

1. INTRODUCTION
(−)-Epigallocatechin-3-gallate (EGCG) is the most abundant polyphenol in green tea (Camellia sinensis, Theaceae). A growing body of data has suggested that both green tea and EGCG have preventive effects against a number of chronic diseases including cancer, cardiovascular diseases, and metabolic syndrome (Chen et al., 2017; Pang et al., 2016; Yang et al., 2016). A number of mechanisms have been proposed to account for these disease preventive effects including induction of endogenous antioxidant systems and mitigation of disease-related oxidative stress (reviewed in (Forester and Lambert, 2011; Yang and Wang, 2016)). Although EGCG has typically been considered an antioxidant, we and others have reported that some of the beneficial biological effects of this compound (i.e. cancer inhibitory activity) may be due to its ability to induce oxidative stress (Lambert and Elias, 2010; Li et al., 2010; Tao et al., 2014; Tao et al., 2015).
One consequence of the growing body of scientific data on the potential beneficial effects of green tea and EGCG has been the proliferation of green tea-based dietary supplements. In 2015, sales of green tea-based dietary supplements through mainstream channels in the United States exceeded USD 48 million (Smith et al., 2016). These supplements can be prepared using different extraction processes and can be standardized to different levels of green tea polyphenols. Some preparations contain greater than 90% EGCG by weight (e.g. TEAVIGO®, www.teavigoinfo.com). The safety of these products is often not directly examined, but is assumed based on the historic safety of green tea beverage and human intervention studies using green tea preparations (Bettuzzi et al., 2006; Chow et al., 2003). A growing number of animal model studies and case-reports suggest that this assumption may not be appropriate.
Since 1999, more than 40 case reports of hepatotoxicity have been associated with use of green tea-based dietary supplements (Mazzanti et al., 2009; Patel et al., 2013; Whitsett et al., 2014). These cases have typically presented with elevated serum liver enzymes (alanine aminotransferase [ALT] or asparatate aminotransferase [AST]), elevated serum bilirubin, abdominal pain, and jaundice. Cessation of supplement use resulted in most cases resulted in resolution of hepatic injury, although in several cases fulminant liver disease developed and required liver transplant (Whitsett et al., 2014; Yellapu et al., 2011). In several cases, re-challenge with the supplement led to re-injury (i.e. hepatic injury returned coincident with subjects resuming use of the supplement). These reports have typically eliminated known hepatotoxic insults (e.g. acetaminophen, alcohol, etc.), however, they are often confounded by the presence of other herbal components. While these case-reports cannot establish a causal relationship between green tea-based supplemented use and liver injury, their increasing number raise significant concerns.
Although a number of controlled human intervention studies have been conducted using high doses of oral green tea extract or EGCG and have not reported widespread significant adverse effects, in many cases liver-related clinical chemistry has not been reported. A recent report from Minnesota Green Tea Trial indicated that women who received green tea extract (containing 843 mg EGCG per dose) for 1 year had increased incidence of elevated plasma ALT levels compared to placebo treated subjects (6.7% vs. 0.07%) (Dostal et al., 2015). In the EGCG-treated group, 1.5% experienced serious ALT-related adverse events (defined as greater than 5 times the upper limit of normal). It remains unclear why EGCG and green tea extracts are related to hepatotoxic events in some subjects, but not others. Further studies are needed to better understand the mechanisms by which EGCG induces hepatotoxicity, and the factors that predispose individuals to heightened risk.
Animal model studies by our laboratory and others have demonstrated the hepatotoxic potential of oral bolus EGCG in both rodents and dogs (Isbrucker et al., 2006; Kapetanovic et al., 2009; Lambert et al., 2010). For example, we have previously reported that a single oral bolus EGCG (500–1500 mg/kg, bw) can induce hepatotoxicity in male CF-1 mice in a dose-dependent fashion. The effects were related to induction of hepatic oxidative stress. The underlying mechanisms of action for these toxicological effects remain unclear, although we have reported that mice receiving toxic doses of EGCG have reduced hepatic expression of genes related to antioxidant response (James et al., 2015). We have previously reported that EGCG can induce mitochondrial dysfunction in oral cancer cells, which lead to induction of intracellular oxidative stress, decreased expression of sirtuin 3 expression and mitochondrial antioxidant response, and cell death (Tao et al., 2014). Based on these results, we hypothesized that at hepatotoxic doses, EGCG might function in a similar manner.
In the present study, we evaluated the dose-dependent effects of EGCG on markers of hepatic oxidative stress (lipid peroxide levels, oxidative DNA damage [measured as phosphorylated histone 2AX], and reduced glutathione concentrations), mitochondrial biogenesis (mitochondrial copy number, expression of electron transport chain components), and mitochondria-related antioxidant response in C57BL/6J mice.
2. MATERIALS AND METHODS
2.1 Chemicals and Reagents
EGCG (93% pure) was purchased from Taiyo Green Power (Wuxi, Jiangsu, China). Rabbit anti-mouse monoclonal antibodies against glutathione peroxidase 1 and superoxide dismutase 2 were purchased from Cell Signaling Technologies (Danvers, MA). PCR primers (Table 1) were synthesized by the Genomics Core Facility at the Pennsylvania State University (University Park, PA). All other reagents were of the highest grade commercially-available.
Table 1.
Primer sequences used for gene expression and mitochondrial DNA copy number analysis in liver tissue.
| Abbr. | Gene Name | Forward Primer | Reverse Primer |
|---|---|---|---|
| MT-Co2 | Cytochrome C Oxidase Subunit II | ATAACCGAGTCGTTCTGCCAAT | TTTCAGAGCATTGGCCTATGAA |
| Foxo3a | Forkhead box O3a | ACAAACGGCTCACTCTGTCCCAG | AGCTCTTGCCAGTTCCCTCATTCTG |
| Gapdh | Glyceraldehyde 3-phosphate Dehydrogenase | TGAAGCAGGCATCTGAGGG | CGAAGGTGGAAGAGTGGGAG |
| Gpx1 | Glutathione peroxidase 1 | GATGGAGCCCATTCCTGAACC | CCCTGTACTTATCCAGGCAGA |
| Ndusf8 | NADH dehydrogenase [ubiquinone] iron-sulfur protein 8 | GTTCATAGGGTCAGAGGTCAAG | TCCATTAAGATGTCCTGTGCG |
| Pgc1a | Peroxisome proliferator-activated receptor γ coactivator 1α | ACAGCCGTAGGCCCAGGTAC | GCCTTTCGTGCTCATAGGCTT |
| Rps18 | 40S ribosomal protein S18 | TGTGTTATTTTACTGGTGGACA | CATCACCCACTTACCCCCAAAA |
| Sirt3 | Sirtuin 3 | ATCCCGGACTTCAGATCCCC | CAACATGAAAAAGGGCTTGGG |
| Sod2 | Superoxide dismutase 2, mitochondrial | CCGAGGAGAAGTACCACGAG | GCTTGATAGCCTCCAGCAAC |
| Uqcrc1 | Cytochrome b-c1 complex subunit 1, mitochondrial | ATCAAGGCACTGTCCAAGG | TCATTTTCCIGCATCTCCCG |
2.2 Animal Care and Treatment
All animal studies were approved by the Institutional Animal Care and Use Committee of the Pennsylvania State University (University Park, PA, IACUC protocol no. 37115). Male C57BL/6J mice (4–5 wks old) were purchased from Jackson Laboratory (Bar Harbor, ME) and maintained on a 12 h light-dark cycle at 21° C and 38% relative humidity. After a 2 wk acclimation period, mice were given ad libitum access to semi-purified, low fat (10 %kcal from fat) diet. After 9 weeks, mice were fasted for 7 h (0700–1400 h) and then given a single oral bolus dose of saline (0 mg/kg) or EGCG (250, 500 or 750 mg/kg, ig.) once daily for 3 days. One hour after administration of the third dose of EGCG or vehicle, mice were anesthetized and blood was collected by cardiac puncture. The doses of EGCG selected are based on our previous studies on EGCG-induced hepatotoxicity, and are equivalent to human doses of 830–2500 mg/d (assuming 70 kg body weight)based on allometric scaling (Schneider et al., 2004). This dose range is equivalent to that delivered by 6–18 servings of typically prepared green tea (Yang et al., 2002). Plasma was prepared by centrifugation at 3200 g for 15 min and frozen at −80°C. The livers were harvested and washed with ice-cold PBS. A 1mm3 section was fixed in modified Karnovsky’s fixative (2.5 % formaldehyde, 1.5 % gluturaldehdye, 0.1 M sodium Cacodylate buffer, 4 % sucrose) for transmission electron microscopy (TEM) analysis. A sample was fixed in 10% buffered formalin for histopathology analysis and the remainder was frozen at −80° for later analysis.
2.3 Analysis of hepatotoxicity
Plasma ALT levels were measured spectrophotometrically using a commercially-available assay (Catachem, Inc, Oxford, CT) as previously described (James et al., 2015; Lambert et al., 2010). Formalin-fixed liver samples were embedded in paraffin, sectioned, and stained with hematoxylin and eosin. Blinded slides were evaluated by a board-certified veterinary pathologist (MJK) and scored for inflammation, necrosis, hemorrhage, and apoptosis (0 = “not present” to 4 = “severe”).
2.4 Transmission Electron Microscopy
The number of mitochondria and mitochondrial area were quantified in mice treated with 0 or 500 mg/kg, ig EGCG using TEM. We elected to examine only this dose of EGCG, because it was the lowest dose used in the studies that caused significant hepatotoxicity without significant mortality. We were concerned that higher doses would produce artefactual changes that were secondary to hepatocellular necrosis. Formaldehyde/glutaraldehyde-fixed samples were washed in 0.1 M cacodylate buffer and fixed with osmium tetroxide (2% in 0.1 M cacodylate buffer) for 1 h. After washing twice with 0.1 M cacodylate buffer and once with deionized water, samples were stained 1 h with 2 % uranyl acetate. Samples were then dehydrated with increasing concentrations of ethanol. Samples were infiltrated with Eponate resin. Samples were then placed in molds and covered with fresh 100% resin and incubated at 60°C overnight to allow resin to polymerize. Blocks containing samples were then trimmed, cut into 50–100 nm thick sections with a Leica UC6 ultramicrotome (Leica Microsystems, Wetzlar, Germany), and stained with lead citrate (3.4 mg/mL) for 12 mins prior to imaging with Transmission electron microscope. TEM images were analyzed using NIH Image J software. Area of mitochondria was determined by tracing each whole mitochondrion in the field, and then averaged per field and number of samples. Density was determined as the number of mitochondria per field and averaged based on number of fields per sample.
2.5 Mitochondrial DNA copy number
Mitochondrial DNA copy number was determined as previously described (Price et al., 2012). In brief, total cellular DNA was extracted from liver tissue and mitochondrial cytochrome oxidase subunit 2 (MT-Co2) was amplified by real-time PCR. 40S ribosomal protein s18 (Rps18) was amplified to quantify nuclear DNA. The ratio of MT-Co2 to RPS18 was used as a proxy for mitochondrial DNA copy number.
2.6 Hepatic glutathione and oxidative stress
Malondialdehyde (MDA) levels were used as a marker of oxidative stress and determined using the thiobarbituric acid reactive substances assay. In brief, liver samples were homogenized in Tissue Protein Extraction Reagent (TPER) containing 1% (v/v) phosphatase and protease inhibitor cocktails (Thermo-Scientific, Rockford, IL). Homogenate samples combined with 6 volume of 100:3 ratio of reagent A (15% trichloroacetic acid and 0.375% thiobarbituric acid (w/v) in 0.25M HCl) and reagent B (2% butylated hydroxytoluene in ethanol (w/v)). The mixture was heated at 95 °C for 15 min and cooled on ice for 10 min. Following centrifugation, the samples were maintained at room temperature for 10 min prior to measuring the absorbance at 532 nm. Quantification was made based on a MDA standard curve. Reduced glutathione levels were determined by HPLC using a modification of a previously reported method (Bayram et al., 2014). Briefly, liver tissue (10–30 mg) was perfused with PBS containing heparin (0.15 mg/mL). Tissue was homogenized in PBS containing 2 mM EDTA, centrifuged at 13, 000 g for 10 mins at 4 °C, and the supernatant was collected. The protein present in the supernatant was precipitated by combination with 1.5 volumes of 1 % metaphosphoric acid and centrifugation at 13, 000 g for 10 mins at 4 °C. The resultant supernatant was analyzed by HPLC with electrochemical detection (ECD). The HPLC system consisted of two LC-20AD pumps (Shimadzu Co, Columbia, MD), an ESA 5600A Coularray detector (Chelmsford, MA), and a Supelcosil LC18 column (4.6 × 150 mm, 5 μm particle size, Supelco, Bellefonte, PA). Samples were resolved using a SUPELCOSIL LC-18 column (15 cm × 4.6 mm × 5 μm) and an isocratic mobile phase of 50 mM NaH2PO4 (pH 3.0). The column was maintained at 30 °C, the flow rate was 0.3 mL/min and the injection volume was 10 μL. GSH in the samples was detected at ECD potential = 600 mV, and quantified by comparison to a standard curve of GSH in metaphosphoric acid. Hepatic levels of phosphorylated histone 2A.X (γH2A.X) were determined as an indirect measure of oxidative DNA damage by western blot analysis as described below (Nikolova et al., 2014).
2.7 Gene expression studies
We examined the expression of genes related to antioxidant response (glutathione peroxidase 1 [Gpx1], superoxide dismutase 2 [Sod2], forkhead box O3a [FOXO3a], and), and the mitochondrial electron transport chain and mitochondrial biogenesis (mitochondrial NADH dehydrogenase [ubiquinone] iron-sulfur protein 8 [Ndufs8], cytochrome b-c1 complex subunit 1 [Uqcrc], peroxisome proliferator activated receptor γ coactivator 1 α [Pgc1a]) using established methods in our laboratory (Gu et al., 2014; James et al., 2015). RNA was isolated from liver tissue using Tri-reagent (Sigma Chemical Co. St. Louis, MO) according to the manufacturer’s instructions. Isolated RNA was quantified using the NanoDrop ND-1000 spectrophotometer. cDNA was synthesized with the RT2HT First Strand Kit (Qiagen, Germany). In brief, 1 μg RNA was incubated at 37°C for 5 mins with genomic DNA elimination buffer. Reverse-transcription mix was added to this mixture, and the reaction was incubated at 42°C for 15 min. The reaction was terminated by incubation at 95°C for 5 min. Following dilution with RNase-free water, SYBR Green PCR Master Mix and forward and reverse primers (sequences shown in Table 1) were added to the diluted cDNA. RT-PCR was performed using Applied Biosystems 7900HT Fast Real-Time PCR System. The thermal cycle was programmed for 10 min at 95°C for initial activation of polymerase, followed by 40 cycles of 15 s at 95°C for denaturation, 1 min at 60°C for annealing. A dissociation step was done at the end of the cycles for 15s at 95°C, then 15s at 60°C, followed by 15s at 95°C. Relative quantification of PCR products were done by SDS software (Applied Biosystems, Foster City, CA). Relative gene expressions were normalized to the house-keeping genes 18S ribosomal RNA gene. Data was represented by fold change in Ct values.
2.8 Western blot analysis
Expression of proteins of interest was determined by western blot. Protein was extracted from liver samples using Tissue Protein Extraction Reagent (TPER) containing 1% (v/v) phosphatase and protease inhibitor cocktails (Thermo-Scientific, Rockford, IL). After homogenization and centrifugation the protein concentration in supernatant was determined by the Bradford assay (Sigma-Aldrich, St.Louis, MO). Lysates were then combined with an equal volume of laemmli sample buffer (Bio-Rad, Hercules, CA) and denatured at 90°C for 5 min. Protein samples (75 μg protein) were resolved by SDS-polyacrylamide (15 %) gel electrophoresis (PAGE) and transferred to a nitrocellulose membrane (Bio-Rad, Hercules, CA). Membranes were probed with primary antibody for protein of interest (1:1000 dilution) overnight at 4 °C. After incubation with a fluorescent-conjugated secondary antibody (1:10,000 dilution) (Li-Cor Co., Lincoln, NE), bands were visualized using a Licor Odyssey Imaging System (LI-COR Corporate). Band density was quantified using Odyssey Application Software Version 3.0. Beta actin was used as a protein loading control.
2.9 Antioxidant enzyme activity
Glutathione peroxidase (Gpx) activity was determined using a commercially available assay kit from Cayman Chemicals (Ann Arbor, MI). In brief, 50 mg of liver tissue was homogenized in 300 μL homogenization buffer (50 mM Tris-HCl, pH 7.5, 5 mM EDTA, and 1 mM DTT) and centrifuged at 10,000 g for 15 mins 4 °C. The supernatant was collected and assayed as per manufacturer’s instructions. Absorbance was measured at 340 nm and quantification was based on a standard curve. Superoxide dismutase (Sod) activity was determined using a previously reported methods (Noori et al., 2009). In brief, 100 mg of tissue was homogenized in 500 μL of homogenization buffer (20 mM HEPES, 1 mM EGTA, 210 mM, 70 mM sucrose; pH 7.2) and centrifuged at 1,500 g for 5 mins at 4 °C. The supernatant was removed diluted 1:50. A volume of 0.1 mL of diluted sample (or standards) was added to a reaction mixture (2.8 mL) containing: 0.05 M Na2CO3 (pH 10.2), 3mM EDTA, 1.5 mg/mL BSA, 0.75 mM NBT, and 3 mM Xanthine was added. The reaction was initiated with 0.1 mL xanthine oxidase (1:200 dilution) and incubated for 20 minutes at room temperature. The reaction was terminated by addition of 6 mM CuCl2. The inhibition of NBT reduction was determined spectrophotometrically at 560 nm.
2.10 Statistical Analysis
All experiments were repeated twice with at least three biological replicates for each treatment. Data are expressed as mean ± SEM. GraphPad Prism 5.0 (San Diego, CA) was used for statistical analysis. The Q test was used to determined outliers. Differences in biochemical markers of hepatotoxicity, gene/protein expression, and histological severity of liver injury were evaluated for statistical significance using one-way ANOVA with Dunnett’s post-test. Differences in the incidence of specific liver lesions were tested for statistical significance using χ-square test. Significance was achieved at p < 0.05.
3. RESULTS
3.1 EGCG-induced hepatotoxicity
Male C57BL/6J mice (aged 14 wks) were treat once daily with EGCG (250–750 mg/kg, ig) for 3 d. Treatment EGCG at 500 or 750 mg/kg caused a significant reduction in final body weight (p < 0.05, Fig. 1A). Plasma ALT levels were analyzed as a marker of hepatotoxicity. Mice treated with 500 or 750 mg/kg, ig EGCG had greater plasma ALT levels than the vehicle-treated controls with the increase at 750 mg/kg reaching statistical significance (p < 0.05, Fig. 1B). Histopathological analysis was performed on liver samples derived from mice treated with 0, 500, and 750 mg/kg, ig (Fig. 1C and Table 2). The incidence and severity of hepatic inflammation, necrosis, hemorrhage, and apoptosis were significantly increased (p < 0.05) at both dose levels of EGCG compared to vehicle-treated controls.
Figure 1.
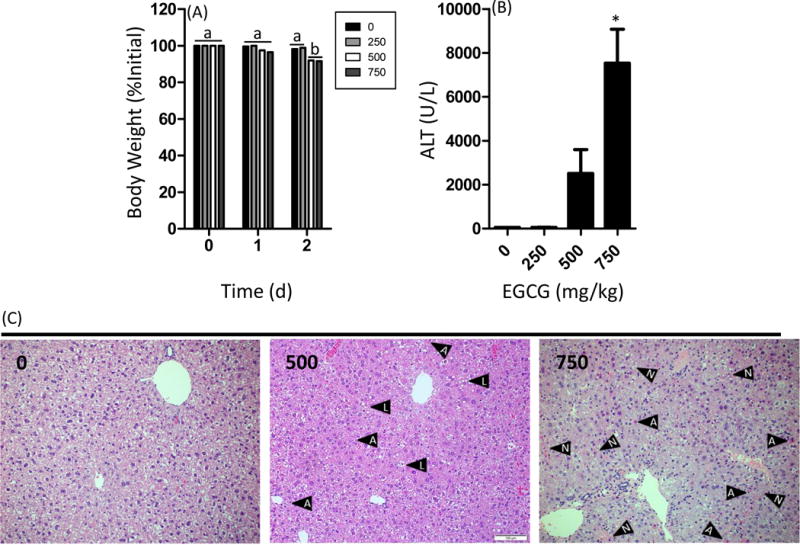
Impact of oral bolus EGCG on body weight gain and plasma alanine aminotransferase levels in C57BL/6J mice. Mice were fasted for 7 h and then treated once daily with EGCG by oral gavage for three days. (A) Mice were weighed daily. Mice were euthanized 1 h after the third dose. (B) Plasma ALT levels were determined using a commercially-available spectrophotometric assay. (C) Histopathological analysis was performed to determine the extent of EGCG-induced hepatotoxicity. Data represent the mean ± SEM for n = 10–18. Body weight changes were compared using two-way ANOVA with Bonferroni post-test. Different superscript letters indicates p < 0.05 between treatment groups. ALT levels were compared using one-way ANOVA with Dunnett’s post-test. An asterisk indicates p < 0.05 compared to control mice. Histologpathological images shown are representative images. Black arrows containing an “A” indicates an apoptotic body, those with an “L” indicate lipid droplets, and those with an “N” indicate areas of necrosis.
Table 2.
Histological evaluation of hepatotoxicity
| EGCG (mg/kg) | Inflammation | Necrosis | Hemorrhage | Apoptosis | ||||
|---|---|---|---|---|---|---|---|---|
|
| ||||||||
| Incidence | Severity1 | Incidence | Severity1 | Incidence | Severity1 | Incidence | Severity1 | |
| 0 | 5/11 | 1 ± 0a | 3/11 | 1 ± 0a | 0/11 | 0 ± 0a | 1/11 | 1 ± 0a |
| 500 | 8/9* | 1.1 ± 0.3a | 7/9* | 2.1 ± 0.9b | 2/9* | 2.5 ± 0.7b | 6/9* | 1.3 ± 0.5b |
| 750 | 10/11* | 1.3 ± 0.6a | 9/11* | 2.4 ± 1.2b | 6/11* | 2.5 ± 1.8b | 7/11* | 1.7 ± 0.9b |
Severity was graded in blinded samples and the values represent the mean ± SD in mice where the damage was present.
p < 0.01 by Chi-square test; different superscript letters indicate difference by one-way ANOVA with Dunnett’s post-hoc test.
3.2 EGCG-induced hepatic oxidative stress
Hepatic lipid peroxide levels and GSH levels were determined as markers of oxidative stress. Treatment with 250 or 500 mg/kg, ig EGCG had no significant effect on hepatic lipid peroxide levels compared to vehicle-treated controls (Fig. 2A). By contrast, treatment with 750 mg/kg, ig EGCG increased hepatic lipid peroxide levels by 2.8-fold (p < 0.05) compared to vehicle-treated controls. Treatment with EGCG significantly decreased hepatic GSH levels at all dose levels (p < 0.05, Fig. 2B). Hepatic γH2AX levels were measured as a marker of oxidative DNA damage (Fig. 2C–D). Both 500 and 750 mg/kg, ig EGCG appeared to increase γH2AX levels, however only the effect of the 750 mg/kg dose-level was statistically significant (p < 0.05).
Figure 2.
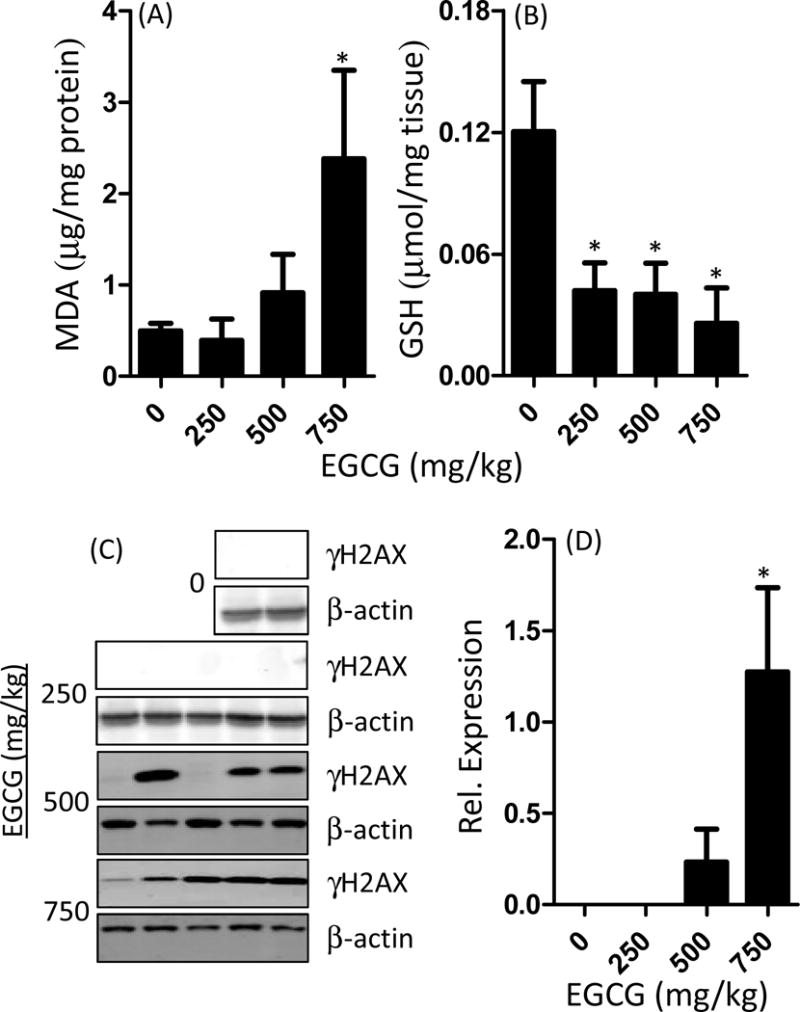
Effect of oral bolus EGCG on markers of hepatic oxidative stress in C57BL/6J mice. Mice were fasted for 7 h and then treated once daily with EGCG by oral gavage for three days. Mice were euthanasized 1 h after the last dose and hepatic malondialdehyde (MDA) levels were determined using the TBARS assay (A). Hepatic GSH levels were determined by HPLC and normalized to wet liver weight (B). Hepatic γH2AX levels were determined by western blot (C). Expression was quantified by densitometry and normalized to β-actin levels. Data represent mean ± SEM for n = 8–10. Data were analyzed using one-way ANOVA with Dunnett’s post-test. An asterisk indicates p < 0.05 compared to control mice. The western blot image shows representative data.
3.3 EGCG-induced changes in hepatic antioxidant enzymes
We examined the hepatic mRNA and protein expression of glutathione peroxidase Gpx1 and superoxide dismutase Sod2 in response to treatment with oral bolus EGCG (Fig. 3). Hepatic Gpx1 mRNA expression was significantly reduced by treatment with both 500 mg/kg and 750 mg/kg EGCG (p < 0.05). Western blot analysis showed that GPX1 protein expression was significantly lower in mice treated with 750 mg/kg EGCG (p < 0.05, Fig. 3). EGCG significantly reduced hepatic mRNA levels of Sod2 at 750 mg/kg, but significantly increased hepatic protein levels at the same dose (p < 0.05, Fig. 3). We determined the effect of EGCG treatment on hepatic Sod and Gpx activity, and found that 750 mg/kg EGCG significantly reduced both activities (p < 0.05, Fig. 3).
Figure 3.
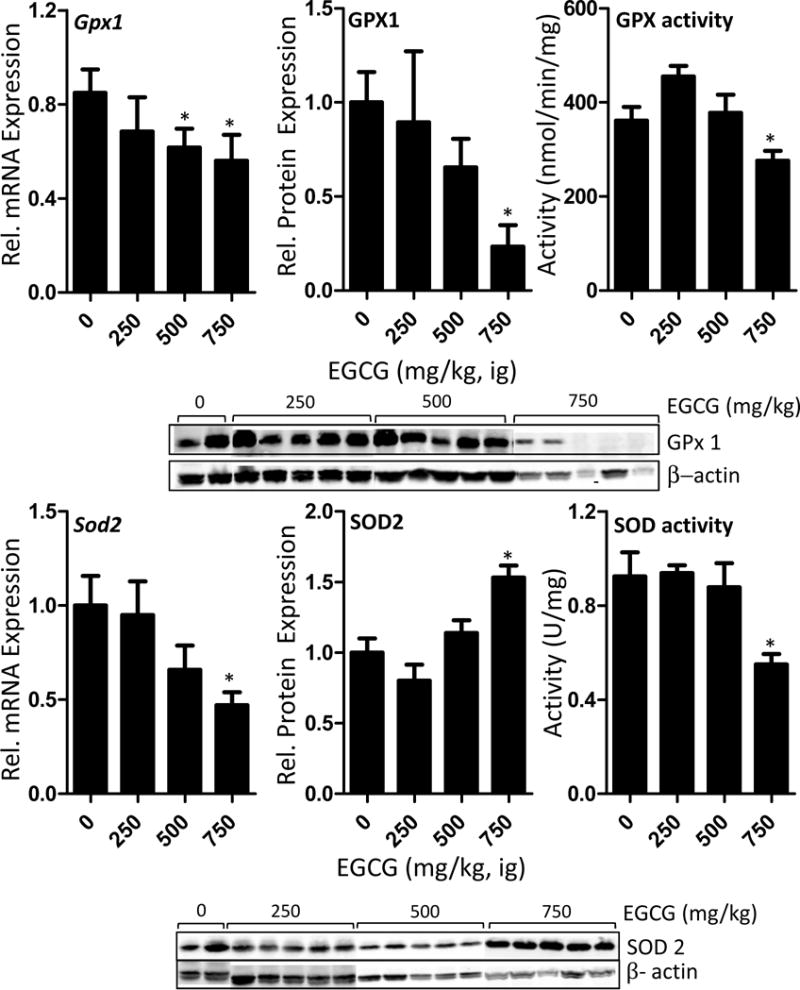
Impact of oral bolus EGCG on the expression and activity of hepatic mitochondria-related antioxidant enzymes in C57BL/6J mice. Mice were fasted for 7 h and then treated once daily with EGCG by oral gavage for three days. Mice were euthanasized 1 h after the last dose and hepatic mRNA levels and protein levels were determined by qRT-PCR and western blot, respectively. Enzyme activities were determined using spectrophotometric assays. Activity was normalized to protein content. Data represent the mean ± SEM of n = 11–18. Data were analyzed using one-way ANOVA with Dunnett’s post-test. An asterisk indicates p < 0.05 compared to control mice. The western blot images show representative data.
3.4 EGCG-induced changes in mitochondrial copy number and related genes
TEM and qPCR were used to determine the impact of EGCG treatment on mitochondrial copy number and structure. We found that treatment with 500 mg/kg, ig EGCG (the only dose assessed) tended to increase hepatic mitochondrial area compared to vehicle-treated control mice, whereas EGCG treatment significantly reduced hepatic mitochondrial density (p < 0.05, Fig. 4). qPCR analysis showed that EGCG treatment dose-dependently decreased mitochondrial copy number with treatment at the 750 mg/kg dose-level reducing copy number by 39% (p < 0.05, Fig. 4). Consistent with this decrease in copy number, we found that EGCG treatment significantly decreased hepatic mRNA levels of the mitochondrial respiratory complex genes Ndusf8 (complex I) and Uqcrc1 (complex III) compared to vehicle-treated control mice (p < 0.05, Fig. 4).
Figure 4.
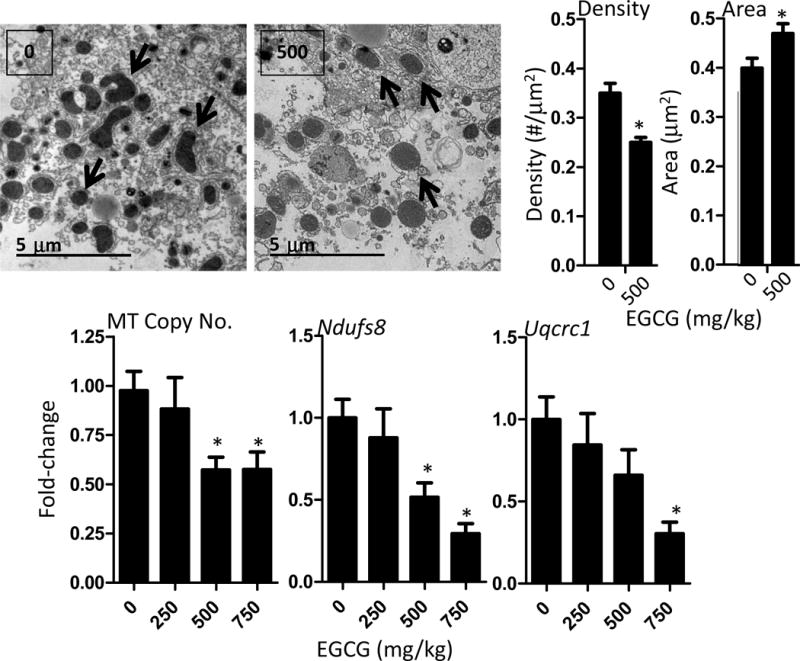
Impact of oral bolus EGCG on hepatic markers of mitochondrial copy number and mitochondrial function in C57BL/6J mice. Transmission electron microscopy was used to determine mitochondria size (as area) and density (number of mitochondria per field) in mice treated with 500 mg/kg, ig EGCG or vehicle. Image J software was used to determine mitochondrial area and the number of mitochondria per field were determined. Representative electron micrographs are shown and black arrows indicate mitochondria. Real-time PCR analysis of the ratio of MT-Co2 (mitochondrial) to Rps18 (genomic) was used as a surrogate of mitochondrial copy number. Data represent mean ± SEM of n = 8–12. The expression of Ndufs8 and Uqcrc1 were determined by qRTPCR and used markers of respiratory complex I and complex III expression, respectively. Data represent mean ± SEM of n = 8–12. Data were analyzed by one-way ANOVA with Dunnett’s post-test. An asterisk indicates p < 0.05 compared to control mice.
3.5 Effect of EGCG on Sirt3 and related transcription factors
SIRT3 plays an important role in coordinating mitochondrial biogenesis and mitochondria-associated antioxidant response (Bause and Haigis, 2013). We found that EGCG treatment significantly decreased both hepatic mRNA (75% decrease) and protein expression (66% decreased) of SIRT3 compared to vehicle-treated control mice (p < 0.05, Fig. 5). We also examined the mRNA expression of two transcription-related factors related to mitochondrial biogenesis and antioxidant response: Pgc1a and Foxo3a. The mRNA levels of Pgc1a and Foxo3a were both significantly reduced at the 500 and 750 mg/kg dose levels (p < 0.05, Fig. 5)
Figure 5.
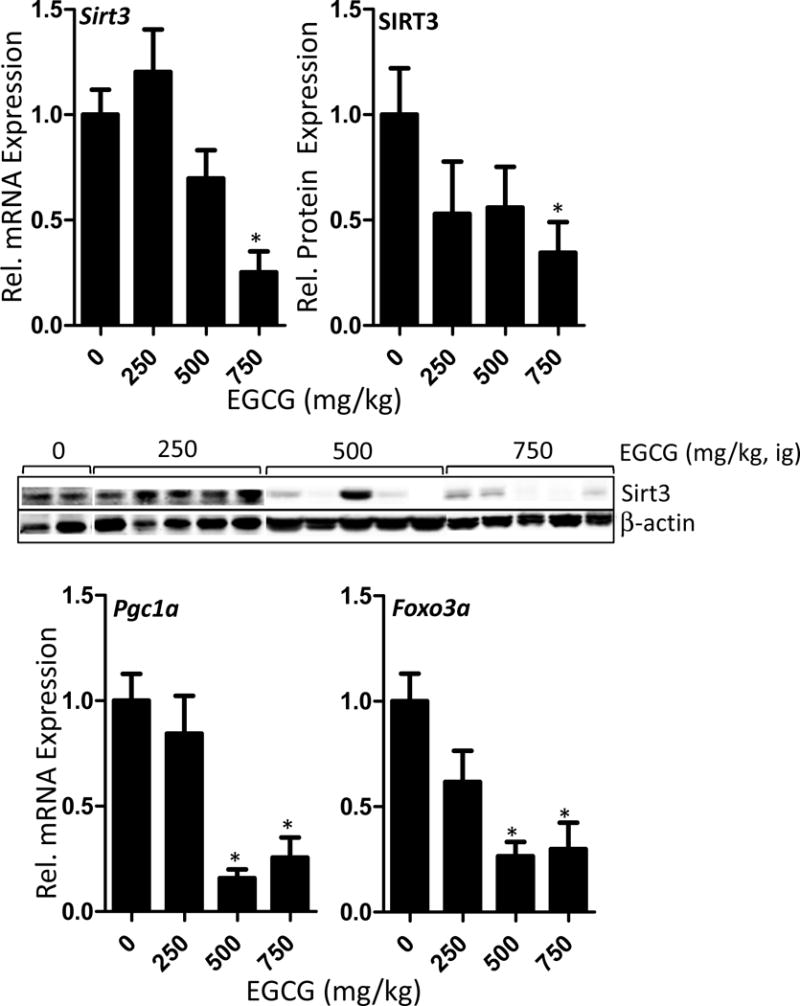
Impact of oral bolus treatment with EGCG on the hepatic expression of mitochondria-related transcription factors and co-factors in C57BL/6J mice. Mice were fasted for 7 h and then treated once daily with EGCG by oral gavage for three days. Mice were euthanasized 1 h after the last dose and hepatic mRNA levels and protein levels were determined by qRT-PCR and western blot, respectively. SIRT3, FOXO3a, and PGC1α play an important role in mitochondrial biogenesis and antioxidant response. Data represent the mean ± SEM of n = 11–18. Data were analyzed using one-way ANOVA with Dunnett’s post-test. An asterisk indicates p < 0.05 compared to control mice. The western blot images show representative data.
4. Discussion
Whereas many human intervention studies have examined the potential health beneficial effects of high doses of green tea extracts and EGCG and have failed to report significant adverse effects, a growing number of case-reports have linked the use of green tea-based dietary supplements with hepatotoxicity in human subjects (Mazzanti et al., 2009; Patel et al., 2013; Whitsett et al., 2014; Yellapu et al., 2011). A recent human intervention study (Minnesota Green Tea Trial) did report that a significantly higher number of participants receiving green tea extract had elevated serum ALT levels compared to subjects receiving placebo (6.7% vs 0.7%) suggesting a treatment-induced adverse effect (Dostal et al., 2015). We and others have demonstrated the EGCG and green tea extracts can induce hepatotoxicity in mice and beagle dogs and that these effects are related to induction of hepatic oxidative stress (Isbrucker et al., 2006; James et al., 2015; Kapetanovic et al., 2009; Lambert et al., 2010). The mechanisms underlying these hepatotoxic effects remain unclear.
In the present study, we examined the dose-dependent hepatotoxic effects of EGCG in mice and evaluated the potential role of the mitochondria as a cellular target for EGCG. We found that once daily dosing with EGCG (250–750 mg/kg, ig) for three days induced significant hepatotoxicity manifested as decreased body weight and increased plasma ALT levels. Histopathological examination showed that treatment with 500 and 750 mg/kg, ig EGCG increased hepatic inflammation, necrosis and hemorrhage compared to vehicle-treated mice. These effects were related to increased hepatic lipid peroxide levels, increased expression of γH2AX, and decreased reduced glutathione levels indicating EGCG-induced oxidative stress. These results are consistent with our previous studies on the hepatotoxicity of EGCG, however the present studies provide greater clarity regarding dose-response effects and histological markers of toxicity (James et al., 2015; Lambert et al., 2010).
The mitochondria is the largest source of endogenous reactive oxygen species and perturbing mitochondrial function and/or redox balance is a known mechanism for toxic compounds including rotenone and antimycin. We have previously shown that EGCG treatment can induce mitochondrial oxidative stress and reduce the expression of mitochondria-related antioxidant response systems in oral cancer cells (Tao et al., 2014). Here we found that mice treated with EGCG had reduced expression of SOD2 and GPX1, which are both mitochondria-related antioxidant enzymes. Interestingly, the mRNA expression and activity of SOD were decreased, whereas protein expression was increased. This could represent a feedback response on the part of the liver in response to some effect of EGCG directly on SOD activity. Specifically, it could indicate EGCG inhibits SOD activity post-translationally, which in turn causes the hepatocyte to compensate by producing more protein. Given that SOD2 transcription and enzyme activity is controlled in part by SIRT3-mediated deacetylation, this result would be consistent with our observation that EGCG reduces the expression of SIRT3. Further studies are needed to more fully investigate this hypothesis.
EGCG treatment also significantly reduced mitochondrial copy number as determined by qPCR analysis of the ratio of MT-CO2 to RPS18 and hepatic density of mitochondria as determined by TEM. EGCG treatment significantly reduced representative genes of mitochondrial respiratory complex I (Ndufs8) and complex III (Ugcrc1).
SIRT3, PGC1α, and FOXO3a play important roles in regulating the expression and activity of mitochondrial antioxidant response systems and in mitochondrial biogenesis (Bause and Haigis, 2013; Kong et al., 2010; Rangarajan et al., 2015). EGCG treatment reduced hepatic mRNA and protein expression of SIRT3, and mRNA expression of PGC1α and FOXO3a. Decreased expression of these factors are consistent with the changes that we observed in SOD2 and GPX1 expression, as well as the changes we observed in mitochondrial copy number. We have previously reported that EGCG can suppress the expression and activity of SIRT3, as well as, the mRNA expression of PGC1α and FOXO3a in human oral cancer cells (Tao et al., 2015). These changes were associated with increased mitochondrial oxidative stress and cell death.
The dose range of EGCG used in the present studies represents a human equivalent dose of 830–2500 mg/d based on allometric scaling (Schneider et al., 2004). This dose range is within the range achievable by consuming normal to slightly elevated doses of many green tea-based dietary supplements. It is, therefore, feasible that effects observed here may be translatable to cases of human hepatotoxicity related to green tea-based supplements.
While our studies do not provide direct information about factors that increase risk of EGCG-related hepatic adverse effects, they do suggest that subjects with polymorphisms which compromise mitochondria-related antioxidant response systems, or who are using pharmaceutical agents that stress the mitochondria (e.g. acetaminophen) might be at greater risk for a hepatotoxic event. Indeed, Salminen et al., have shown that the combination of EGCG and acetaminophen under certain dosing conditions can lead to greater hepatotoxicity in mice than either compound alone (Salminen et al., 2012). Further studies are needed to better understand the factors that increase potential risk of EGCG-mediated hepatotoxicity.
The present study has some limitations. Most notably, we did not examine mitochondrial function or levels of mitochondrial reactive oxygen species directly. Such studies are needed to ultimately understand the impact of EGCG on the mitochondria. In addition, several markers that were examined (e.g. FOXO3a, NDUFS8, UQCRC1) were only measured at the mRNA level. It is possible that the gene expression changes observed do not correlate with changes in protein and activity. Our confidence in our conclusions is strengthened by the fact that data are internally consistent across different markers and each marker seems to support a key role for the mitochondria as an important mechanistic target.
5. CONCLUSIONS
In summary, we report here the results of our on-going studies on the potential hepatotoxicity of oral bolus EGCG. We demonstrate for the first time the potential role of the mitochondria as a mechanistic target related to EGCG-induced hepatotoxicity. Future studies will focus on directly characterizing the effects of EGCG on mitochondrial function, and determining the potential toxicological interactions between EGCG and other hepatotoxic compounds that are known to affect the mitochondria (e.g. rotenone) or induce oxidative stress (e.g. acetaminophen).
HIGHLIGHTS.
EGCG induced dose-dependent hepatotoxicity in male C57BL/6J mice
EGCG increased hepatic oxidative stress in male C57BL/6J mice
EGCG decreased mitochondrial DNA copy number, density, and complex expression
EGCG inhibited sirtuin 3 expression and related antioxidant response genes
Acknowledgments
The authors thank Drs. Yeyi Gu, Sudathip Sae-tan, Zachary Bitzer, and Sarah Forester for technical assistance. The authors thank Ms. Roberta Horner and the Penn State Animal Diagnostic Laboratory for preparation of liver samples for histopathological analysis. The authors thank Ms. Missy Hazen for technical assistance in performing TEM and interpreting the results of that analysis. This study was supported in part by grant no. AT004678 from the National Center for Complementary and Integrative Health (to JDL), a Penn State College of Agricultural Sciences Graduate Student Research grant (to KDJ), the Azzara Family Bioactives Award (to JDL), and USDA Hatch Project no. 4565.
Abbreviations
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- MT-Co2
cytochrome oxidase subunit II
- EGCG
(−)epigallocatechin-3-gallate
- FOXO3a
forkhead box O3a
- GAPDH
glyceraldehyde-3 phosphate dehydrogenase
- γH2AX
phosphorylated histone 2AX
- GSH
reduced glutathione
- GPX1
glutathione peroxidase 1
- HPLC-ECD
high performance liquid chromatography with electrochemical detection
- MDA
malondialdehyde
- NDUFS8
NADH dehydrogenase [ubiquinone] iron-sulfur protein 8, mitochondrial
- PGC1α
peroxisome proliferator-activated receptor γ coactivator 1
- qPCR
quantitative PCR
- qRT-PCR
quantitative reverse-transcriptase PCR
- RPS18
40S ribosomal protein s18
- SIRT3
sirtuin 3
- SOD2
Superoxide dismutase 2, mitochondrial
- TEM
transmission electron microscopy
- Uqcrc
cytochrome b-c1 complex subunit 1, mitochondrial
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bause AS, Haigis MC. SIRT3 regulation of mitochondrial oxidative stress. Exper Gerontol. 2013;48:634–639. doi: 10.1016/j.exger.2012.08.007. [DOI] [PubMed] [Google Scholar]
- Bayram B, Rimbach G, Frank J, Esatbeyoglu T. Rapid method for glutathione quantitation using high-performance liquid chromatography with coulometric electrochemical detection. J Agric Food Chem. 2014;62:402–408. doi: 10.1021/jf403857h. [DOI] [PubMed] [Google Scholar]
- Bettuzzi S, Brausi M, Rizzi F, Castagnetti G, Peracchia G, Corti A. Chemoprevention of human prostate cancer by oral administration of green tea catechins in volunteers with high-grade prostate intraepithelial neoplasia: a preliminary report from a one-year proof-of-principle study. Cancer Res. 2006;66:1234–1240. doi: 10.1158/0008-5472.CAN-05-1145. [DOI] [PubMed] [Google Scholar]
- Chen Y, Wu Y, Du M, Chu H, Zhu L, Tong N, Zhang Z, Wang M, Gu D, Chen J. An inverse association between tea consumption and colorectal cancer risk. Oncotarget. 2017;8:37367–37376. doi: 10.18632/oncotarget.16959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow HH, Cai Y, Hakim IA, Crowell JA, Shahi F, Brooks CA, Dorr RT, Hara Y, Alberts DS. Pharmacokinetics and safety of green tea polyphenols after multiple-dose administration of epigallocatechin gallate and polyphenon E in healthy individuals. Clin Cancer Res. 2003;9:3312–3319. [PubMed] [Google Scholar]
- Dostal AM, Samavat H, Bedell S, Torkelson C, Wang R, Swenson K, Le C, Wu AH, Ursin G, Yuan JM, Kurzer MS. The safety of green tea extract supplementation in postmenopausal women at risk for breast cancer: results of the Minnesota Green Tea Trial. Food Chem Toxicol. 2015;83:26–35. doi: 10.1016/j.fct.2015.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Forester SC, Lambert JD. The role of antioxidant versus pro-oxidant effects of green tea polyphenols in cancer prevention. Mol Nutr Food Res. 2011;55:844–854. doi: 10.1002/mnfr.201000641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Yu S, Lambert JD. Dietary cocoa ameliorates obesity-related inflammation in high fat-fed mice. Eur J Nutr. 2014;53:149–158. doi: 10.1007/s00394-013-0510-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isbrucker RA, Edwards JA, Wolz E, Davidovich A, Bausch J. Safety studies on epigallocatechin gallate (EGCG) preparations. Part 2: dermal, acute and short-term toxicity studies. Food Chem Toxicol. 2006;44:636–650. doi: 10.1016/j.fct.2005.11.003. [DOI] [PubMed] [Google Scholar]
- James KD, Forester SC, Lambert JD. Dietary pretreatment with green tea polyphenol, (−)-epigallocatechin-3-gallate reduces the bioavailability and hepatotoxicity of subsequent oral bolus doses of (−)-epigallocatechin-3-gallate. Food Chem Toxicol. 2015;76:103–108. doi: 10.1016/j.fct.2014.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapetanovic IM, Crowell JA, Krishnaraj R, Zakharov A, Lindeblad M, Lyubimov A. Exposure and toxicity of green tea polyphenols in fasted and non-fasted dogs. Toxicol. 2009;260:28–36. doi: 10.1016/j.tox.2009.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong X, Wang R, Xue Y, Liu X, Zhang H, Chen Y, Fang F, Chang Y. Sirtuin 3, a new target of PGC-1alpha, plays an important role in the suppression of ROS and mitochondrial biogenesis. PLoS One. 2010;5:e11707. doi: 10.1371/journal.pone.0011707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JD, Elias RJ. The antioxidant and pro-oxidant activities of green tea polyphenols: a role in cancer prevention. Arch Biochem Biophys. 2010;501:65–72. doi: 10.1016/j.abb.2010.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambert JD, Kennett MJ, Sang S, Reuhl KR, Ju J, Yang CS. Hepatotoxicity of high oral dose (−)-epigallocatechin-3-gallate in mice. Food Chem Toxicol. 2010;48:409–416. doi: 10.1016/j.fct.2009.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li GX, Chen YK, Hou Z, Xiao H, Jin H, Lu G, Lee MJ, Liu B, Guan F, Yang Z, Yu A, Yang CS. Pro-oxidative activities and dose-response relationship of (−)-epigallocatechin-3-gallate in the inhibition of lung cancer cell growth: a comparative study in vivo and in vitro. Carcinogenesis. 2010;31:902–910. doi: 10.1093/carcin/bgq039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazzanti G, Menniti-Ippolito F, Moro PA, Cassetti F, Raschetti R, Santuccio C, Mastrangelo S. Hepatotoxicity from green tea: a review of the literature and two unpublished cases. Eur J Clin Pharmacol. 2009;65:331–341. doi: 10.1007/s00228-008-0610-7. [DOI] [PubMed] [Google Scholar]
- Nikolova T, Dvorak M, Jung F, Adam I, Kramer E, Gerhold-Ay A, Kaina B. The gammaH2AX assay for genotoxic and nongenotoxic agents: comparison of H2AX phosphorylation with cell death response. Toxicol Sci. 2014;140:103–117. doi: 10.1093/toxsci/kfu066. [DOI] [PubMed] [Google Scholar]
- Noori S, Nasir K, Mahboob T. Effects of cocoa powder on oxidant/antioxidant status in liver, heart and kidney tissues of rats. J Anim Plant Sci. 2009;19:174–178. [Google Scholar]
- Pang J, Zhang Z, Zheng TZ, Bassig BA, Mao C, Liu X, Zhu Y, Shi K, Ge J, Yang YJ, Dejia H, Bai M, Peng Y. Green tea consumption and risk of cardiovascular and ischemic related diseases: A meta-analysis. Int J Cardiol. 2016;202:967–974. doi: 10.1016/j.ijcard.2014.12.176. [DOI] [PubMed] [Google Scholar]
- Patel SS, Beer S, Kearney DL, Phillips G, Carter BA. Green tea extract: a potential cause of acute liver failure. World J Gastroenterol. 2013;19:5174–5177. doi: 10.3748/wjg.v19.i31.5174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price NL, Gomes AP, Ling AJY, Duarte FV, Martin-Montalvo A, North BJ, Agarwal B, Ye L, Ramadori G, Teodoro JS, Hubbard BP, Varela AT, Davis JG, Varamini B, Hafner A, Moaddel R, Rolo AP, Coppari R, Palmeira CM, de Cabo R, Baur JA, Sinclair DA. SIRT1 Is Required for AMPK Activation and the Beneficial Effects of Resveratrol on Mitochondrial Function. Cell Metab. 2012;15:675–690. doi: 10.1016/j.cmet.2012.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rangarajan P, Karthikeyan A, Lu J, Ling EA, Dheen ST. Sirtuin 3 regulates Foxo3a-mediated antioxidant pathway in microglia. Neurosci. 2015;311:398–414. doi: 10.1016/j.neuroscience.2015.10.048. [DOI] [PubMed] [Google Scholar]
- Salminen WF, Yang X, Shi Q, Greenhaw J, Davis K, Ali AA. Green tea extract can potentiate acetaminophen-induced hepatotoxicity in mice. Food Chem Toxicol. 2012;50:1439–1446. doi: 10.1016/j.fct.2012.01.027. [DOI] [PubMed] [Google Scholar]
- Schneider K, Oltmanns J, Hassauer M. Allometric principles for interspecies extrapolation in toxicological risk assessment–empirical investigations. Regul Toxicol Pharmacol. 2004;39:334–347. doi: 10.1016/j.yrtph.2004.03.001. [DOI] [PubMed] [Google Scholar]
- Smith T, Kawa K, Eckl V, Johnson J. Sales of herbal dietary supplements in US increased 7.5% in 2015 consumers spent $6.92 billion on herbal supplements in 2015, marking the 12th consecutive year of growth HerbalGram. 2016;111:67–73. [Google Scholar]
- Tao L, Forester SC, Lambert JD. The role of the mitochondrial oxidative stress in the cytotoxic effects of the green tea catechin, (−)-epigallocatechin-3-gallate, in oral cells. Mol Nutr Food Res. 2014;58:665–676. doi: 10.1002/mnfr.201300427. [DOI] [PubMed] [Google Scholar]
- Tao L, Park JY, Lambert JD. Differential prooxidative effects of the green tea polyphenol, (−)-epigallocatechin-3-gallate, in normal and oral cancer cells are related to differences in sirtuin 3 signaling. Mol Nutr Food Res. 2015;59:203–211. doi: 10.1002/mnfr.201400485. [DOI] [PubMed] [Google Scholar]
- Whitsett M, Marzio DH, Rossi S. SlimQuick-Associated Hepatotoxicity Resulting in Fulminant Liver Failure and Orthotopic Liver Transplantation. ACG case reports journal. 2014;1:220–222. doi: 10.14309/crj.2014.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CS, Maliakal P, Meng X. Inhibition of carcinogenesis by tea. Annu Rev Pharmacol Toxicol. 2002;42:25–54. doi: 10.1146/annurev.pharmtox.42.082101.154309. [DOI] [PubMed] [Google Scholar]
- Yang CS, Wang H. Cancer Preventive Activities of Tea Catechins. Molecules. 2016;21:E1679. doi: 10.3390/molecules21121679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang CS, Zhang JS, Zhang L, Huang JB, Wang YJ. Mechanisms of body weight reduction and metabolic syndrome alleviation by tea. Mol Nutr Food Res. 2016;60:160–174. doi: 10.1002/mnfr.201500428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yellapu RK, Mittal V, Grewal P, Fiel M, Schiano T. Acute liver failure caused by ‘fat burners’ and dietary supplements: a case report and literature review. Canadian J Gastroenterol. 2011;25:157–160. doi: 10.1155/2011/174978. [DOI] [PMC free article] [PubMed] [Google Scholar]