

Abstract
Background
Significant advances in understanding brain development and behavior have not been accompanied by revisions of traditional academic structure. Disciplinary isolation and a lack of meaningful interdisciplinary opportunities are persistent barriers in academic medicine. To enhance clinical practice, research, and training for the next generation, academic centers will need to take bold steps that challenge traditional departmental boundaries. Such change is not only desirable but, in fact, necessary to bring about a truly innovative and more effective approach to treating disorders of the developing brain.
Methods
I focus on developmental disorders as a convergence point for transcending traditional academic boundaries. First, the current taxonomy of developmental disorders is described with emphasis on how current diagnostic systems inadvertently hinder research progress. Second, I describe the clinical features of autism, a phenomenologically defined condition, and Rett and fragile X syndromes, neurogenetic diseases that are risk factors for autism. Finally, I describe how the fields of psychiatry, psychology, neurology, and pediatrics now have an unprecedented opportunity to promote an interdisciplinary approach to training, research, and clinical practice and, thus, advance a deeper understanding of developmental disorders.
Results
Research focused on autism is increasingly demonstrating the heterogeneity of individuals diagnosed by DSM criteria. This heterogeneity hinders the ability of investigators to replicate research results as well as progress towards more effective, etiology-specific interventions. In contrast, fragile X and Rett syndromes are ‘real’ diseases for which advances in research are rapidly accelerating towards more disease-specific human treatment trials.
Conclusions
A major paradigm shift is required to improve our ability to diagnose and treat individuals with developmental disorders. This paradigm shift must take place at all levels – training, research and clinical activity. As clinicians and scientists who are currently constrained by disciplinary-specific history and training, we must move towards redefining ourselves as clinical neuroscientists with shared interests and expertise that permit a more cohesive and effective approach to improving the lives of patients.
Keywords: Autism, fragile X syndrome, Rett syndrome, interdisciplinary training, developmental disorder, brain development, genetic risk factor, neurogenetic disorder, academic medicine, clinical neuroscience, disciplinary boundaries
The year is 2009 and the parents of a two-year-old child call the central referral line of a major academic medical center. They ask to have their child evaluated for language delay and a variety of behavior problems possibly consistent with the diagnosis of autism. The professional staff in the departments of Psychiatry, Neurology, Pediatrics, and Clinical Psychology of this medical center all have some expertise in evaluating children with developmental delay and or disability, so to whom should the child be referred? The child would receive a thorough neurological work-up in Neurology and a general developmental and medical assessment in Pediatrics. Clinicians in Psychiatry may focus on some aspects of each of these domains but likely would also concentrate more on an evaluation of behavior and the family. Finally, Psychology personnel, depending on their orientation and training, might focus on various aspects of development, behavior or assessment of cognition. Depending on the individual training of practitioners in each of these disciplines, the child may or may not receive an assessment for common genetic causes of developmental disability and treatment options are likely to vary considerably. In the end, disciplinary overlaps and differences in expertise and services offered could easily lead to confusion for the parents and the referring health care practitioner, perhaps leading to suboptimal clinical evaluation.
Jump to the year 2025 – a call with the same parental concerns. In this case, a referral is made to the ‘Clinical Neuroscience Section’ of the University, a trans-disciplinary program that serves as the hub for evaluation and treatment of children with developmental disabilities. Though clinical practitioners comprising the Section represent broad and complementary areas of expertise in the clinical neurosciences, all have sufficient training and skill to initiate an evaluation that focuses on putative biological and environmental factors (and their interaction) that contribute to the child’s neurodevelopmental problems. Treatment options offered to the family are based on the results of the evaluation and are dependent on identification of identifiable risk factors contributing to suboptimal development and maladaptive behavior in the patient.
Is the future scenario described above plausible? At first blush, it seems unlikely. Though significant progress in basic and behavioral sciences has occurred over the past two decades, these advances have not been accompanied by concurrent revisions of the traditional academic structure or departmental boundaries within universities that sponsor programs in psychiatry, neurology, and psychology. Continuing disciplinary programmatic isolation and a lack of meaningful interdisciplinary structures are common barriers that persist in all major domains of academic medicine: research, training, and, in particular, clinical service.
Reasons for the existence and maintenance of hardened disciplinary boundaries are numerous. For example, academic programs within psychiatry, neurology, and psychology have very different traditions and histories (Reiss, 2005). Compounding this problem is the fact that disciplinary boundaries are often reinforced by self-contained explanatory systems that attempt to accommodate all observed and unobserved behavioral phenomena. Such self-contained systems not only discourage disciplinary integration, but also act as impediments to scientific progress as they leave little or no room for discovery or divergence. Examples include traditional psychodynamic theory in psychiatry, lesion-based theories of cognition and behavior in neurology, and Gestalt theory in psychology. Though not a theoretical framework per se, our current taxonomy of mental disorders, the Diagnostic and Statistical Manual (DSM) (APA, 2000), may inadvertently turn out to be the most damaging self-contained system of all with respect to impeding scientific progress. As discussed in greater detail below, the phenomenological, categorical approach incorporated by the DSM has led to the increasing realization that virtually all designated ‘disorders’ are greatly heterogeneous with respect to risk factors and likely pathophysiological mechanisms (e.g., Happe, Ronald, & Plomin, 2006; Pardo & Eberhart, 2007).
The potential advantages of incorporating a new interdisciplinary clinical neurosciences approach into our academic institutions of the future clearly outweigh the challenges. For example, graduate training associated within these programs of the future have the potential to produce new kinds of investigators and clinicians, fluent in key aspects of several disciplines, and able to effectively incorporate interdisciplinary knowledge into a cohesive approach to research and clinical care. New programs such as these also will be particularly well positioned to attract funding earmarked for innovative interdisciplinary research such as the NIH Roadmap initiative (nihroadmap.nih.gov).
To conceptualize and enhance clinical practice, research, and training in the clinical neurosciences for the next generation, academic centers will need to take bold steps that challenge traditional departmental boundaries and bring about a host of ‘hot button’ issues including those related to regulatory oversight of training and clinical practice, and competition for limited financial and institutional resources. However, I argue that such change is not only desirable but, in fact, necessary to bring about a truly innovative and more effective approach to studying, diagnosing, and treating disorders of the brain in the future.
In this paper, I will focus on developmental disorders as a particularly fruitful point of convergence where traditional academic boundaries can be transcended. In the first section of this paper, the current taxonomy of developmental disorders will briefly be described; emphasis is placed on how this taxonomy, while providing advantages over previous methods of classification, may also hinder progress in the creation of a new interdisciplinary infrastructure in the clinical neurosciences. Second, I will summarize what is presently known about the clinical features, risk factors, and effective treatments of three specific developmental disorders. The first disorder, autism, is phenomenologically defined by the presence or absence of behavioral and developmental features. The second and third disorders, Rett and fragile X syndromes, are actual neurogenetic diseases that are, themselves, risk factors for autism. Finally, I will describe how clinical psychology, neurology, pediatrics, and psychiatry now have an unprecedented opportunity to promote an interdisciplinary approach to training, research, and clinical practice and, thus, advance a deeper understanding of developmental disorders in general, as well as these three specific conditions in particular.
The taxonomy and definition of developmental disorders
General categories and features of developmental disorders
Developmental disabilities are defined by limitations in core functional domains (e.g., motor, communication, social, academic) resulting from aberrant development of the nervous system. These limitations can manifest during infancy or childhood as delays in reaching developmental milestones, and as qualitative abnormalities or lack of function in one or multiple domains. Deficits that define these disorders cross multiple disciplinary boundaries, making them of potential interest to psychiatrists, neurologists, and psychologists as well as practitioners from other health disciplines such as developmental-behavioral pediatrics, physical and occupational therapists, and speech and language pathologists. Clinical features of developmental disabilities are often variable in severity as well as in the specific areas of dysfunction. Children with developmental disabilities also are frequently affected in multiple domains of function because of the non-specific nature and extent of insults during brain maturation or increased susceptibility to other causes of disability (e.g., malnutrition, trauma, infection).
While abnormal development in one or more domains of function during childhood is the hallmark of developmental disorders, the causes of other brain disorders not currently designated in this manner may also have their origin in early aberrant neurodevelopment. For example, schizophrenia, which often manifests as core symptoms of delusions and hallucinations in late adolescence or early adulthood, is one such disorder thought to have earlier roots in childhood (Raedler, Knable, & Weinberger, 1998). Early manifestations can include subtle sensory-motor deficits, abnormalities of socialization, learning disorders and non-specific behavioral problems. Thus, as more sensitive biological markers and selective early diagnosis and treatments begin to emerge in the field of clinical neuroscience, disorders such as these will increasingly come to be thought of as neurodevelopmental in nature.
Differential diagnosis of developmental disorders
The overarching concept of ‘developmental disorders’ is highly heterogeneous with respect to etiology and pathogenic mechanisms leading to aberrant function; further, this concept is comprised of highly heterogeneous major sub-divisions that include intellectual disability (mental retardation), learning disorders, communication disorders, motor disorders, and pervasive developmental disorders. Guidelines and manuals for the diagnosis of these disorders, such as the DSM and the International Classification of Diseases (ICD), have improved the reliability of diagnosis and communication among clinical providers and researchers. Yet, with the exception of Rett syndrome, which I will discuss below, current taxonomies fall significantly short of defining disease entities with respect to biological construct validity. Without valid biological markers, clinicians and investigators from fields with dissimilar disciplinary backgrounds will tend to define the boundaries of typical and atypical development, behavior, and cognition differently. This includes, for example, varying perspectives on whether developmental disorders (such as reading disability) represent the farthest end of the ‘normal distribution’ of function or whether they represent a unique disease category (Grigorenko, 2001; Olson, 2002; Shaywitz, Gruen, & Shaywitz, 2007). Despite decades of research on phenomenologically defined developmental disorders, questions such as this have not been (and probably cannot) be fully answered.
Why are there unanswered fundamental questions that underlie how we define and view developmental disorders? Traditional research efforts aimed at understanding the pathogenesis of phenomenologically defined childhood-onset developmental disorders, such as reading disability or autism, may be impeded by the etiological heterogeneity of individuals meeting the widely accepted diagnostic criteria defining these disorders (Figure 1). Without reliable and valid biological markers for the presence of a pathological condition or state, investigators often are left to ponder a circular approach to research methodology. We may initially define or categorize a disorder according to a consensus of experts in the field agreeing to a diagnostic algorithm that includes the presence or absence of observed symptoms or signs. Such definitions may even have reasonable psychometric properties from the standpoints of diagnostic reliability and discriminative validity (from other phenomenologically defined disorders). However, the logic underlying this process is, inherently, at risk for circular arguments. The fact that we can reliably diagnose a disorder does not necessarily confer pathogenetic or etiologic homogeneity.
Figure 1.
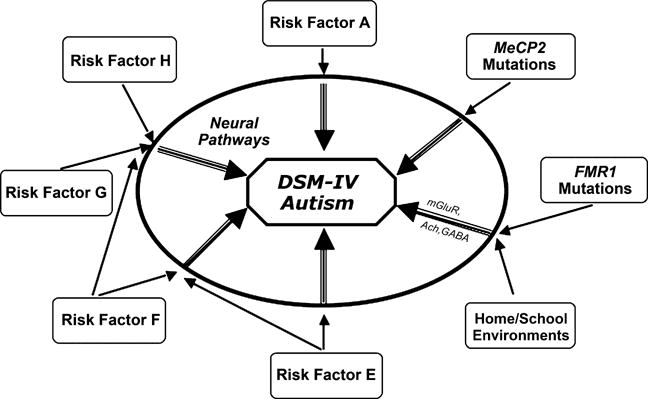
A conceptualization of the current state of phenomenologically defined (e.g., DSM or ICD) disorders. In the center is a DSM-IV defined diagnosis, here shown with autism as the example. Multiple biological and environmental factors (shown around the periphery) modify risk for the development of aberrant ‘neural pathways’ during brain development. Some of these risk factors are of moderate or greater influence (e.g., MeCP2 mutations that lead to Rett syndrome shown here), and thus are able to increase the likelihood of brain dysfunction with relatively less influence from other genetic or environmental factors. Other factors, such as FMR1 mutations associated with fragile X syndrome (also shown in the figure), may contribute moderately increased risk for autistic behavior. However, this risk can be moderated by measurable environmental factors related to the home and school (Hessl et al., 2001). ‘Neural pathways’ leading to manifestations of DSM defined disorders (shown as the intermediate step between risk factors and diagnosis) also would be expected to vary for a given DSM diagnosis, even though they might result in a (somewhat) similar phenotype. Examples of neural pathways influenced by FMR1 mutations are shown in the figure (mGluR-metabotropic glutamate receptor, Ach-Acetylcholine system, GABA-gamma-aminobutyic acid). Given the lack of scientific precision of such phenomenologically based diagnostic taxonomies, alternatives to the DSM should be strongly considered for future research studies focused on elucidating the pathogenesis of (currently) idiopathic developmental disorders
When biological markers for disease are discovered, this has often dramatically changed the way we view disorders previously defined by phenomenological criteria. For example Neurology’s previously held conceptualization of two distinct disorders within the category of muscular dystrophy (Duchenne and Becker) has been altered by recognition that different mutations within the same gene (dystrophin) result in differing phenotypes (Muntoni, Torelli, & Ferlini, 2003). Similarly, as I will describe below, mutations in the gene associated with Rett syndrome (MeCP2), a DSM-IV defined pervasive developmental disorder, can increase risk for neuropsychiatric or neurodevelopmental phenotypes other than those associated with this diagnostic category (Chahrour & Zoghbi, 2007). Conversely, intensive research over the past two decades has uncovered over 25 known diseases comprising the clinically defined disorder, spinocerebellar ataxia (Costa Lima & Pimentel, 2004).
Accordingly, the process of revamping or parsing behaviorally or phenomenologically defined disorders into more etiologically or pathophysiologically meaningful subgroups (or dimensions) is essential before we can begin to understand fundamental neurodevelopmental processes leading to brain dysfunction, and how these processes are influenced by genetic and environmental factors. To this end, it will be important to adopt a true interdisciplinary approach, focused on explicating multiple levels of inquiry to more fully encompass quantitative assessments of neurocognitive, affective, neurological, and neurobehavioral functioning, genetic influences, brain structure and function, as well as environmental factors.
DSM disorder versus disease
Autism
The category of pervasive developmental disorders, which includes the diagnosis of autism, has attracted increasing interest from both researchers and the lay public. Children with autism can present with a broad range of clinical features including qualitative abnormalities of development, neurological problems such as sensory/motor symptoms and epilepsy, cognitive dysfunction, serious impairments in adaptive behavior, and aberrant regulation of emotion.
From the lay and media perspective, autism can appear as a particularly enigmatic disorder of childhood development. Severe deficits in social and communicative abilities in individuals with this disorder conflict with fundamental tenets of human behavior – in particular, that children are naturally endowed with the motivation and ability to be reasonably effective in their social world. Though originally viewed as a disorder arising from parenting defects, it is now clear that autism is caused by aberrant brain development and function and has strong genetic influences.
Increased interest in autism also has been generated because of recent reports of increased prevalence of the disorder. Although various theories have been espoused to account for this increased frequency of autism, it is widely accepted that the majority of this increase is due to changes in diagnostic criteria, detection and reporting strategies, and a lowered threshold for making the diagnosis (Wing & Potter, 2002). Although inclusion and exclusion criteria are well defined in the DSM-IV (and ICD-10), diagnostic algorithms allow for a fairly broad spectrum and topography of possible deficits within the population of individuals meeting diagnostic criteria. Thus, the phenomenological nature of our current taxonomy potentially contributes to a ‘moving’ diagnostic target for autism and related pervasive developmental disorders. As such, the nature of this taxonomy also likely plays a role in reinforcing disciplinary specific approaches to diagnosis and treatment of affected children. These disciplinary specific approaches can be observed, for example, by the lack of consensus amongst experts in the field as to what constitutes appropriate evaluation and treatment of the autistic child (Filipek et al., 2000; Matson, 2007; Shattuck & Grosse, 2007; Shea, 2004).
Core symptoms associated with autism include disturbances in reciprocal social interaction with caregivers and/or peers, motor abnormalities (e.g., stereotyped movements, hand-flapping, toe walking), qualitative abnormalities of language and communication (echolalia, perseveration, dysprosody), atypical processing of parts and wholes, ritualistic and compulsive behavior, and often, aberrant emotional expression such as anxiety and depression. Although not a core symptom as such, individuals with autism also manifest increased risk for late-onset seizure disorder. Given that these symptoms clearly cross multiple clinical disciplines, many have stressed the need for comprehensive multi-disciplinary evaluations including psychiatric, neurologic, pediatric, genetic, special educational and psychological examinations of the autistic child.
Though it is unlikely that a single cause or pathophysiological mechanism will be described that applies to most individuals currently diagnosed with autism, recent discoveries in genetic research, brain imaging, and early identification and treatment of autistic children have produced new findings (and questions) of mutual interest to clinicians and researchers from multiple disciplinary backgrounds. For example, an aberrant trajectory of early brain overgrowth has been detected in some young autistic children (Stanfield et al., 2007), though how this anatomical finding relates to symptoms and signs of the disorder is not yet well understood. Similarly, recent studies have begun to identify and describe genetic risk factors (e.g., associated with neurogenetic diseases) that increase the likelihood for autistic behavior (Gupta & State, 2007). Two of these risk factors – associated with fragile X and Rett syndromes – are described below. Finally, intervention studies with young, more mildly affected children with autism show greater potential for improvement in some individuals than was thought possible 20 years ago (Landa, 2007).
Recent findings in autism research are of obvious relevance to psychiatrists, psychologists, neurologists, and developmental-behavioral pediatricians alike, as they have revealed new avenues for research and discovery that have important diagnostic and treatment implications. However, disciplinary boundaries continue to shape the focus and scope of much of the autism research that takes place today, despite shared interests and expertise across academic domains. As representative of these boundaries, autism programs and research staff are housed within multiple separate institutes of the NIH, including extramural programs within the NIMH, NICHD, NINDS, and NIDCD. If we are to someday discover the multifactorial risk factors, neural substrates, early-warning signs, and disease-specific treatments for children who currently receive the diagnosis of autism, the research of the future will require a concerted interdisciplinary approach that melds and implements expertise from the medical, basic, and behavioral sciences. As I will emphasize throughout this paper, this new research also can serve as the core knowledge base for designing innovative training programs in the clinical setting.
Rett syndrome
Rett syndrome (RS), occurring in 1 in 10,000 live female births, is an important etiology for intellectual disability and developmental regression in young girls. The disorder manifests in symptoms that chiefly involve the expertise of physicians, particularly neurologists, developmental-behavioral pediatricians, and psychiatrists. Psychologists with clinical, particularly behavioral, training also play a substantive role in helping the child and her family once the disorder has manifest. Because RS also has a genetic basis, medical geneticists also are increasingly involved in diagnosis and family genetic counseling.
The earliest symptoms of RS are likely to first come to the attention of a pediatrician who may then refer the child to a neurologist or other developmental specialist for further work-up and diagnosis. The chief signs and symptoms include slow development from birth, although some patients show normal or near normal attainment of early developmental milestones in the early weeks or months of life. Suboptimal head growth may be noted in infancy, while stagnation or regression in hand use and communication classically occurs at around 1–2 years, with hand stereotypy, other involuntary movements, and irregular breathing. Muscle tone is reduced initially, with increasing impairment as time goes on. Accompanying developmental plateau and regression, the child with RS may lose previously developed interest and skills in social and communicative behaviors. It is at this time that the diagnosis of autism is often considered and psychologists or psychiatrists are consulted. Following the period of developmental regression described above, the course of RS is more stable, with gradual establishment of a significantly slowed trajectory of developmental and cognitive growth. Generalized motor or partial seizures are present in about 50% of affected individuals.
The medical genetic diagnosis of RS, previously based on DSM phenomenology alone, is now established by the clinical presentation and confirmation, in most cases, by identification of a mutation in the methyl-CpG binding protein 2 (MeCP2) gene located on the X chromosome (Chahrour & Zoghbi, 2007). The MeCP2 gene, present in about 95% of females diagnosed with ‘classical’ RS, codes for a protein that is essential for neurons to mature normally. Specifically, the MeCP2 protein binds to DNA and interacts with other proteins to silence other genes through transcriptional suppression. Analysis of genotype–phenotype associations reveals that the spectrum of manifestations from MeCP2 mutations in humans is broader than initially suspected and can, for example, occur in males without the classical symptoms observed in affected females; mutations have been discovered in RS variants, individuals with nonspecific intellectual disability, autistic children and even normal females (Chahrour & Zoghbi, 2007; Naidu et al., 2003; Neul & Zoghbi, 2004). A variety of factors may account for the wide range of severity and manifestations of MeCP2 mutation associated phenotypes, including the type and location of the MeCP2 mutation, mosaicism, and X chromosome inactivation (in females).
Though research has not yet determined all of the genes regulated by MeCP2, such genes can be presumed to be important for normal development and function of the central nervous system, in particular, the development and modification of synapses (Chao, Zoghbi, & Rosenmund, 2007). With the discovery of a sensitive and specific genetic marker, RS transcended the DSM taxonomy designed for disorders of unknown etiology (such as autism) and moved into the realm of ‘neurogenetic disease’. In fact, given that Rett syndrome is the only DSM-IV diagnosis with a valid genetic marker, it will be intriguing to observe how the next iteration of the DSM handles this situation.
New discoveries about the range of symptom manifestations associated with MeCP2 mutations have led to further revision in thought and practice as to what actually constitutes neurodevelopmental disease in children. Though clearly distinct from the construct of autism by virtue of its association with an identifiable genetic marker and molecular pathophysiology, RS also represents fertile ground for the intersection of multiple clinical disciplines comprising the domain of medical neuroscience as well as with psychologists, geneticists, and neurobiologists. Enhancement of this type of interaction is particularly exciting when viewed in the context that animal models for Rett syndrome now exist and that treatments under development are increasingly thought to be viable in preventing or reducing disability from this currently devastating condition (Chahrour & Zoghbi, 2007).
Fragile X syndrome
Fragile X syndrome (FXS), an X-linked semi-dominant disorder, is the most common heritable cause of neurodevelopmental dysfunction, with prevalence of up to 1 in 4,000 males and 1 in 8,000 females (Crawford, Acuna, & Sherman, 2001). FXS is most often caused by an abnormal expansion of CGG trinucleotide repeats within the promoter region of the FMR1 gene located on the long arm of the X chromosome. Repeat lengths up to approximately 40–45 triplet repeats are considered normal with a modal number of 29–30 found in humans. Trinucleotide expansions beyond 50 repeats are associated with increased risk for developmental, cognitive, behavioral, and/or neurological symptoms.
The profile of clinical symptoms associated with abnormal repeat expansion of FMR1 varies according to the size of the expanded repeat (Visootsak, Warren, Anido, & Graham, 2005). From 50 to approximately 200 repeats is labeled as the ‘premutation’ while over 200 repeats is referred to as the ‘full mutation’. Unlike unexpanded or most premutation alleles, the full mutation is also defined by the presence of hypermethylation of FMR1, which results in reduced transcription of mRNA and, accordingly, FMR1 protein (FMRP) (Figure 2). Reduction in FMRP from the full mutation causes abnormal brain development and function, in turn leading to a cascade of cognitive, behavioral, emotional, and neurological problems associated with the diagnosis of FXS (Reiss & Dant, 2003). Because random X inactivation occurs in females, at least 50% of females (and nearly all males) who have the full mutation exhibit readily identifiable cognitive symptoms. Some males and females with FXS have a mixture of cells with different ranges of repeats (i.e., repeat mosaicism) and therefore, a large range of phenotypic features may be observed in affected individuals. Before puberty, boys with fragile X can have somewhat large heads but few other distinguishing features; after puberty, the features may be more distinctive – long face with prominent jaw and forehead, large ears, and macroorchidism. However, the presence or absence of physical features in both males and females is not sufficient to make or rule out the diagnosis.
Figure 2.
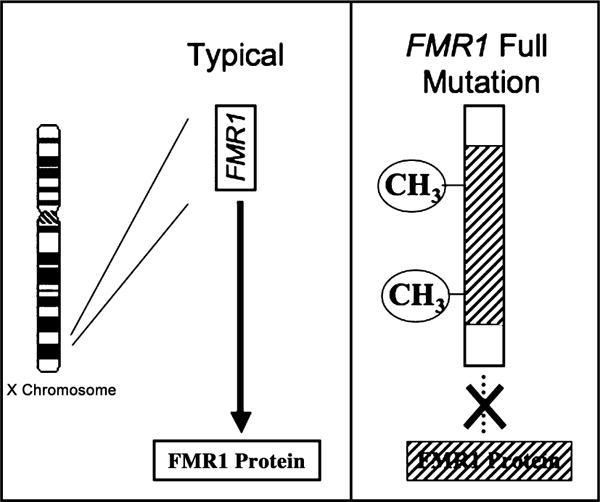
The genetic basis for the fragile X full mutation. FMR1 gene from X chromosome shown on left under typical conditions. Modal trinucleotide (CGG) repeat length is 29–30. Significantly expanded CGG repeats in the FMR1 full mutation (right) lead to hypermethylation (CH3), transcriptional repression and reduced levels of FMRP. Reduced levels of FMRP lead to neurobiological dysfunction and the cognitive-behavioral phenotype (also see Figure 3)
Both female and male children and adolescents with the full mutation are at significantly increased risk for developing a characteristic profile of behavioral, cognitive, emotional, and neurological problems beginning in infancy that is qualitatively similar between the sexes, but quantitatively different (Hooper et al., 2008; Reiss & Dant, 2003; Reiss & Hall, 2007). Within the first year of life most boys with fragile X and some affected girls show delays in developmental milestones, particularly expressive language. However, most children with fragile X are not diagnosed until their second or third year of life, usually as a result of persistent speech delays or behavioral abnormalities. As boys with FXS reach preschool age, there is variability in cognitive and adaptive behavioral development, but overall, their rate of development ranges from one-third to one-half that expected for typically developing boys (Bailey, Hatton, & Skinner, 1998; Hatton et al., 2003). Expressive language is typically more adversely affected than receptive language, while scores for motor and adaptive function are relatively higher compared with communication and cognitive functioning.
Beginning in the preschool years, and extending into the school and adolescent years, boys with fragile X show pervasive deficits in conversational language skills with increasing discrepancy between language level and chronological age. Patterns of behavioral, social, and developmental abnormalities that emerge in many young boys with the FMR1 full mutation suggest that the fragile X full mutation increases the risk for autistic behavior (Hall, Lightbody, & Reiss, 2008; Hatton et al., 2006). These behaviors include significant social deficits and avoidance, particularly with peers, qualitative abnormalities of communication such as perseverative speech and gaze aversion, and stereotypic motor behavior.
Girls with FXS are more variable in their development – while those with the full mutation may show mildly to moderately severe quantitative and qualitative abnormalities (Reiss & Dant, 2003). In preschool and school-aged girls, the presence of social anxiety, shyness, attentional problems, and avoidant behavior may predispose girls with the full mutation to the emergence of depression in adolescence and young adulthood. Executive functioning, particularly involving working memory, inhibition, and planning, also fail to develop at expected rates (Lightbody, Hall, & Reiss, 2006).
FMRP functions as an mRNA binding protein, transporting messenger ribonucleoprotein complexes between nucleus and cytoplasm of the neuron. These FMRP-associated mRNAs, which have been identified as important to neuronal plasticity and development, synaptic maturation, and axon pathfinding, translate proteins in dendrites during critical developmental periods of activity-dependent synaptic function, maturation, and plasticity (Garber, Visootsak, & Warren, 2008). When FMRP levels are reduced or absent, as occurs in FXS or the mouse and Drosophila knockout model of this condition, abnormal morphologies of neuronal dendritic processes are observed. The resultant disorganization in neuronal circuitry of subjects with FXS is thought to contribute to the observable profile of cognitive, emotional, and behavioral abnormalities in this disorder.
Neuroimaging studies have begun to identify anatomical and functional differences of the brain in individuals with FXS (Gothelf et al., 2008; Hessl, Rivera, & Reiss, 2004). Though a number of anatomical abnormalities have been observed, the most robust morphological finding is the presence of significant volume increase of the caudate nucleus throughout the age span in the context of normal or slightly enlarged overall brain size. Enlargement of the caudate is correlated with measures of cognition and behavior in FXS (Gothelf et al., 2008; Reiss, Abrams, Greenlaw, Freund, & Denckla, 1995), and is thought to be associated with abnormalities of prefrontalcaudate neural activity observed with functional MRI (Hoeft et al., 2007; Menon, Leroux, White, & Reiss, 2004), and abnormal prefrontal-caudate white matter development seen with diffusion tensor imaging (Barnea-Goraly et al., 2003). Interestingly, enlarged overall brain size and caudate volume are neuroanatomical findings reported in some individuals with autism as well (Stanfield et al., 2007).
Though this paper has primarily focused on the traditional categorical taxonomy associated with FMR1 mutations (i.e., full mutation, premutation), recent research suggests that ‘blurring’ of these boundaries and other FMR1 associated conditions occur in humans. Though adequate research into possible phenotypic manifestations associated with the premutation in children or young adults has not been performed, preliminary evidence suggests that individuals with higher end premutation repeat sizes (e.g., 100–200 repeats) may be vulnerable to mild cognitive and behavioral symptoms (Johnston et al., 2001). It has been established, however, that some older men with the premutation are at increased risk for the development of a tremor-ataxia syndrome (Berry-Kravis et al., 2007) and that women with the premutation of child-bearing age are at increased risk for premature ovarian insufficiency/failure (Martin & Arici, 2008). Given the wide range of phenotypic manifestations associated with FMR1 mutations and the fact that the cognitive status of individuals with these mutations can range from completely normal to the very impaired, this author recently proposed that the definition of the ‘FMR1’ gene label change from the originally designated ‘fragile X mental retardation 1’ (Verkerk et al., 1991) to ‘fragile (X) marker related 1’ or ‘fragile (X) mutation related 1’.
Like Rett syndrome, the earliest symptoms of FXS (i.e., full mutation), typically language delay, are usually first brought to the attention of a pediatrician. Whether children with FXS are referred to specialists in neurology, psychiatry, psychology, and medical genetics, and the timing of such a referral, is likely to depend on recognition, quality and severity of developmental and cognitive delay, intellectual disability or learning dysfunction, behavioral and emotional abnormalities, and presence of physical and neurological symptoms of the disorder. Current treatments for affected individuals are largely symptom based (such as SSRIs for anxiety, behavior therapy for aggression) although the confluence of new knowledge from recent neuroscience, genetic, imaging, and neuropsychological investigations have led to preliminary trials of new pharmacological and behavioral treatments targeted to deleterious downstream effects associated from reduced FMRP (Berry-Kravis et al., 2006; Reiss & Hall, 2007; Kesler et al., in press) Fig 3.
Figure 3.
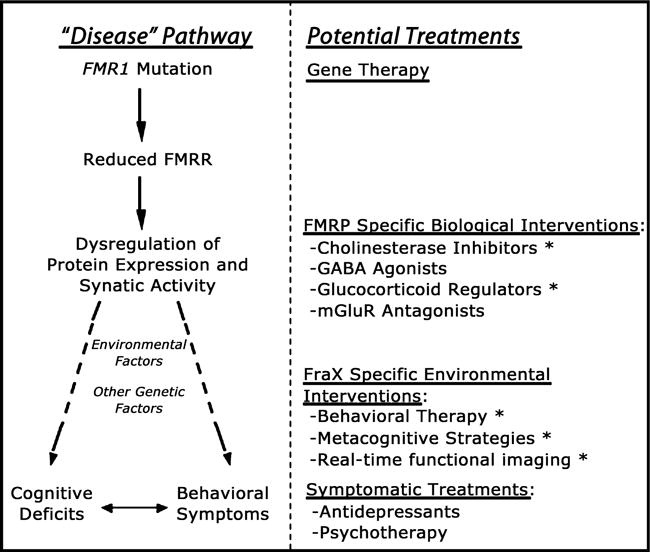
A hierarchical model for planning treatment strategies in fragile X syndrome. The left-hand side of the diagram shows the genetic and neurobiological mechanisms leading to brain dysfunction in this condition. Specifically, the FMR1 mutation causes reduction in FMRP, leading to transcriptional dysregulation of FMRP mRNA targets (involved in synapse maturation and function). Other (non-FMR1 related) genetic and environmental factors interact with this genetic-neurobiological pathway, ultimately culminating in the cognitive-behavioral phenotype associated with fragile X. The right-hand side of the diagram shows potential treatment approaches matched to the corresponding genetic and pathophysiological level on the left. Given that gene therapy approaches are not feasible at the present time, the most promising current approaches are at the level of pathways ‘downstream’ to reduced levels of FMRP (shown under the category of ‘FMRP-specific Biological Interventions). Abbreviations: FraX: fragile X syndrome; FMRP: fragile X mental retardation protein; mGlurR: metabotropic glutamate receptor; GABA: gammaaminobutyric acid. Asterisks (*) next to treatment approaches indicate ongoing trials in the Center for Interdisciplinary Brain Sciences Research at Stanford University
Though a vastly improved understanding of the molecular and neurobiological basis of FXS has increased the possibility of developing a disease-specific pharmacological treatment to reduce brain dysfunction in the near future, it also is clear that cognitive and behavioral interventions will be critical factors in optimizing outcome in affected individuals (Reiss & Hall, 2007). Thus, from the standpoint of enhancing future progress in understanding and treating FXS, dismantling disciplinary specific boundaries between neurology, psychiatry, and psychology will serve this process well.
Developmental disorders as a convergence point for increasing interdisciplinary collaboration
Each of the three disorders described in the previous section is comprised of symptom elements or clusters that might be considered fundamental to different disciplines within the medical and behavioral sciences. Fragile X syndrome and Rett syndrome are associated with specific genetic risk factors and biological markers; yet different components of symptom expression associated with these disorders clearly fall within the historical disciplinary boundaries of psychiatry, neurology, and psychology. Developmental-behavioral pediatrics, a clinical area of pediatrics that was recently granted board-certified subspecialty status, also has a large stake in these and related disorders. It is recognized that considerable individual variability exists in the severity of developmental manifestations and symptoms in these conditions, and therefore, it is important to elucidate the full range of this variability and to identify factors, other than reduced FMRP or MeCP2 mutation type, that contribute to phenotypic variation.
Though research indicates that autism has strong genetic influences, no single risk factor has yet been identified to explain the preponderance of affected individuals. Though it is possible that such etiologies will be discovered in the future, it is more likely that phenomenologically defined conditions such as autism will, ultimately, be found to be comprised of many subgroups, as well as heterogeneous and multifactorial with respect to genetic and environmental influences and symptom pathophysiology (Happé et al., 2006).
One of the major lessons learned from investigation of disorders with identifiable genetic risk such as FXS and RS is that neither the quality nor the breadth of phenotypic expression of these disorders neatly corresponds to discrete DSM-defined diagnoses. In FXS, affected individuals may demonstrate symptoms of autism, ADHD, social phobia, generalized anxiety disorder, depression, mental retardation, or learning disorders (Reiss & Dant, 2003). As noted above, MeCP2 mutations also can result in a broad range of developmental problems with ‘classical’ RS seeming to represent the higher end of the spectrum of severity. Thus, new interdisciplinary approaches for investigation of behavioral, cognitive, and emotional dysfunction of developmental origin might consider alternatives to the DSM or ICD systems for studying and grouping affected individuals. Such approaches include behavioral neurogenetics methods (Reiss & Dant, 2003), research focusing on dimensional characteristics of behavior or development, and the use of ‘endophenotypic’ markers such as findings obtained from brain imaging studies.
Another important lesson from recent research in genetics, neuroscience, and the associated clinical disciplines is that any further discussion invoking the mutual exclusivity of ‘nature’ versus ‘nurture’ is moot. Genetic factors influence the creation of an individual’s environment while environmental factors can clearly affect the expression of genes, on both a long-term and short-term basis. Similarly, discussion of ‘functional’ versus ‘organic’ with respect to the etiology of brain disorders or dysfunction is plainly outdated. While Franz Joseph Gall’s 19th-century tenets of phrenology may have been to some extent excessive or misguided, his first principle, ‘the brain is the organ of the mind,’ has never been more compelling than today. This principle may, in fact, be the most important rationale for new efforts to join and merge the multiple medical and behavioral disciplines together in a new constellation of training, research, and clinical applications. For example, it is self-evident that any extrinsic environmental or biological intervention designed to affect behavior (broadly defined) must be considered a form of ‘brain therapy.’ This would include many varieties of psychotherapeutic interventions, speech and language therapy, pharmacotherapy, occupational and physical therapy and even patient education and guidance. Our challenge as a field is to understand which of these interventions are most effective, to identify the particular components of the intervention that account for the most beneficial effects, and to learn how to optimize the benefit with respect to prevention, early identification, and rapid response.
Within the clinical domain, historically disparate disciplines must increasingly recognize the need for each other’s skills and knowledge. For example, in the evaluation of autism, a psychiatrist may be called upon to assess the patient for the presence of OCD-like symptoms or ADHD. The same patient may be referred to a neurologist for consultation regarding the evaluation of seizures as well as to exclude other causes of symptoms such as Landau-Kleffner syndrome (Stefanatos, Kinsbourne, & Wasserstein, 2002). A psychiatrist, developmental-behavioral pediatrician, or neurologist might prescribe medication if symptoms are severe while a psychologist may support treatment with family or behavioral therapy and a specialized educational plan, if needed. Yet, even as the interdisciplinary nature of developmental disorders is increasingly recognized, the existence of clinics where both evaluation and treatment from multiple clinical disciplines is a standard feature generally remains the exception to the rule. This shortfall in clinical practice, while arising from disciplinary specific boundaries, has also been propagated and maintained as a result of increasing economic challenges to clinical reimbursement, particularly for disorders classified within the domain of ‘mental health’.
Synthesis and summary
For much of the past century, the medical disciplines and behavioral sciences, particularly neurology, psychiatry, pediatrics, and psychology, have been largely independent in their approach to developmental disorders. These disciplinary boundaries were created and maintained by divergence of histories, philosophical approaches, as well as research and treatment methods. Scientific advances in recent decades have made it clearer, however, that these disciplinary boundaries are mostly artificial and arbitrary. Thus, for brain disorders in general and developmental disorders in particular, it is essential that both scientists and clinicians begin to dismantle disciplinary specific barriers in order for meaningful progress in diagnosis and treatment to take place.
Some training programs have begun preparing clinicians and researchers to understand and appreciate multidisciplinary translational approaches from basic genetic, neuroscience, behavioral, and cognitive areas to applied clinical research. However, training programs of the future will need to adopt a more integrated multi-level scientific approach to bridge traditional, between-discipline gaps in methodology and to take advantage of continuing advances. In support of this approach will be the NIH Roadmap, which prioritizes interdisciplinary research in an effort to remove roadblocks to collaboration.
What does the future hold for collaboration between and integration among the currently disparate disciplines? In the late 1800s, Sigmund Freud, unable to build a coherent physiologic model of the human brain, nevertheless believed that future scientific advancements would some day allow psychiatrists to construct such a model. Today, over 100 years later, revolutionary advances in neuroimaging, human genetics, and molecular biology have allowed scientists across many disciplines to begin to construct a dynamic model of the human brain that considers the complex and changing interactions between neurophysiological, neurodevelopmental, environmental, and genetic influences. Scientists and clinicians of the future will need to consider these interactions to more fully understand childhood neurodevelopmental disorders. This will require bold changes in training, research, and cooperation between the disciplines, changes that will challenge the regulatory, hierarchical, and economic status quo of the present.
Key points.
Phenomenologically (symptomatically) defined developmental disorders such as autism are common, yet often poorly understood conditions for which we lack precision in diagnosis and effective treatments.
Rett and fragile X syndromes are specific neurogenetic diseases that are risk factors for the development of autism and other developmental anomalies.
Despite shared interest in developmental disorders across the fields of psychiatry, psychology, neurology and pediatrics, current disciplinary boundaries and diagnostic taxonomies (such as the DSM) are impediments to meaningful progress in research and treatment.
A major paradigm shift is required to improve our ability to more specifically diagnose and treat individuals with developmental brain disorders.
Acknowledgments
I wish to sincerely thank Drs. Amy Lightbody and Scott Hall for contributing to the work described in this manuscript. I also would like to thank the many patients, research subjects and families with whom I have had the privilege to interact over many years of clinical practice and research.
Abbreviations
- DSM
Diagnostic and Statistical Manual
- FMR1
fragile X mental retardation-1 gene
- FMRP
FMR1 protein
- FXS
fragile X syndrome
- MeCP2
methyl-CpG binding protein 2
- RS
Rett syndrome
Footnotes
Conflict of interest statement: No conflicts declared.
References
- APA. Diagnostic and statistical manual of mental disorders, fourth edition – text revision (DSMIV-TR) Arlington, VA: American Psychiatric Associaiton; 2000. [Google Scholar]
- Bailey DB, Jr, Hatton DD, Skinner M. Early developmental trajectories of males with fragile X syndrome. American Journal of Mental Retardation. 1998;103:29–39. doi: 10.1352/0895-8017(1998)103<0029:EDTOMW>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Barnea-Goraly N, Eliez S, Hedeus M, Menon V, White CD, Moseley M, et al. White matter tract alterations in fragile X syndrome: Preliminary evidence from diffusion tensor imaging. American Journal of Medical Genetics B: Neuropsychiatric Genetetics. 2003;118B:81–88. doi: 10.1002/ajmg.b.10035. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Abrams L, Coffey SM, Hall DA, Greco C, Gane LW, et al. Fragile X-associated tremor/ataxia syndrome: Clinical features, genetics, and testing guidelines. Movement Disorders. 2007;22:2018–2030. doi: 10.1002/mds.21493. quiz 2140. [DOI] [PubMed] [Google Scholar]
- Berry-Kravis E, Krause SE, Block SS, Guter S, Wuu J, Leurgans S, et al. Effect of CX516, an AMPA-modulating compound, on cognition and behavior in fragile X syndrome: A controlled trial. Journal of Child and Adolescent Psychopharmacology. 2006;16:525–540. doi: 10.1089/cap.2006.16.525. [DOI] [PubMed] [Google Scholar]
- Chahrour M, Zoghbi HY. The story of Rett syndrome: From clinic to neurobiology. Neuron. 2007;56:422–437. doi: 10.1016/j.neuron.2007.10.001. [DOI] [PubMed] [Google Scholar]
- Chao HT, Zoghbi HY, Rosenmund C. MeCP2 controls excitatory synaptic strength by regulating glutamatergic synapse number. Neuron. 2007;56:58–65. doi: 10.1016/j.neuron.2007.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa Lima MA, Pimentel MM. Dynamic mutation and human disorders: The spinocerebellar ataxias (review) International Journal of Molecular Medicine. 2004;13:299–302. [PubMed] [Google Scholar]
- Crawford DC, Acuna JM, Sherman SL. FMR1 and the fragile X syndrome: Human genome epidemiology review. Genetics in Medicine. 2001;3:359–371. doi: 10.1097/00125817-200109000-00006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filipek PA, Accardo PJ, Ashwal S, Baranek GT, Cook EH, Jr, Dawson G, et al. Practice parameter: Screening and diagnosis of autism: Report of the Quality Standards Subcommittee of the American Academy of Neurology and the Child Neurology Society. Neurology. 2000;55:468–479. doi: 10.1212/wnl.55.4.468. [DOI] [PubMed] [Google Scholar]
- Garber KB, Visootsak J, Warren ST. Fragile X syndrome. European Journal of Human Genetics. 2008;16:666–672. doi: 10.1038/ejhg.2008.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gothelf D, Furfaro JA, Hoeft F, Eckert MA, Hall SS, O’Hara R, et al. Neuroanatomy of fragile X syndrome is associated with aberrant behavior and the fragile X mental retardation protein (FMRP) Annals of Neurology. 2008;63:40–51. doi: 10.1002/ana.21243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigorenko EL. Developmental dyslexia: An update on genes, brains, and environments. Journal of Child Psychology and Psychiatry. 2001;42:91–125. [PubMed] [Google Scholar]
- Gupta AR, State MW. Recent advances in the genetics of autism. Biological Psychiatry. 2007;61:429–437. doi: 10.1016/j.biopsych.2006.06.020. [DOI] [PubMed] [Google Scholar]
- Hall SS, Lightbody AA, Reiss AL. Compulsive, self-injurious, and autistic behavior in children and adolescents with fragile X syndrome. American Journal of Mental Retardation. 2008;113:44–53. doi: 10.1352/0895-8017(2008)113[44:CSAABI]2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Happé F, Ronald A, Plomin R. Time to give up on a single explanation for autism. Nature Neuroscience. 2006;9:1218–1220. doi: 10.1038/nn1770. [DOI] [PubMed] [Google Scholar]
- Hatton DD, Sideris J, Skinner M, Mankowski J, Bailey DB, Jr, Roberts J, et al. Autistic behavior in children with fragile X syndrome: Prevalence, stability, and the impact of FMRP. American Journal of Medical Genetics A. 2006;140A:1804–1813. doi: 10.1002/ajmg.a.31286. [DOI] [PubMed] [Google Scholar]
- Hatton DD, Wheeler AC, Skinner ML, Bailey DB, Sullivan KM, Roberts JE, et al. Adaptive behavior in children with fragile X syndrome. American Journal of Mental Retardation. 2003;108:373–390. doi: 10.1352/0895-8017(2003)108<373:ABICWF>2.0.CO;2. [DOI] [PubMed] [Google Scholar]
- Hessl D, Dyer-Friedman J, Glaser B, Wisbeck J, Barajas RG, Taylor A, et al. The influence of environmental and genetic factors on behavior problems and autistic symptoms in boys and girls with fragile X syndrome. Pediatrics. 2001;108:E88. doi: 10.1542/peds.108.5.e88. [DOI] [PubMed] [Google Scholar]
- Hessl D, Rivera SM, Reiss AL. The neuroanatomy and neuroendocrinology of fragile X syndrome. Mental Retardation and Developmental Disabilities Research Reviews. 2004;10:17–24. doi: 10.1002/mrdd.20004. [DOI] [PubMed] [Google Scholar]
- Hoeft F, Hernandez A, Parthasarathy S, Watson CL, Hall SS, Reiss AL. Fronto-striatal dysfunction and potential compensatory mechanisms in male adolescents with fragile X syndrome. Human Brain Mapping. 2007;28:543–554. doi: 10.1002/hbm.20406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hooper SR, Hatton D, Sideris J, Sullivan K, Hammer J, Schaaf J, et al. Executive functions in young males with fragile X syndrome in comparison to mental age-matched controls: Baseline findings from a longitudinal study. Neuropsychology. 2008;22:36–47. doi: 10.1037/0894-4105.22.1.36. [DOI] [PubMed] [Google Scholar]
- Johnston C, Eliez S, Dyer-Friedman J, Hessl D, Glaser B, Blasey C, et al. Neurobehavioral phenotype in carriers of the fragile X premutation. American Journal of Medical Genetics. 2001;103:314–319. [PubMed] [Google Scholar]
- Kesler SR, Lightbody AA, Reiss AL. Cholinergic dysfunction in males with fragile X syndrome and potential pharmacological intervention: A preliminary 1H MRS study. American Journal of Medical Genetics A. doi: 10.1002/ajmg.a.32697. (in press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landa R. Early communication development and intervention for children with autism. Mental Retardation and Developmental Disabilities Research Reviews. 2007;13:16–25. doi: 10.1002/mrdd.20134. [DOI] [PubMed] [Google Scholar]
- Lightbody AA, Hall SS, Reiss AL. Chronological age, but not FMRP levels, predicts neuropsychological performance in girls with fragile X syndrome. American Journal of Medical Genetics B: Neuropsychiatric Genetics. 2006;141B:468–472. doi: 10.1002/ajmg.b.30307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JR, Arici A. Fragile X and reproduction. Current Opinion in Obstetrics and Gynecology. 2008;20:216–220. doi: 10.1097/GCO.0b013e3282fe7254. [DOI] [PubMed] [Google Scholar]
- Matson JL. Determining treatment outcome in early intervention programs for autism spectrum disorders: A critical analysis of measurement issues in learning based interventions. Research in Developmental Disabilities. 2007;28:207–218. doi: 10.1016/j.ridd.2005.07.006. [DOI] [PubMed] [Google Scholar]
- Menon V, Leroux J, White CD, Reiss AL. Frontostriatal deficits in fragile X syndrome: Relation to FMR1 gene expression. Proceedings of the National Academy of Sciences U S A. 2004;101:3615–3620. doi: 10.1073/pnas.0304544101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muntoni F, Torelli S, Ferlini A. Dystrophin and mutations: One gene, several proteins, multiple phenotypes. Lancet Neurology. 2003;2:731–740. doi: 10.1016/s1474-4422(03)00585-4. [DOI] [PubMed] [Google Scholar]
- Naidu S, Bibat G, Kratz L, Kelley RI, Pevsner J, Hoffman E, et al. Clinical variability in Rett syndrome. Journal of Child Neurology. 2003;18:662–668. doi: 10.1177/08830738030180100801. [DOI] [PubMed] [Google Scholar]
- Neul JL, Zoghbi HY. Rett syndrome: A prototypical neurodevelopmental disorder. Neuroscientist. 2004;10:118–128. doi: 10.1177/1073858403260995. [DOI] [PubMed] [Google Scholar]
- Olson RK. Dyslexia: Nature and nurture. Dyslexia. 2002;8:143–159. doi: 10.1002/dys.228. [DOI] [PubMed] [Google Scholar]
- Pardo CA, Eberhart CG. The neurobiology of autism. Brain Pathology. 2007;17:434–447. doi: 10.1111/j.1750-3639.2007.00102.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raedler TJ, Knable MB, Weinberger DR. Schizophrenia as a developmental disorder of the cerebral cortex. Current Opinion in Neurobiology. 1998;8:157–161. doi: 10.1016/s0959-4388(98)80019-6. [DOI] [PubMed] [Google Scholar]
- Reiss A. Childhood developmental disorders. Paper presented at the The Convergence of Neuroscience, Behavioral Science, Neurology, and Psychiatry 2005 [Google Scholar]
- Reiss AL, Abrams MT, Greenlaw R, Freund L, Denckla MB. Neurodevelopmental effects of the FMR-1 full mutation in humans. Nature Medicine. 1995;1:159–167. doi: 10.1038/nm0295-159. [DOI] [PubMed] [Google Scholar]
- Reiss AL, Dant CC. The behavioral neurogenetics of fragile X syndrome: Analyzing gene–brain–behavior relationships in child developmental psychopathologies. Development and Psychopathology. 2003;15:927–968. doi: 10.1017/s0954579403000464. [DOI] [PubMed] [Google Scholar]
- Reiss AL, Hall SS. Fragile X syndrome: Assessment and treatment implications. Child and Adolescent Psychiatric Clinics of North America. 2007;16:663–675. doi: 10.1016/j.chc.2007.03.001. [DOI] [PubMed] [Google Scholar]
- Shattuck PT, Grosse SD. Issues related to the diagnosis and treatment of autism spectrum disorders. Mental Retardation and Developmental Disabilities Research Reviews. 2007;13:129–135. doi: 10.1002/mrdd.20143. [DOI] [PubMed] [Google Scholar]
- Shaywitz SE, Gruen JR, Shaywitz BA. Management of dyslexia, its rationale, and underlying neurobiology. Pediatric Clinics of North America. 2007;54:609–623, viii. doi: 10.1016/j.pcl.2007.02.013. [DOI] [PubMed] [Google Scholar]
- Shea V. A perspective on the research literature related to early intensive behavioral intervention (Lovaas) for young children with autism. Autism. 2004;8:349–367. doi: 10.1177/1362361304047223. [DOI] [PubMed] [Google Scholar]
- Stanfield AC, McIntosh AM, Spencer MD, Philip R, Gaur S, Lawrie SM. Towards a neuroanatomy of autism: A systematic review and meta-analysis of structural magnetic resonance imaging studies. European Psychiatry. 2007;23:289–299. doi: 10.1016/j.eurpsy.2007.05.006. [DOI] [PubMed] [Google Scholar]
- Stefanatos GA, Kinsbourne M, Wasserstein J. Acquired epileptiform aphasia: A dimensional view of Landau-Kleffner syndrome and the relation to regressive autistic spectrum disorders. Child Neuropsychology. 2002;8:195–228. doi: 10.1076/chin.8.3.195.13498. [DOI] [PubMed] [Google Scholar]
- Verkerk AJ, Pieretti M, Sutcliffe JS, Fu YH, Kuhl DP, Pizzuti A, et al. Identification of a gene (FMR-1) containing a CGG repeat coincident with a breakpoint cluster region exhibiting length variation in fragile X syndrome. Cell. 1991;65:905–914. doi: 10.1016/0092-8674(91)90397-h. [DOI] [PubMed] [Google Scholar]
- Visootsak J, Warren ST, Anido A, Graham JM., Jr Fragile X syndrome: An update and review for the primary pediatrician. Clinical Pediatrics. 2005;44:371–381. doi: 10.1177/000992280504400501. [DOI] [PubMed] [Google Scholar]
- Wing L, Potter D. The epidemiology of autistic spectrum disorders: Is the prevalence rising? Mental Retardation and Developmental Disabilities Research Reviews. 2002;8:151–161. doi: 10.1002/mrdd.10029. [DOI] [PubMed] [Google Scholar]