

Engineering the ubiquitin transfer cascades by phage display enables an efficient way to profile E3 substrates.
Abstract
E3 ubiquitin (UB) ligases E4B and carboxyl terminus of Hsc70-interacting protein (CHIP) use a common U-box motif to transfer UB from E1 and E2 enzymes to their substrate proteins and regulate diverse cellular processes. To profile their ubiquitination targets in the cell, we used phage display to engineer E2-E4B and E2-CHIP pairs that were free of cross-reactivity with the native UB transfer cascades. We then used the engineered E2-E3 pairs to construct “orthogonal UB transfer (OUT)” cascades so that a mutant UB (xUB) could be exclusively used by the engineered E4B or CHIP to label their substrate proteins. Purification of xUB-conjugated proteins followed by proteomics analysis enabled the identification of hundreds of potential substrates of E4B and CHIP in human embryonic kidney 293 cells. Kinase MAPK3 (mitogen-activated protein kinase 3), methyltransferase PRMT1 (protein arginine N-methyltransferase 1), and phosphatase PPP3CA (protein phosphatase 3 catalytic subunit alpha) were identified as the shared substrates of the two E3s. Phosphatase PGAM5 (phosphoglycerate mutase 5) and deubiquitinase OTUB1 (ovarian tumor domain containing ubiquitin aldehyde binding protein 1) were confirmed as E4B substrates, and β-catenin and CDK4 (cyclin-dependent kinase 4) were confirmed as CHIP substrates. On the basis of the CHIP-CDK4 circuit identified by OUT, we revealed that CHIP signals CDK4 degradation in response to endoplasmic reticulum stress.
INTRODUCTION
E3 ubiquitin (UB) ligases C terminus of Hsc70-interacting protein (CHIP) and E4B are on the front line of defense against misfolded or damaged proteins in the cell by tagging them with UB and channeling them to the proteasome for degradation (1–3). The two E3s use a signature U-box domain to mediate UB transfer from an E1-E2 relay to their target proteins (4, 5). They play a key role in protein quality control; inadequate degradation of their substrates such as tau, huntingtin, and α-synuclein triggers Alzheimer’s disease, Huntington’s disease, and Parkinson’s disease, respectively (6, 7). Both E3s can elongate UB chains appended to cellular proteins by other E3s, a property termed “E4 ligation” (8, 9). For examples, the E3-E4 pair of parkin-CHIP ubiquitinates Pael-R (Pael receptor) and signals its degradation to relieve endoplasmic reticulum (ER) stress in neuronal cells, and the E3-E4 pair of mouse double minute 2 (Mdm2)–E4B induces p53 degradation and promotes the survival of cancer cells (10, 11). The multifaceted roles of CHIP and E4B demand an investigation of their substrate profiles to reveal their roles in cell regulation.
CHIP and E4B run the last leg of the E1-E2-E3 relay in transferring UB to the cellular proteins. The large diversity of E3s (>600 in human cells) makes it a significant challenge to identify the direct substrates of a specific E3 in the cell. We developed a method termed “orthogonal UB transfer (OUT)” to profile E3 substrate specificity (12). OUT is used to transfer an engineered UB (xUB) through an engineered cascade (xE1-xE2-xE3) to the substrate proteins of a specific E3 (“x” designates engineered UB or UB transferring enzymes). To implement OUT, we need to engineer xUB-xE1, xE1-xE2, and xE2-xE3 pairs that are orthogonal (free of cross-reactivity) with native UB and the E1, E2, and E3 enzymes. We previously showed that we could generate xUB-xE1 and xE1-xE2 pairs with yeast E1 and E2 (Uba1 and Ubc1) (12). Here, we report that we can engineer xE2-xE3 pairs with E4B and CHIP to assemble full-length OUT cascades with the two E3s. We used the OUT cascades to identify the ubiquitination targets of E4B and CHIP in human embryonic kidney 293 (HEK293) cells. On the basis of the substrate profiles generated by OUT, we found that CHIP plays an important role in regulating cyclin-dependent kinase 4 (CDK4) activity during ER stress.
RESULTS
Constructing an xUba1-xUbcH5b pair for the OUT cascade
We previously reported that an engineered yeast E1 (xUba1) could activate a UB mutant (xUB) and transfer it to an engineered yeast E2 (xUbc1) (Table 1 and fig. S1). We could thus use xUba1 and xUbc1 to assemble a two-step OUT cascade for xUB transfer (12). On the basis of the sequence homology between the human and yeast E1s, we generated human xUba1 by incorporating mutations Q608R, S621R, and D623R into the adenylation domain and mutations E1037K, D1047K, and E1049K into the UB fold domain (UFD) of human Uba1 (Table 1). This human xUba1 activates xUB to form xUB~xUba1 thioester conjugates (fig. S2A). We also introduced mutations K4E and K8E into UbcH5b based on the mutations in yeast xUbc1 and found that the UbcH5b mutant (xUbcH5b) could accept xUB from xUba1 (figs. S1D and S2A). This human xUba1-xUbcH5b pair did not cross-react with either native protein complement—Western blots of the crossover reaction showed that xUB could not be activated by the wild-type (wt) human Uba1, and vice versa, wt UB could not be activated by human xUba1. Furthermore, because of the mutations in the UFD domain, xUB could not be transferred from xUba1 to wt UbcH5b (fig. S2A). Thus, the activity of the human xUba1-xUbcH5b pair is completely orthogonal to the native E1-E2 pair in xUB transfer. In a recent report, we incorporated adenylation domain mutations into the two human E1s (Uba1 and Uba6) to activate xUB. We then paired xUB with either Uba1 or Uba6 mutant to differentiate the downstream ubiquitination targets of the two E1 enzymes (13).
Table 1. Mutants of UB, E1, E2, and E3 for the assembly of the OUT cascade with E4B and CHIP.
| xUB (human) | R42E, R72E |
| xE1 | |
| xUba1 (yeast) | Q576R, S589R, D591R, E1004K, D1014K, E1016K |
| xUba1 (human) | Q608R, S621R and D623R, E1037K, D1047K, E1049K |
| xUba1(UFD) (human) | E1037K, D1047K, E1049K |
| xE2 | |
| xUbc1 (yeast) | K5D, R6E, K9E, E10Q, Q12L |
| xUbcH5b (human) | K4E, K8E |
| xE3 | |
| xE4B (fE4B-KB2) | R1233K, L1236I, D1238H |
| xE4B (fE4B-KB12) | R1233K, L1236I, D1238R |
| xCHIP (CHIP-KB2) | C213K, G214D, K215P, S217M, F218H, E219T |
| xCHIP (CHIP-KB12) | C213K, G214D, K215P, S217M, F218R, E219T |
Engineering an xUbaH5b-xE4B pair for OUT
E4B and CHIP engage UB~E2 with their C-terminal U-box domains (5). U-boxes are 70-residue domains that share a similar fold to the RING domains found in most of the E3s (4, 14, 15). Both RING and U-box domains bind the N-terminal helix of the E2 with residues in loop1 (fig. S3) (16–20). The key difference between the two is that the RING domain relies on the chelation with two Zn2+ atoms to stabilize its fold, whereas the U-box domain relies on multiple hydrogen bonding and salt bridge interactions to fold properly (15). We found that xUB could not be transferred to the U-box domain of E4B, full-length CHIP, or the RING domains of E3s c-Cbl, Cbl-b, or Traf6 through the xUba1-xUbcH5b pair (fig. S2B) (17, 19, 21). Presumably, the mutations in the N-terminal helix of xUbcH5b (K4E and K8E) block the interaction of E2 with the wt U-box. To confirm this, we generated a Uba1 mutant, xUba1(UFD), which has a wt adenylation domain to activate wt UB but a mutated UFD domain to enable UB transfer to xUbcH5b (Table 1 and fig. S1). We found that xUba1(UFD) could load wt UB onto xUbcH5b but that xUbcH5b could not transfer wt UB to CHIP or the U-box of E4B (fig. S2C). This proves that the K4E and K8E mutations in the N-terminal helix of xUbcH5b blocked UB transfer from xE2 to U-box E3s.
The crystal structures of the E4B U-box in complex with UbcH5c and the CHIP U-box in complex with UbcH5a both suggest an important role for E2 residues K4 and K8 in binding the U-box loop1 residues (fig. S3) (16, 18). Because the E2-E3 interface is dynamic, the crystal structures serve as guidelines for interactions that may form in the solution (22, 23). In the complex of UbcH5c with the E4B U-box, K4 and K8 of E2 mainly engage loop1 residues R1233, L1236, M1237, D1238, and T1239 of the U-box (fig. S3, A and B) (16). We thus surmised that a U-box library of E4B with randomized residues at these sites could be used in combination with phage display to identify U-box mutants with restored UB transfer from xUbcH5b.
The E4B U-box functions as a monomer; hence, a library of this domain could be displayed on T7 phage to select for enhanced interaction with wt UbcH5c based on UB transfer (14, 24). Encouraged by these reports, we adopted a similar strategy to select for U-box mutants based on UB transfer from xUbcH5b. In the selection scheme, we used xUba1(UFD) to load biotin-labeled wt UB (biotin-UB) onto xUbcH5b (Fig. 1A). We then reacted the biotin-UB~xUbcH5b conjugate with the E4B U-box library displayed on the phage surface. U-box mutants that were recognized by xUbcH5b would be covalently conjugated with biotin-UB and the corresponding phage particles selected by affinity binding with streptavidin. We found that the E4B U-box could be displayed well on M13 phage and that it was highly active in autoubiquitination with wt UbcH5b (fig. S4A). We also used model selection to confirm that wt U-box displayed on phage could be selected with high efficiency (fig. S4, B and C).
Fig. 1. Phage selection for the orthogonal xUbcH5b-xU-box pair for E4B.
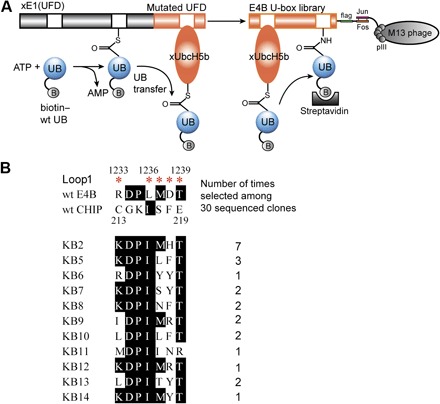
(A) wt UB labeled with biotin (biotin–wt UB) was activated by xUba1(UFD) and loaded on xUbcH5b. The U-box library of E4B was displayed on the surface of M13 phage. Phage displaying U-box mutants reactive with xUbcH5b were conjugated with biotin-UB and selected by binding to streptavidin. ATP, adenosine 5′-triphosphate; AMP, adenosine 5′-monophosphate. (B) Sequence alignment of U-box mutants of E4B from phage selection. Sites of randomized residues in the loop1 of the E4B U-box are marked by red asterisks. After five rounds of selection, 30 phage clones were sequenced; the alignment of the sequences showed that the phage library was converged to KB2 and other highly homologous clones.
We carried out five rounds of phage selection on the E4B U-box library. As the selection proceeded, we observed a gradual increase in the number of phage eluted from the biotin-UB loading reaction compared to the controls with either xUba1(UFD) or xUbcH5b missing from the reaction (fig. S4D). Sequencing of the U-box clones from the fifth round revealed a clear pattern of convergence (Fig. 1B). Among the 30 clones sequenced, KB2 appeared seven times, and KB5, KB7, and KB8 homologous to KB2 appeared two or three times. The most significant change in the selected clones was that the negatively charged D1238 in wt U-box was replaced with positively charged His or Arg, a change that matched K4E and K8E mutations in xUbcH5b by restoring charge complementarity at the E2–U-box interface. In addition, R1233 was frequently converted to Lys, and L1236 was almost uniformly converted to Ile. The same L1236I mutation was enriched by T7 phage selection to enhance UB transfer from wt UbcH5c to E4B U-box (24). In the other two randomized positions, M1236 and T1239, wt residues dominated in the selected pool. T1239 was almost never changed, suggesting that its interaction with D1254 on the opposing helix might be important for U-box structure and activity (fig. S3B). We prepared phage with selected clones KB2 and KB12 and found that the U-box mutants displayed on phage could be efficiently conjugated with biotin-UB transferred through the xUba1(UFD)-xUbcH5b pair (Fig. 2A). This implies that the U-box mutants were selected on the basis of catalytic UB transfer to the phage library.
Fig. 2. Confirmation of the activity of the selected E4B U-box mutants in mediating UB transfer with xUbcH5b.
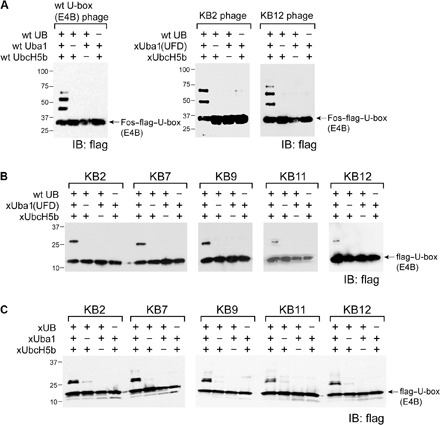
(A) wt UB could be efficiently transferred through the xUba1(UFD)-xUbcH5b pair to the KB2 and KB12 mutants displayed on the phage surface. wt UB transfer through the wt Uba1-UbcH5b pair to wt U-box of E4B on phage surface was set up as a control. IB, immunoblotting. (B) U-box mutants KB2, KB7, KB9, KB11, and KB12 from phage selection were expressed as fusions with a flag tag, and their autoubiquitination with wt UB though the xUba1(UFD)-xUbcH5b pair was analyzed by Western blotting. (C) Similar to (B), U-box mutants were subjected to xUB transfer through the xUba1-xUbcH5b pair. In both cases, efficient modification of the U-box by wt UB and xUB was observed. Reactions were stopped at the stage of monoubiquitination to compare the reaction efficiency among various U-box mutants.
Activity of the selected U-box mutants of E4B
We expressed the U-box mutants KB2, KB7, KB9, KB11, and KB12 fused to a flag tag and reacted them with wt UB in the presence of the xUba1(UFD)-xUbcH5b pair. We stopped the reaction at monoubiquitination to compare the reactivity of U-box variants (Fig. 2B). All the U-box mutants were active with the xUba1(UFD)-xUbcH5b pair in modification with wt UB. They were also active with the xUba1-xUbcH5b pair that transferred xUB to the U-box (Fig. 2C). Next, we cloned the U-box mutants into the full-length E4B (fE4B) to assay if they could mediate xUB transfer to fE4B and then to its substrate p53 (11). We chose KB2 and KB12 for incorporation into fE4B because KB2 is the most abundant U-box clone from phage selection, and KB12 only differed from KB2 by an Arg instead of His replacing D1238. fE4B is a large protein of 1137 residues (1). We found that it could be expressed in Escherichia coli with a pET vector and that its activity could be enhanced by ammonium sulfate precipitation after eluting the protein from the nickel–nitrilotriacetic acid (Ni-NTA) column. wt fE4B could be efficiently ubiquitinated with wt UB through the wt Uba1-UbcH5b pair, yet it could not be modified by xUB through the xUba1-xUbcH5b pair (Fig. 3A). In contrast, fE4B with U-box mutants of KB2 and KB12 (fE4B-KB2 and fE4B-KB12) could be efficiently ubiquitinated with xUB through the xUba1-xUbcH5b pair. We have thus constructed an OUT cascade for xUB transfer to fE4B-KB2 or fE4B-KB12. We also found that xUB could be transferred to p53 through xUba1-xUbcH5b relaying with either fE4B-KB2 or fE4B-KB12 and that, with a similar efficiency, wt UB could be transferred through wt Uba1-UbcH5b-fE4B to p53 (Fig. 4A). The crossover cascade of xUba1-xUbcH5b-wt fE4B was incapable of transferring xUB to p53, suggesting the orthogonality of the OUT cascade with the native UB transfer cascade. Hence, either fE4B-KB2 or fE4B-KB12 could be used as an xE4B to construct the OUT cascade for profiling E4B substrates.
Fig. 3. Activity of engineered fE4B and CHIP mutants in autoubiquitination with xUB.
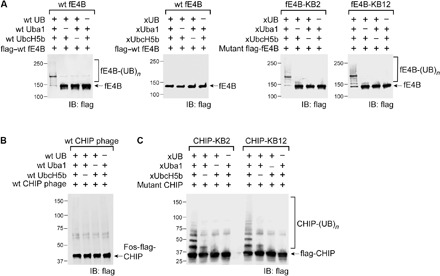
(A) fE4B-KB2 and fE4B-KB12 are fE4B with mutated U-box domains KB2 and KB12. They could be autoubiquitinated by xUB through the xUba1-xUbcH5b pair. The activity of mutant E4B autoubiquitination was similar to wt fE4B autoubiquitination. In contrast, wt fE4B could not be ubiquitinated by xUB through the xUba1-xUbcH5b pair, suggesting the orthogonality of the OUT cascade and the native cascade of E4B. (B) wt CHIP displayed on the surface of M13 phage lost activity in autoubiquitination by wt UB and the wt Uba1-UbcH5b pair. (C) CHIP-KB2 and CHIP-KB12 were constructed by replacing the loop1 of the CHIP U-box with corresponding sequences in the KB2 and KB12 mutants of the E4B U-box. This enabled the engineered CHIP to be ubiquitinated by xUB through the xUba1-xUbcH5b pair. The efficiency of CHIP-KB2/12 autoubiquitination with xUB was similar to that of wt CHIP ubiquitination by wt UB through the wt Uba1-UbcH5b pair (fig. S2B).
Fig. 4. xUB transfer through the OUT cascade of E4B and CHIP to p53.
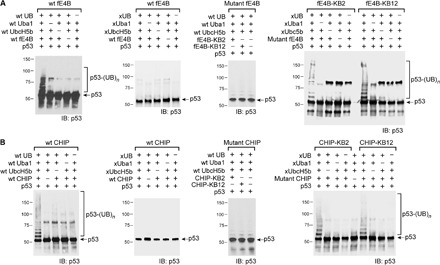
(A) fE4B-KB2 and fE4B-KB12 could assemble an OUT cascade with xUba1 and xUbcH5b to mediate xUB transfer to p53. The efficiency of p53 ubiquitination by xUB and the OUT cascade was similar to p53 ubiquitination with wt UB and the wt Uba1-UbcH5b-fE4B cascade. In contrast, wt E4B could not pair with xUba1-xUbcH5b to transfer xUB to p53, suggesting the orthogonality between the OUT cascade and native E3s. Mutant fE4B KB2 or KB12 could not pair with wt Uba1–wt UbcH5b to transfer wt UB to p53. (B) Similar to E4B OUT cascade, CHIP-KB2 and CHIP-KB12 could relay with xUba1-xUbcH5b to transfer xUB to p53. The efficiency of xUB modification of p53 by the CHIP OUT cascades was similar to that of p53 modification by wt UB going through the wt Uba1-UbcH5b-CHIP cascade. xUB could not be transferred to p53 with the crossover cascade of xUba1-xUbcH5b–wt CHIP. wt UB could not be transferred to p53 with the crossover cascade of wt Uba1–wt UbcH5b–mutant CHIP (KB2 or KB12).
Constructing an OUT cascade with CHIP
We set out to use phage selection to identify U-box mutants of CHIP with restored UB transfer from xUbcH5b. However, although the full-length CHIP including the U-box domain could be displayed on the phage surface, it was not active in autoubiquitination reactions with wt UB transferred through the wt Uba1-UbcH5b pair (Fig. 3B). CHIP functions as a dimer, so the lack of activity was attributed to the inability of CHIP to form suitable dimers when displayed on phage (fig. S3C) (20, 25). To address this challenge, we took an alternative approach by transplanting the mutated loop1 residues from the KB2 and KB12 variants of the E4B U-box into CHIP. We reasoned that this might restore CHIP interaction with xUbcH5b because the U-box domains of CHIP and E4B are highly homologous in structure, although their loop1 sequences do not align well (Fig. 1B and fig. S3). To this end, loop1 residues 213CGKISFE219 in the CHIP U-box were replaced with corresponding residues from the KB2 U-box (1233KDPIMHT1239) and KB12 U-box (1233KDPIMRT1239), generating CHIP-KB2 and CHIP-KB12, respectively (Table 1). The success of this design was confirmed in xUB autoubiquitination reactions with the xUba1-xUbcH5b pair (Fig. 3C). Furthermore, both CHIP mutants could transfer xUB to p53, a known CHIP substrate (26), at an efficiency comparable to wt UB transfer by wt CHIP (Fig. 4B). Moreover, xUB could not be transferred to p53 through the crossover cascade of xUba1-xUbcH5b–wt CHIP. These results demonstrated that either CHIP-KB2 or CHIP-KB12 could be used as an xCHIP in an OUT cascade to profile its substrates.
Profiling E4B and CHIP substrates by OUT
We decided to use fE4B-KB2 as xE4B and CHIP-KB12 as xCHIP to assemble the OUT cascades of the two U-box E3s and profile their substrates in the cell (Table 1). To assay the orthogonality of the OUT cascades with the native UB transferring enzymes in the cell, we expressed xUba1 or wt Uba1 in HEK293 cells with the coexpression of xUB or wt UB with a tandem 6×His tag and a biotin tag at the N terminus (HBT-xUB or HBT–wt UB) (27). Coimmunoprecipitation proves the formation of the xUB~xUba1 conjugate but not the xUB~wt Uba1 or wt UB~xUba1 conjugates in the cell (fig. S5A). This suggests that xUB is specifically transferred to xUba1 of the OUT cascade. Similarly, we found that xUB was transferred to xUbcH5b or xE4B or xCHIP expressed in the cell but not the native E2 or E3 enzymes (fig. S5, B to D). This proves that the OUT cascade exclusively delivered xUB to xE3 in the cell.
We generated stable HEK293 cell lines expressing the OUT cascades of E4B and CHIP. We transiently transfected the cell lines to express HBT-xUB (fig. S6A). To isolate the substrate proteins, cells were lysed and xUB-conjugated proteins were purified by tandem affinity chromatography with Ni-NTA and streptavidin resin under denaturing conditions. The substrates bound to the streptavidin resin were digested by trypsin and identified by mass spectrometry (MS) proteomics. To filter away proteins bound nonspecifically to the resin or conjugated to xUB independent of xE3, HBT-xUB was expressed in stable cell lines that expressed the xUba1-xUbcH5b pair without xE4B or xCHIP. xUB-conjugated proteins were purified from the control cells and identified by MS. Comparison of the profiles of xUB-conjugated proteins from cells expressing the OUT cascade and the control cells identified proteins that were dependent on xE4B or xCHIP for modification by xUB. These proteins are candidates for the direct ubiquitination targets of E4B and CHIP in the cell.
Following this protocol, we generated profiles of xUB-conjugated proteins in the cells expressing the E4B OUT cascade and in control cells expressing the xUba1-xUbcH5b pair without xE4B (fig. S6, B and C). To compare the two profiles, we calculated the ratio of peptide spectrum matches (PSMs) of each protein purified from cells with the OUT cascade versus the control cells. We performed tandem purification and proteomic profiling on the two cell lines three times. Proteins with a PSM ratio equal to or greater than 2 in all three repeats are listed in table S1. With the same approach, we generated a profile of CHIP substrates from the OUT screen (fig. S6E and table S2).
Among a few E4B substrates reported, we found p53 as an E4B target identified by OUT (table S1). Bioinformatics analysis of the E4B substrates with the Ingenuity Pathway Analysis (IPA) revealed that a protein network controlling DNA replication, recombination, and repair had the most significant association with potential E4B substrates—p53 and 31 other E4B substrates from the OUT screen were associated with this network (table S3). The CHIP substrates identified by OUT included a number of known CHIP targets such as p53, protein Arg N-methyltransferase 5 (PRMT5), filamin-A, and Hsp70 chaperones (28). IPA suggested that CHIP substrates were associated most significantly with a regulatory network of cell death and survival and DNA replication, recombination, and repair, which included p53, protein phosphatase 3 catalytic subunit alpha (PPP3CA), and 30 other substrates from the OUT screen (table S4). We found that several kinases and methyltransferases are potential targets of E4B and CHIP; their ubiquitination by the U-box E3s might underlie new mechanisms of cell regulation. For example, protein PRMT1 (protein arginine N-methyltransferase 1), kinase MAPK3 (mitogen-activated protein kinase 3), and phosphatase PPP3CA are potentially shared substrates of E4B and CHIP; deubiquitinase OTUB1 (ovarian tumor domain containing ubiquitin aldehyde binding protein 1) and phosphatase PGAM5 (phosphoglycerate mutase 5) could be E4B substrates; and transcription factor β-catenin and kinase CDK4 are CHIP substrates (tables S1 and S2). We carried out ubiquitination assays in vitro and in the cell to verify whether these proteins were bona fide targets of E4B or CHIP.
Verification of E4B and CHIP substrates
We expressed PRMT1, MAPK3, PPP3CA, PGAM5, OTUB1, β-catenin, and CDK4 in E. coli and set up in vitro ubiquitination reactions with wt fE4B and wt CHIP. Substrates expressed from the E. coli might not have the proper posttranslational modification such as phosphorylation to mediate recognition by an E3, or adaptor proteins could be missing to mediate UB transfer. Nevertheless, we observed polyubiquitination of PRMT1, MAPK3, and OTUB1 when wt UB was transferred through the wt Uba1-UbcH5b-fE4B cascade. PPP3CA and PGAM5 mainly gave monoubiquitinated species after reaction with the UB transfer cascade of E4B (Fig. 5A). We also found that CHIP could polyubiquitinate MAPK3, β-catenin, and CDK4 and that CHIP ubiquitination of PRMT1 and PPP3CA generated monoubiquitinated species (Fig. 5B).
Fig. 5. In vitro assay to verify that E4B and CHIP catalyzed the ubiquitination of the substrates.
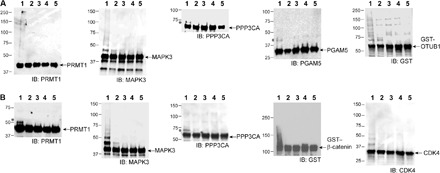
(A) Ubiquitination of PRMT1, MAPK3, PPP3CA, PGAM5, and OTUB1 by wt UB through the wt Uba1-UbcH5b-fE4B cascade (lane 1). In lanes 2, 3, 4, and 5, wt E4B, Uba1, UbcH5b, and wt UB were missing from the reaction mixture, respectively. GST, glutathione S-transferase. (B) Ubiquitination of PRMT1, MAPK3, PPP3CA, β-catenin, and CDK4 by wt UB through the wt Uba1-UbcH5b-CHIP cascade (lane 1). In lanes 2, 3, 4, and 5, wt CHIP, Uba1, UbcH5b, and wt UB were missing from the reaction mixture, respectively. Bands marked with asterisks were likely generated by monoubiquitinated substrates.
To verify the ubiquitination of the identified substrate proteins by E4B and CHIP in the cell, we transfected HEK293 cells with short hairpin RNA (shRNA) against E4B or CHIP and screened them for stable cell lines with inhibited expression of the E3s (Fig. 6, A and C). HEK293 cells overexpressing E4B or CHIP or cells overexpressing an E3 and, at the same time, stably transfected with shRNA against the same E3 were also screened. Substrate-specific antibodies were used to immunoprecipitate each substrate protein from the cells, and an anti-UB antibody was used to probe their ubiquitination levels. We found a decreased level of ubiquitination of PRMT1, MAPK3, and PPP3CA in the cells expressing shRNA against E4B or CHIP. Overexpression of either E4B or CHIP in cells stably transfected with shE4B or shCHIP significantly increased ubiquitination of the substrates (Fig. 6, B and D). These results suggest that PRMT1, MAPK3, and PPP3CA are likely shared ubiquitination targets of E4B and CHIP; inhibiting the expression of either E3 would significantly affect the ubiquitination of these substrates in the cell. Using the same assay, we confirmed that ubiquitination of PGAM5 and OTUB1 was dependent on E4B—their ubiquitination was significantly decreased in shE4B cells, and the ubiquitination level was restored by overexpressing E4B in shE4B cells (Fig. 6B). Similarly, we confirmed CHIP-dependent ubiquitination of β-catenin and CDK4 in the cell (Fig. 6D).
Fig. 6. E4B and CHIP-dependent ubiquitination of substrate proteins in HEK293 cells.
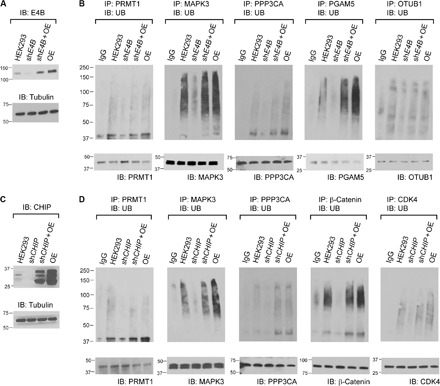
(A) Inhibition of E4B expression in HEK293 cells by shE4B was confirmed by Western blotting probed with an antibody against E4B. (B) Ubiquitination of PRMT1, MAPK3, PPP3CA, PGAM5, and OTUB1 in HEK293 cells was assayed by immunoprecipitation with antibodies against each substrate proteins and probing their ubiquitination levels with an anti-UB antibody on the Western blots. The cells were treated with 10 μM MG132 for 1.5 hours before they were lysed. The ubiquitination of each substrate protein was compared among the control HEK293 cell (HEK293), HEK293 expressing shE4B (shE4B), HEK293 expressing both shE4B and recombinant E4B complementary DNA (cDNA) (shE4B + OE), and HEK293 overexpressing E4B from recombinant cDNA (OE). IgG, immunoglobulin G. (C) Similar to (A) to confirm the inhibition of CHIP expression in HEK293 cells by shCHIP. (D) Similar to (B) to confirm CHIP-dependent ubiquitination of PRMT1, MAPK3, PPP3CA, β-catenin, and CDK4 in HEK293 cell (HEK293) and its derivatives expressing shCHIP (shCHIP), shCHIP and recombinant CHIP (shCHIP + OE), and recombinant CHIP (OE).
To measure the effect of E4B and CHIP expression on the stability of the identified substrate proteins, we expressed increasing amounts of the E3 enzymes in HEK293 cells and assay the level of substrate proteins by immunoblotting. We found that expression of E4B significantly decreased the level of PRMT1, PPP3CA, and OTUB1 in the cell and that synthesis of CHIP significantly decreased the level of PRMT1, MAPK3, PPP3CA, β-catenin, and CDK4 in the cell (Fig. 7). We also inhibited protein synthesis by cycloheximide (CHX) and compared the rate of substrate degradation in HEK293 cells with the overexpression of E4B and CHIP and with the decreased expression of the two E3s by shRNAs (Figs. 8 and 9). We found that overexpression of E4B accelerated the degradation of PRMT1, MAPK3, PPP3CA, and OTUB1 in HEK293 cells. Decreased E4B expression by shRNA extended the stability of PRMT1, MAPK3, PPP3CA, PGAM5, and OTUB1 in the cell (Fig. 8). We also found that overexpression of CHIP accelerated the degradation of PRMT1, MAPK3, PPP3CA, β-catenin, and CDK4 in HEK293 cells and that decreased CHIP expression by shRNA stabilized these proteins in the cell (Fig. 9). These results suggest a direct regulatory relationship between the E4B and CHIP and their ubiquitination targets in the cell.
Fig. 7. Expression of E4B and CHIP accelerated the degradation of the identified substrates.
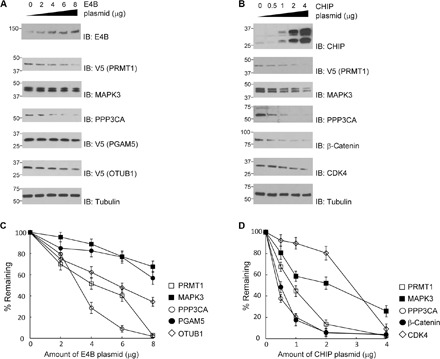
(A) E4B reduced the steady-state levels of the substrate proteins in the HEK293 cells. Cells were transfected with an increasing amount of E4B plasmid. Levels of the E4B substrates were assayed with immunoblots of the cell lysate probed with substrate-specific antibodies. Approximately 5 × 106 cells were used for each transfection of the E4B plasmid. (B) CHIP reduced the steady-state levels of the substrate proteins in the HEK293 cells. Assays were performed with a similar protocol to (A). (C and D) Quantitative analysis of substrate levels in correlation with E4B and CHIP expression, respectively. Intensity of the bands in (A) and (B) was plotted against the amount of pLenti E4B or CHIP plasmid used for transfection, assuming 100% substrate protein when an empty plasmid was used for mock transfection. Results were the average of three repeats.
Fig. 8. Degradation of E4B substrate proteins assayed by CHX chase.
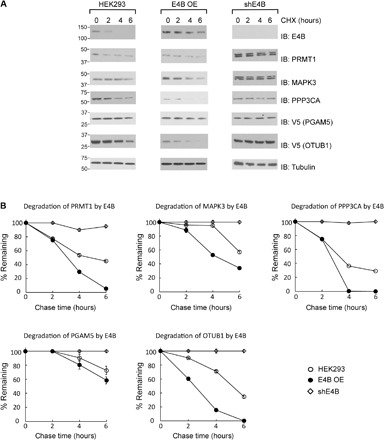
(A) Comparison of the rate of substrate degradation in HEK293 cells with E4B overexpressed (E4B OE) or inhibition of E4B expression by shRNA (shE4B). A total of 5 × 106 cells were transfected with various plasmids, and the cells were treated with CHX (100 mg/ml) 48 hours after transfection. Cell extracts were collected at 0, 2, 4, and 6 hours after incubation with CHX, followed by immunoblotting with substrate-specific antibodies. (B) Quantitative analysis of the levels of the substrate proteins in the cells after CHX chase. Data are representative of three independent trials.
Fig. 9. Degradation of CHIP substrate proteins assayed by CHX chase.
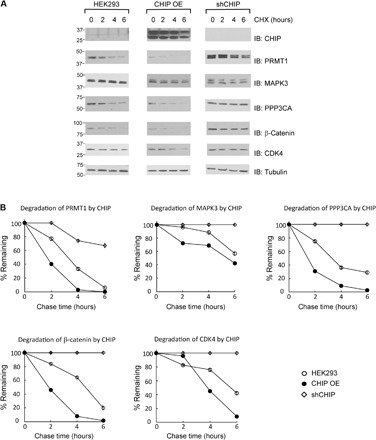
(A) Comparison of the rate of substrate degradation in HEK293 cells with CHIP overexpressed (CHIP OE) or inhibition of CHIP expression by shRNA (shCHIP). A total of 5 × 106 cells were transfected with various plasmids, and the cells were treated with CHX (100 mg/ml) 48 hours after transfection. Cell extracts were collected at 0, 2, 4, and 6 hours after incubation with CHX, followed by immunoblotting with substrate-specific antibodies. (B) Quantitative analysis of the levels of the substrate proteins in the cells after CHX chase. Data are representative of three independent trials.
The role of CHIP in ER stress–induced degradation of CDK4
We then assessed the biological significance of CHIP-mediated ubiquitination of CDK4, one of the newly identified substrates. Ubiquitination of CDK4 has not been studied previously, and no E3 ligase has been identified to mediate CDK4 ubiquitination. However, CDK4 is known to associate with Hsp90 and its co-chaperone, CDC37, which are thought to form a protein quality control mechanism, ensure proper folding, and maintain the stability of many protein kinase clients (29, 30). CHIP has been shown to be part of the unfolded protein response (UPR) stimulated by ER stress (10, 31, 32). Thus, to determine whether CHIP-mediated polyubiquitination plays a role in the control of CDK4 under ER stress, we treated control HEK293 cells and cells stably expressing shCHIP with the ER stress inducer tunicamycin (Fig. 10A). CDK4 protein levels were decreased in control cells by tunicamycin in a concentration-dependent manner, whereas tunicamycin did not significantly alter CDK4 levels in shCHIP cells (Fig. 10B). We detected robust interaction between CDK4 and Hsp90 or CDC37 in control HEK293 cells treated with MG132, and these interactions were significantly down-regulated after tunicamycin treatment even under proteasome inhibition (Fig. 10, C and E). In shCHIP cells, the levels of CDK4-Hsp90 and CDK4-CDC37 complexes were significantly lower than those in control cells, and tunicamycin minimally affected these complexes. In contrast, CDK4 interaction with Hsp70 was affected by tunicamycin or shCHIP only modestly (Fig. 10D). These observations suggest that ER stress induced by tunicamycin inhibits CDK4 association with the Hsp90-CDC37 complex and targets CDK4 to CHIP-mediated polyubiquitination and proteasomal degradation.
Fig. 10. ER stress–induced degradation of CDK4 is mediated by CHIP.
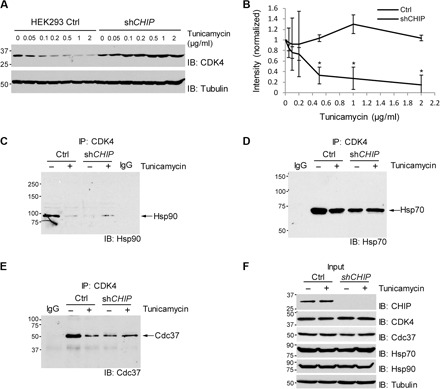
(A) shCHIP abrogates down-regulation of CDK4 protein in HEK293 cells after treatment with tunicamycin. Cells were incubated for 72 hours with tunicamycin at the indicated concentrations and harvested for immunoblotting. (B) Quantification of CDK4 protein levels after normalization by the levels of tubulin. Data are means ± SEM from three biological replicates, and the asterisks indicate statistical significance (P < 0.05). (C) ER stress decreases association of CDK4 with Hsp90 in HEK293 control cells but not in shCHIP cells. (D) ER stress does not significantly affect CDK4 association with Hsp70. (E) ER stress decreases association of CDK4 with Cdc37 in HEK293 control cells but not in shCHIP cells. (C to E) Cells were incubated with tunicamycin (1 μg/ml) for 24 hours and then 10 μM MG132 was added to the medium, followed by incubation for an additional 1.5 hours. Cell lysates were immunoprecipitated (IP) with CDK4 antibody, followed by immunoblotting for the indicated proteins. IgG, negative control for IP with normal IgG. (F) The levels of the indicated proteins determined by direct immunoblotting using the cell lysates before IP.
DISCUSSION
Here, we constructed OUT cascades for U-box E3s E4B and CHIP and used the OUT screen to identify hundreds of proteins as their potential substrates. Previously, only a few E4B substrates were reported in the literature including p53, ataxin-3, FEZ1 (fasciculation and elongation protein zeta 1), and EGFR (epidermal growth factor receptor) (11, 33–35). We found p53 among the E4B substrates identified by OUT (table S1) (36). We verified that kinase MAPK3, methyltransferase PRMT1, protein phosphatases PPP3CA and PGAM5, and deubiquitinase OTUB1 were ubiquitinated by E4B in vitro and in HEK293 cells (37–40). The crucial roles of these enzymes in cell cycle, DNA repair, gene regulation, etc. render E4B a key position in certain cell signaling networks.
In contrast to the limited knowledge about E4B substrates, more CHIP substrates have been identified including kinases Akt, Src, and MEKK2 (MAP3K2) and ASK1 (MAP3K5) of the MAPK pathway (41–44). From the OUT screen, we identified MAPK3, another kinase in the MAPK pathway, as a CHIP substrate. Two protein Arg methyltransferases PRMT1 and PRMT5 were found in the OUT screen as CHIP substrates. Ubiquitination of PRMT1 by CHIP was confirmed in our work, whereas ubiquitination of PRMT5 by CHIP was recently reported (45). We have also confirmed CHIP ubiquitination of protein phosphatase PPP3CA and β-catenin, the master transcriptional regulator in the Wnt signaling pathway. It is well established that the Skp1–Cullin–F-box E3 complex including β-TrCP as an F-box component polyubiquitinates cytoplasmic β-catenin under basal conditions without the Wnt signal (46–49). In addition to the canonical β-TrCP–dependent E3 activity, several other E3s have been shown to ubiquitinate β-catenin in the literature. For example, Mule/Huwe1 could down-regulate β-catenin under hyperactive Wnt signaling, whereas c-Cbl and TRIM33 may specifically target nuclear β-catenin (50–52). Our identification of CHIP as another E3 for β-catenin awaits further investigations on physiological cellular contexts that require CHIP-mediated degradation of β-catenin and possible involvement of the β-catenin control in the understudied roles of CHIP in tumorigenesis (53).
The OUT screen identified a number of chaperone proteins as CHIP targets, such as Hsp70, CDC37, DNAJ (DNAJA1 and DNAJC7), and co-chaperones BAG2 and BAG6 (table S2). It is known that CHIP is a co-chaperone of Hsp70 and Hsp90 and can ubiquitinate these proteins when they do not carry cargos (3, 54). The OUT screen with CHIP suggests an even broader association of CHIP with molecular chaperones in the protein quality control cycle, where it serves in triage between refolding and degradation. Furthermore, our study demonstrated that ER stress triggers CHIP-mediated polyubiquitination of CDK4. In the absence of proteotoxicity, the CDC37-Hsp90 complex binds to and stabilizes a substantial population of CDK4 (29, 30). Our data showed that upon ER stress induced by tunicamycin, the interaction between CDK4 and CDC37-Hsp90 is down-regulated and CDK4 actively undergoes CHIP-dependent proteasomal degradation. Activation of UPR in response to ER stress leads to cell cycle arrest in the G1 phase. The transient cessation of proliferation is thought to contribute to cellular adaptation and allow cells to attempt to reestablish homeostatic conditions or commit to death. A previous study suggested the involvement of translational repression of the CDK4-regulatory subunit cyclin D1 in the process of ER stress–induced arrest (55). Our data on the CHIP-CDK4 axis suggest that ubiquitination also plays roles in proteotoxicity-induced cell cycle arrest. Consistently, a recent study showed that parkin (PARK2) ubiquitinates and degrades cyclins D and E (56). Thus, proteasome-mediated degradation of G1-S regulators is likely to function as a translation-independent mechanism of cell cycle control in response to proteostasis. Hsp90 inhibitors have been developed and tested as anticancer drugs (57), and further investigations are necessary to elucidate the roles of the chaperone-associated E3s and their substrates in tumor suppression and sensitivity to anti-Hsp90 therapies.
The verification of E4B and CHIP substrates from the OUT screen proves that OUT is an effective platform for profiling the substrate specificities of the U-box E3s. Because E3s play key roles in cell regulation, many methods have been developed to identify the substrates of E3s to reveal their biological functions. The transient interactions between E3s and their substrates and many auxiliary proteins associated with E3 such as adaptors or regulators make it a significant challenge to identify E3 substrates. Still, conventional coimmunoprecipitation methods have been useful in defining the scope of E3 substrates based on binding affinity (58, 59). A reasonable readout of UB conjugation is the proteasomal degradation of the ubiquitinated proteins. By correlating the change in protein stability with the expression of an E3, a powerful method known as “global protein stability profiling” has been developed to assign E3 substrates (60, 61). The recent development of anti-diGly antibody and advanced proteomic technology allows the quantitative comparison of protein ubiquitination levels in the cell (62, 63). This enables the assignment of E3 substrates by following the changes of protein ubiquitination levels upon the perturbation of E3 expression (64, 65). To identify the direct ubiquitination targets of an E3, researchers have developed ingenious methods such as “UBAIT” and “Ubiquitin Ligase Substrate Trapping” by constructing fusions of E3 with UB or with UB binding proteins to capture E3 substrates after UB transfer (66, 67). In another creative approach, E2 for Nedd8 transfer is fused with E3 to turn E3 into a “NEDDylator.” E3 substrates can then be identified among neddylated proteins in the cell (68). An E. coli–based selection system to screen E2-E3 and E3-substrate relationships has also been reported recently (69).
The advantage of OUT is that it identifies the substrate of an E3 directly based on the catalytic transfer of UB from a designated E3 to its ubiquitination targets. The OUT cascade reenacts the action of native UB transfer cascades in the cell except that it exclusively delivers an affinity-tagged xUB to the substrates of a specific E3. The disadvantage of OUT is that the OUT cascade needs to be engineered with each E3 under investigation. We have shown that we can generate xUB-xE1 and xE1-xE2 pairs by mutating key residues in E1 and E2 based on sequence homology with xUba1 and xUbc1 (fig. S1). Here, we also generated xE2-xCHIP pairs based on the homology of the loop1 residues between CHIP and E4B (fig. S3). We expect that the strategy of loop grafting to engineer xE2-xE3 pairs could be used on other U-box E3s including UIP5, CYC4, and Prp19, each of which is poorly characterized (4, 5). U-box domains share significant structural homology with the RING domains (4). As for U-box domains, the loop1 region in the RING is a key site to engage E2s (17, 19). Thus, it would be of interest to test whether mutations in the U-box can be transplanted to RING domains to generate xE2-xE3 pairs. If transplantation of mutations is not effective, then the phage selection platform we developed for U-box could be used to engineer xE2-xE3 pairs with RING domains to further extend the reach of the OUT cascade.
It should be noted that in the xUB-expressing stable cells, both xUB and wt UB are present. xUB contains all seven lysine residues, each of which can act as a receptor for a wt UB, resulting in the formation of wt UB–xUB chains. In addition, xUB can function as a donor (with xE1-xE2-xE3) to react with the wt UB to form wt UB–xUB chains. A large body of evidence indicates that different pairs of E2-E3 can cooperate to form poly-UB chains that include branched structures. We found that both xUB and wt UB were present in the sample collected by tandem affinity purification with the HBT tag on xUB (Fig. 11 and Table 2). Thus, among the substrates identified as xUB-conjugated proteins, the xE1-xE2-xE3 cascade may add xUB either directly to the substrate or to a wt UB preconjugated to the substrate. The recognition of the substrate by xE3 would be a prerequisite in either case. Nevertheless, it is important to verify the results of the OUT screen by independent approaches such as the effect of silencing the designed E3 gene (Fig. 6).
Fig. 11. Peptide sequence of wt UB and sites of trypsin digestion.
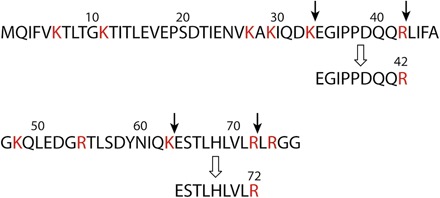
Trypsin digests peptides immediately after Lys (K) or Arg (R) residues. Because of the mutations of R42E and R72E in xUB, trypsin would not cleave the xUB peptide at residues 42 and 72. Peptides with the sequence “EGIDPPDQQR” and “ESTLHLVLR” are unique to trypsin digestion of wt UB.
Table 2. Peptides unique to wt UB identified by proteomics.
Peptides with the sequence “EGIDPPDQQR” and “ESTLHLVLR” would originate from wt UB but not from xUB (Fig. 11). The two peptides were present in all data sets collected with the OUT cascades of E4B and CHIP. This suggests that wt UB was conjugated to E4B or CHIP substrates that were purified by binding to HBT-xUB.
| Peptide | Screen with the OUT cascade of E4B | Screen with the OUT cascade of CHIP | ||||
|
No. of PSMs (screen 1) |
No. of PSMs (screen 2) |
No. of PSMs (screen 3) |
No. of PSMs (screen 1) |
No. of PSMs (screen 2) |
No. of PSMs (screen 3) |
|
| EGIDPPDQQR | 11 | 1 | 7 | 19 | 20 | 9 |
| ESTLHLVLR | 1 | 2 | 1 | 2 | 4 | 2 |
MATERIALS AND METHODS
Materials
The following antibodies were purchased from Santa Cruz Biotechnology: anti–β-catenin (sc-7963), anti-CHIP (sc-66830), anti–c-Myc (sc-40), anti-GST (sc-138), anti–hemagglutinin (HA) (sc-7392), anti-MAPK3 (sc-94), anti-OTUB1 (sc-130458), anti-p53 (sc-126), anti-PGAM5 (sc-161156), anti–PP2B-A (sc-9070), anti-PRMT1 (sc-59648), anti-tubulin (sc-23948), anti-UB (sc-8017), anti-V5 (sc-271944), and goat anti-rabbit IgG (sc-2004). Goat anti-mouse IgG antibody (31438) was from Thermo Fisher Scientific. Penta-His antibody (34660) was from Qiagen. Anti-flag (M2) antibody (F3165) was from Sigma-Aldrich. p53 protein (SP-452-020) was from R&D Systems. Biotin-conjugated wt UB was from Boston Biochem. Polymerase chain reaction (PCR) primers were from Integrated DNA Technologies and are listed in table S5.
Constructing the expression plasmids for xUba1 and xUbcH5b
To incorporate mutations Q608R, S621R, and D623R into human Uba1, primer pairs Bo184-Bo185 and Bo186-Bo187 were used to amplify the human Uba1 gene. Overlap extension of the fragments would afford the mutated Uba1 gene that was cloned into the pET vector to generate pET-xUba1(A). To incorporate mutations E1037K, D1047K, and E1049K into the UFD domain of human Uba1, the primer pair Bo13-Bo73 was used to amplify the human Uba1 gene to generate pET-xUba1(UFD). Combination of the mutations in the two plasmids afforded the plasmid pET-xUba1 for the expression of the xUba1 protein. Primers Kar64 and Kar65 were used to amplify human UbcH5b gene to incorporate the K4E and K8E mutations to generate pET-xUbcH5b.
Displaying the E4B U-box domain on M13 phage and verification of the catalytic activity
Primers Kar77 and Kar78 were used to clone the U-box domain of E4B into the phagemid vector pJF3H to display the U-box on phage with a C-terminal flag tag (70). Phage preparation was performed following the protocol reported previously (12). The display of the U-box domain on phage was confirmed by Western blotting probed with an anti-flag antibody. To confirm the catalytic activity of the U-box domain displayed on phage surface, ubiquitination reactions were set up with 2 × 1010 phage particles, 0.5 μM Uba1, 6 μM Ubch5b, and 14 μM HA–wt UB in buffer with 50 mM tris-HCl (pH 7.5), 10 mM MgCl2, 3 mM ATP, and 50 μM dithiothreitol (DTT). After 1-hour incubation at room temperature, the reaction mixture was analyzed by SDS–polyacrylamide gel electrophoresis (PAGE) and Western blotting probed with an anti-HA antibody. To analyze the ubiquitination of the U-box protein on phage by enzyme-linked immunosorbent assay, the reaction was set up with similar conditions with the use of 0.2 μM biotin–wt UB replacing HA-UB. After the reaction, phage particles were bound to a 96-well plate coated with streptavidin, and the reaction was diluted 10-fold across the plate. After incubation, the plate was washed with TBST [25 mM tris (pH 7.5), 150 mM NaCl, 0.05% Triton X-100, and 0.05% Tween 20] and TBS [25 mM tris (pH 7.5) and 150 mM NaCl]. Phage bound to the plate was revealed by an anti-M13 antibody–horseradish peroxidase conjugate.
Selecting the E4B U-box library by phage display
Detailed procedures for library construction and selection are documented in the study by Bhuripanyo (71). Residues to be randomized in the E4B U-box (R1233, L1236, M1237, D1238, and T1239) were first mutated to Ala to generate phagemid pJF-E4B U-box 5Ala. For mutagenesis, the U-box gene was amplified by two sets of primers Kar77-Kar203 and Kar78-Kar204. The PCR fragments were assembled by overlap extension for cloning into the pJF phagemid. For randomization of the five residues, pJF-E4B U-box 5Ala was used as the template for PCR reactions with primer pairs Jun13-Karlib2 and Karlib1-Kar78. The overlap extension of the amplified fragments was cloned into pJF phagemid to generate the library. The library DNA was transformed into SS320 cells (Agilent) by electroporation. The cells were plated on LB-agar plates containing ampicillin (100 μg/ml) and 2% glucose and incubated overnight at 37°C. The phagemid DNA for the library was prepared with a DNA Maxiprep kit (Qiagen). The library phage was prepared as reported previously (12).
For the first round of phage selection, 1010 phage were reacted with 1 μM xUba1(UFD), 10 μM xUbcH5b, and 0.5 μM biotin–wt UB for 1 hour. Phage were diluted 10-fold into 0.1% bovine serum albumin (BSA)–TBST and bound to a streptavidin plate for 1 hour at room temperature. The plate was washed 30 times with 0.1% BSA-TBST. One hundred microliters of TBS containing 100 mM DTT was added to each well to elute the phage particles. Phage eluted was added to a culture of XL1-Blue cells, and the culture was shaken slowly for 2 hours at 37°C. After incubation, the cells were plated onto LB-agar plates containing carbenicillin (100 μg/ml) and 2% glucose. After overnight incubation at 37°C, the colonies on the plates were collected, and the phagemid DNA was extracted with a DNA Miniprep kit. The library phagemid was then used for the next round of phage preparation. The concentration of enzymes and biotin-UB was decreased in each round of phage selection. For the fifth round of selection, 0.06 μM xUba1(UFD), 5 μM xUbcH5b, and 0.1 μM biotin–wt UB were used, and the reaction time was shortened to 10 min.
Assaying the activity of the selected U-box mutants of E4B
The U-box mutants from phage selection were cloned into the pET vector to express the proteins with a C-terminal flag tag. Ubiquitination of the U-box was set up with 5 μM U-box mutant, 0.1 μM xUba1(UFD), 2 μM xUbcH5b, and 5 μM HA-UB in buffer with 50 mM tris-HCl (pH 7.5), 10 mM MgCl2, and 1.5 mM ATP. The reaction was incubated at 37°C for 10 min and was subject to SDS-PAGE and Western blotting with an anti-flag antibody.
The fE4B gene was cloned into the pET30a plasmid for protein expression with an N-terminal flag tag. To incorporate mutations of KB2 into the fE4B gene, primer set Kar239-Kar241 was used to amplify the U-box gene. The fragment was assembled with the PCR fragment generated with the Kar238-Kar242 pair and cloned into the pET30a-wt E4B plasmid for expression of the fE4B-KB2 protein. Similarly, primer set Kar240-Kar241 was used to incorporate the mutations of KB12 into the E4B gene to express the fE4B-KB12 protein. The pET30a expression vector for fE4B-KB2 and fE4B-KB12 would express fE4B mutants with an N-terminal flag.
To express the fE4B protein and the KB2 and KB12 mutants, the pET30a plasmids were transformed into ArcticExpress (DE3) cells (Agilent). The cells were grown in terrific broth supplemented with kanamycin (70 mg/ml) at 37°C. When the culture reached an optical density of 1.0, the culture was cooled down to 13°C, and IPTG (isopropyl-β-d-thiogalactopyranoside) was added to a concentration of 4 mM. The culture was shaken at 13°C for 18 hours. The cells were then harvested, and the proteins were purified with a Ni-NTA affinity column (Qiagen) according to the vendor’s protocol. Protein eluted from the column was dialyzed overnight at 4°C in a precipitation buffer containing 50 mM tris-HCl (pH 7.5), 1 M ammonium sulfate, 1 M KCl, 5 mM DTT, and 10% glycerol. The protein precipitation was collected by centrifugation and was dissolved in TBS supplemented with 10 mM MgCl2.
Ubiquitination reactions were set up with 5 μM wt fE4B, fE4B-KB2, or fE4B-KB12; 2 μM wt Uba1 or xUba1; 10 μM wt UbcH5b or xUbcH5b; and 60 μM wt UB or xUB. The reaction was allowed to proceed at 37°C for 1.5 hours and analyzed by SDS-PAGE and Western blotting with an anti-flag antibody. p53 ubiquitination was set up with 4 μM wt fE4B, fE4B-KB2, or fE4B-KB12; 0.4 μM p53; 0.2 μM wt Uba1 or xUba1; 0.2 μM wt UbcH5b or xUbcH5b; and 30 μM wt UB or xUB. The reaction was incubated at 30°C for 1 hour and analyzed by Western blotting with an anti-p53 antibody.
Cloning and assaying the activity of xCHIP
The loop-transplanting mutants of xCHIP-KB2 and xCHIP-KB12 were constructed by PCR amplification of the CHIP gene with Kar232-Kar306 or Kar233-Kar306 primer pairs. The fragments were assembled with CHIP gene fragment amplified with the Kar234-Kar306 primer pair to generate the mutant CHIP genes. They were then cloned into the pET vector for protein expression. Ubiquitination of CHIP and CHIP-catalyzed p53 ubiquitination were set up similar to the reaction with E4B. Ubiquitination of CHIP was analyzed by Western blotting probed with an anti-CHIP antibody.
In vitro ubiquitination of substrate proteins
Genes of potential substrate proteins PRMT1, MAPK3, PPP3CA, and PGAM5 were cloned into pET vector for protein expression. Genes of OTUB1 and β-catenin were cloned into pGEX vector. The plasmids were transformed into BL21 (DE3) or ArcticExpress (DE3) cells to express the protein. To assay ubiquitination by E4B or CHIP, 5 to 10 μM substrate proteins were incubated with 1 μM wt Uba1, 2 μM wt UbcH5b, and 50 μM wt UB in TBS supplemented with 10 mM MgCl2 and 1.5 mM ATP. After 2-hour reaction at room temperature, the ubiquitination of substrates was analyzed by Western blotting probed with either substrate-specific antibodies or antibodies against the GST tag fused to the substrates.
Lentiviral vector construction and selection for stable cell lines
pLenti6-hygromycin-HBT-xUB plasmids for the expression of HBT-xUB were constructed by PCR-amplifying the xUB gene and the His-biotin tag from the plasmid pQCXIP HBT-UB (27). The two fragments were overlap-extended and cloned into the pLenti vector with a hygromycin-resistant gene. Flag-xUba1 gene was amplified with primers WY15 and WY16 and cloned into pLenti6-blasticdin vector. V5-xUbcH5b gene was amplified with primers WY101 and WY102 and cloned into pLenti4-zeocin vector. flag-xE4B and flag-xCHIP genes were amplified with primers Kar301-Kar302 and Kar303-Kar304 pairs, respectively, and cloned into pLenti4-puromycin plasmid. Virus packaging of the pLenti plasmids, infection, and selection of stable cell lines were performed according to the protocol for the ViraPower Lentiviral Expression System. Stable HEK293 cell lines expressing Flag-xUba1 and V5-xUbcH5a were selected with blasticidin (10 μg/ml) and zeocin (100 μg/ml), respectively. Stable cell lines for myc-xE4B and myc-xCHIP were selected with puromycin (1 μg/ml). Expression of transfected genes was induced by the addition of doxycycline (1 μg/ml) to the medium.
Tandem affinity purification of xUB-conjugated proteins
Tandem purification of HBT-xUB–conjugated proteins was performed as previously described (27). Briefly, 30 dishes (10 cm in diameter) of HEK293 cells stably expressing the xUba1-xUbch5b-xE4B/xCHIP cascade were acutely infected with lentivirus HBT-xUB for 72 hours. To inhibit proteasome activity, cells were treated with 10 μM MG132 for 4 hours at 37°C. Cells were then washed twice with ice-cold 1× phosphate-buffered saline (PBS) (pH 7.4) and harvested by a cell scraper with buffer A [8 M urea, 300 mM NaCl, 50 mM tris, 50 mM NaH2PO4, 0.5% NP-40, 1 mM phenylmethylsulfonyl fluoride, and benzonase (125 U/ml) (pH 8.0)]. For Ni-NTA purification, cell lysates were centrifuged at 15,000g for 30 min at room temperature. Thirty-five microliters of Ni2+ Sepharose beads (GE Healthcare) for each 1 mg of protein lysates was added to the clarified supernatant. After incubation overnight at room temperature in buffer A with 10 mM imidazole on a rocking platform, Ni2+ Sepharose beads were pelleted by centrifugation at 100g for 3 min and washed sequentially with a 20-bead volume of buffer A (pH 8.0), buffer A (pH 6.3), and buffer A (pH 6.3) with 10 mM imidazole. After washing the beads, proteins were eluted twice with a 5-bead volume of buffer B [8 M urea, 200 mM NaCl, 50 mM Na2HPO4, 2% SDS, 10 mM EDTA, 100 mM tris, and 250 mM imidazole (pH 4.3)]. For streptavidin purification, the elution solution was adjusted to pH 8.0. Fifty microliters of streptavidin Sepharose beads (Thermo Fisher Scientific) was added to the elution to bind HBT-xUB–conjugated proteins. After incubation on a rocking platform overnight at room temperature, streptavidin beads were pelleted and washed sequentially with 1.5 ml of buffer C [8 M urea, 200 mM NaCl, 2% SDS, and 100 mM tris (pH 8.0)], buffer D [8 M urea, 1.2 M NaCl, 0.2% SDS, 100 mM tris, 10% EtOH, and 10% isopropanol (pH 8.0)], and buffer E [8 M urea and 100 mM NH4HCO3 (pH 8)].
Sample digestion and LC-MS/MS analysis
After washing, the streptavidin beads were spun down, residual urea was removed, and liquid chromatography coupled to tandem MS (LC-MS/MS) on an Orbitrap Fusion mass spectrometer (Thermo Fisher Scientific) was performed at the Emory Integrated Proteomics Core (EIPC) according to previously published methods (72, 73). Collected spectra were searched using Proteome Discoverer 2.0 against human UniProt database (90,300 target sequences). Searching parameters included fully tryptic restriction and a parent ion mass tolerance (±20 parts per million). Methionine oxidation (+15.99492 Da), asparagine and glutamine deamidation (+0.98402 Da), lysine ubiquitination (+114.04293 Da), and protein N-terminal acetylation (+42.03670) were variable modifications (up to three were allowed per peptide); cysteine was assigned a fixed carbamidomethyl modification (+57.021465 Da). Percolator was used to filter the PSMs to a false discovery rate of 1%.
Bioinformatics analysis
IPA software (www.ingenuity.com) was used to map and identify the biological networks and molecular pathways with a significant proportion of genes having E4B or CHIP ubiquitination targets. Fisher’s exact test in IPA software was used to calculate the P values for pathways and networks. The level of statistical significance was set at P < 0.05. IPA was also used to visualize the identified biological networks.
Lentiviral silencing of E4B and CHIP
Lentiviral GPIZ plasmids encoding shRNAs against E4B and CHIP were obtained from GE Dharmacon, and lentiviruses were produced using the manufacturer’s lentivirus packaging system and 293FT cells. HEK293 cells were infected with each lentivirus, followed by selection with puromycin for stable cell populations. The efficiency of gene silencing in each shRNA group was determined by immunoblotting using stable cell populations. For functional restoration, HEK293 cell population stably expressing anti-E4B shRNA#2 and anti-CHIP shRNA#2 was infected with the lentivirus packaged with pLenti6-Myc-wt E4B and wtCHIP, respectively.
Coimmunoprecipitation and in vivo assay to confirm E4B and CHIP substrates
Transfection of pLenti-E4B and CHIP into the HEK293 cells was conducted with the Lipofectamine 2000 according to the manufacturer’s protocol. To immunoprecipitate the substrate proteins, cells were treated with 10 μM MG132 (American Peptide) for 90 min at 72 hours after transfection. HEK293 cells (80 to 90% confluent monolayer in 75-cm2 cell culture flask) expressing control plasmid, shE4B/CHIP, shE4B/CHIP + E4B/CHIP cDNA, and E4B/CHIP cDNA were washed twice with ice-cold PBS (pH 7.4). One milliliter of ice-cold radioimmunoprecipitation assay (RIPA) buffer was added to the cell monolayer and incubated with the cell at 4°C for 10 min. The cells were disrupted by repeated aspiration through a 21-gauge needle. The cell lysate was transferred to a 1.5-ml tube. The cell debris was pelleted by centrifugation at 13,000 rpm for 20 min at 4°C, and the supernatant was transferred to a 1.5-ml centrifuge tube and precleared by adding 1.0 μg of the appropriate control IgG (normal mouse or rabbit IgG corresponding to the host species of the primary antibody). Twenty microliters of resuspended volume of Protein A/G PLUS-Agarose was added to the supernatant, and incubation was continued for 30 min at 4°C. The agarose beads were pelleted by centrifugation at 2500 rpm for 5 min at 4°C. From the cleared cell lysate, a volume containing 2 mg of the total protein was transferred to a new tube. Thirty microliters (that is, 6 μg) of primary antibody against a specific substrate protein was then added, and incubation was continued for 1 hour at 4°C. After incubation, 50 μl of resuspended volume of Protein A/G PLUS-Agarose was added. The tubes were capped and incubated at 4°C overnight on a rocker platform. The agarose beads were pelleted by centrifugation at 2500 rpm for 5 min at 4° C. The beads were then washed four times each time with 1.0 ml of PBS. After the final wash, the beads were resuspended in 40 μl of 1× Laemmli buffer with β-mercaptoethanol. The samples were boiled for 5 min and analyzed by SDS-PAGE and Western blotting probed with antibodies against UB.
Protein degradation assays
To examine the effect of E4B or CHIP on steady-state levels of the substrates, HEK293 cells (5 × 106 cells) were transiently transfected with varying amounts of pLenti plasmid of E4B or CHIP with Lipofectamine 2000. Cells were harvested at 48 hours after transfection, and the amount of substrate proteins in the cell lysate was assayed by immunoblotting with substrate-specific antibodies. For CHX chase assays, HEK293 cells (5 × 106 cells) were transiently transfected with 4 μg of empty pLenti, 8 μg of pLenti-E4B, or 4 μg of pLenti-CHIP plasmids. After 48 hours, cells were treated with CHX (100 μg/ml) to block de novo protein synthesis, and the cells were harvested after varying lengths of incubation time with CHX. The amount of substrate proteins in the cell was assayed by immunoblotting with antibodies against each substrate protein. Protein levels were normalized to tubulin. Alternatively, CHX chase assays were performed on HEK293 cells stably expressing anti-E4B or anti-CHIP shRNA to measure the effect of decreased expression of E4B or CHIP on substrate stability.
Assays for ER stress
HEK293 cells were obtained from the American Type Culture Collection (ATCC) and cultured under standard conditions recommended by the ATCC. HEK293 cells were cultured on 60-mm dishes to 40 to 50% confluence, treated with tunicamycin for 72 hours at the indicated concentrations to induce ER stress, and harvested for immunoblotting with anti-CDK4 antibody.
For immunoprecipitation with anti-CDK4, HEK293 Ctrl or shCHIP cells were incubated with tunicamycin (1 μg/ml) for 24 hours and then treated with 10 μM MG132 for 1.5 hours. The cells were lysed by sonication in RIPA lysis buffer as described previously (13). Four milligrams of protein lysates was incubated with 20 μl of anti-CDK4 agarose (#sc-260, Santa Cruz Biotechnology) overnight at 4°C. The beads were washed with RIPA buffer three times, and 60 μl of 1× SDS-PAGE loading buffer was added. The sample was boiled for 5 min. Half of the immunoprecipitates or 50 μg of the total protein lysate was loaded for SDS-PAGE and assayed by Western blotting. The membranes were probed with anti-HSP70 (SC-24, Santa Cruz Biotechnology), anti-HSP90 (SC-13119, Santa Cruz Biotechnology), anti-CDC37 (SC-13129, Santa Cruz Biotechnology), anti-CHIP (SC-133066, Santa Cruz Biotechnology), and anti–α-tubulin antibody (T6199, Sigma-Aldrich).
Supplementary Material
Acknowledgments
We thank W. Harper, R. E. Klevit, and C. Patterson for providing the gene constructs of Uba1, U-box of E4B, and CHIP, respectively. Funding: This work was supported by grants from the NIH [GM104498 (to J.Y. and H.K.), CA112282 (to H.K.), and GM075156 and GM118089 (to W.J.C.)], the NSF [1420193 and 1710460 (to J.Y.)], the Chicago Biomedical Consortium [Catalyst-026 (to H.K. and J.Y.) and PDR-010 (to X.L.)], and the National Natural Science Foundation of China [31770921 (to B.Z.)] and by Project 985 start-up grants [WF220417001 and WF114117001/004 (to B.Z.)], the Lynn Sage Breast Cancer Research Foundation, and the fund from the Department of Pharmacology at Northwestern University. This study was supported in part by the EIPC, which is subsidized by the Emory University School of Medicine. Author contributions: J.Y. and H.K. conceived the idea. K.B. engineered E3 enzymes and characterized their activities in vitro. Y.W. did all the proteomics and cell biology experiments. X.L. developed the initial scheme for cell line construction, tandem affinity purification, and target validation and conducted ER stress–related experiments. L.Z., R.L., and B.Z. contributed to cloning and substrate ubiquitination and stability assays. D.D. and N.T.S. performed proteomic analysis. Y.B. performed bioinformatics analysis. W.J.C. constructed the fE4B gene for expression. J.Y. wrote the manuscript with K.B., Y.W., W.J.C., and H.K. All authors approved the final version. Author responsibility for the figures are as follows: K.B. and J.Y. for Figs. 1, 3, and 5, and figs. S3 and S4; K.B., L.Z., and J.Y. for Fig. 2 and fig. S2; K.B., L.Z., R.L., and J.Y. for Fig. 4; Y.W. and J.Y. for Figs. 6 and 7 and figs. S5 and S6; Y.W., L.Z., R.L., and J.Y. for Figs. 8 and 9; X.L. and H.K. for Fig. 10; N.T.S. and J.Y. for Fig. 11; and K.B., B.Z., and J.Y. for fig. S1. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors. The proteomics data for identification of E4B and CHIP substrates are available at the ProteomeXchange Consortium via the PRoteomics IDEntifications partner repository (http://proteomecentral.proteomexchange.org).
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/e1701393/DC1
fig. S1. Design of the xUB-xUba1 and xUba1-xUbcH5b pairs for the OUT cascade.
fig. S2. xUB transfer through the human xUba1-xUbcH5b pair.
fig. S3. Structure analysis of the U-box domains of E4B and CHIP interacting with the UbcH5c and UbcH5a, respectively.
fig. S4. Model selection of the E4B U-box domain displayed on M13 phage.
fig. S5. Orthogonality of the OUT cascades of E4B and CHIP with the native UB transferring enzymes.
fig. S6. Affinity purification of xUB-modified proteins to identify the substrates of E4B and CHIP.
table S1. Potential E4B substrates identified by OUT.
table S2. Potential CHIP substrates identified by OUT.
table S3. Top networks associated with the E4B substrates identified by the OUT screen.
table S4. Top networks associated with the CHIP substrates identified by the OUT screen.
table S5. Primers used in this study.
REFERENCES AND NOTES
- 1.Hatakeyama S., Yada M., Matsumoto M., Ishida N., Nakayama K.-i., U box proteins as a new family of ubiquitin-protein ligases. J. Biol. Chem. 276, 33111–33120 (2001). [DOI] [PubMed] [Google Scholar]
- 2.Meacham G. C., Patterson C., Zhang W., Younger J. M., Cyr D. M., The Hsc70 co-chaperone CHIP targets immature CFTR for proteasomal degradation. Nat. Cell Biol. 3, 100–105 (2001). [DOI] [PubMed] [Google Scholar]
- 3.Qian S.-B., McDonough H., Boellmann F., Cyr D. M., Patterson C., CHIP-mediated stress recovery by sequential ubiquitination of substrates and Hsp70. Nature 440, 551–555 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Aravind L., Koonin E. V., The U box is a modified RING finger—A common domain in ubiquitination. Curr. Biol. 10, R132–R134 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Hatakeyama S., Nakayama K.-i., U-box proteins as a new family of ubiquitin ligases. Biochem. Biophys. Res. Commun. 302, 635–645 (2003). [DOI] [PubMed] [Google Scholar]
- 6.Zeinab R. A., Wu H., Sergi C., Leng R., UBE4B: A promising regulatory molecule in neuronal death and survival. Int. J. Mol. Sci. 13, 16865–16879 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pratt W. B., Gestwicki J. E., Osawa Y., Lieberman A. P., Targeting Hsp90/Hsp70-based protein quality control for treatment of adult onset neurodegenerative diseases. Annu. Rev. Pharmacol. Toxicol. 55, 353–371 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hoppe T., Multiubiquitylation by E4 enzymes: ‘One size’ doesn’t fit all. Trends Biochem. Sci. 30, 183–187 (2005). [DOI] [PubMed] [Google Scholar]
- 9.Koegl M., Hoppe T., Schlenker S., Ulrich H. D., Mayer T. U., Jentsch S., A novel ubiquitination factor, E4, is involved in multiubiquitin chain assembly. Cell 96, 635–644 (1999). [DOI] [PubMed] [Google Scholar]
- 10.Imai Y., Soda M., Hatakeyama S., Akagi T., Hashikawa T., Nakayama K.-i., Takahashi R., CHIP is associated with Parkin, a gene responsible for familial Parkinson’s disease, and enhances its ubiquitin ligase activity. Mol. Cell 10, 55–67 (2002). [DOI] [PubMed] [Google Scholar]
- 11.Wu H., Pomeroy S. L., Ferreira M., Teider N., Mariani J., Nakayama K. I., Hatakeyama S., Tron V. A., Saltibus L. F., Spyracopoulos L., Leng R. P., UBE4B promotes Hdm2-mediated degradation of the tumor suppressor p53. Nat. Med. 17, 347–355 (2011). [DOI] [PubMed] [Google Scholar]
- 12.Zhao B., Bhuripanyo K., Zhang K., Kiyokawa H., Schindelin H., Yin J., Orthogonal ubiquitin transfer through engineered E1-E2 cascades for protein ubiquitination. Chem. Biol. 19, 1265–1277 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Liu X., Zhao B., Sun L., Bhuripanyo K., Wang Y., Bi Y., Davuluri R. V., Duong D. M., Nanavati D., Yin J., Kiyokawa H., Orthogonal ubiquitin transfer identifies ubiquitination substrates under differential control by the two ubiquitin activating enzymes. Nat. Commun. 8, 14286 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Nordquist K. A., Dimitrova Y. N., Brzovic P. S., Ridenour W. B., Munro K. A., Soss S. E., Caprioli R. M., Klevit R. E., Chazin W. J., Structural and functional characterization of the monomeric U-box domain from E4B. Biochemistry 49, 347–355 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ohi M. D., Vander Kooi C. W., Rosenberg J. A., Chazin W. J., Gould K. L., Structural insights into the U-box, a domain associated with multi-ubiquitination. Nat. Struct. Biol. 10, 250–255 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Benirschke R. C., Thompson J. R., Nominé Y., Wasielewski E., Juranić N., Macura S., Hatakeyama S., Nakayama K. I., Botuyan M. V., Mer G., Molecular basis for the association of human E4B U box ubiquitin ligase with E2-conjugating enzymes UbcH5c and Ubc4. Structure 18, 955–965 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dou H., Buetow L., Hock A., Sibbet G. J., Vousden K. H., Huang D. T., Structural basis for autoinhibition and phosphorylation-dependent activation of c-Cbl. Nat. Struct. Mol. Biol. 19, 184–192 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu Z., Kohli E., Devlin K. I., Bold M., Nix J. C., Misra S., Interactions between the quality control ubiquitin ligase CHIP and ubiquitin conjugating enzymes. BMC Struct. Biol. 8, 26 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yin Q., Lin S.-C., Lamothe B., Lu M., Lo Y.-C., Hura G., Zheng L., Rich R. L., Campos A. D., Myszka D. G., Lenardo M. J., Darnay B. G., Wu H., E2 interaction and dimerization in the crystal structure of TRAF6. Nat. Struct. Mol. Biol. 16, 658–666 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhang M., Windheim M., Roe S. M., Peggie M., Cohen P., Prodromou C., Pearl L. H., Chaperoned ubiquitylation—Crystal structures of the CHIP U box E3 ubiquitin ligase and a CHIP-Ubc13-Uev1a complex. Mol. Cell 20, 525–538 (2005). [DOI] [PubMed] [Google Scholar]
- 21.Dou H., Buetow L., Sibbet G. J., Cameron K., Huang D. T., Essentiality of a non-RING element in priming donor ubiquitin for catalysis by a monomeric E3. Nat. Struct. Mol. Biol. 20, 982–986 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pruneda J. N., Littlefield P. J., Soss S. E., Nordquist K. A., Chazin W. J., Brzovic P. S., Klevit R. E., Structure of an E3:E2~Ub complex reveals an allosteric mechanism shared among RING/U-box ligases. Mol. Cell 47, 933–942 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Soss S. E., Klevit R. E., Chazin W. J., Activation of UbcH5c~Ub is the result of a shift in interdomain motions of the conjugate bound to U-box E3 ligase E4B. Biochemistry 52, 2991–2999 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Starita L. M., Pruneda J. N., Lo R. S., Fowler D. M., Kim H. J., Hiatt J. B., Shendure J., Brzovic P. S., Fields S., Klevit R. E., Activity-enhancing mutations in an E3 ubiquitin ligase identified by high-throughput mutagenesis. Proc. Natl. Acad. Sci. U.S.A. 110, E1263–E1272 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Nikolay R., Wiederkehr T., Rist W., Kramer G., Mayer M. P., Bukau B., Dimerization of the human E3 ligase CHIP via a coiled-coil domain is essential for its activity. J. Biol. Chem. 279, 2673–2678 (2004). [DOI] [PubMed] [Google Scholar]
- 26.Esser C., Scheffner M., Höhfeld J., The chaperone-associated ubiquitin ligase CHIP is able to target p53 for proteasomal degradation. J. Biol. Chem. 280, 27443–27448 (2005). [DOI] [PubMed] [Google Scholar]
- 27.Tagwerker C., Flick K., Cui M., Guerrero C., Dou Y., Auer B., Baldi P., Huang L., Kaiser P., A tandem affinity tag for two-step purification under fully denaturing conditions: Application in ubiquitin profiling and protein complex identification combined with in vivocross-linking. Mol. Cell. Proteomics 5, 737–748 (2006). [DOI] [PubMed] [Google Scholar]
- 28.Paul I., Ghosh M. K., A CHIPotle in physiology and disease. Int. J. Biochem. Cell Biol. 58, 37–52 (2015). [DOI] [PubMed] [Google Scholar]
- 29.Verba K. A., Wang R. Y.-R., Arakawa A., Liu Y., Shirouzu M., Yokoyama S., Agard D. A., Atomic structure of Hsp90-Cdc37-Cdk4 reveals that Hsp90 traps and stabilizes an unfolded kinase. Science 352, 1542–1547 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Stepanova L., Leng X., Parker S. B., Harper J. W., Mammalian p50Cdc37 is a protein kinase-targeting subunit of Hsp90 that binds and stabilizes Cdk4. Genes Dev. 10, 1491–1502 (1996). [DOI] [PubMed] [Google Scholar]
- 31.McDonough H., Patterson C., CHIP: A link between the chaperone and proteasome systems. Cell Stress Chaperones 8, 303–308 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edkins A. L., CHIP: A co-chaperone for degradation by the proteasome. Subcell. Biochem. 78, 219–242 (2015). [DOI] [PubMed] [Google Scholar]
- 33.Matsumoto M., Yada M., Hatakeyama S., Ishimoto H., Tanimura T., Tsuji S., Kakizuka A., Kitagawa M., Nakayama K. I., Molecular clearance of ataxin-3 is regulated by a mammalian E4. EMBO J. 23, 659–669 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Okumura F., Hatakeyama S., Matsumoto M., Kamura T., Nakayama K. I., Functional regulation of FEZ1 by the U-box-type ubiquitin ligase E4B contributes to neuritogenesis. J. Biol. Chem. 279, 53533–53543 (2004). [DOI] [PubMed] [Google Scholar]
- 35.Sirisaengtaksin N., Gireud M., Yan Q., Kubota Y., Meza D., Waymire J. C., Zage P. E., Bean A. J., UBE4B protein couples ubiquitination and sorting machineries to enable epidermal growth factor receptor (EGFR) degradation. J. Biol. Chem. 289, 3026–3039 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ross O. A., Rutherford N. J., Baker M., Soto-Ortolaza A. I., Carrasquillo M. M., DeJesus-Hernandez M., Adamson J., Li M., Volkening K., Finger E., Seeley W. W., Hatanpaa K. J., Lomen-Hoerth C., Kertesz A., Bigio E. H., Lippa C., Woodruff B. K., Knopman D. S., White C. L. III, Van Gerpen J. A., Meschia J. F., Mackenzie I. R., Boylan K., Boeve B. F., Miller B. L., Strong M. J., Uitti R. J., Younkin S. G., Graff-Radford N. R., Petersen R. C., Wszolek Z. K., Dickson D. W., Rademakers R., Ataxin-2 repeat-length variation and neurodegeneration. Hum. Mol. Genet. 20, 3207–3212 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cobb M. H., Hepler J. E., Cheng M., Robbins D., The mitogen-activated protein kinases, ERK1 and ERK2. Semin. Cancer Biol. 5, 261–268 (1994). [PubMed] [Google Scholar]
- 38.Komander D., Clague M. J., Urbe S., Breaking the chains: Structure and function of the deubiquitinases. Nat. Rev. Mol. Cell Biol. 10, 550–563 (2009). [DOI] [PubMed] [Google Scholar]
- 39.Shi Y., Serine/threonine phosphatases: Mechanism through structure. Cell 139, 468–484 (2009). [DOI] [PubMed] [Google Scholar]
- 40.Tang J., Frankel A., Cook R. J., Kim S., Paik W. K., Williams K. R., Clarke S., Herschman H. R., PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. J. Biol. Chem. 275, 7723–7730 (2000). [DOI] [PubMed] [Google Scholar]
- 41.Dickey C. A., Koren J., Zhang Y.-J., Xu Y.-f., Jinwal U. K., Birnbaum M. J., Monks B., Sun M., Cheng J. Q., Patterson C., Bailey R. M., Dunmore J., Soresh S., Leon C., Morgan D., Petrucelli L., Akt and CHIP coregulate tau degradation through coordinated interactions. Proc. Natl. Acad. Sci. U.S.A. 105, 3622–3627 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hwang J. R., Zhang C., Patterson C., C-terminus of heat shock protein 70–interacting protein facilitates degradation of apoptosis signal-regulating kinase 1 and inhibits apoptosis signal-regulating kinase 1–dependent apoptosis. Cell Stress Chaperones 10, 147–156 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Maruyama T., Kadowaki H., Okamoto N., Nagai A., Naguro I., Matsuzawa A., Shibuya H., Tanaka K., Murata S., Takeda K., Nishitoh H., Ichijo H., CHIP-dependent termination of MEKK2 regulates temporal ERK activation required for proper hyperosmotic response. EMBO J. 29, 2501–2514 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yang M., Wang C., Zhu X., Tang S., Shi L., Cao X., Chen T., E3 ubiquitin ligase CHIP facilitates Toll-like receptor signaling by recruiting and polyubiquitinating Src and atypical PKCζ. J. Exp. Med. 208, 2099–2112 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang H.-T., Zeng L.-F., He Q.-Y., Tao W. A., Zha Z.-G., Hu C.-D., The E3 ubiquitin ligase CHIP mediates ubiquitination and proteasomal degradation of PRMT5. Biochim. Biophys. Acta 1863, 335–346 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kitagawa M., Hatakeyama S., Shirane M., Matsumoto M., Ishida N., Hattori K., Nakamichi I., Kikuchi A., Nakayama K.-i., Nakayama K., An F-box protein, FWD1, mediates ubiquitin-dependent proteolysis of β-catenin. EMBO J. 18, 2401–2410 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hart M., Concordet J. P., Lassot I., Albert I., del los Santos R., Durand H., Perret C., Rubinfeld B., Margottin F., Benarous R., Polakis P., The F-box protein β-TrCP associates with phosphorylated β-catenin and regulates its activity in the cell. Curr. Biol. 9, 207–211 (1999). [DOI] [PubMed] [Google Scholar]
- 48.Latres E., Chiaur D. S., Pagano M., The human F box protein β-Trcp associates with the Cul1/Skp1 complex and regulates the stability of β-catenin. Oncogene 18, 849–854 (1999). [DOI] [PubMed] [Google Scholar]
- 49.Winston J. T., Strack P., Beer-Romero P., Chu C. Y., Elledge S. J., Harper J. W., The SCFβ-TRCP–ubiquitin ligase complex associates specifically with phosphorylated destruction motifs in IκBα and β-catenin and stimulates IκBα ubiquitination in vitro. Genes Dev. 13, 270–283 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xue J., Chen Y., Wu Y., Wang Z., Zhou A., Zhang S., Lin K., Aldape K., Majumder S., Lu Z., Huang S., Tumour suppressor TRIM33 targets nuclear β-catenin degradation. Nat. Commun. 6, 6156 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chitalia V., Shivanna S., Martorell J., Meyer R., Edelman E., Rahimi N., c-Cbl, a ubiquitin E3 ligase that targets active β-catenin: A novel layer of Wnt signaling regulation. J. Biol. Chem. 288, 23505–23517 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Dominguez-Brauer C., Khatun R., Elia A. J., Thu K. L., Ramachandran P., Baniasadi S. P., Hao Z., Jones L. D., Haight J., Sheng Y., Mak T. W., E3 ubiquitin ligase Mule targets β-catenin under conditions of hyperactive Wnt signaling. Proc. Natl. Acad. Sci. U.S.A. 114, E1148–E1157 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sun C., Li H.-L., Shi M.-L., Liu Q.-H., Bai J., Zheng J.-N., Diverse roles of C-terminal Hsp70-interacting protein (CHIP) in tumorigenesis. J. Cancer Res. Clin. Oncol. 140, 189–197 (2014). [DOI] [PubMed] [Google Scholar]
- 54.Morales J. L., Perdew G. H., Carboxyl terminus of hsc70-interacting protein (CHIP) can remodel mature aryl hydrocarbon receptor (AhR) complexes and mediate ubiquitination of both the AhR and the 90 kDa heat-shock protein (hsp90) in vitro. Biochemistry 46, 610–621 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Brewer J. W., Diehl J. A., PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. U.S.A. 97, 12625–12630 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Gong Y., Zack T. I., Morris L. G., Lin K., Hukkelhoven E., Raheja R., Tan I.-L., Turcan S., Veeriah S., Meng S., Viale A., Schumacher S. E., Palmedo P., Beroukhim R., Chan T. A., Pan-cancer genetic analysis identifies PARK2 as a master regulator of G1/S cyclins. Nat. Genet. 46, 588–594 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Barrott J. J., Haystead T. A. J., Hsp90, an unlikely ally in the war on cancer. FEBS J. 280, 1381–1396 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Merbl Y., Kirschner M. W., Large-scale detection of ubiquitination substrates using cell extracts and protein microarrays. Proc. Natl. Acad. Sci. U.S.A. 106, 2543–2548 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tan M.-K., Lim H.-J., Bennett E. J., Shi Y., Harper J. W., Parallel SCF adaptor capture proteomics reveals a role for SCFFBXL17 in NRF2 activation via BACH1 repressor turnover. Mol. Cell 52, 9–24 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Emanuele M. J., Elia A. E. H., Xu Q., Thoma C. R., Izhar L., Leng Y., Guo A., Chen Y.-N., Rush J., Hsu P. W.-C., Yen H.-C., Elledge S. J., Global identification of modular cullin-RING ligase substrates. Cell 147, 459–474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yen H.-C., Elledge S. J., Identification of SCF ubiquitin ligase substrates by global protein stability profiling. Science 322, 923–929 (2008). [DOI] [PubMed] [Google Scholar]
- 62.Xu G., Jaffrey S. R., The new landscape of protein ubiquitination. Nat. Biotechnol. 29, 1098–1100 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu P., Duong D. M., Seyfried N. T., Cheng D., Xie Y., Robert J., Rush J., Hochstrasser M., Finley D., Peng J., Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kim W., Bennett E. J., Huttlin E. L., Guo A., Li J., Possemato A., Sowa M. E., Rad R., Rush J., Comb M. J., Harper J. W., Gygi S. P., Systematic and quantitative assessment of the ubiquitin-modified proteome. Mol. Cell 44, 325–340 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Sarraf S. A., Raman M., Guarani-Pereira V., Sowa M. E., Huttlin E. L., Gygi S. P., Harper J. W., Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496, 372–376 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mark K. G., Loveless T. B., Toczyski D. P., Isolation of ubiquitinated substrates by tandem affinity purification of E3 ligase–polyubiquitin-binding domain fusions (ligase traps). Nat. Protoc. 11, 291–301 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.O’Connor H. F., Lyon N., Leung J. W., Agarwal P., Swaim C. D., Miller K. M., Huibregtse J. M., Ubiquitin-activated interaction traps (UBAITs) identify E3 ligase binding partners. EMBO Rep. 16, 1699–1712 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhuang M., Guan S., Wang H., Burlingame A. L., Wells J. A., Substrates of IAP ubiquitin ligases identified with a designed orthogonal E3 ligase, the NEDDylator. Mol. Cell 49, 273–282 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Levin-Kravets O., Tanner N., Shohat N., Attali I., Keren-Kaplan T., Shusterman A., Artzi S., Varvak A., Reshef Y., Shi X., Zucker O., Baram T., Katina C., Pilzer I., Ben-Aroya S., Prag G., A bacterial genetic selection system for ubiquitylation cascade discovery. Nat. Methods 13, 945–952 (2016). [DOI] [PubMed] [Google Scholar]
- 70.Crameri R., Suter M., Display of biologically active proteins on the surface of filamentous phages: A cDNA cloning system for selection of functional gene products linked to the genetic information responsible for their production. Gene 137, 69–75 (1993). [DOI] [PubMed] [Google Scholar]
- 71.K. Bhuripanyo, thesis, University of Chicago (2016); https://knowledge.uchicago.edu/handle/11417/136.
- 72.Wingo T. S., Duong D. M., Zhou M., Dammer E. B., Wu H., Cutler D. J., Lah J. J., Levey A. I., Seyfried N. T., Integrating next-generation genomic sequencing and mass spectrometry to estimate allele-specific protein abundance in human brain. J. Proteome Res. 16, 3336–3347 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Comstra H. S., McArthy J., Rudin-Rush S., Hartwig C., Gokhale A., Zlatic S. A., Blackburn J. B., Werner E., Petris M., D’Souza P., Panuwet P., Barr D. B., Lupashin V., Vrailas-Mortimer A., Faundez V., The interactome of the copper transporter ATP7A belongs to a network of neurodevelopmental and neurodegeneration factors. eLife 6, e24722 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lee I., Schindelin H., Structural insights into E1-catalyzed ubiquitin activation and transfer to conjugating enzymes. Cell 134, 268–278 (2008). [DOI] [PubMed] [Google Scholar]
- 75.Yin J., Mills J. H., Schultz P. G., A catalysis-based selection for peroxidase antibodies with increased activity. J. Am. Chem. Soc. 126, 3006–3007 (2004). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/4/1/e1701393/DC1
fig. S1. Design of the xUB-xUba1 and xUba1-xUbcH5b pairs for the OUT cascade.
fig. S2. xUB transfer through the human xUba1-xUbcH5b pair.
fig. S3. Structure analysis of the U-box domains of E4B and CHIP interacting with the UbcH5c and UbcH5a, respectively.
fig. S4. Model selection of the E4B U-box domain displayed on M13 phage.
fig. S5. Orthogonality of the OUT cascades of E4B and CHIP with the native UB transferring enzymes.
fig. S6. Affinity purification of xUB-modified proteins to identify the substrates of E4B and CHIP.
table S1. Potential E4B substrates identified by OUT.
table S2. Potential CHIP substrates identified by OUT.
table S3. Top networks associated with the E4B substrates identified by the OUT screen.
table S4. Top networks associated with the CHIP substrates identified by the OUT screen.
table S5. Primers used in this study.