

Abstract
Background/Aim: Severe sepsis is associated with high morbidity and mortality rates. Inflammation and coagulation play pivotal roles in the pathogenesis of sepsis leading to multiple organ failure, especially in the liver. The aim of the present study was to assess the mechanism from sepsis to liver damage in a mouse model. Materials and Methods: We created a sepsis model by injecting lipopolysaccharide (LPS) intraperitoneally in mice. At 0, 6, 12, and 24 h following intraperitoneal injection of LPS, mice were euthanised and analyzed. Primary antibodies against myeloperoxidase (MPO), hepatic sinusoidal endothelial cells (SE-1), and P-selectin (CD62p) were used. Expression and localization in neutrophil, sinusoidal endothelial, and platelet cells were assessed by immunohistochemistry. Results: Immunohistochemical analyses revealed a positive staining for MPO, most abundantly in neutrophil granulocytes, within the hepatic sinusoids immediately after injection. Neutrophil extracellular trap (NET)-like structures stained for MPO, indicating the presence of neutrophils undergoing NETosis, were confirmed at 6 h after LPS administration. SE-1 staining for liver sinusoidal endothelial cells was significantly reduced at 12 h post-LPS administration through sinusoidal endothelial injury or detachment. Furthermore, the presence of extravasated platelets was confirmed in the space of Disse at 24 h after LPS administration. Blood sample analyses showed that white blood cell counts and platelet counts decreased gradually, while MPO amounts increased until 12 h after LPS administration. Conclusion: We conclude that NET formation and intravasated platelet aggregation are the first steps from sepsis to liver damage, and that extravasated platelet aggregation promoted by NET-facilitated detachment of sinusoidal endothelial cells is the origin of sepsis-induced liver dysfunction.
Keywords: Sepsis, multiple organ failure (MOF), neutrophil extracellular traps (NETs), extravasated platelet aggregation
Sepsis is a clinical syndrome of systemic inflammatory responses arising from an infectious process with a presumed or known focus (1,2). Severe sepsis, defined as sepsis with acute organ dysfunction, is associated with high morbidity and mortality rates (3). Inflammation and coagulation play pivotal roles in the pathogenesis of sepsis (4,5). Sepsis-induced multiple organ failure (MOF) has numerous causes, such as various types of shock, adult respiratory distress syndrome (ARDS), and disseminated intravascular coagulation (DIC) (6). Gando et al. (7) reported that DIC is frequently associated with systemic inflammatory response syndrome (SIRS; 83%) and that such patients have a high mortality rate (63%). Ogura et al. (4) evaluated coagulation activity, organ dysfunction, and SIRS in critically-ill patients with thrombocytopenia and examined the balance between coagulopathy and systemic inflammation. In critically-ill patients with thrombocytopenia, they found that coagulopathy and organ dysfunction progressed with a significant mutual correlation, depending on the increase in SIRS scores. Thus, SIRS-associated coagulopathy may play a critical role in inducing organ dysfunction after severe insult.
Crosstalk between inflammation and coagulation is essential, because coagulation can be activated by inflammation, as observed in SIRS-associated coagulopathy (8).
Previous studies showed that the generation of neutrophil extracellular traps (NETs) is correlated with the severity of organ dysfunction in sepsis (9,10). Released extracellularly from activated neutrophils in response to both infection and the sterile inflammatory process, NETs are fibrous structures that trap and kill pathogens within their matrices as part of the host’s defence mechanism. The principal components of NETs are DNA, granule protein myeloperoxidase (MPO), nuclear protein high mobility group box 1 (HMGB1), and histones, which cause endothelial damage when released (11). It was recently reported that NETs induce the formation of thrombi through a process termed immunothrombosis and that this response helps to prevent the systemic dissemination and tissue invasion of pathogens (12). Plasma from severely septic patients induced Toll-like receptor 4 (TLR4)-dependent platelet-neutrophil interactions, leading to the production of NETs (13). However, the same research demonstrated that NETs can deviate from the role of infection control, namely that immunothrombosis can induce damage to the host tissue through abnormal activation of the coagulation and immune systems, which can lead to MOF (14). In addition, NETs have components containing proteins that lead to tissue and endothelial damage as damage-associated molecular patterns (DAMPs) (15).
However, the mechanism of organ dysfunction resulting from sepsis is unclear. Shedding light on this mechanism may reveal whether management of NET formation could be a new therapeutic strategy for sepsis prevention.
The aim of the present study was to assess the mechanism from sepsis to organ dysfunction in a mouse sepsis model.
Materials and Methods
Experimental animals. Animals were treated in accordance with the Fundamental Guidelines for Proper Conduct of Animal Experiments and Related Activities in Academic Research Institutions, under the jurisdiction of the Ministry of Education, Culture, Sports, Science and Technology of Japan. All animal experiments were approved by the Committee on Animal Experimentation of Kanazawa University (AP 111868; Kanazawa, Japan). We used male BALB/c mice (aged 6-10 weeks; Charles River Laboratories, Kanagawa, Japan) as a sepsis model to observe the effects of NETs and extravasated platelets.
Sepsis model. Lipopolysaccharide (LPS) (Sigma Chemical, St. Louis, MO, USA) was dissolved in saline and diluted to 0.0003 g/ml. The solution was administered at a rate of 1 mg/kg to each mouse according to its mass and injected into the abdominal cavity.
Specimen extraction. At 0, 6, 12, and 24 h following intraperitoneal injection of LPS, mice were administered isoflurane anaesthesia and exsanguinated. Blood samples were collected and liver tissues were removed after euthanasia.
Pathological assessment. Excised organs were washed with 10% formalin. After fixation in 10% formalin solution for at least 24 h, the liver was sliced into 3-mm sections and embedded in paraffin. Each paraffin block was further sliced into 0.5-l-mm-thick sections and mounted on slides. Some sections from each mouse were prepared for histological analysis with hematoxylin and eosin (H&E) staining.
Immunohistochemistry. The remaining sections were immunostained with anti-MPO antibody (Santa Cruz Biotechnology, Santa Cruz, CA, USA), anti-hepatic sinusoidal endothelial cell antibody (SE-1) (Abcam, San Francisco, CA, USA), and anti-CD62p antibody (Abcam, San Francisco, CA, USA). Deparaffinized sections were pre-treated by autoclaving in 10% citric acid buffer (pH 8.0) at 120˚C for 15 min. Following treatment with protein block serum (Dako Cytomation, Kyoto, Japan) for 10 min and incubation with 2% skim milk for 30 min to block nonspecific reactions, sections were incubated with a primary antibody at 4˚C overnight. The EnVision polymer solution (horseradish peroxidase; Dako Cytomation) was then applied for 1 h. Signals were developed in 0.02% 3,3’-diaminobenzidine tetrahydrochloride solution containing 0.1% H2O2. Sections were lightly counterstained with hematoxylin and examined using a fluorescence microscope (Olympus, Tokyo, Japan). As negative controls, sections were incubated with Tris(hydroxymethyl)aminomethane-buffered saline containing either non-immune mouse IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA) or non-immune rabbit IgG (Santa Cruz Biotechnology, Santa Cruz, CA, USA). Sections were then lightly counterstained with hematoxylin and examined under the fluorescence microscope.
Blood samples. Plasma samples were immediately separated from the blood samples by centrifugation at 1,000 × g for 10 min. The plasma samples were then frozen and stored at −80˚C until analysis.
White blood cell (WBC) and platelet (PLT) counts were measured with an MEK-6458 (NIHON KODEN, Tokyo, Japan) using 500 μL of sampled blood (n=6). Direct bilirubin (D-Bil), aspartate aminotransferase (AST), and alanine aminotransferase (ALT) were also evaluated (n=6).
Enzyme-linked immunosorbent assay (ELISA). The concentration of MPO in the plasma was measured by ELISA using an MPO Mouse ELISA Kit (Abcam, San Francisco, CA, USA) according to the manufacturer’s instructions (n=6).
Statistical analysis. We investigated differences among the data by one-way analysis of variance or two-sided Student’s t-test with the SPSS 10.0 software package (SPSS, Chicago, IL, USA). Values of p<0.05 were considered to indicate statistically significant differences.
Results
Immunohistochemical analysis for MPO expression. Immunohistochemical analysis revealed positive staining for MPO, which was most abundantly expressed in neutrophil granulocytes, within the hepatic sinusoids immediately after LPS injection (Figure 1A). NET-like structures stained for MPO, indicating the presence of neutrophils undergoing NETosis, were confirmed at 6 h post-LPS administration (Figure 1B). This phenomenon was even more readily apparent at 12 and 24 h (Figures 1C, D).
Figure 1. Immunohistochemical analysis of MPO for shape changes after LPS administration in hepatic tissues. Immunostaining for MPO immediately after LPS administration. Neutrophils are stained as spheres (A). Immunostaining for MPO at 6 h after LPS administration. Neutrophils are stained in hepatic sinusoids as a moderately indistinct belt (B). Immunostaining for MPO at 12 h after LPS administration. Neutrophils are stained in NET-like structures (C). Immunostaining for MPO at 24 h after LPS administration. Neutrophils are more clearly stained in NET-like structures (D). Black arrows: MPO staining.

Hematoxylin-eosin staining. Liver sinusoids were lined with fenestrated endothelial cells and flanked by plates of hepatocytes in zone 3 immediately after LPS injection (Figure 2A). However, sinusoidal dilatation and congestion occurred in zone 3 at 12 h post-LPS injection (Figure 2B).
Figure 2. H&E and immunohistochemical analysis for the presence and localization of platelet aggregation and sinusoidal endothelial disorder after LPS administration. Liver sinusoids are lined with fenestrated endothelial cells and flanked by plates of hepatocytes in zone 3 immediately after LPS injection. Black arrows: Normal sinusoid (A). Sinusoidal dilatation and congestion occur in zone 3 at 12 h post-LPS injection. White arrows: Dilated sinusoid (B). Immunostaining with SE-1 immediately after LPS administration. Sinusoidal endothelial cells are stained (C). Immunostaining with SE- 1 at 12 h after LPS administration. Sinusoidal endothelial cells are not stained. Black arrows: Hepatic sinusoidal endothelial cells. White arrows: Lack of SE-1 staining in zone 3 (D). Immunostaining for CD62p immediately after LPS administration. Sinusoidal endothelial cells are stained. Black arrows: Hepatic sinusoidal endothelial cells (E). Immunostaining for CD62p at 24 h after LPS administration. Sinusoidal endothelial cells are not stained, and platelets are aggregated in the space of Disse outside the sinusoids. White arrows: Extravasated platelets (F).

Immunohistochemical analysis for SE-1 expression. We examined liver sinusoidal endothelial cells using SE-1 staining. Endothelial cells were confirmed along the sinus immediately after LPS injection (Figure 2C). However, staining was significantly reduced at 12 h post-LPS injection through sinusoidal endothelial injury or detachment (Figure 2D).
Immunohistochemical analysis for CD62p expression. We examined the presence of platelets using CD62p staining. CD62p is expressed in platelets and hepatic sinusoids endothelial cells. Immediately following LPS administration, CD62p expression was observed in hepatic sinusoid endothelial cells, but not in platelets (Figure 2E). CD62p expression was confirmed in the space of Disse at 24 h following LPS administration, indicating platelet extravasation, but there was no expression of CD62p in the hepatic sinusoidal endothelium (Figure 2F).
Blood sample analysis: Changes in WBC, PLT, and MPO. WBC, PLT, and MPO were quantified in blood samples immediately after LPS administration, and at 6, 12, and 24 h after LPS administration, and the change in each item was compared with the initial value. The mean WBC count immediately after LPS administration was 53×102/μl, followed by a temporary decrease to 11×102/μl at 6 h (p<0.001), before returning to 55×102/μl at 12 h post-LPS administration. No significant difference was found between the latter value and the value immediately after administration (p=0.412). At 24 h post-LPS administration, the WBC count had marginally decreased to 50×102/μl (p=0.037; Figure 3A). For the PLT counts, the mean value was 61×104/μl immediately after LPS administration, followed by gradual decreases to 51×104/μl at 6 h (p=0.030), 45×104/μl at 12 h (p<0.001), and 33×104/μl at 24 h (p<0.001; Figure 3B). Conversely, the mean MPO amount was 16 ng/mL immediately after LPS administration, but suddenly increased to 229 ng/mL at 6 h before peaking at 356 ng/mL at 12 h (p<0.001; Figure 3C).
Figure 3. Sequential changes in blood sample values. At 6 h following LPS administration, decreases in WBC (A) and PLT (B) counts and an increase in MPO amount (C) were confirmed. WBC: White blood cells (×102/μl); PLT: platelets (×104/μl); MPO: myeloperoxidase (ng/ml).

Analysis for liver damage: Changes in AST, ALT, and D-Bil. In this LPS-administered mouse model, we measured the levels of AST, ALT, and D-Bil in blood samples. At 12 h, the levels of AST, ALT, and D-Bil were all slightly elevated. However, at 24 h, the levels were significantly elevated: AST to a mean of 1133 IU/l (p<0.001), ALT to 935 IU/l (p=0.002), and D-Bil to 0.145 mg/dL (p=0.004) compared with the levels at 0 h (Table I).
Table I. Analysis for liver damage.

Plasma samples from mice were assessed for liver ALT, AST, and D-Bil levels. At 24 h post-LPS administration, all levels were significantly elevated. AST: Aspartate aminotransferase (IU/l); ALT: alanine aminotransferase (IU/l); D-Bil: direct bilirubin (mg/dl).
Discussion
Severe sepsis is associated with high morbidity and mortality rates. Inflammation and coagulation play pivotal roles in the pathogenesis of sepsis (8). It has been thought that DIC is characterized by the systemic activation of coagulation, which results in generation and deposition of fibrin. In turn, this process leads to microvascular thrombi in various organs, significantly contributing to MOF (16).
Neutrophils internalize pathogens through phagocytosis (17). Bacteria are destroyed within phagosomes when neutrophils’ various antimicrobial proteins are released, a process called degranulation. However, independent of phagocytic uptake, activation of neutrophils also releases chromatin and granule proteins that together form extracellular fibrillar matrices (18). Called NETs, these matrices degrade virulence factors and bind, disarm, and kill Gram-positive and Gram-negative bacteria (19). Through this active process of trapping and killing pathogens, NETs play an important role in host defence. However, recent reports have shown that NETs also interact with platelets and leukocytes, and act as three-dimensional frameworks for both deposition of fibrin and later stabilization of thrombi (20). Subsequent platelet activation induces injury to the endothelium, leading to thrombosis and impaired blood flow (21). Specifically, activated platelets stimulate neutrophils to release NETs, resulting in increased endothelial permeability (10,22).
Other recent reports have shown that excessive formation of NETs increases the risk of damage to the host (11). The principal components of NETs are DNA, granule protein MPO, nuclear protein HMGB1, and histones. Such proteins risk causing tissue and endothelial damage as DAMPs (12,23). Furthermore, this damage associated with NETs contributes to denuding of the endothelium or loss of the fenestrations and endothelial detachment (10,11,24). In the liver tissue, sinusoidal endothelial cell damage allows platelets to enter the space of Disse, which contains collagen type 3 (24,25). Within the space of Disse, platelets bind to and form aggregates with the collagen type 3 (26). Such platelet aggregation in the space of Disse has been termed extravasated platelet aggregation.
The present study revealed the process for sepsis-induced liver damage from its point of origin with NETs and resultant extravasated platelet aggregation. In this study, immunohistochemical analysis revealed positive staining for MPO, that is most abundantly expressed in neutrophil granulocytes (27), within hepatic sinusoids immediately after LPS injection. NET-like structures stained for MPO, indicating the presence of neutrophils undergoing NETosis, were confirmed at 6 h following LPS administration and thereafter. Liver sinusoidal endothelial cells stained with SE-1 were significantly reduced at 12 h post-LPS administration, the result of denuding of the endothelium, loss of the fenestrations, or endothelial detachment. Furthermore, the presence of extravasated platelets was confirmed in the space of Disse at 24 h following LPS administration.
As a consequence of unscheduled cell death, HMGB1 is released with MPO, and represents a crucial signal in the inflammatory response to tissue injury (28). To date, it has been thought that extracellular HMGB1 is a late mediator of sepsis (29); however, it is also upregulated by activated platelets in the early phase of sepsis (30). HMGB1 acts as an early and late mediator of septic lethality. The early major source of HMGB1 is derived from intravasated or extravasated platelet aggregation, and the later source is derived through the development of tissue injury.
In addition, by measuring the WBC and PLT counts in peripheral blood, as well as the quantity of MPO, the present study revealed a sudden decrease in WBC and an increase in MPO at 6 h after LPS administration. These findings were caused by rupture of neutrophils and subsequent release of MPO leading to NET formation. It is thought that the WBC count rose again temporarily because immature neutrophils from bone marrow waiting to attack foreign intruders were mobilized soon afterwards. Meanwhile, the PLT count decreased gradually, reflecting the consumption of intravasated or extravasated platelet aggregation.
It has been thought that organ dysfunction caused by sepsis arises because DIC leads to microclots that occur simultaneously and systemically (31). However, we must consider the influence of extravasated platelet aggregation wherein platelets adhere to the outside of blood vessels. In the present study, the existence of extravasated platelet aggregation in the liver was confirmed at 24 h following intraperitoneal injection of LPS. Concurrently, the AST, ALT, and D-Bil levels were also significantly elevated. The present study revealed that the combination of NET and intravasated platelet aggregation induced the early liver damage.
In our previous study, we classified the progression from sepsis to lung dysfunction (e.g. acute lung injury/ARDS) into three phases, by characterizing the interactions between neutrophils and platelets (10): Phase 1, NET formation and intravasated platelet aggregation; Phase 2, extravasated platelet aggregation, promoted by NET-facilitated detachment of endothelial cells; and Phase 3, organ failure. The present study on this phenomenon in the liver tended to confirm our previous supposition with respect to liver dysfunction arising from sepsis (Figure 4).
Figure 4. Mechanism for the progression from sepsis to liver dysfunction, characterized by neutrophil extracellular traps (NETs) and platelet aggregation. Extravasated platelet aggregation in the space of Disse, initiated by damage to the endothelium, is induced by the formation of NETs or intravasated platelet aggregation in sepsis. Various growth factors released by extravasated platelet aggregation, including TXA2, 5-HT, PAI-1, and TGF-β, induce portal hypertension and promote the progression of liver fibrosis. Furthermore, EPA-derived VEGF-A, TSP-1, s-CD40L, and TGF-β may also contribute to immunoparalysis.
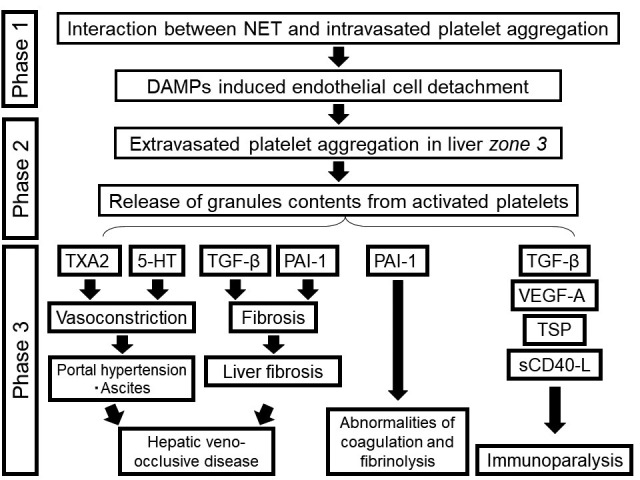
Regarding the promotion of liver damage, we suggest the following: thromboxane A2 (TXA2) (32), vascular endothelial growth factor-A (VEGF-A) (33), and 5-hydroxytryptamine (5-HT) (34) released from extravasated platelet aggregation induce vasoconstriction in the liver’s central venules and lead to portal hypertension (Figure 4). We also suggest the possibility of matter exchange obstruction between sinusoids and hepatocytes through the existence of platelets that have leaked into the space of Disse.
In addition, the release of superabundant plasminogen activator inhibitor-1 (PAI-1) (35) and transforming growth factor-β (TGF-β) (36) from platelets leads to plasmin activity inhibition and fibrinolysis, which are both involved in the development of liver failure.
Recently, a new phenotype of MOF has emerged: persistent inflammatory, immunosuppressed, catabolic syndrome (PICS). It is well known that TGF-β and VEGF-A are potent immunosuppressive factors that drive the expansion of regulatory T-cells and myeloid-derived suppressor cells (37). Moreover, platelets also contain proteins such as thrombospondin-1 (TSP-1) and CD40L. TSP-1 is a potent suppressor of T-cell activation via its receptor CD47 (38). The CD40–CD40L co-stimulatory pathway has been shown to play a crucial role in the production of cytokines, including interleukin (IL)-10, a known immunosuppressive cytokine that inhibits macrophage-dependent antigen presentation, T-cell proliferation, and Th1 cytokine secretion of IL-2, interferon-γ, and tumor necrosis factor-α (39). We suggest that the release of s-CD40L, TSP-1, and VEGF-A by the surface membrane of activated platelets leads to a state of immunoparalysis (Figure 4). Therefore, we consider that extravasated platelet aggregation plays an essential role in the progression of PICS.
We conclude that NET formation and intravasated platelet aggregation comprise the first steps from sepsis to liver dysfunction. We further conclude that extravasated platelet aggregation promoted by NET-facilitated detachment of sinusoidal endothelial cells is most likely the origin of sepsis-induced liver dysfunction.
In the initial management of sepsis, treatment basics should first and foremost include a reduction in the release of surplus NETs, reduction in platelet aggregation, and protection of the blood vessel endothelium. Moreover, inhibition of platelet activation will reduce NET release and tissue injury. It goes without saying that it is vital to investigate and treat sepsis early to prevent its progression to Phase II and Phase III.
Conflicts of Interest
The Authors declare that no financial or other conflicts of interest exist in relation to the content of this article.
References
- 1.Nathens AB, Marshall JC. Sepsis, SIRS, and MODS: what’s in a name. World J Surg. 1996;20:386–391. doi: 10.1007/s002689900061. [DOI] [PubMed] [Google Scholar]
- 2.Poole GV, Muakkassa FF, Griswold JA. The role of infection in outcome of Multiple Organ Failure. Am Surg. 1993;59:727–732. [PubMed] [Google Scholar]
- 3.Levy MM, Dellinger RP, Townsend SR, Linde-Zwirble WT, Marshall JC, Bion J, Schorr C, Artigas A, Ramsay G, Beale R, Parker MM, Gerlach H, Reinhart K, Silva E, Harvey M, Regan S, Angus DC. The Surviving Sepsis Campaign: results of an international guideline-based performance improvement program targeting severe sepsis. Crit Care Med. 2010;38:367–374. doi: 10.1097/CCM.0b013e3181cb0cdc. [DOI] [PubMed] [Google Scholar]
- 4.Ogura H, Gando S, Iba T, Eguchi Y, Ohtomo Y, Okamoto K, Koseki K, Mayumi T, Murata A, Ikeda T, Ishikura H, Ueyama M, Kushimoto S, Saitoh D, Endo S, Shimazaki S. SIRS-associated coagulopathy and organ dysfunction in critically ill patients with thrombocytopenia. Shock. 2007;28:411–417. doi: 10.1097/shk.0b013e31804f7844. [DOI] [PubMed] [Google Scholar]
- 5.Levi M, van der Poll T. Inflammation and coagulation. Crit Care Med. 2010;38:S26–34. doi: 10.1097/CCM.0b013e3181c98d21. [DOI] [PubMed] [Google Scholar]
- 6.Hardaway RM, Vasquez Y. A shock toxin that produces disseminated intravascular coagulation and multiple organ failure. Am J Med Sci. 2001;322:222–228. doi: 10.1097/00000441-200110000-00013. [DOI] [PubMed] [Google Scholar]
- 7.Gando S, Kameue T, Nanzaki S, Nakanishi Y. Disseminated intravascular coagulation is a frequent complication of systemic inflammatory response syndrome. Thromb Haemost. 1996;75:224–228. [PubMed] [Google Scholar]
- 8.van der Poll T, Levi M. Crosstalk between inflammation and coagulation: the lessons of sepsis. Curr Vas Pharmacol. 2012;10:632–638. doi: 10.2174/157016112801784549. [DOI] [PubMed] [Google Scholar]
- 9.Brinkmann V, Reichard U, Goosmann C, Fauler B, Uhlemann Y, Weiss DS, Weinrauch Y, Zychlinsky A. Neutrophil extracellular traps kill bacteria. Science. 2004;303:1532–1535. doi: 10.1126/science.1092385. [DOI] [PubMed] [Google Scholar]
- 10.Miyashita T, Ahmed AK, Nakanuma S, Okamoto K, Sakai S, Kinoshita J, Makino I, Nakamura K, Hayashi H, Oyama K, Tajima H, Takamura H, Ninomiya I, Fushida S, Harmon JW, Ohta T. A Three-phase Approach for the Early Identification of Acute Lung Injury Induced by Severe Sepsis. In Vivo. 2016;30:341–349. [PubMed] [Google Scholar]
- 11.Brinkmann V, Zychlinsky A. Neutrophil extracellular traps: is immunity the second function of chromatin. J Cell Biol. 2012;198:773–783. doi: 10.1083/jcb.201203170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kambas K, Mitroulis I, Ritis K. The emerging role of neutrophils in thrombosis-the journey of TF through NETs. Front Immunol. 2012;3:385. doi: 10.3389/fimmu.2012.00385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Clark SR, Ma AC, Tavener SA, McDonald B, Goodarzi Z, Kelly MM, Patel KD, Chakrabarti S, McAvoy E, Sinclair GD, Keys EM, Allen-Vercoe E, Devinney R, Doig CJ, Green FH, Kubes P. Platelet TLR4 activates neutrophil extracellular traps to ensnare bacteria in septic blood. Nat Med. 2007;13:463–469. doi: 10.1038/nm1565. [DOI] [PubMed] [Google Scholar]
- 14.Vazquez-Garza E, Jerjes-Sanchez C Navarrete A, Joya-Harrison J, Rodriguez D. Venous thromboembolism: thrombosis, inflammation, and immunothrombosis for clinicians. J Thromb Thrombolysis 20. 2017 doi: 10.1007/s11239-017-1528-7. doi: 10.1007/s11239-017-1528-7. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 15.Chen R, Kang R, Fan XG, Tang D. Release and activity of histone in diseases. Cell Death Dis. 2014;5:e1370. doi: 10.1038/cddis.2014.337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Vincent JL, De Backer D. Does disseminated intravascular coagulation lead to multiple organ failure. Crit Care Clin. 2005;21:469–477. doi: 10.1016/j.ccc.2005.04.002. [DOI] [PubMed] [Google Scholar]
- 17.Mayadas TN, Cullere X, Lowell CA. The multifaceted functions of neutrophils. Annu Rev Pathol Mech. 2014;9:181–218. doi: 10.1146/annurev-pathol-020712-164023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thomas MP, Whangbo J, McCrossan G, Deutsch AJ, Martinod K, Walch M, Lieberman J. Leukocyte protease binding to nucleic acids promotes nuclear localization and cleavage of nucleic acid binding proteins. J Immunol. 2014;192:5390–5397. doi: 10.4049/jimmunol.1303296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wartha F, Beiter K, Normark S, Henriques-Normark B. Neutrophil extracellular traps: casting the NET over pathogenesis. Curr Opin Microbiol. 2007;10:52–56. doi: 10.1016/j.mib.2006.12.005. [DOI] [PubMed] [Google Scholar]
- 20.Russwurm S, Vickers J, Meier-Hellmann A, Spangenberg P, Bredle D, Reinhart K, Lösche W. Platelet and leukocyte activation correlate with the severity of septic organ dysfunction. Shock. 2002;17:263–268. doi: 10.1097/00024382-200204000-00004. [DOI] [PubMed] [Google Scholar]
- 21.Katz JN, Kolappa KP, Becker RC. Beyond thrombosis: the versatile platelet in critical illness. Chest. 2011;139:658–668. doi: 10.1378/chest.10-1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Caudrillier A, Kessenbrock K, Gilliss BM, Nguyen JX, Marques MB, Monestier M, Toy P, Werb Z, Looney MR. Platelets induce neutrophil extracellular traps in transfusion-related acute lung injury. J Clin Invest. 2012;122:2661–2671. doi: 10.1172/JCI61303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yipp BG, Kubes P. NETosis: how vital is it. Blood. 2013;122:2784–2794. doi: 10.1182/blood-2013-04-457671. [DOI] [PubMed] [Google Scholar]
- 24.Miyashita T, Nakanuma S, Ahmed AK, Makino I, Hayashi H, Oyama K, Nakagawara H, Tajima H, Takamura H, Ninomiya I, Fushida S, Harmon JW, Ohta T. Ischemia reperfusion-facilitated sinusoidal endothelial cell injury in liver transplantation and the resulting impact of extravasated platelet aggregation. Eur Surg Journal ISSN. 2016;48:92–98. doi: 10.1007/s10353-015-0363-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nakanuma S, Miyashita T, Hayashi H, Tajima H, Takamura H, Tsukada T, Okamoto K, Sakai S, Makino I, Kinoshita J, Nakamura K, Oyama K, Inokuchi M, Nakagawara H, Ninomiya I, Kitagawa H, Fushida S, Fujimura T, Ohta T. Extravasated platelet aggregation in liver zone 3 may correlate with the progression of sinusoidal obstruction syndrome following living donor liver transplantation: A case report. Exp Ther Med. 2015;9:1119–1124. doi: 10.3892/etm.2015.2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Monnet E, Fauvel-Lefeve F. A new platelet receptor specific to type III collagen – Type III collagen-binding protein. J Biol Chem. 2000;275:10912–10917. doi: 10.1074/jbc.275.15.10912. [DOI] [PubMed] [Google Scholar]
- 27.Klebanoff SJ. Myeloperoxidase: friend and foe. J Leukoc Biol. 2005;77:598–625. doi: 10.1189/jlb.1204697. [DOI] [PubMed] [Google Scholar]
- 28.Andersson U, Tracey KJ. HMGB1 is a therapeutic target for sterile inflammation and infection. Annu Rev Immunol. 2011;29:139–162. doi: 10.1146/annurev-immunol-030409-101323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wang H, Zhu S, Zhou R, Li W, Sama AE. Therapeutic potential of HMGB1-targeting agents in sepsis. Expert Rev Mol Med. 2008;10:e32. doi: 10.1017/S1462399408000884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Maugeri N, Campana L, Gavina M, Covino C, De Metrio M, Panciroli C, Maiuri L, Maseri A, D’Angelo A, Bianchi ME, Rovere-Querini P, Manfredi AA. Activated platelets present high mobility group box 1 to neutrophils, inducing autophagy and promoting the extrusion of neutrophil extracellular traps. J Thromb Haemost. 2014;12:2074–2088. doi: 10.1111/jth.12710. [DOI] [PubMed] [Google Scholar]
- 31.Asakura H. Classifying types of disseminated intravascular coagulation: clinical and animal models. J Intensive Care. 2014;2:20. doi: 10.1186/2052-0492-2-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Li P, Ferrario CM, Brosnihan KB. Losartan inhibits thromboxane A2-induced platelet aggregation and vascular constriction in spontaneously hypertensive rats. J Cardiovasc Pharmacol. 1998;32:198–205. doi: 10.1097/00005344-199808000-00005. [DOI] [PubMed] [Google Scholar]
- 33.Parenti A, Brogelli L, Filippi S, Donnini S, Ledda F. Effect of hypoxia and endothelial loss on vascular smooth muscle cell responsiveness to VEGF-A: role of flt-1/VEGF-receptor-1. Cardiovasc Res. 2002;55:201–212. doi: 10.1016/s0008-6363(02)00326-7. [DOI] [PubMed] [Google Scholar]
- 34.Moerland M, Kemme M, Dijkmans A, Bergougnan L, Burggraaf J. Modulation of vasoactivity and platelet aggregation by selective 5-HT receptor antagonism in humans. J Cardiovasc Pharmacol. 2011;58:575–580. doi: 10.1097/FJC.0b013e31822f6b8d. [DOI] [PubMed] [Google Scholar]
- 35.Lee JH, Lee KH, Lee JH, Kim S, Seol M, Park CJ, Chi HS, Kang W, Kim ST, Kim WK, Lee JS. Plasminogen activator inhibitor-1 is an independent diagnostic marker as well as severity predictor of hepatic veno-occlusive disease after allogeneic bone marrow transplantation in adults conditioned with busulphan and cyclophosphamide. Br J Haematol. 2002;118:1087–1094. doi: 10.1046/j.1365-2141.2002.03748.x. [DOI] [PubMed] [Google Scholar]
- 36.Pohlers D, Brenmoehl J, Löffler I, Müller CK, Leipner C, Schultze-Mosgau S, Stallmach A, Kinne RW, Wolf G. TGF-beta and fibrosis in different organs – molecular pathway imprints. Biochim Biophys Acta. 2009;1792:746–756. doi: 10.1016/j.bbadis.2009.06.004. [DOI] [PubMed] [Google Scholar]
- 37.Terme M, Tartour E, Taieb J. VEGFA/VEGFR2-targeted therapies prevent the VEGFA-induced proliferation of regulatory T cells in cancer. Oncoimmunology. 2013;2:e25156. doi: 10.4161/onci.25156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Miller TW, Kaur S, Ivins-O’Keefe K, Roberts DD. Thrombospondin-1 is a CD47-dependent endogenous inhibitor of hydrogen sulfide signaling in T cell activation. Matrix Biol. 2013;32:316–324. doi: 10.1016/j.matbio.2013.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santin AD, Hermonat PL, Ravaggi A, Bellone S, Pecorelli S, Roman JJ, Parham GP, Cannon MJ. Interleukin-10 increases Th1 cytokine production and cytotoxic potential in human papillomavirus-specific CD8(+) cytotoxic T lymphocytes. J Virol. 2000;74:4729–4737. doi: 10.1128/jvi.74.10.4729-4737.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]