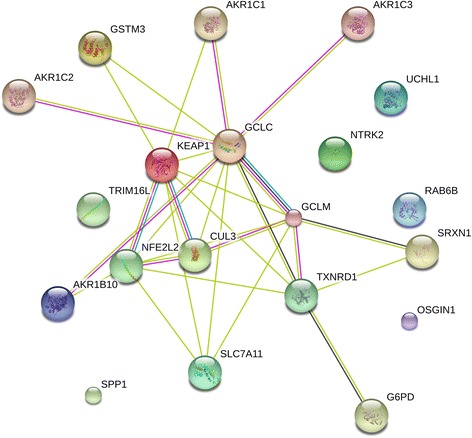
Protein-protein interaction network analysis of the 17-gene signature predicting the functional correlation of the KEAP1-NRF2-CUL3 axis with genes involved in drug metabolism and glutathione metabolic pathways in HNSCC
Apart from known NRF2-regulated genes, we found 4 putative KEAP1-NRF2-CUL3 axis-regulated genes: NTRK2, RAB6B, TRIM16L, and UCHL1. NTRK2, also known as tropomyosin receptor kinase B, is a neurotrophin-binding protein that phosphorylates members of the MAPK pathway. This receptor plays a major role in cell differentiation, specifically neuronal proliferation, differentiation, and survival, through its kinase signaling cascade [44]. Emerging evidence suggests that NTRK2 plays an important role in different cancers. For instance, it has been reported to be highly expressed in non-small cell lung cancer A549 cells [45] and is associated with a worse outcome in patients with Wilms’ tumor [46].
Although NRF2-ARE sequences were not found in the NTRK2 promoter region, we looked into why NTRK2 was highly upregulated in altered samples. Surprisingly, a recent report revealed that NTRK2 inhibits KEAP1 expression in breast cancer cells and is involved in cancer proliferation, survival, and metastasis [47]. Thus, the overexpression of NTRK2 in altered samples clearly suggests that NTRK2 inhibits the expression of KEAP1, initiates the hyperactivation of genes downstream of NRF2, and is involved in HNSCC tumorigenesis. Another putative KEAP1-NRF2-CUL3 gene, UCHL1 (ubiquitin C-terminal hydrolase L1), has also been implicated in different types of human cancer such as breast [48, 49], melanoma [50], ovarian [51], colorectal [52], osteosarcoma [53], and gastric [54, 55] cancers, and multiple myeloma [56]. Most of the cancer studies on UCHL1 have revealed that overexpression and promoter methylation of UCHL1 are key reasons for UCHL1-mediated metastasis. Due to the adverse effect of overexpression of UCHL1, it is considered to be a biomarker and a therapeutic target in many cancers. The exact functions of the other two putative KEAP1-NRF2-CUL3axis-regulated genes, RAB6B and TRIM16L, are unknown in cancer cells and therefore are under investigation in our lab. Altogether, the above evidence suggests an oncogenic role of the 17-gene signature in many cancers.
Conclusions
In conclusion, we have identified a comprehensive gene signature of the KEAP1-NRF2-CUL3 axis, increased expression of which predicts poor survival in HNSCC. Moreover, the components of this 17-gene signature can be used as potential biomarkers to identify genetic alterations of the NRF2 pathway in HNSCC. Furthermore, the development of combined inhibitors for this 17-gene signature, along with NRF2, could pave the way for the development of personalized/precision medicine to suppress NRF2-mediated tumor growth and drug resistance.
Additional files
List of differentially expressed genes obtained from the RNA-Seq analysis of altered versus normal samples. (XLS 48 kb)
List of differentially expressed genes obtained from the RNA-Seq analysis of altered versus wild-type samples. (XLS 29 kb)
List of differentially expressed genes obtained from the RNA-Seq analysis of wild-type versus normal samples. (XLS 49 kb)
List of NRF2-AREs identified in the -5 kb promoter regions of RAB6B, UCHL1 and TRIM16L genes. (XLS 38 kb)
Multivariate analysis of 17-gene signature in 4 independent cohorts (XLS 25 kb)
Kaplan-Meier plots showing the survival analysis of TCGA-HNSCC cohort clinical variables: tumor grades G2 (A) and G3 (B); pathologic T stagesT1 (C) and T2 (D); and pathologic disease stages II (E) and III (F). (TIFF 798 kb)
Acknowledgements
The authors would like to thank the TCGA network for providing publicly available NGS data.
Funding
This work was supported by the National Natural Science Foundation of China to XT (31,170,743 and 81,172,230).
Availability of data and materials
The TCGA dataset and other patients microarray datas utilized in this study are publicly available and mentioned in the article.
Abbreviations
- AKR1B10
Aldo-keto reductase family 1 member B10
- AKR1C1
Aldo-keto reductase family 1 member C1
- AKR1C2
Aldo-keto reductase family 1 member C2
- AKR1C3
Aldo-keto reductase family 1 member C3
- ARE
Antioxidant responsive element
- BH
Benjamini-Hochberg
- CUL3
Cullin-dependent E3 ligase
- DAVID
Database for annotation visualization and integrated discovery
- DEG
Differential Expression Genes
- DFS
Disease free survival
- edgeR
Empirical Analysis of Digital Gene Expression Data in R
- FDR
False discovery rate
- G6PD
Glucose-6-phosphate dehydrogenase
- GCLC
Glutamate-cysteine ligase catalytic subunit
- GCLM
Glutamate-cysteine ligase modifier subunit
- GDAC
Genome Data Analysis Center
- GO
Gene ontology
- GSH
glutathione
- GSTM3
Glutathione S-transferase mu 3
- HNSCCC
Head and Neck Squamous Cell Cancer
- KEAP1
Kelch like-ECH-associated protein 1
- KEGG
Kyoto Encyclopedia of Genes and Genomes
- MFS
Metastasis-free survival
- NRF2
Nuclear factor erythroid 2-related factor
- NTRK2
Neurotrophic receptor tyrosine kinase 2
- OSGIN1
Oxidative stress induced growth inhibitor 1
- PPI
Protein-Protein interaction
- RAB6B
RAB6B, member RAS oncogene family
- RBX1
Ring-Box 1
- RT-qPCR
Reverse transcription–quantitative polymerase chain reaction
- SLC7A11
Solute carrier family 7 member 11
- SPP1
Secreted phosphoprotein 1
- SRXN1
Sulforedoxin 1
- TCGA
The Cancer Genome Atlas
- TRIM16L
Tripartite motif containing 16-like
- TrkB
Tropomyosin receptor kinase B
- Trx
Thioredoxin
- TXNRD1
Thioredoxin reductase 1
- UCHL1
Ubiquitin C-terminal hydrolase L1
Authors’ contributions
XT and AN conceived the project; AN, Md-MR, MC and XT analyzed the data and drafted the manuscript; MC had critically read the manuscript; XT edited and reviewed the manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to participate
Not required.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Footnotes
Electronic supplementary material
The online version of this article (10.1186/s12885-017-3907-z) contains supplementary material, which is available to authorized users.
Contributor Information
Akhileshwar Namani, Email: akhileshwar_namani@yahoo.co.in.
Md. Matiur Rahaman, Email: matiur.stat@gmail.com
Ming Chen, Email: mchen@zju.edu.cn.
Xiuwen Tang, Phone: +0086-(0)571-88208266, Email: xiuwentang@zju.edu.cn.
References
- 1.Mehanna H, Paleri V, West CM, Nutting C. Head and neck cancer--part 1: epidemiology, presentation, and prevention. BMJ. 2010;341:c4684. doi: 10.1136/bmj.c4684. [DOI] [PubMed] [Google Scholar]
- 2.Ferlay J, Soerjomataram I, Dikshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Forman D, Bray F. Cancer incidence and mortality worldwide: sources, methods and major patterns in GLOBOCAN 2012. Int J Cancer. 2015;136(5):E359–E386. doi: 10.1002/ijc.29210. [DOI] [PubMed] [Google Scholar]
- 3.Pai SI, Westra WH. Molecular pathology of head and neck cancer: implications for diagnosis, prognosis, and treatment. Annu Rev Pathol. 2009;4:49–70. doi: 10.1146/annurev.pathol.4.110807.092158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Marron M, Boffetta P, Zhang ZF, Zaridze D, Wunsch-Filho V, Winn DM, Wei Q, Talamini R, Szeszenia-Dabrowska N, Sturgis EM, et al. Cessation of alcohol drinking, tobacco smoking and the reversal of head and neck cancer risk. Int J Epidemiol. 2010;39(1):182–196. doi: 10.1093/ije/dyp291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Leemans CR, Braakhuis BJ, Brakenhoff RH. The molecular biology of head and neck cancer. Nat Rev Cancer. 2011;11(1):9–22. doi: 10.1038/nrc2982. [DOI] [PubMed] [Google Scholar]
- 6.Namani A, Li Y, Wang XJ, Tang X. Modulation of NRF2 signaling pathway by nuclear receptors: implications for cancer. Biochim Biophys Acta. 2014;1843(9):1875–1885. doi: 10.1016/j.bbamcr.2014.05.003. [DOI] [PubMed] [Google Scholar]
- 7.Ahmed SM, Luo L, Namani A, Wang XJ, Tang X. Nrf2 signaling pathway: pivotal roles in inflammation. Biochim Biophys Acta. 2017;1863(2):585–597. doi: 10.1016/j.bbadis.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 8.Jaramillo MC, Zhang DD. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev. 2013;27(20):2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang H, Liu K, Geng M, Gao P, Wu X, Hai Y, Li Y, Luo L, Hayes JD, Wang XJ, et al. RXRalpha inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 2013;73(10):3097–3108. doi: 10.1158/0008-5472.CAN-12-3386. [DOI] [PubMed] [Google Scholar]
- 10.Tang X, Wang H, Fan L, Wu X, Xin A, Ren H, Wang XJ. Luteolin inhibits Nrf2 leading to negative regulation of the Nrf2/ARE pathway and sensitization of human lung carcinoma A549 cells to therapeutic drugs. Free Radic Biol Med. 2011;50(11):1599–1609. doi: 10.1016/j.freeradbiomed.2011.03.008. [DOI] [PubMed] [Google Scholar]
- 11.Suzuki T, Motohashi H, Yamamoto M. Toward clinical application of the Keap1-Nrf2 pathway. Trends Pharmacol Sci. 2013;34(6):340–346. doi: 10.1016/j.tips.2013.04.005. [DOI] [PubMed] [Google Scholar]
- 12.Stacy DR, Ely K, Massion PP, Yarbrough WG, Hallahan DE, Sekhar KR, Freeman ML. Increased expression of nuclear factor E2 p45-related factor 2 (NRF2) in head and neck squamous cell carcinomas. Head Neck. 2006;28(9):813–818. doi: 10.1002/hed.20430. [DOI] [PubMed] [Google Scholar]
- 13.Huang CF, Zhang L, Ma SR, Zhao ZL, Wang WM, He KF, Zhao YF, Zhang WF, Liu B, Sun ZJ. Clinical significance of Keap1 and Nrf2 in oral squamous cell carcinoma. PLoS One. 2013;8(12):e83479. doi: 10.1371/journal.pone.0083479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.The Cancer Genome Atlas Network (348 collaborators). Comprehensive genomic characterization of head and neck squamous cell carcinomas. Nature. 2015;517(7536):576–82. [DOI] [PMC free article] [PubMed]
- 15.Cescon DW, She D, Sakashita S, Zhu CQ, Pintilie M, Shepherd FA, Tsao MS. NRF2 pathway activation and adjuvant chemotherapy benefit in lung Squamous cell carcinoma. Clin Cancer Res. 2015;21(11):2499–2505. doi: 10.1158/1078-0432.CCR-14-2206. [DOI] [PubMed] [Google Scholar]
- 16.Qian Z, Zhou T, Gurguis CI, Xu X, Wen Q, Lv J, Fang F, Hecker L, Cress AE, Natarajan V, et al. Nuclear factor, erythroid 2-like 2-associated molecular signature predicts lung cancer survival. Sci Rep. 2015;5:16889. doi: 10.1038/srep16889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.MacLeod AK, Acosta-Jimenez L, Coates PJ, McMahon M, Carey FA, Honda T, Henderson CJ, Wolf CR. Aldo-keto reductases are biomarkers of NRF2 activity and are co-ordinately overexpressed in non-small cell lung cancer. Br J Cancer. 2016;115(12):1530–1539. doi: 10.1038/bjc.2016.363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Namani A, Cui QQ, Wu Y, Wang H, Wang XJ, Tang X. NRF2-regulated metabolic gene signature as a prognostic biomarker in non-small cell lung cancer. Oncotarget. 2017;8(41):69847–69862. doi: 10.18632/oncotarget.19349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Martinez VD, Vucic EA, Thu KL, Pikor LA, Lam S, Lam WL. Disruption of KEAP1/CUL3/RBX1 E3-ubiquitin ligase complex components by multiple genetic mechanisms: association with poor prognosis in head and neck cancer. Head Neck. 2015;37(5):727–734. doi: 10.1002/hed.23663. [DOI] [PubMed] [Google Scholar]
- 20.Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discovery. 2012;2(5):401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6(269):pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Saintigny P, Zhang L, Fan YH, El-Naggar AK, Papadimitrakopoulou VA, Feng L, Lee JJ, Kim ES, Ki Hong W, Mao L. Gene expression profiling predicts the development of oral cancer. Cancer Prev Res (Phila) 2011;4(2):218–229. doi: 10.1158/1940-6207.CAPR-10-0155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Jung AC, Job S, Ledrappier S, Macabre C, Abecassis J, de Reynies A, Wasylyk B. A poor prognosis subtype of HNSCC is consistently observed across methylome, transcriptome, and miRNome analysis. Clin Cancer Res. 2013;19(15):4174–4184. doi: 10.1158/1078-0432.CCR-12-3690. [DOI] [PubMed] [Google Scholar]
- 24.Cohen EE, Zhu H, Lingen MW, Martin LE, Kuo WL, Choi EA, Kocherginsky M, Parker JS, Chung CH, Rosner MR. A feed-forward loop involving protein kinase Calpha and microRNAs regulates tumor cell cycle. Cancer Res. 2009;69(1):65–74. doi: 10.1158/0008-5472.CAN-08-0377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Goldstein LD, Lee J, Gnad F, Klijn C, Schaub A, Reeder J, Daemen A, Bakalarski CE, Holcomb T, Shames DS, et al. Recurrent loss of NFE2L2 exon 2 is a mechanism for Nrf2 pathway activation in human cancers. Cell Rep. 2016;16(10):2605–2617. doi: 10.1016/j.celrep.2016.08.010. [DOI] [PubMed] [Google Scholar]
- 26.Morris VK, Lucas FA, Overman MJ, Eng C, Morelli MP, Jiang ZQ, Luthra R, Meric-Bernstam F, Maru D, Scheet P et al: Clinicopathologic characteristics and gene expression analyses of non-KRAS 12/13, RAS-mutated metastatic colorectal cancer. Ann Oncol 2014, 25(10):2008-2014. [DOI] [PMC free article] [PubMed]
- 27.Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 2004;5(10):R80. doi: 10.1186/gb-2004-5-10-r80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Babicki S, Arndt D, Marcu A, Liang Y, Grant JR, Maciejewski A, Wishart DS. Heatmapper: web-enabled heat mapping for all. Nucleic Acids Res. 2016;44(W1):W147–W153. doi: 10.1093/nar/gkw419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wickham H. ggplot2: elegant graphics for data analysis. New York: Springer-Verlag; 2009. [Google Scholar]
- 31.Huang d W, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc. 2009;4(1):44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 32.Szklarczyk D, Morris JH, Cook H, Kuhn M, Wyder S, Simonovic M, Santos A, Doncheva NT, Roth A, Bork P, et al. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lee C, Huang CH. LASAGNA-search 2.0: integrated transcription factor binding site search and visualization in a browser. Bioinformatics. 2014;30(13):1923–1925. doi: 10.1093/bioinformatics/btu115. [DOI] [PubMed] [Google Scholar]
- 34.Aguirre-Gamboa R, Gomez-Rueda H, Martinez-Ledesma E, Martinez-Torteya A, Chacolla-Huaringa R, Rodriguez-Barrientos A, Tamez-Pena JG, Trevino V. SurvExpress: an online biomarker validation tool and database for cancer gene expression data using survival analysis. PLoS One. 2013;8(9):e74250. doi: 10.1371/journal.pone.0074250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chorley BN, Campbell MR, Wang X, Karaca M, Sambandan D, Bangura F, Xue P, Pi J, Kleeberger SR, Bell DA. Identification of novel NRF2-regulated genes by ChIP-Seq: influence on retinoid X receptor alpha. Nucleic Acids Res. 2012;40(15):7416–7429. doi: 10.1093/nar/gks409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hayes JD, Dinkova-Kostova AT. The Nrf2 regulatory network provides an interface between redox and intermediary metabolism. Trends Biochem Sci. 2014;39(4):199–218. doi: 10.1016/j.tibs.2014.02.002. [DOI] [PubMed] [Google Scholar]
- 37.Shibata T, Saito S, Kokubu A, Suzuki T, Yamamoto M, Hirohashi S. Global downstream pathway analysis reveals a dependence of oncogenic NF-E2-related factor 2 mutation on the mTOR growth signaling pathway. Cancer Res. 2010;70(22):9095–9105. doi: 10.1158/0008-5472.CAN-10-0384. [DOI] [PubMed] [Google Scholar]
- 38.Ma J, Luo DX, Huang C, Shen Y, Bu Y, Markwell S, Gao J, Liu J, Zu X, Cao Z, et al. AKR1B10 overexpression in breast cancer: association with tumor size, lymph node metastasis and patient survival and its potential as a novel serum marker. Int J Cancer. 2012;131(6):E862–E871. doi: 10.1002/ijc.27618. [DOI] [PubMed] [Google Scholar]
- 39.Jung KA, Choi BH, Nam CW, Song M, Kim ST, Lee JY, Kwak MK. Identification of aldo-keto reductases as NRF2-target marker genes in human cells. Toxicol Lett. 2013;218(1):39–49. doi: 10.1016/j.toxlet.2012.12.026. [DOI] [PubMed] [Google Scholar]
- 40.Lou H, Du S, Ji Q, Stolz A. Induction of AKR1C2 by phase II inducers: identification of a distal consensus antioxidant response element regulated by NRF2. Mol Pharmacol. 2006;69(5):1662–1672. doi: 10.1124/mol.105.019794. [DOI] [PubMed] [Google Scholar]
- 41.Penning TM, Drury JE. Human aldo-keto reductases: function, gene regulation, and single nucleotide polymorphisms. Arch Biochem Biophys. 2007;464(2):241–250. doi: 10.1016/j.abb.2007.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Trachootham D, Alexandre J, Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat Rev Drug Discov. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 43.Roh JL, Jang H, Kim EH, Shin D. Targeting of the glutathione, Thioredoxin, and Nrf2 antioxidant Systems in Head and Neck Cancer. Antioxid Redox Signal. 2017;27(2):106–14. [DOI] [PubMed]
- 44.Minichiello L. TrkB signalling pathways in LTP and learning. Nat Rev Neurosci. 2009;10(12):850–860. doi: 10.1038/nrn2738. [DOI] [PubMed] [Google Scholar]
- 45.Zhang S, Guo D, Luo W, Zhang Q, Zhang Y, Li C, Lu Y, Cui Z, Qiu X. TrkB is highly expressed in NSCLC and mediates BDNF-induced the activation of Pyk2 signaling and the invasion of A549 cells. BMC Cancer. 2010;10:43. doi: 10.1186/1471-2407-10-43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Eggert A, Grotzer MA, Ikegaki N, Zhao H, Cnaan A, Brodeur GM, Evans AE. Expression of the neurotrophin receptor TrkB is associated with unfavorable outcome in Wilms' tumor. J Clin Oncol Off J Am Soc Clin Oncol. 2001;19(3):689–696. doi: 10.1200/JCO.2001.19.3.689. [DOI] [PubMed] [Google Scholar]
- 47.Kim MS, Lee WS, Jin W. TrkB promotes breast cancer metastasis via suppression of Runx3 and Keap1 expression. Molecules Cells. 2016;39(3):258–265. doi: 10.14348/molcells.2016.2310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Schroder C, Milde-Langosch K, Gebauer F, Schmid K, Mueller V, Wirtz RM, Meyer-Schwesinger C, Schluter H, Sauter G, Schumacher U. Prognostic relevance of ubiquitin C-terminal hydrolase L1 (UCH-L1) mRNA and protein expression in breast cancer patients. J Cancer Res Clin Oncol. 2013;139(10):1745–1755. doi: 10.1007/s00432-013-1496-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jin Y, Zhang W, Xu J, Wang H, Zhang Z, Chu C, Liu X, Zou Q. UCH-L1 involved in regulating the degradation of EGFR and promoting malignant properties in drug-resistant breast cancer. Int J Clin Exp Pathol. 2015;8(10):12500–12508. [PMC free article] [PubMed] [Google Scholar]
- 50.Wulfanger J, Biehl K, Tetzner A, Wild P, Ikenberg K, Meyer S, Seliger B. Heterogeneous expression and functional relevance of the ubiquitin carboxyl-terminal hydrolase L1 in melanoma. Int J Cancer. 2013;133(11):2522–2532. doi: 10.1002/ijc.28278. [DOI] [PubMed] [Google Scholar]
- 51.Suong DN, Thao DT, Masamitsu Y, Thuoc TL. Ubiquitin carboxyl hydrolase L1 significance for human diseases. Protein Peptide Letters. 2014;21(7):624–630. doi: 10.2174/0929866521666140403125959. [DOI] [PubMed] [Google Scholar]
- 52.Heitzer E, Artl M, Filipits M, Resel M, Graf R, Weissenbacher B, Lax S, Gnant M, Wrba F, Greil R, et al. Differential survival trends of stage II colorectal cancer patients relate to promoter methylation status of PCDH10, SPARC, and UCHL1. Modern Pathol. 2014;27(6):906–915. doi: 10.1038/modpathol.2013.204. [DOI] [PubMed] [Google Scholar]
- 53.Zheng S, Qiao G, Min D, Zhang Z, Lin F, Yang Q, Feng T, Tang L, Sun Y, Zhao H, et al. Heterogeneous expression and biological function of ubiquitin carboxy-terminal hydrolase-L1 in osteosarcoma. Cancer Lett. 2015;359(1):36–46. doi: 10.1016/j.canlet.2014.12.001. [DOI] [PubMed] [Google Scholar]
- 54.Wang G, Zhang W, Zhou B, Jin C, Wang Z, Yang Y, Chen Y, Feng X. The diagnosis value of promoter methylation of UCHL1 in the serum for progression of gastric cancer. Biomed Res Int. 2015;2015:741030. doi: 10.1155/2015/741030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.YY G, Yang M, Zhao M, Luo Q, Yang L, Peng H, Wang J, Huang SK, Zheng ZX, Yuan XH, et al. The de-ubiquitinase UCHL1 promotes gastric cancer metastasis via the Akt and Erk1/2 pathways. Tumor Biol. 2015;36(11):8379–8387. doi: 10.1007/s13277-015-3566-0. [DOI] [PubMed] [Google Scholar]
- 56.Hussain S, Bedekovics T, Chesi M, Bergsagel PL, Galardy PJ. UCHL1 is a biomarker of aggressive multiple myeloma required for disease progression. Oncotarget. 2015;6(38):40704–40718. doi: 10.18632/oncotarget.5727. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
List of differentially expressed genes obtained from the RNA-Seq analysis of altered versus normal samples. (XLS 48 kb)
List of differentially expressed genes obtained from the RNA-Seq analysis of altered versus wild-type samples. (XLS 29 kb)
List of differentially expressed genes obtained from the RNA-Seq analysis of wild-type versus normal samples. (XLS 49 kb)
List of NRF2-AREs identified in the -5 kb promoter regions of RAB6B, UCHL1 and TRIM16L genes. (XLS 38 kb)
Multivariate analysis of 17-gene signature in 4 independent cohorts (XLS 25 kb)
Kaplan-Meier plots showing the survival analysis of TCGA-HNSCC cohort clinical variables: tumor grades G2 (A) and G3 (B); pathologic T stagesT1 (C) and T2 (D); and pathologic disease stages II (E) and III (F). (TIFF 798 kb)
Data Availability Statement
The TCGA dataset and other patients microarray datas utilized in this study are publicly available and mentioned in the article.