

Abstract
Human cytomegalovirus, a member of the herpesvirus family, can cause significant morbidity and mortality in immune compromised patients resulting from either primary lytic infection or reactivation from latency. Latent infection is associated with a restricted viral transcription programme compared to lytic infection which consists of defined protein coding RNAs but also includes a number of virally encoded microRNAs (miRNAs). One of these, miR-UL112-1, is known to target the major lytic IE72 transcript but, to date, a functional role for miR-UL112-1 during latent infection has not been shown. To address this, we have analysed latent infection in myeloid cells using a virus in which the target site for miR-UL112-1 in the 3′ UTR of IE72 was removed such that any IE72 RNA present during latent infection would no longer be subject to regulation by miR-UL112-1 through the RNAi pathway. Our data show that removal of the miR-UL112-1 target site in IE72 results in increased levels of IE72 RNA in experimentally latent primary monocytes. Furthermore, this resulted in induction of immediate early (IE) gene expression that is detectable by IE-specific cytotoxic T-cells (CTLs); no such CTL recognition of monocytes latently infected with wild-type virus was observed. We also recapitulated these findings in the more tractable THP-1 cell line model of latency. These observations argue that an important role for miR-UL112-1 during latency is to ensure tight control of lytic viral immediate early (IE) gene expression thereby preventing recognition of latently infected cells by the host's potent pre-existing anti-viral CTL response.
Keywords: latency, human cytomegalovirus (HCMV), viral miRNAs, immune survelliance
Introduction
Human cytomegalovirus (HCMV) is a species-specific herpesvirus carried by 60–80 % of human populations, depending upon demographics (Ross & Boppana, 2005). In the immune competent, primary infection rarely results in clinical problems. However, in the immune compromised and the neonate, primary HCMV infection can cause significant disease sequelae (Ho, 1990; Zaia, 1990).
Like all herpesviruses, HCMV is never cleared after primary infection but maintains a life-long persistence which is underpinned by the ability of the virus to undergo latent infection (Sinclair & Sissons, 2006). As with primary infection, reactivation of virus from latency in the immune compromised or during pregnancy can also result in severe disease to the immunocompromised individual or the fetus in utero (Adler, 1983; Griffiths & Walter, 2005; Rubin, 1990; Sissons & Carmichael, 2002).
One established site of latent carriage, in vivo, is in cells of the myeloid lineage, including CD14+ monocytes and their CD34+ progenitors (Hahn et al., 1998; Khaiboullina et al., 2004; Mendelson et al., 1996; Taylor-Wiedeman et al., 1991). However, differentiation of these cells to dendritic cells (DCs) or macrophages results in full virus reactivation (Reeves et al., 2005b; Soderberg-Naucler et al., 2001; Taylor-Wiedeman et al., 1994).
During latent infection of monocytes or CD34+ cells, carriage of latent viral genomes is associated with the expression of a restricted pattern of viral genes compared to lytically infected cells (Slobedman et al., 2010); the absence of infectious virion production during this life cycle mitigates the need for expression of much of the virus genome and probably, in itself, reduces the ability of the host's immune response to detect and clear virus-infected cells. Instead, viral gene expression during latency is probably reduced to those that play an important role in establishment and maintenance of the latent life cycle, and functions for proteins encoded by many of these latency-associated transcripts are now beginning to be elucidated (Keyes et al., 2012b; O'Connor & Murphy, 2012; Poole et al., 2014, 2015; Tarrant-Elorza et al., 2014; Weekes et al., 2013). In addition to these, it is also becoming clear that known viral non-coding RNAs are also expressed during latency (Rossetto et al., 2013) and these include viral microRNAs (miRNAs) (Fu et al., 2014; Meshesha et al., 2016; Shen et al., 2014). Although the functional targets of a number of viral miRNAs expressed during lytic infection have been described (for reviews see: Dhuruvasan et al., 2011; Goldberger & Mandelboim, 2014; Hook et al., 2014a), little is known about the role of HCMV-encoded miRNAs during latency.
One viral miRNA expressed during latency in primary myeloid cells is miR-UL112-1 (Meshesha et al., 2016) (B. Lau and others unpublished observations). A number of cellular and viral targets of this miRNA have been validated during lytic infection (Grey, 2015; Huang et al., 2013; Lee et al., 2012); these include multiple components of the host secretory pathway (Hook et al., 2014b), the NK cell activatory ligand MICB (Stern-Ginossar et al., 2008), cellular transcription factor BclAF1 (Lee et al., 2012) as well as the viral IE72 immediate early (IE) gene and viral UL114 (Grey et al., 2007; Murphy et al., 2008; Stern-Ginossar et al., 2009). Furthermore, the targeting of MICB and BclAF1 by miR-UL112-1 was shown to aid immune evasion and virus spreading, respectively, during lytic infection (Lee et al., 2012; Stern-Ginossar et al., 2008). Despite a relative wealth of information about the role of this viral miRNA during lytic infection, nothing is known about the role of miR-UL112-1 during latency. On the basis that miR-UL112-1 targets IE72 expression during lytic infection, we reasoned that an even more crucial role for this viral miRNA could be its ability to target IE72 during latent infection, thereby helping to ensure tight negative regulation of lytic gene expression, and hence virus production, during latent carriage – a known hallmark of latency.
Here, we have analysed whether the ability of miR-UL112-1 to target IE72, specifically, is important for latent carriage. Since miR-UL112-1 is known to have other cellular targets (Hook et al., 2014b; Huang et al., 2013; Lee et al., 2012) besides viral IE72, we approached this by generating recombinant virus in which the miR-UL112-1 target site in the 3′ UTR of IE72 was removed (Δ112-1 target virus) rather than by deleting the miR-UL112-1 miRNA coding sequence itself – so ensuring that any phenotype we observed during latency was a result of lack of targeting of IE72 by miR-UL112-1 rather than its other known or unknown targets.
Our results show that, as expected, latent infection with wild-type (WT) virus results in very low levels of IE72 RNA expression. In contrast, latent infection with Δ112-1 target virus results in latently infected cells expressing substantial levels of IE72 RNA. Importantly, as opposed to cells latently infected with WT virus, cells latently infected with Δ112-1 target virus now become overt targets for IE72-specific CD8+ cytotoxic T-cells (CTLs).
Finally, the difficulty in using primary cells from healthy donors led us to interrogate whether cell line models of HCMV latency could be used for similar immunological analyses of HCMV latent infection. Using the THP-1 cell line model of latency, we have recapitulated our observations on the effects of latent infection in the absence of miR-UL112-1; as with primary cells, THP-1 cells latently infected with Δ112-1 target virus also become overt targets for IE72-specific CD8+ CTLs.
Taken together, our data define an important role for miR-UL112-1 in the maintenance of latency as well as demonstrating the applicability of the THP-1 model system for intricate analysis of HCMV latency at the molecular and immunological levels. We believe that expression of miR-UL112-1 during latency is important to ‘mop up’ leaky IE RNA expression to prevent recognition of latently infected cells by the robust IE72-specific CTL response known to be present in normal HCMV carriers (Gillespie et al., 2000; Kern et al., 1998).
Results
Deletion of the miR-UL112-1 target site in the IE72 3′ UTR results in low-level IE expression in primary monocytes but not full reactivation of virus production
The viral miR-UL112-1 miRNA is expressed during latency in a number of experimental latent models including experimentally latent primary monocytes and CD34+ haemopoietic progenitor cells (Meshesha et al., 2016) (B. Lau and others unpublished observations). Besides the documented ability of miR-UL112-1 to target IE72, this viral miRNA is also known to target cellular MICB (Stern-Ginossar et al., 2008), multiple components of the host secretory pathway (Hook et al., 2014b), BclAF1 (Lee et al., 2012) and IL-32 (Huang et al., 2013), and is likely to have other unidentified viral and cellular target RNAs as many biochemical analyses suggest that miRNAs may typically target dozens of targets. As we wanted to specifically address the effect of lack of IE72 RNA regulation by miR-UL112-1, without potential confounding effects of miR-UL112-1 on other targets, we decided to construct a recombinant virus (∆112-1 target) in which the miR-UL112-1 target binding site was removed (Fig. 1a), so allowing an analysis of the effect of miR-UL112-1 on IE72 expression, in isolation.
Fig. 1.

Removal of the miR-UL112-1 target site leads to induction of immediate early (IE) gene expression in the absence of virion production. (a) The target site of miR-UL112-1 in the 3′ UTR of IE72 was deleted to generate a recombinant virus in which IE72 is no longer targeted by miR-UL112-1. (b) Monocytes (mono) were either uninfected, infected with wild-type TB40E (WT) or the miR-UL112-1 target site mutant virus (∆112-1) and latency was established for 4 days before harvesting and analysing mRNA levels for viral products UL138, IE and pp28 relative to cellular gene glyceraldehyde 3-phosphate dehydrogenase (GAPDH). Alternatively, these gene products were analysed following reactivation from latency by differentiation to DCs (reactivation). (c) Both latent monocytes and reactivating DCs, described in (a), were also co-cultured with indicator fibroblasts and supernatants were harvested and seeded onto fresh fibroblasts for 24 h before fixing and immunofluorescence staining for IE protein, which was used to determined the IE foci forming units. Error bars shown in (b) and (c) denote the standard error of two independent experiments, with each containing three technical repeats.
We initially analysed the establishment of latency after 4 days of infection in CD14+ monocytes as we have done previously (Krishna et al., 2016; Weekes et al., 2013) with both WT or ∆112-1 target virus which resulted in similar numbers of GFP+ latently infected cells (approximately 10–20 %, data not shown) as well as similar levels of expression of the latency-associated gene UL138 (Fig. 1b). However, the suppression of IE gene expression in comparison to levels of UL138 expression (which is a hallmark of latent infection) was only observed with the WT virus; perhaps not unexpectedly, cells latently infected with ∆112-1 target virus resulted in a discernible increase in levels of IE72 RNA (Fig. 1b). Importantly, although cells latently infected with ∆112-1 target virus did show less suppressed IE72 expression, there was no evidence of virus production from these cells or, as expected, from cells latently infected with WT virus (Fig. 1c). Consistent with this, no late viral gene expression (pp28) was observed in cells latently infected with either virus (Fig. 1b). In contrast, reactivation of cells latently infected with WT or ∆112-1 target virus showed similar levels of viral gene IE and late gene expression (Fig. 1b) and both produced similar levels of infectious virions (Fig. 1c).
Expression of IE by cells otherwise latently infected with the miR-UL112-1 target mutant virus can be detected by IE-specific T-cells
Given that cells latently infected with ∆112-1 target virus expressed detectable levels of IE72 RNA, we reasoned that, if translated, this would make these cells vulnerable to surveillance by the well-characterized IE72-specific CD8+ CTL response known to be present in the peripheral blood of many healthy HCMV carriers; up to 10 % of all circulating CTLs in healthy virus carriers are IE72 specific (Gillespie et al., 2000; Kern et al., 1998). Consequently, we next tested whether cells latently infected with ∆112-1 target virus were differentially recognized by IE72-specific CTLs compared to cells latently infected with WT virus.
To do this, we generated a T-cell line which recognized the previously mapped IE72 peptide VLE (Khan et al., 2002), presented by MHC Class I HLA-A2. Using Fluorospot analysis, we first checked that the T-cell line produced IFNγ when cultured with both peptide pulsed monocytes and DCs. These T-cells were then used against latently infected monocyte targets as well as reactivating DC targets. As expected, the T-cell line recognized monocytes latently infected with WT or ∆112-1 target virus which had been induced to reactivate by their differentiation to DCs (Fig. 2). In contrast, monocytes latently infected with WT virus showed little IE72-specific CTL recognition, whereas monocytes latently infected with ∆112-1 target virus were clearly recognized by IE-specific CTLs (Fig. 2). Taken together, these data suggest that miR-UL112-1 is important for reinforcing the suppression of IE gene expression during latency to help prevent T-cell recognition of latently infected cells by IE72-specific CTLs.
Fig. 2.
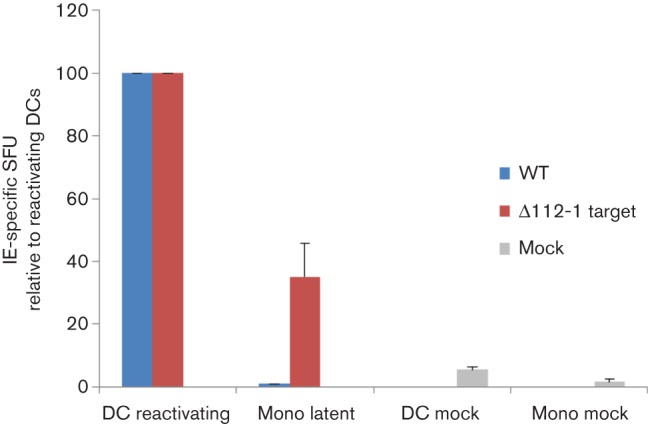
Monocytes latently infected with HCMV ∆112-1 target site mutant are recognized specifically by IE-specific CD8+ T-cells. Monocytes (Mono) were latently infected with either wild-type HCMV (WT) or miR-UL112-1 target site mutant virus (∆112-1) and a proportion of cells from each population were also differentiated to DCs by IL-4 and granulocyte-macrophage colony-stimulating factor treatment to induce reactivation. Both the undifferentiated and the differentiated cells were then co-cultured with IE- (VLE) specific CD8 T-cells and then assayed for IFNγ secretion using Fluorospot assays. Mock-infected monocytes (Mono mock) and dendritic cells (DC mock) were also co-cultured with IE- (VLE) specific CD8 T-cells. The level of T-cell recognition is shown relative to the level of T-cell recognition of reactivating DCs. Error bars denote standard deviation of three biological replicates. T-cells alone showed no background spots and reactivating DCs routinely showed in the region of 40–50 spot-forming units (SFU).
Validation of the THP-1 cell model system
All the analyses, so far, were carried out using latently infected primary human monocytes obtained from healthy blood donors; this requires local ethical approval and access to clinical services, which are not always readily available. On this basis, we wanted to determine if the effects of a lack of the miR-UL112-1 target site on T-cell surveillance could be recapitulated in other, more tractable, models of HCMV latency.
A number of models of HCMV latency that use established myeloid cell lines have been described and these include myeloid cells expressing the CD34+ marker, such as Kasumi-3 (O'Connor & Murphy, 2012), or CD14+ monocytic cells, such as THP-1s (Keyes et al., 2012a), which can recapitulate many aspects of HCMV latency and reactivation observed in primary cells (Albright & Kalejta, 2013). The ability to use such established cell line model systems in the analysis of HCMV latency has a number of advantages, which include their ease of use compared with primary cells, the lack of a requirement to recruit donors and, importantly, there would be no need to tissue type match each individual donor as with, for example, immunological assays. Such cell lines are also homogenous and more easily to manipulate by, for example, transfection.
As observed by others (Keyes et al., 2012a), THP-1 cells acted as a tractable model of latent infection. However, we found that growing cells in 2 % FCS, rather than 10 % FCS (as is standard), after infection reduced large-scale expansion of the cell population and the associated loss of viral genomes (see Fig. S1, available in the online Supplementary Material). Fig. 3(a) shows that latent infection of THP-1 cells resulted in cells which express good levels of viral UL138 RNA in a background of much reduced expression of viral IE72 RNA, compared to levels of IE72 RNA expressed upon reactivation (e.g. see Fig. 3a, tracks labelled +PMA). Consistent with this low level of IE72 RNA expression, we observed extremely low levels of IE72 protein expression using indirect immunofluorescence (Fig. 3b, left panel). In contrast, differentiation of these latently infected cells resulted in induction of IE72 expression at the level of RNA (Fig. 3a) and detection of numerous IE72-positive cells (Fig. 3b, right panel). Perhaps most importantly, on the basis that lack of infectious virus is a hallmark of latency, co-culture of the undifferentiated latently infected THP-1 cells (shown in the left panel of Fig. 3b) with fully permissive indicator fibroblasts showed no infectious virus production (Fig. 3c, left panel). In contrast, reactivated virus was clearly detectable (Fig. 3c, right panel) after co-culture of these differentiated latently infected THP-1 cells (shown in the right panel of Fig. 3b) with indicator fibroblasts. Taken together, these data show that infection of THP-1 cells results in latent HCMV infection, which can be reactivated by differentiation.
Fig. 3.

Characterization of the THP-1 latency and reactivation model in low serum. (a) THP-1 cells were infected with TB40/E at an m.o.i. of 5 and left for 3 days to obtain a latently infected population. Phorbol 12-myristate 13-acetate (PMA) was added on day 3 and left on the cells for 24, 48 or 72 h; cDNA was produced and amplified using IE, UL138 and GAPDH primers. Reverse transcriptase (RT) minus control samples (no RT) were run in parallel to exclude genomic DNA contamination. A representative experiment of three independent replicates is shown. Lane 1: 100 bp DNA ladder (Life Technologies); Lane 2: mock infected, untreated; Lane 3: mock infected, 100 nM PMA; Lane 4: TB40/E infected, untreated; Lane 5: TB40/E infected, 100 nM PMA; Lane 6: TB40/E infected, untreated; Lane 7: TB40/E infected, 100 nM PMA; Lane 8: TB40/E infected, untreated; Lane 9: TB40/E infected, 100 nM PMA. (b) Micrographs of TB40/E-infected, undifferentiated THP-1 cells (latency) (left panel) and TB40/E latently infected THP-1 cells, differentiated on day 3 post-infection with 100 nM PMA (reactivation) (right panel). (c) Cells from (b), left panel or right panel, were co-cultured with fibroblasts for 5 days (left panel or right panel, respectively). For all panels in (b) and (c), IE expression was visualized using immunofluorescence staining with anti-IE72/86 antibody. Arrows indicate IE-expressing cells.
THP-1 cells latently infected with ∆112-1 target virus also show de-repressed IE expression and become targets for IE-specific CTLs
Fig. 2 clearly showed that removal of the miR-UL112-1 target site in IE72 prevented the repression of IE gene expression during HCMV latency in primary monocytes. Consequently, we tested whether latently infected THP-1 cells also showed the same effect. Fig. 4(a) shows that infection of THP-1 cells with both WT virus and ∆112-1 target virus resulted in equivalent levels of UL138 expression; this was not the case for expression of IE72. Although infection with WT virus did result in some detectable levels of IE72 expression, consistent with the less robust control of expression of IE72 known to occur upon latent infection of THP-1 cells (Keyes et al., 2012a) compared to monocytes, infection with ∆112-1 target virus resulted in substantially increased levels of IE72 RNA (Fig. 4b). This argues that, during latent infection of THP-1 cells, the ∆112-1 target virus is less able to suppress IE gene expression during latency in the absence of the miR-UL112-1 target site; this is exactly as observed for primary monocytes.
Fig. 4.
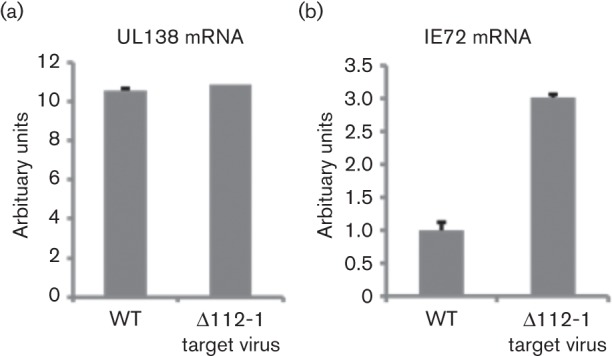
Latent infection with Δ112-1 target site mutant virus results in increased levels of IE72 RNA expression compared with WT virus. THP-1 cells were infected at an m.o.i. of 5 with the parental WT virus or the Δ112-1 target site mutant. Latently infected cells were sorted for GFP expression at 2 days post-infection, before mRNA expression was analysed by quantitative reverse transcriptase PCR at 3 days post-infection. The level of UL138 and IE72 normalized to the mRNA level of housekeeping gene GAPDH is shown in (a) and (b), respectively. Data shown is representative of three independent repeats, whilst error bars shown mark standard deviation of technical replicates.
To determine if THP-1 cells latently infected with ∆112-1 target virus were also more susceptible to surveillance by IE72-specific CTLs, we needed to determine whether THP-1 cells could act as targets for IE-specific T-cells in an MHC Class I restricted manner. THP-1 cells are known to express high levels of HLA-A2 and, consequently, when loaded with the HLA-A2-restricted VLE polypeptide of IE72 should present and be recognized by VLE-specific CTLs (Khan et al., 2002). Fig. 5(a) shows that, as expected, THP-1 cells express high levels of HLA-A2 and, when pulsed with a VLE polypeptide, are recognized in a dose-dependent manner by a VLE-specific CTL line using IFNγ ELISpot analysis (Fig. 5b). Given that THP-1 cells can act as IE-specific CTL targets in this setting, we next tested whether these IE-specific T-cells recognized latently infected THP-1 cells, lytically infected differentiated THP-1 cells or latently infected THP-1 induced to reactivate by differentiation. Fig. 5(c) shows that differentiated THP-1 cells infected with HCMV, which undergo lytic infection, are recognized by IE-specific CTLs, as expected. In contrast, THP-1 cells latently infected with HCMV are poorly recognized by VLE-specific CTLs but do become targets when these latently infected cells are induced to reactivate. This is consistent with the targeting of infected THP-1 by IE72-specific T-cells only occurring when IE is fully expressed, i.e. during lytic infection and reactivation, but not latency.
Fig. 5.

IE- (VLE) specific CD8+ T-cells recognize lytically but not latently infected THP-1 cells. (a) THP-1 cells were stained with monoclonal antibody specific for MHC Class I HLA-A2 (filled plot) or an isotype-matched control antibody (open plot). (b) THP-1 cells were pulsed with IE (VLE) peptide (pulsed) and assayed with control THP-1 cells (unpulsed) for their ability to be recognized by increasing numbers of IE- (VLE) specific CD8+ T-cells, as measured by IFNγ production in ELISPOT assays. (c) Latently infected THP-1 cells (latent), lytically infected differentiated THP-1 cells (lytic) or latently infected THP-1 cells differentiated with PMA to induce HCMV reactivation (reactivated) were co-cultured with increasing numbers of IE- (VLE) specific CD8+ T-cells (as detailed in the figure) and assayed for IFNγ using ELISPOT assay. Data shown is representative of three independent experiments. (d) The ∆112-1 target mutant allows recognition and removal of latently infected cells. THP-1 cells were infected at an m.o.i. of 5 with the parental WT virus (WT) or the Δ112-1 target virus before latently infected cells were sorted for GFP expression at 2 days post-infection. At 3 days post-infection, the cells were co-cultured with CD8+ T-cells as indicated for a further 24 h before the numbers of GFP-expressing cells were enumerated. Changes in GFP+ cell numbers are shown as fold change over the number of GFP+ cells present in WT virus-infected THP-1 cells cultured in the absence of T-cells. Statistical analysis was performed using a t-test, with an asterisk indicating significant difference (P<0.05). Error bars denote standard deviation of technical replicates.
Finally, we analysed whether THP-1 cells latently infected with ∆112-1 target virus were also targets for IE-specific CTLs, as we had observed using primary monocytes. To do this, we analysed the ability of IE-specific CTLs to reduce the number of GFP-tagged latently infected cells after latent infection with WT or ∆112-1 target virus. Fig. 5(d) shows that, as expected, irrelevant Epstein–Barr virus (EBV)-specific T-cells had no effect on the number of GFP-tagged latently infected THP-1 cells regardless of whether latency was established with WT or ∆112-1 target virus. In contrast, if IE-specific CTLs were used in the assay, the number of GFP-tagged latently infected THP-1 cells was reduced almost threefold when latency was established using ∆112-1 target virus compared with cells latently infected with WT virus.
Formally, whilst it is possible that this removal of these otherwise latently infected THP-1 cells expressing IE antigen results from CTL-mediated repression of IE gene expression and that these THP-1 cells still carry reactivatable virus, we think this is unlikely as cells that are induced to transiently express IE72 by treatment of latently infected cells with histone deacetylase inhibitors (HDACi) are killed by IE-specific CTLs (Krishna et al., 2016).
Collectively, these data show that THP-1 cells provide a tractable experimental model of latency in which we were able to reproduce the observations with regards to the down-regulation of IE72 by miR-UL112-1 during latency.
Discussion
The inability of HCMV latently infected cells to be cleared by the host after primary infection, even though host T-cell responses to HCMV are profound and long term, argues that latent reservoirs are unable to be surveilled and targeted by the extremely large HCMV-specific CD8+ and CD4+ T-cell response known to be present in normal healthy HCMV carriers (Wills et al., 2015). Indeed, up to 10 % of the CD8+T-cell compartment in peripheral blood of some healthy HCMV-seropositive carriers recognizes IE72 antigen (Gillespie et al., 2000; Kern et al., 1998). Consequently, robust repression of IE72 expression during latent carriage is likely to be of critical importance in order to prevent latently infected cells from being recognized and cleared by the well-established IE72-specific antiviral T-cell response.
We and others have shown that latent infection of both experimentally and naturally latent myeloid lineage cells results in a latency-associated transcription programme in which IE gene expression is profoundly suppressed; this results from repression of the viral major immediate early promoter (MIEP) mediated by concerted actions of cellular transcriptional suppressor proteins (Murphy et al., 2002; Rauwel et al., 2015; Reeves et al., 2005b) and viral regulators of IE expression (Lee et al., 2015) reflected in repressive histone post-translational modifications around the MIEP (Murphy et al., 2002; Reeves et al., 2005a, b). Our view is that, as with transcription in general, such repression is likely to be leaky, resulting in incomplete silencing of IE gene expression and, hence, requiring additional levels of IE suppression. The viral miR-UL112-1 miRNA has been shown to attenuate IE72 expression during lytic infection at low multiplicities of infection (Grey et al., 2007; Murphy et al., 2008). This led us to analyse the role of miR-UL112-1 during latent infection when, we predict, such fine tuning of IE72 expression may be particularly important. Consistent with this, experimental latent infection of CD14+ monocytes with the ∆112-1 target virus consistently resulted in a significant increase in IE72 RNA compared with latency established with WT virus. Despite this, the increase in IE72 RNA in cells latently infected with ∆112-1 target did not result in virus reactivation as determined by virus co-culture analysis. It did, however, result in the ability of these cells latently infected with ∆112-1 target virus to be detected by IE72-specific CTLs, resulting in their killing; again, no such killing of cells latently infected with WT virus was observed.
We also addressed whether we could recapitulate these findings in an established cell line model of HCMV latency, as there are a number of advantages associated with using such cell lines over primary cells for HCMV latency studies: their ease of use, the lack of requirement for ethical approval, the ability to manipulate them by, for example, transfection as well as their homogeneity as a cell population. Using these cells, we are able to recapitulate our key observations using primary monocytes and, therefore, believe that the THP-1 latency model represents a tractable model for also addressing immune control of HCMV latency and reactivation.
Taking all these observations together, we posit that miR-UL112-1 has a key role during latency which is to ensure robust control of expression of viral IE72 by minimizing translation of any low levels of IE72 transcription which escapes latency-associated repression of the MIEP; if left uncontrolled, expression of this lytic antigen would otherwise result in latently infected cells becoming targets for the robust IE72 antiviral response routinely observed in healthy HCMV carriers.
Methods
Cells and viruses.
For latent infections, primary monocytes were isolated from venous blood of an HLA-A2 HCMV-seronegative donor by CD14+ MACS microbeads positive selection (Miltenyi Biotec) as instructed by the manufacturer. Briefly, PBMCs were extracted from venous blood by Lymphoprep density gradient centrifugation (Axis-Shield), as previously described (Mason et al., 2013). PBMCs were then incubated with CD14 direct microbeads before application to a magnetized LS column, and subsequently eluting the bound cells after washing the column. The purity of isolated cells was analysed by flow cytometry which routinely showed 98.1 % cells expressed CD14 (range 97.4–98.9 %). Primary monocytes were cultured in X-Vivo-15 (Lonza) supplemented with 2.5 mM l-glutamine (GE Healthcare).
THP-1 cells (TIB-202; ATCC) were propagated in RPMI (Lonza) supplemented with 10 % heat-inactivated FCS (F7524; Sigma-Aldrich) and 0.04 % gentamicin (Gibco – Life Technologies). During latency and reactivation experiments, THP-1 cells were cultured in RPMI supplemented with 2 % FCS.
ARPE-19 cells (CRL-2302; ATCC) and neonatal normal human dermal fibroblasts (CC-2509; Lonza) were grown respectively in Dulbecco's modified Eagle's medium (DMEM):F12 with l-glutamine (733-1713; Biowhittaker) and minimum essential medium (MEM) (11095-098; Gibco – Life Technologies) containing 10 % FCS and 0.04 % gentamicin. Stocks of HCMV TB40/E (Chou & Scott, 1988) were generated on ARPE-19 cells to obtain highly endothelial tropic virus. Virus yield was determined by an immune-fluorescence assay adapted from Chou & Scott (1988). After scoring, the virus titre (TCID50/ml) was calculated based on the Spearman-Kärber method.
TB40E-GFP ∆112-1 target virus was derived from TB40E-eGFP (O'Connor & Murphy, 2012), based on the HCMV bacterial artificial chromosome (BAC)-derived strain TB40/E (clone 4) and which was previously engineered to express eGFP. TB40-eGFP was used as the WT virus where indicated and was used to generate the TB40/Egfp Δ112-1 mutant using galK BAC recombineering protocols. The TB40/Egfp Δ112-1 mutant contains 12 single base pair substitutions that abrogate expression of miR-112-1, while maintaining the coding potential of the UL114 ORF located on the opposite strand. Mutagenesis was performed on the BAC clone of TB40/Egfp, using the galK selection and counter selection recombineering protocols as previously described (Warming et al., 2005). Briefly, the TB40/Egfp-BAC was transformed into Escherichia coli SW102, which has the heat inducible recombination genes required for BAC-mediated recombineering. PCR products were generated with primers that contain 50 bp of HCMV flanking sequence targeting recombination of the galK expression cassette to the location of interest. The forward and reverse primers to insert galK into the miR-UL112-1 sequence were 5′-GGCGCTCTGACAGCCTCCGGATCACATGGTTACTCAGCGTCTGCCAGCCcctgttgacaattaatcatcggcga-3′ and 5′-GCGACGCGGCGTGCTGCTGCTCAACACCGTGTTCACCGTGGTGCACGGACtcagcactgtcctgctcctt-3′, respectively (capitalized nucleotides correspond to the HCMV sequence). TB40-eGFP-BAC-containing SW102 cells were made competent and transformed with the HCMV flanking galK PCR product. Transformed cells were plated on M63 minimal salt agar plates supplemented with chloramphenicol (12.5 µg ml−1) and 0.2 % galactose. TB40-eGFP-BAC clones that underwent homologous recombination were additionally screened for growth on McConkey's medium supplemented with galactose. Positive colonies (red) were picked and further screened by PCR to confirm the proper location of the galK insert. Positive colonies were made electrocompetent and transformed with 80 bp dsDNA with HCMV sequences that flank the galK integrated sequence surrounding the sequence to be substituted into the viral genome. The dsDNA used to mutate the mature miRNA sequence of miR-UL112-1 was generated by annealing the following oligonucleotides: 5′-GTGCTGCTGCTCAACACCGTGTTCACCGTGGTGCACGGACAACCCGGGAGCCATAGGCATCTGG GCTGGCAGACGCTGAGTAACCATGTGATCCGGAGGCTG-3′ and 5′-CAGCCTCCGGATCACATGGTTACTCAGCGTCTGCCAGCCCA GATGCCTATGGCTCCCGGGTTGTCCGTGCACCACGGTGAACAC GGTGTTGAGCAGCAGCAC-3′. Positive recombinants are selected by growth on m63 minimal salt agar plates containing 0.2 % 2-deoxy-galactose (2-DOG) and 0.2 % glucose. All mutations were validated by sequencing the recombinant viruses. Virus was generated by electroporation of primary fibroblasts with BAC DNA (20 µg) plus an HCMV pp71-expressing plasmid (pCGNpp71). After a 100 % cytopathic effect was achieved, virus was harvested by ultracentrifugation through a 20 % sorbitol cushion. Viral pellets were resuspended in X-VIVO 15 medium (Lonza) supplemented with 1.5 % BSA. Aliquots were flash frozen in liquid nitrogen and stored at −80 °C until further use. Stock titres were assessed by 50 % tissue culture infective dose assays on primary fibroblasts.
THP-1 latency and reactivation model.
Twenty-four hours prior to infection, THP-1 cells were pre-incubated in RPMI with 2 % heat-inactivated FCS (RPMI/2 % FCS). This reduced large-scale expansion of the cell population and the associated loss of viral genomes (see Fig. S1). To obtain latently infected cells, undifferentiated THP-1 cells were infected at an m.o.i. of 5 with HCMV for 3–4 h at 37 °C in 0.5 ml RPMI/2 % FCS per 6×105 cells with occasional agitation to avoid settling. After infection, the cells were washed once with 2 ml RPMI/2 % FCS and spun down at 300 g (5 min). The cells were resuspended in RPMI/2 % FCS, plated in a 24-well plate and incubated for 3 days at 37 °C/5 % CO2. To induce reactivation, the cells were differentiated to THP-1 macrophages using 100 nM PMA(Sigma-Aldrich).
A similar protocol was used for immunological assays with small adaptations. After the establishment of latency (72 h post-infection), the medium was replaced by fresh medium and 24 h later cells were used as stimulator cells in IFNγ ELISPOT assays and in T-cell co-culture experiments (see below). To obtain reactivated cells for IFNγ ELISPOT assays, latently infected cells were treated with PMA as described above and incubated for 96 h. As controls, lytically infected cells were obtained by pre-treatment with PMA (100 nM) for 48 h. Subsequently, the THP-1 macrophages were infected with TB40/E virus at an m.o.i. of 5, incubated for a further 96 h and then used as stimulator cells in IFNγ ELISPOT assays.
IE antigen immunofluorescence assay.
On the day of read-out, undifferentiated THP-1 cells in suspension were collected by centrifugation at 300 g for 5 min. The cells were washed once with PBS, resuspended in 200 µl PBS and adhered to a glass adhesion slide (0901000; Mariënfeld Laboratory Glassware) as described by the manufacturer. Differentiated adherent THP-1 cells were stained directly in the 24-well plate.
Fixation of the cells was carried out with 100 % ice-cold methanol (10 min at −20 °C). The slides or dishes were washed twice with PBS and incubated for 20 min at room temperature with anti-IE antigen antibody (11-003; Argene) diluted 1:100 in PBST (PBS containing 0.05 % Tween 20). Before adding the secondary antibody (1:100, Alexa Fluor 488 goat anti-mouse IgG, A-11001; Life Technologies), the slides or dishes were washed twice with PBS. After 20 min of incubation with the secondary antibody, the cells were washed once with PBS. Positive cells were manually counted under an Axiovision fluorescence microscope.
Fluorescence images were taken using an Axiovision fluorescence microscope (brightfield: 5–9 ms exposure; pseudo colour green fluorescence at a laser intensity of 25 %: 500 ms exposure). Overlays were made automatically using the Axiovision software.
Analysis of viral RNA and DNA.
DNA and RNA samples were prepared from monocytes obtained from one well or three wells, respectively, of a 24-well plate. Non-adherent cells were collected by centrifugation (5 min, 300 g) to remove the medium and were subsequently washed once with PBS. The cells were collected again by centrifugation (300 g, 5 min) and lysed in 500 µl lysis buffer (Invisorb Spin Cell Mini Kit) or 600 µl RLT Plus buffer (Qiagen RNeasy Plus kit) to obtain DNA and RNA samples, respectively. After removing the medium, adherent cells were washed and lysed in the well. DNA was prepared according to the manufacturer’s instructions. RNA samples were prepared using a Qiagen RNeasy Plus kit with an additional on-column DNase digestion step according to the manufacturer’s instructions.
Reverse transcription was carried out with oligo-dTs using the Superscript III kit (both from Life Technologies) according to the manufacturer’s instructions with an RNA input of 250–500 ng. Gene-specific primers were used to amplify glyceraldehyde 3-phosphate dehydrogenase (GAPDH), IE1/UL123 and UL138 by PCR using the Expand High Fidelity PCR system (Roche) with the following cycling conditions: 95 °C (5 min), 30 cycles of 94 °C (40 s), 55 °C (40 s) and 72 °C (60 s), and a final extension at 72 °C for 5 min (for primers see Table 1). PCR products were analysed using agarose gel electrophoresis (2 % E-gel; Life Technologies). Images were taken using an ImageQuant LAS 4000 imager.
Table 1. Primer sets for PCR and qPCR used in this study .
| Gene | Sequence | Reference |
|---|---|---|
| Conventional PCR | ||
| IE Fwd | CATCCACATCTCCCGCTTAT | Reeves et al. (2012) |
| IE Rev | CACGACGTTCCTGCAGACTATG | |
| GAPDH Fwd | GAGTCAACGGATTTGGTCGT | |
| GAPDH Rev | TTGATTTTGGAGGGATCTCG | |
| UL138 Fwd | TGCGCATGTTTCTGAGCTAC | Goodrum et al. (2007) |
| UL138 Rev | ACGGGTTTCAACAGATCGAC | |
| Probe-based quantitative PCR | ||
| IE72 Fwd | CAAGAACTCAGCCTTCCCTAAGAC | Visconti et al. (2004) |
| IE72 Rev | TGAGGCAAGTTCTCGAATGC | |
| IE72 probe | [6FAM]CCAATGGCTGCAGTCAGGCCATG[TAM] | |
| GAPDH Fwd | GGAAGCTTGTCATCAATG | Poole et al. (2014) |
| GAPDH Rev | CCCCACTTGATTTTGGAG | |
| GAPDH probe | [JOE]ATCACCATCTTCCAGGAGCGAG[BHQ1] | |
| UL138 Fwd | CGCTGTTTCTCTGGTTAG | Krishna et al. (2016) |
| UL138 Rev | CAGACGATACCGTTTCTC | |
| UL138 probe | [Cy5]CCGACGACGAAGACGATGAAC[BHQ2] | |
| pp28 Fwd | CGAACTCTGCAAACGAATA | Krishna et al. (2016) |
| pp28 Rev | GAGGGATGTTGTCGTAGG | |
| pp28 probe | [Cy3]CGTAGAGACACCTGGCGACC[BHQ2] | |
Reverse transcriptase quantitative PCR (RT-qPCR) analyses were performed using the QuantiTect virus kit (Qiagen) as per the manufactuer's instructions, using the primers and probes listed in Table 1. The reaction mixtures consisted of 1× QuantiTect Virus NR Master Mix, 1× ROX Dye solution, 0.4 µM of each forward and reverse primer, 0.2 µM probe, 1× QuantiTect Virus RT Mix and 20–50 ng sample RNA. The RT-qPCRs were performed on a 7500 Real-Time PCR System (Applied Biosystems) with cycling conditions as follows: 20 min at 50 °C, 5 min at 95 °C, and then 50 cycles of 15 s at 95 °C and 45 s at 60 °C. All qPCRs were performed in duplicate with minus template controls and minus reverse transcriptase controls. Viral transcript levels were normalized to the housekeeping transcript GAPDH. The sensitivities of the qPCRs for UL138 and IE1/UL123 were approximately equivalent with both having a detection limit of about 10 copies of the HCMV genome (B. Lau, unpublished observations). In contrast, the conventional RT-PCRs with products detected by agarose gel electrophoresis were not quantitative and detect UL138 more efficiently than IE72 (E. Poole, data not shown).
Preparation of PBMCs.
Venous blood was obtained from an HLA-A2 HCMV-seropositive individual and collected in sterile 50 ml tubes containing 4 ml heparin sodium (100 IU ml−1) in PBS. The blood was diluted 1 : 2 with RPMI-1640 containing no serum (PAA laboratories) supplemented with 105 IU penicillin ml−1, 100 mg streptomycin ml−1 and 2 mM l-glutamine. PBMCs were isolated by Ficoll-Hypaque density gradient centrifugation, and 25 ml blood was layered onto 12.5 ml Lymphoprep (Axis-Shield) and centrifuged at 800 g for 15 min without the brake. PBMCs were washed twice in PBS and resuspended in RPMI supplemented with 10 % FCS (RPMI-10). Autologous serum was also collected and incubated at 56 °C for 30 min in a water bath to inactivate complement. Following incubation the serum was UV-irradiated for 30 min and aliquoted for freezing at −20 °C.
Expansion of HCMV-specific CD8+ T-cells.
CD8+ T-cells were isolated from the PBMCs of an HLA-A2 HCMV-seropositive individual using a magnetic CD8+ T-cell enrichment kit (StemCell technologies) and then resuspended in RPMI containing 10 % FCS (Invitrogen) and 10 % heat-inactivated autologous donor serum. Cells were stimulated with irradiated autologous PBMCs which had been pulsed with HCMV IE72 peptide VLE (Khan et al., 2002) or EBV-specific GLC peptide (Steven et al., 1996). T-cell cultures were made in the presence of 2.5 IU ml−1 human recombinant IL-2 (UK National Institute for Biological Standards and Control) in 96-well plates at 37 °C and a humidified CO2atmosphere for 10–14 days with feeding of the culture with the same media every 5 days. The specificity of expanded CD8+ cultures for mapped peptides was assessed by VLE HLA A2-specific pentamer staining. Briefly, cells were harvested and washed and then stained with the specific unlabelled pentamer (ProImmune), washed and then further stained with pentamer-specific PE fluorophore (ProImmune) and CD8 and CD3 antibodies conjugated to PerCP-Cy5.5 and FITC, respectively. After this, cells were fixed and acquired on a FACS Sort device using CellQuest software (BD Biosciences). Data were analysed using FlowJo software. All expanded cell lines were tested for specificity using IFNγ ELISpot or Fluorospot assays against THP-1 cells, monocytes and DCs. Antigen-specific CD8+ T-cells were frozen in liquid nitrogen at 2×106 cells ml−1.
Detection of cytokine production by ELISPOT and Fluorospot.
Human IFNγ ELISPOT (Ready-SET-Go!; eBioscience) assays were performed according to the manufacturer’s instructions using 96-well PVDF membrane plates (Millipore). VLE-specific CD8+ T-cells were rapidly defrosted at 37 °C and diluted into 25 ml of warm RPMI-10, washed once and incubated at 37 °C for 2 h before being counted and then added to wells at 12 500, 25 000 or 50 000 T-cells per well in triplicate. The T-cells were stimulated with 50 000 THP-1 cells that had been latently or lytically infected with HCMV or latently infected cells that had been reactivated. Positive controls were THP-1 cells pulsed with cognate peptide and negative controls were unpulsed and uninfected THP-1 cells.
Plates were incubated for 48 h at 37 °C and then developed according to the manufacturer’s instructions. Images of each well were taken using an ELISPOT plate scanner (ELISPOT Reader System; AID) and spots were enumerated using ImageJ (National Institutes of Health).
Human IFNγ Fluorospot plate (Mabtech AB) assays were performed according to the manufacturer’s instructions. Monocytes from an HLA-A2 HCMV-seronegative donor were latently infected for 6 days with WT or ∆112-1 target virus and then left untreated or treated with IL-4/GM-CSF for an additional 6 days and then matured with lipopolysaccharide for 1 day to reactivate latent virus. These were then co-cultured with HLA-A2 restricted VLE-specific CD8+ T-cells at 37 °C in a humidified CO2 atmosphere for 48 h. The cells and medium were decanted from the plate and the assay was developed following the manufacturer’s instructions. Developed plates were read using an AID iSpot reader (AID) and counted using AID EliSpot v7 software.
THP-1 co-culture.
THP-1 cells were plated at 5×104 cells per well in 48-well plates. At 3 days post-infection, VLE- or EBV- (GLC) specific CD8+ T-cells were rapidly defrosted at 37 °C and diluted into 25 ml of warm RPMI-10, washed once and incubated at 37 °C for 2 h before being counted and then added at an effector/target ratio of 3 : 4. The numbers of GFP-expressing cells across entire wells were enumerated 24 h later.
Ethical approval.
All human samples were obtained under ethical approval and after approval of protocols from the Cambridgeshire 2 Research Ethics Committee (REC reference 97/092) and these protocols were conducted in accordance with the Declaration of Helsinki. Informed written consent was obtained from all of the volunteers included in this study prior to providing blood samples and all experiments were carried out in accordance with the approved guidelines.
Acknowledgements
We gratefully acknowledge funding from the UK Medical Research Council (J. H. S. and M. R. W. G:0701279) which supports the current research in our laboratory and also the support of NIHR UK Biomedical Research Centre (J. H. S. and M. R. W.).
Supplementary Data
References
- Adler S. P.(1983). Transfusion-associated cytomegalovirus infections. Rev Infect Dis 5977–993. 10.1093/clinids/5.6.977 [DOI] [PubMed] [Google Scholar]
- Albright E. R., Kalejta R. F.(2013). Myeloblastic cell lines mimic some but not all aspects of human cytomegalovirus experimental latency defined in primary CD34+ cell populations. J Virol 879802–9812. 10.1128/JVI.01436-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou S. W., Scott K. M.(1988). Rapid quantitation of cytomegalovirus and assay of neutralizing antibody by using monoclonal antibody to the major immediate-early viral protein. J Clin Microbiol 26504–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhuruvasan K., Sivasubramanian G., Pellett P. E.(2011). Roles of host and viral microRNAs in human cytomegalovirus biology. Virus Res 157180–192. 10.1016/j.virusres.2010.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu M., Gao Y., Zhou Q., Zhang Q., Peng Y., Tian K., Wang J., Zheng X.(2014). Human cytomegalovirus latent infection alters the expression of cellular and viral microRNA. Gene 536272–278. 10.1016/j.gene.2013.12.012 [DOI] [PubMed] [Google Scholar]
- Gillespie G. M., Wills M. R., Appay V., O'Callaghan C., Murphy M., Smith N., Sissons P., Rowland-Jones S., Bell J. I., Moss P. A.(2000). Functional heterogeneity and high frequencies of cytomegalovirus-specific CD8(+) T lymphocytes in healthy seropositive donors. J Virol 748140–8150. 10.1128/JVI.74.17.8140-8150.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberger T., Mandelboim O.(2014). The use of microRNA by human viruses: lessons from NK cells and HCMV infection. Semin Immunopathol 36659–674. 10.1007/s00281-014-0447-3 [DOI] [PubMed] [Google Scholar]
- Goodrum F., Reeves M., Sinclair J., High K., Shenk T.(2007). Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110937–945. 10.1182/blood-2007-01-070078 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey F., Meyers H., White E. A., Spector D. H., Nelson J.(2007). A human cytomegalovirus-encoded microRNA regulates expression of multiple viral genes involved in replication. PLoS Pathog 3e163. 10.1371/journal.ppat.0030163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grey F.(2015). Role of microRNAs in herpesvirus latency and persistence. J Gen Virol 96739–751. 10.1099/vir.0.070862-0 [DOI] [PubMed] [Google Scholar]
- Griffiths P. D., Walter S.(2005). Cytomegalovirus. Curr Opin Infect Dis 18241–245. 10.1097/01.qco.0000168385.39390.1b [DOI] [PubMed] [Google Scholar]
- Hahn G., Jores R., Mocarski E. S.(1998). Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc Natl Acad Sci U S A 953937–3942. 10.1073/pnas.95.7.3937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho M.(1990). Epidemiology of cytomegalovirus infections. Rev Infect Dis 12S701–710. 10.1093/clinids/12.Supplement_7.S701 [DOI] [PubMed] [Google Scholar]
- Hook L., Hancock M., Landais I., Grabski R., Britt W., Nelson J. A.(2014a). Cytomegalovirus microRNAs. Curr Opin Virol 740–46. 10.1016/j.coviro.2014.03.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook L. M., Grey F., Grabski R., Tirabassi R., Doyle T., Hancock M., Landais I., Jeng S., McWeeney S., et al. (2014b). Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 15363–373. 10.1016/j.chom.2014.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y., Qi Y., Ma Y., He R., Ji Y., Sun Z., Ruan Q.(2013). The expression of interleukin-32 is activated by human cytomegalovirus infection and down regulated by hcmv-miR-UL112-1. Virol J 1051. 10.1186/1743-422X-10-51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kern F., Surel I. P., Brock C., Freistedt B., Radtke H., Scheffold A., Blasczyk R., Reinke P., Schneider-Mergener J., et al. (1998). T-cell epitope mapping by flow cytometry. Nat Med 4975–978. 10.1038/nm0898-975 [DOI] [PubMed] [Google Scholar]
- Keyes L. R., Bego M. G., Soland M., St Jeor S.(2012a). Cyclophilin A is required for efficient human cytomegalovirus DNA replication and reactivation. J Gen Virol 93722–732. 10.1099/vir.0.037309-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keyes L. R., Hargett D., Soland M., Bego M. G., Rossetto C. C., Almeida-Porada G., St. Jeor S.(2012b). HCMV protein LUNA is required for viral reactivation from latently infected primary CD14+ cells. PLoS One 7e52827 10.1371/journal.pone.0052827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khaiboullina S. F., Maciejewski J. P., Crapnell K., Spallone P. A., Dean Stock A., Pari G. S., Zanjani E. D., Jeor S. S.(2004). Human cytomegalovirus persists in myeloid progenitors and is passed to the myeloid progeny in a latent form. Br J Haematol 126410–417. 10.1111/j.1365-2141.2004.05056.x [DOI] [PubMed] [Google Scholar]
- Khan N., Cobbold M., Keenan R., Moss P. A.(2002). Comparative analysis of CD8+ T cell responses against human cytomegalovirus proteins pp65 and immediate early 1 shows similarities in precursor frequency, oligoclonality, and phenotype. J Infect Dis 1851025–1034. 10.1086/339963 [DOI] [PubMed] [Google Scholar]
- Krishna B. A., Lau B., Jackson S. E., Wills M. R., Sinclair J. H., Poole E.(2016). Transient activation of human cytomegalovirus lytic gene expression during latency allows cytotoxic T cell killing of latently infected cells. Sci Rep 624674. 10.1038/srep24674 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H., Kalejta R. F., Kerry J., Semmes O. J., O'Connor C. M., Khan Z., Garcia B. A., Shenk T., Murphy E.(2012). BclAF1 restriction factor is neutralized by proteasomal degradation and microRNA repression during human cytomegalovirus infection. Proc Natl Acad Sci U S A 1099575–9580. 10.1073/pnas.1207496109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S. H., Albright E. R., Lee J. H., Jacobs D., Kalejta R. F.(2015). Cellular defense against latent colonization foiled by human cytomegalovirus UL138 protein. Sci Adv 1e1501164. 10.1126/sciadv.1501164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason G. M., Jackson S., Okecha G., Poole E., Sissons J. G., Sinclair J., Wills M. R.(2013). Human cytomegalovirus latency-associated proteins elicit immune-suppressive IL-10 producing CD4+ T cells. PLoS Pathog 9e1003635. 10.1371/journal.ppat.1003635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendelson M., Monard S., Sissons P., Sinclair J.(1996). Detection of endogenous human cytomegalovirus in CD34+ bone marrow progenitors. J Gen Virol 773099–3102. 10.1099/0022-1317-77-12-3099 [DOI] [PubMed] [Google Scholar]
- Meshesha M. K., Bentwich Z., Solomon S. A., Avni Y. S.(2016). In vivo expression of human cytomegalovirus (HCMV) microRNAs during latency. Gene 575101–107. 10.1016/j.gene.2015.08.040 [DOI] [PubMed] [Google Scholar]
- Murphy E., Vanícek J., Robins H., Shenk T., Levine A. J.(2008). Suppression of immediate-early viral gene expression by herpesvirus-coded microRNAs: implications for latency. Proc Natl Acad Sci U S A 1055453–5458. 10.1073/pnas.0711910105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy J. C., Fischle W., Verdin E., Sinclair J. H.(2002). Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J 211112–1120. 10.1093/emboj/21.5.1112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Connor C. M., Murphy E. A.(2012). A myeloid progenitor cell line capable of supporting human cytomegalovirus latency and reactivation, resulting in infectious progeny. J Virol 869854–9865. 10.1128/JVI.01278-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole E., Avdic S., Hodkinson J., Jackson S., Wills M., Slobedman B., Sinclair J.(2014). Latency-associated viral interleukin-10 (IL-10) encoded by human cytomegalovirus modulates cellular IL-10 and CCL8 secretion during latent infection through changes in the cellular microRNA hsa-miR-92a. J Virol 8813947–13955. 10.1128/JVI.02424-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poole E., Lau J. C., Sinclair J.(2015). Latent infection of myeloid progenitors by human cytomegalovirus protects cells from FAS-mediated apoptosis through the cellular IL-10/PEA-15 pathway. J Gen Virol 962355–2359. 10.1099/vir.0.000180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauwel B., Jang S. M., Cassano M., Kapopoulou A., Barde I., Trono D.(2015). Release of human cytomegalovirus from latency by a KAP1/TRIM28 phosphorylation switch. eLife 4e06068 10.7554/eLife.06068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves M. B., Lehner P. J., Sissons J. G., Sinclair J. H.(2005a). An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J Gen Virol 862949–2954. 10.1099/vir.0.81161-0 [DOI] [PubMed] [Google Scholar]
- Reeves M. B., MacAry P. A., Lehner P. J., Sissons J. G. P., Sinclair J. H.(2005b). Latency, chromatin remodeling, and reactivation of human cytomegalovirus in the dendritic cells of healthy carriers. Proc Natl Acad Sci U S A 1024140–4145. 10.1073/pnas.0408994102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves M. B., Breidenstein A., Compton T.(2012). Human cytomegalovirus activation of ERK and myeloid cell leukemia-1 protein correlates with survival of latently infected cells. Proc Natl Acad Sci U S A 109588–681. 10.1073/pnas.1114966108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross S. A., Boppana S. B.(2005). Congenital cytomegalovirus infection: outcome and diagnosis. Semin Pediatr Infect Dis 1644–49. 10.1053/j.spid.2004.09.011 [DOI] [PubMed] [Google Scholar]
- Rossetto C. C., Tarrant-Elorza M., Pari G. S.(2013). Cis and trans acting factors involved in human cytomegalovirus experimental and natural latent infection of CD14 (+) monocytes and CD34 (+) cells. PLoS Pathog 9e1003366. 10.1371/journal.ppat.1003366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin R. H.(1990). Impact of cytomegalovirus infection on organ transplant recipients. Rev Infect Dis 12S754–766. 10.1093/clinids/12.Supplement_7.S754 [DOI] [PubMed] [Google Scholar]
- Shen Z. Z., Pan X., Miao L. F., Ye H. Q., Chavanas S., Davrinche C., McVoy M., Luo M. H.(2014). Comprehensive analysis of human cytomegalovirus microRNA expression during lytic and quiescent infection. PLoS One 9e88531. 10.1371/journal.pone.0088531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinclair J., Sissons P.(2006). Latency and reactivation of human cytomegalovirus. J Gen Virol 871763–1779. 10.1099/vir.0.81891-0 [DOI] [PubMed] [Google Scholar]
- Sissons J. G., Carmichael A. J.(2002). Clinical aspects and management of cytomegalovirus infection. J Infect 4478–83. 10.1053/jinf.2001.0949 [DOI] [PubMed] [Google Scholar]
- Slobedman B., Cao J. Z., Avdic S., Webster B., McAllery S., Cheung A. K., Tan J. C., Abendroth A.(2010). Human cytomegalovirus latent infection and associated viral gene expression. Future Microbiol 5883–900. 10.2217/fmb.10.58 [DOI] [PubMed] [Google Scholar]
- Stern-Ginossar N., Gur C., Biton M., Horwitz E., Elboim M., Stanietsky N., Mandelboim M., Mandelboim O.(2008). Human microRNAs regulate stress-induced immune responses mediated by the receptor NKG2D. Nat Immunol 91065–1073. 10.1038/ni.1642 [DOI] [PubMed] [Google Scholar]
- Stern-Ginossar N., Saleh N., Goldberg M. D., Prichard M., Wolf D. G., Mandelboim O.(2009). Analysis of human cytomegalovirus-encoded microRNA activity during infection. J Virol 8310684–10693. 10.1128/JVI.01292-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steven N. M., Leese A. M., Annels N. E., Lee S. P., Rickinson A. B.(1996). Epitope focusing in the primary cytotoxic T cell response to Epstein–Barr virus and its relationship to T cell memory. J Exp Med 1841801–1813. 10.1084/jem.184.5.1801 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Söderberg-Nauclér C., Streblow D. N., Fish K. N., Allan-Yorke J., Smith P. P., Nelson J. A.(2001). Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J Virol 757543–7554. 10.1128/JVI.75.16.7543-7554.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarrant-Elorza M., Rossetto C. C., Pari G. S.(2014). Maintenance and replication of the human cytomegalovirus genome during latency. Cell Host Microbe 1643–54. 10.1016/j.chom.2014.06.006 [DOI] [PubMed] [Google Scholar]
- Taylor-Wiedeman J., Sissons J. G., Borysiewicz L. K., Sinclair J. H.(1991). Monocytes are a major site of persistence of human cytomegalovirus in peripheral blood mononuclear cells. J Gen Virol 722059–2064. 10.1099/0022-1317-72-9-2059 [DOI] [PubMed] [Google Scholar]
- Taylor-Wiedeman J., Sissons P., Sinclair J.(1994). Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J Virol 681597–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visconti M. R., Pennington J., Garner S. F., Allain J. P., Williamson L. M.(2004). Assessment of removal of human cytomegalovirus from blood components by leukocyte depletion filters using real-time quantitative PCR. Blood 1031137–1139. 10.1182/blood-2003-03-0762 [DOI] [PubMed] [Google Scholar]
- Warming S., Costantino N., Court D. L., Jenkins N. A., Copeland N. G.(2005). Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33e36. 10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weekes M. P., Tan S. Y., Poole E., Talbot S., Antrobus R., Smith D. L., Montag C., Gygi S. P., Sinclair J. H., Lehner P. J.(2013). Latency-associated degradation of the MRP1 drug transporter during latent human cytomegalovirus infection. Science 340199–202. 10.1126/science.1235047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wills M. R., Poole E., Lau B., Krishna B., Sinclair J. H.(2015). The immunology of human cytomegalovirus latency: could latent infection be cleared by novel immunotherapeutic strategies? Cell Mol Immunol 12128–138. 10.1038/cmi.2014.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaia J. A.(1990). Epidemiology and pathogenesis of cytomegalovirus disease. Semin Hematol 275–10, discussion 28–19. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.