

Abstract
Despite induction immunosuppression and the use of aggressive maintenance immunosuppressive regimens, acute allograft rejection following lung transplantation is still a problem with important diagnostic and therapeutic challenges. As well as causing early graft loss and mortality, acute rejection also initiates the chronic alloimmune responses and airway-centred inflammation that predispose to bronchiolitis obliterans syndrome (BOS), also known as chronic lung allograft dysfunction (CLAD), which is a major source of morbidity and mortality after lung transplantation. Cellular responses to human leukocyte antigens (HLAs) on the allograft have traditionally been considered the main mechanism of acute rejection, but the influence of humoral immunity is increasingly recognised. As with other several other solid organ transplants, antibody-mediated rejection (AMR) is now a well-accepted and distinct clinical entity in lung transplantation. While acute cellular rejection (ACR) has defined histopathological criteria, transbronchial biopsy is less useful in AMR and its diagnosis is complicated by challenges in the measurement of antibodies directed against donor HLA, and a determination of their significance. Increasing awareness of the importance of non-HLA antigens further clouds this issue. Here, we review the pathophysiology, diagnosis, clinical presentation and treatment of ACR and AMR in lung transplantation, and discuss future potential biomarkers of both processes that may forward our understanding of these conditions.
Keywords: Lung transplant, cellular rejection, acute antibody rejection, humoral rejection
Introduction
Acute rejection of the pulmonary allograft remains a potential cause of acute graft failure but more commonly presents with sub-acute and often subtle graft dysfunction. While arguments continue regarding the risk-benefit of a scheduled surveillance lung biopsy program and indications for therapeutic intervention it is timely to focus on techniques to improve the operating characteristics of surveillance and clinically mandated procedures as well as the kappa scores of the reporting histopathologists. The brave beating of a distant drum heralds the potential utility of gene transcript analysis technology to supplant our current “gold standard” but while theoretically appealing has not been validated in the crucible of real time clinical practice. Until such time as it is, we are wise to continue quality control measures which improve the safety of the procedure and the adequacy of samples provided to the histopathologists with expert clinical input into final diagnoses and therapies according to our consensus guideline statements.
Acute cellular rejection (ACR)
ACR of the pulmonary allograft is a serious complication in lung transplantation not only because of potential acute graft dysfunction or failure, but because it is a major established risk factor for the development of chronic lung allograft dysfunction (CLAD) particularly the bronchiolitis obliterans syndrome (BOS). Early detection and treatment of acute rejection is thus of critical importance for lung transplant recipients. Antibody mediated rejection (AMR) may occur concurrently or separately from ACR and requires additional diagnostic and therapeutic manoeuvres and is covered later in the manuscript.
Epidemiology and risk factors
Registry data from the International Society for Heart and Lung Transplantation (ISHLT) show that about a third of patients will have at least one episode of treated rejection in the first year after transplantation (1). Furthermore, acute rejection is responsible for 3.6% of deaths among adult lung transplant recipients in the first 30 days, and 1.8% in the period from 1 to 12 months post-transplant (1). ACR is the most common form of acute lung transplant rejection and is most likely to be diagnosed within the first 6 months following lung transplantation (2).
Compared to the large body of data on the risk factors for CLAD, recipient, immunological and environmental factors predicting ACR are less well studied. Reported risk factors for ACR include the degree of human leukocyte antigens (HLA) mismatching (with discrepancies at certain loci—such as at HLA-DR, HLA-B and HLA-A of greater significance (3), and genetically-determined differences in innate and adaptive recipient immunological responses to the allograft (4,5). Younger age is also associated with a higher rate of rejection in the first year after transplantation, but whether this association is confounded by the indication for transplantation and the underlying disease process is unclear (1). The impact of both induction and maintenance immunosuppression on the incidence of ACR has been studied. Evidence from ISHLT registry data suggests a protective effect of the interleukin-2 receptor antagonists (basiliximab and daclizumab) when compared to no induction, or the use of the CD52 antagonist, alemtuzumab (1). However, there is a paucity of randomised trial data to support this observation, and a systematic review could not find a difference between induction agents (6). With maintenance immunosuppression, tacrolimus is associated with lower rates of BOS than cyclosporine, but the evidence supporting its use over cyclosporine for ACR is less conclusive. Despite Registry data and a single randomized trial (7) supporting its use, a systematic review could not conclusively demonstrate a benefit (8).
Lastly, infections, particularly community-acquired viral infections like respiratory syncytial virus (RSV), coronavirus, rhinovirus and the influenza and parainfluenza viruses, potentially via stimulation of innate immunity, and exposure of cryptic antigens by epithelial injury, may stimulate alloimmune responses and precipitate ACR.
Mechanisms of ACR
ACR is a response driven by T lymphocytes that recognises foreign major histocompatibility complex (MHC) antigens, also called HLA on allogeneic tissue. The function of MHC is to bind foreign antigenic peptides and display them on the surface of antigen presenting cells (APCs) for presentation to T cells. MHC genes are coded on the short arm of chromosome 6, and are highly polymorphic, resulting in extreme diversity between individuals. This difference in MHC between individuals is the major immunological barrier to transplantation, as the MHC molecules themselves act as antigens and permit rapid recognition of self from non-self.
In cellular rejection of the allograft, two distinct pathways of allorecognition are implicated. In the direct pathway, donor dendritic cells migrate from the graft to the secondary lymphoid tissue (“passenger leukocytes”) to present MHC directly to recipient T cells (9). In the indirect pathway, recipient dendritic cells process and present alloantigens derived from dying donor APCs to T cells, either in the secondary lymphoid organs or in the allograft itself (Figure 1). Other non-immunological processes, like ischaemia and reperfusion injury or pulmonary infection may cause non-specific activation of local innate immunity which stimulates acute rejection through mechanisms that have not been completely elucidated.
Figure 1.

The direct and indirect pathways of allo-sensitisation. In the direct pathway, donor-derived MHC complexes, presented by donor-derived ‘passenger’ dendritic cells (antigen-presenting cells or APCs), are recognised directly by recipient T cells. In the indirect pathway, host APCs take up immunogenic proteins from apoptotic graft cells and present donor-derived processed peptides on host MHC to host T cells. MHC, major histocompatibility complex; APC, antigen presenting cell.
Once recipient T-cells are activated, they undergo clonal expansion and differentiate into alloreactive killer T cells (also called cytotoxic T lymphocytes) which migrate to the graft and dock with the allograft’s MHC molecule and initiate tissue destruction (10). Effective blockade of T cell activation and proliferation is required to prevent rejection, and the advent of calcineurin inhibitors was a revolutionary advance in this area.
Clinical manifestations
Patients with acute rejection present with non-specific features that may mimic the many other infectious and post-infectious complications affecting lung transplant recipients. Shortness of breath, cough with or without sputum production and even low-grade fever can be observed in ACR, and there are no signs or symptoms that can reliably be used to distinguish from opportunistic pulmonary infection. Increasing grade of rejection is associated with more severe symptoms (11). Conversely, many episodes of ACR may be diagnosed on routine surveillance trans-bronchial biopsies in the early post-transplant period in asymptomatic patients. The physical examination may be normal, or reveal squeaks or crackles or features of a pleural effusion.
Laboratory findings
There are no specific laboratory findings in ACR. Increasing peripheral blood eosinophil count can precede clinically significant ACR in both lung and heart transplants, but the diagnostic accuracy and positive predictive value of this finding is modest at best (12). A recent review of peripheral blood findings in ACR has suggested that the differential count can be used to guide diagnosis, with an absolute eosinophil count greater than 0.4×109 cells, absolute lymphocyte greater than 1.5×109 cells, and a relative basophil count greater than 2% favouring rejection, and a neutrophil count greater than 9×109 favouring infection (13); however, the diagnostic accuracy of such an algorithm requires formal evaluation in clinical practice.
Pulmonary function tests
Spirometry is an easily performed, non-invasive, safe and repeatable test, routinely performed at follow-up visits or even by patients themselves at home, to screen recipients at risk for rejection. The usual spirometric abnormality in ACR is airflow limitation with reduced forced expiratory volume in one second (FEV1). A decline in FEV1 of 10% from a stable baseline that persists for more than 48 hours should generally trigger the need for further investigations (14,15). The utility of spirometry may be less helpful in single lung transplants, where changes may be confounded by progression of disease in the native lung (16). However, stable spirometry does not exclude ACR, and the specificity of spirometry is also low (17). Other airway-centric processes such as acute infection and airway anastomotic problems can cause a similar reduction in FEV1.
Pulmonary imaging
A chest X-ray is routinely obtained in the investigation of a lung transplant recipient with new respiratory symptoms. In the early post-transplant period, recipients with ACR may demonstrate bilateral pulmonary infiltrates on plain radiography (18), but the sensitivity and discriminant value of a plain radiograph is poor (19). High-resolution computed tomography (HRCT) findings are generally also non-specific (20); however, a recent study found that the presence of bilateral ground-glass opacities with a lower lobe predominance, in association with interlobular septal thickening, and in the absence of features of fluid overload (cardiomegaly), consolidation and atelectasis, had a positive predictive value of ~90% for acute rejection (21).
Bronchoscopy and transbronchial biopsy
The importance of a histological gold standard for the diagnosis of ACR is clear from the non-specific nature and poor diagnostic accuracy of the clinical parameters or non-invasive investigations listed above. A presumptive diagnosis of ACR requires intensification of immunosuppression, which could potentially be hazardous in the presence of an alternative diagnosis, particularly undiagnosed opportunistic infection. The need to make a confident tissue diagnosis of ACR and to reliably exclude intrapulmonary infection cannot thus be overemphasised. Both may occur simultaneously. Bronchoscopy allows for invasive sampling for histopathological determination, as well as the collection of bronchoalveolar lavage fluid and tissue for cytological and microbiological analysis. The usual practice is to only biopsy one lung in heart-lung or bilateral lung recipients. In cases of radiographic patchy disease, transbronchial biopsies can be directed to the involved areas (22). In the absence of radiological abnormalities or with diffuse disease, trans-bronchial biopsies are routinely obtained from the lower lobe (and, in many centres, also the ipsilateral right middle lobe or lingula, depending on the side biopsied whereas upper lobes are less commonly biopsied); the geographic distribution of rejection grades amongst the lung lobes has been shown to be similar (22), and avoidance of upper lobe biopsies reduces the risk of iatrogenic pneumothorax. The Lung Rejection Study Group (LRSG) recommends a minimum of 5 pieces of evaluable, well-expanded alveolar parenchyma to provide adequate sensitivity for diagnosing ACR (23): a study has shown that this amount can reliably be obtained by performing 10 to 12 biopsy specimens, with a resultant low complication rate (2). Complication rates in lung transplant recipients are low and include, in order of frequency, desaturation (10.5%), haemorrhage more than 100 mL (4%), pneumothorax (0.6–2.5%), post-procedural pneumonia (2%), arrhythmia (0.57%), and the need for mechanical ventilation (0.32%) (2,24,25). Mortality from the procedure has not been reported (Figure 2).
Figure 2.

Trans-bronchial biopsies at high-power field demonstrating the different grades of rejection.
Pathology
The histopathological hallmark of ACR is the demonstration on trans-bronchial lung biopsies of a mononuclear inflammatory cell infiltrate centred around small vessel and capillaries, and/or small airways (26). The LRSG of the ISHLT has developed a “working formulation of the standardization of the nomenclature in the diagnosis of lung rejection”, last revised in 2007, which establishes the diagnostic criteria for ACR and objectively grades its severity (Table 1) (23). Perivascular inflammation, termed “A-grade”, evaluates the presence and extent of mononuclear cell invasion—composed chiefly of T cells, although B-cells and eosinophils have also been described (27)—around the blood vessels, surrounding submucosal interstitium and alveolar walls. The grades range from A0 (no rejection) to A4 (severe). Airway inflammation, termed “B-grade” rejection, evaluates the lymphocytic response in the submucosa of bronchioles, which may extend through the basement membrane at higher grades. The interpretation of B-grade rejection is problematic as airway tissue is often not represented on a trans-bronchial biopsy specimen, and airway-centric mononuclear inflammation is not specific and can also be seen during episodes of acute infection. The finding of significant airway neutrophilia, necrosis, granulomas and viral cytopathic effect together perivascular or peribronchiolar inflammation may favour infection over rejection, although it is important to remember that the two processes are not mutually exclusive and can occur contemporaneously. In general, the LRSG advises grading both A- and B-grade rejection only after the rigorous exclusion of infection (28,29). Other processes that can confound the histological diagnosis of ACR include drug toxicity, aspiration, ischaemia/reperfusion injury, AMR and recurrent primary disease (26).
Table 1. Pathological grading of ACR*.
| Grade | Meaning | Appearance |
|---|---|---|
| A-GRADE: perivascular inflammation | ||
| 0 | None | Normal lung parenchyma |
| 1 | Minimal | Scattered, infrequent small mononuclear perivascular infiltrates; no eosinophils |
| 2 | Mild | More frequent perivascular infiltrates identifiable at low magnification; eosinophils may be present |
| 3 | Moderate | Dense perivascular infiltrates, eosinophils and neutrophils common. Pathognomonic feature is extension into alveolar septae and airspaces |
| 4 | Severe | Diffuse perivascular, interstitial and air-space infiltrates with pneumocyte damage and features of acute lung injury |
| B-GRADE: airway-associated inflammation | ||
| 0 | None | No evidence of bronchiolar inflammation |
| 1R | Low grade | Single-layer mononuclear cells in bronchiolar submucosa |
| 2R | High grade | Larger infiltrates of larger and activated lymphocytes in bronchiolar submucosa, with potential involvement of eosinophils and plasmacytoid cells |
| X | Ungradable | No bronchiolar tissue available |
*, adapted from the 2007 Working Formulation of the ISHLT (23); “R” in B-grade inflammation denotes “revised”. ACR, acute cellular rejection.
The sensitivity of trans-bronchial lung biopsy for diagnosing ACR is only ~70%, and is highly dependent on the experience of the histopathologist (30). Despite attempts to standardise the histological interpretation of ACR, inter-observer agreement has been modest. The 2007 revision has attempted to improve this, but follow-up studies have not shown increased concordance (31).
A promising technique being studied to increase the diagnostic yield in ACR by obtaining more lung tissue is cryobiopsy (32). Cryobiopsy is a relatively new interventional bronchoscopic technique that yields larger specimens with better representation of airways, alveoli and vascular structures. In the cryobiopsy procedure, a flexible cryoprobe delivers a compressed gas at the tip of the probe which freezes tissue on the probe’s tip. Disadvantages are the need for intubation, and there are theoretical concerns about higher risks of pneumothorax and haemorrhage. A recent study of cryobiopsy in lung transplant recipients, however, found more representative specimens, with less crush artefact, and without a higher rate of complications when compared to conventional biopsies (33).
Surveillance bronchoscopy in asymptomatic recipients
The role of surveillance trans-bronchial biopsies in asymptomatic lung transplant recipients, at regular intervals is controversial and is not universally practised amongst lung transplant centres. Schedules for surveillance bronchoscopies vary, but generally involve monthly biopsies in the immediate post-transplant period (when the incidence of ACR is the highest), and continue at less regular intervals for the period from 3 to 12 months after transplant. The rationale for surveillance is the high incidence of asymptomatic ACR (~25%) observed (25), and the suboptimal diagnostic accuracy of non-invasive tests (described above). Prospective cohort studies have shown that the incidence of high-grade perivascular or airway inflammation on routine surveillance biopsies is 16% and 14%, respectively (25,34). However, despite this finding, the evidence that early detection of ACR with subsequent augmentation of immunosuppression alters outcomes in lung transplantation is lacking. A small study randomising patients to either surveillance bronchoscopy or clinically-indicated biopsy did not find a difference in acute rejection, the development of CLAD or survival (35).
Other biomarkers
The identification of an objective, repeatable and specific non-invasive test that could replace trans-bronchial biopsy—which, as has been discussed, is an imperfect gold standard with modest inter-observer reliability—for the diagnosis of ACR remains an attractive but elusive goal. Furthermore, in the absence of a suitable biomarker, the current practice has also been to use the serum drug levels of calcineurin inhibitor as a proxy for the degree of immunosuppression, the intensity of which has been shown to correlate with a lower incidence of acute rejection (11,36). This latter approach has obvious drawbacks, not least the pharmacokinetic and pharmacogenetic variation between individuals, and the future identification of a biomarker more indicative of the underlying state of immune activation and/or rejection would be a much better guide to inform ongoing immunosuppression prescription. Several potential biomarkers are currently being evaluated. Exhaled nitric oxide (FeNO) as a marker of underlying lung inflammation in lung transplant recipients has been well studied, and levels are significantly elevated compared to baseline values in patients with ACR (37) (especially in those with lymphocytic airway inflammation), but the specificity is poor, as pulmonary infections also increase FeNO (38,39).
Quantification of donor-derived cell free DNA (ddcfDNA) from the allograft in circulating plasma (detectable by next-generation sequencing) is an exciting blood-based biomarker in solid organ transplantation. Preliminary work suggests that increased levels of ddcfDNA are associated with both acute and chronic rejection, correlate closely with histopathological features of ACR, and are associated with declines in lung function (40). However, validation studies are still required and optimal cut-off values are yet to be determined.
There are also several direct functional tests of immune function, some of which are commercially available. The Cylex Immune Cell Function Assay (ImmuKnow; Cylex, Inc., Columbia, MD, USA) measures ATP synthesis in activated CD4 T cells after exposure to a stimulant (phytohemagglutinin) as a measure of cell-mediated immunity, but its sensitivity for ACR seems to be low (41). The alloreactive T-cell frequency assay can demonstrate increased CD4, CD8 and Treg proliferation by mixed lymphocyte reaction to donor antigens in ACR, but its clinical application is limited by the complexity of the assay and other alloimmune responses (42).
Lastly, transcriptome signatures of ACR can be studied. MicroRNAs (miRNA) are small non-coding RNA molecules that are critical controllers of post-transcriptional regulation of gene expression. Several studies have implicated their involvement in renal and heart transplant rejection (43,44), and a small study in lung transplant recipients identified a distinct miRNA signature in airway epithelium that distinguished patients with ACR from those without rejection (45). Even more promising is that miRNA profiles that detect cardiac ACR have been found in peripheral blood, and the potential of miRNA as a blood-based marker of ACR is an exciting future field of study.
Treatment of ACR
Treatment decisions are dependent on institutional practice and the histological grade of rejection. Whilst there is consensus about treating higher grades of acute perivascular rejection (symptomatic grade A2 and above), the management of minimal or mild rejection (A1, asymptomatic grade A2, or isolated B-grade rejection) is more controversial. Some centres will elect not treat in the latter situations, rather repeating the biopsies in 4–6 weeks’ time. However, considering that minimal rejection has been shown to be associated with an increased risk of CLAD comparable to higher grades of rejection (46), and that lymphocytic bronchiolitis (B-grade rejection) has also been shown to be independently associated with increased risk of CLAD and death (34), both types and all grades of ACR should probably be strongly considered for treatment, especially in patients with a clinical syndrome suggestive of rejection. A special consideration may be the asymptomatic patient with minimal A1 rejection (and no B-grade rejection) detected on surveillance biopsies in whom augmentation of immunosuppression may considerably jeopardise the treatment of a current and serious opportunistic viral or fungal infection—in this situation, deferment of treatment and re-biopsy may be a safer strategy.
The cornerstone of treatment for ACR is the steroid pulse. However, there are no study data to guide the dose or duration of therapy. Most centres advocate pulsed-dose methylprednisolone for ~3 days with transition to a tapering oral steroid wean. Some clinicians will use only oral prednisolone (0.5–1.0 mg/kg) for milder grades of rejection, although evidence for this practice is lacking. Most centres which perform surveillance bronchoscopies will also perform a follow-up bronchoscopy 4–6 weeks after an episode of ACR. This practice is supported by the finding of significant rates of persistent ACR that are often clinically occult (47,48).
The management of persistent or refractory ACR is not well established. Failure to respond to treatment for ACR should trigger investigations for concomitant AMR. Together with a repeat steroid pulse, other changes to therapy in cases of persistent ACR may include a switch to tacrolimus (if on a cyclosporine-based regimen) (49,50) or the addition of a mammalian target of rapamycin (mTOR) inhibitor, such as everolimus. Other potential treatments that can be considered, if available, include alemtuzumab (an antibody to CD52, a peptide present on the surface of mature lymphocytes, which causes antibody-dependent lysis of lymphocytes) (51), and extracorporeal photophoresis (ECP). ECP is thought to reduce immune responses in transplant recipients by stimulating and expanding the number of peripheral regulatory T-cells, and has been shown to stabilise recurrent ACR and ameliorate lung function decline in CLAD (52).
AMR
We have discussed acute rejection post lung transplantation (LTX) as a cellular driven immune response, which has historically been considered the main mechanism of acute lung allograft rejection. Over the past decade however, the role played by antibodies in acute lung allograft, as well as CLAD has stimulated growing interest (53-56) and AMR has evolved from a hypothetical and controversial concept (23), to an important diagnostic consideration in patients with acute allograft dysfunction and a well-recognised clinical entity post lung transplantation (56).
Much of our initial understanding of pulmonary AMR has been extrapolated from the renal and heart transplant literature, where AMR has been recognised for some time (57,58). One of the biggest difficulties facing clinicians in reaching a diagnosis of AMR and choosing suitable management strategies is the lack of standardised data available, with different studies using various criteria for its diagnosis. A significant development has been the publication of the first consensus document on pulmonary AMR by the ISHLT. This consensus document has provided much needed standardisation of the criteria used to define AMR, which will facilitate universal interpretation of future research. It is generally accepted that this is an active document and ongoing research as well as increasing numbers of recognised pulmonary AMR diagnoses will generate information to help us understand the pathophysiology of pulmonary AMR and address numerous questions, which as yet remain unanswered (56).
In this section we will address the pathophysiology, diagnosis, clinical presentation, classification, and treatment of AMR, and explore some of the diagnostic and management challenges.
Pathophysiology of AMR
AMR results from the recipient’s immunological system’s ability to recognize donor HLA present in the lung allograft thereby inducing an allogeneic immune response resulting in the production of donor-specific antibodies (DSA) by B cells and plasma cells. These DSA target donor HLA expressed on donor organ capillary endothelial cells. HLA antigens exist as two distinct classes. The HLA class I antigens (HLA-A, HLA-B, HLA-C) are expressed in both T Cells and B cells, whereas HLA class II antigens (HLA-DP, HLA DQ, HLA-DR) are only expressed in B cells.
The polymorphic nature of HLA antigens provides up to 2×104 different endogenous antigens that can be presented to T Cells with subsequent activation of B cells and plasma cells to produce DSA that target the graft (59). These DSA may be formed prior to transplant in patients who have been previously sensitized following exposure to allogeneic tissue via previous pregnancy, blood transfusions, or transplantation (60,61), or they may also develop de novo post LTX. The rate of detection of DSA by single antigen bead (SAB) testing in various studies ranges from 10–61% of patients within 1 year post lung transplant (62-66), and their detection in post-transplant serum is associated with a significantly increased risk of AMR (67-69) and CLAD (62,65,66,68,70-73).
The significance of pre-transplant DSA has been well established in kidney and heart transplants, and in lungs they have been associated with acute and chronic effects on the lung allograft including refractory acute rejection, lymphocytic bronchiolitis (74), AMR (75), and CLAD (72,76,77). Pre-formed DSA have also been reported to promote de novo DSA development early after transplant and impact patient survival (64,78,79).
Graft injury as a result of DSA can occur via complement-dependent and complement-independent mechanisms. The initial activating step is the formation of the antigen-antibody complex, which results in an amplified immune response with numerous downstream effects (56). In the complement-dependent pathway, otherwise known as the classical pathway, antibody binds to its corresponding HLA-antigen on the airway epithelium and the resulting antigen-antibody complex activates the complement cascade. However, complement is not always necessary for antibodies to cause graft damage, as they are able to directly act on inflammatory cells such as macrophages, natural killer cells, and neutrophils, via interaction with the Fc receptor portion on the antibody, and induce pro-inflammatory cytokine production and microangiopathy (78,80). Not all patients with DSA present in their serum exhibit other features of AMR. This could be due to the variable pathogenicity of the IgG subclass itself. There are 4 different IgG subclasses—IgG1, IgG2, IgG3, and IgG4, each with variable affinity for binding to Fc receptors. IgG4 and IgG2 are usually considered to be non-complement binding and IgG3 and IgG1 bind complement with the former having the strongest affinity to the Fc receptor. However, when there is high antigen, epitope level, or increased complement concentration, all IgG subclasses have the ability to activate complement (81). This may therefore be a case of a “wolf in sheep’s clothing”, where DSA that may previously have been considered relatively benign, can become pathogenic depending on changes within immunological environment such as inflammatory responses to certain triggers (82,83).
DSAs to HLAs have traditionally been considered responsible for AMR lung allograft injury. However some patients meet criteria for AMR but have no evidence of HLA-DSA and there is growing evidence to support the role of non-HLA DSA or tissue self-antigens (SAGs) which are directed towards epithelial antigens, such as K-alpha 1 tubulin (Kα1T) and Collagen V (Col-V). These may account for the unmeasurable HLA-DSA in some patients with AMR (84-89). Similarly, there are patients in whom DSA are clearly present yet they do not exhibit other manifestations of AMR, which suggests alternative factors may influence the susceptibility or risk of rejection in the presence of antibodies that can bind the graft (90).
Significant work has been done by Bharat and his colleagues on SAGs. Their work has demonstrated that up to 30% of patients undergoing lung transplantation have pre-existing antibodies against Col-V and Kα1T, which strongly predispose patients to primary graft dysfunction (PGD), development of de novo alloimmunity, and CLAD (86,88). Their group have introduced the concept of the ‘two hit’ hypothesis for the development SAGs. The initial ‘hit’ involves an initial lung injury as a result of triggers such as infections with community acquired respiratory viruses (CARV), gastro-oesophageal reflux disease (GORD), ischaemia-reperfusion injury, or even the presence of HLA-DSA (91,92). The resulting local inflammatory response exposes SAGs such as Col-V and Kα1T which stimulate up-regulation of self-reactive lymphocytes and non-HLA antibody production. The second ‘hit’ is a result of down-regulation of regulatory T cells (TREGS) with loss of inhibition of self-reactive lymphocytes against SAGs which would usually induce T cell tolerance to SAGs. This ultimately leads to SAG autoimmunity Bharat (91,93). These SAGs may be a significant contributing factor to ongoing CLAD in patients who clear DSA post AMR treatment using commercially available kits that do not measure SAGs. A study by Hachem et al. has previously shown that more than 96% of LTX recipients with pre-existing HLA-DSA developed de novo SAGs within three years of LTX and were strongly predisposed to development of CLAD. However, AMR treatment was only effective in reducing the increased risk of CLAD if SAGs were cleared as well as HLA-DSA. Patients who cleared HLA-DSA but had persistent SAGs shared an equal risk of CLAD as those with both HLA-DSA and SAGs (34). Ongoing inflammation in patients post LTX recipients and those with end-stage lung disease provides an ideal environment for the above hypothesis, particularly in patients with cystic fibrosis (CF) who have ongoing infection and patients with idiopathic pulmonary fibrosis (IPF) who have ongoing inflammation. Studies have demonstrated a higher prevalence of SAGs pre-transplant in patients with CF and IPF compared to patients with other conditions such as COPD, whereas other studies have shown this can lead to the expansion of auto-reactive lymphocytes. Individuals with these diseases have the highest prevalence of SAGs pre-LTX compared to patients with other diseases such as COPD and alpha-1 anti-trypsin deficiency (89,94-96). Future research will no doubt help clarify the association between the pathophysiology of AMR (67-69) and that of CLAD (62,65,66,69-72). However, more data are needed to prove a link between these phenomena (97) .
Histopathological features of AMR
Trans-bronchial biopsies, when safe to perform, are an essential component of the diagnostic algorithm for patients presenting with a drop in lung function of more than 10% from baseline. It is considered the ‘gold standard’ for the diagnosis of ACR, however the appearance of ‘capillary injury’ with neutrophil margination, neutrophil capillaritis, and arteritis are considered by pathologists to be suggestive of AMR (23) and help clinicians make a more confident diagnosis by addressing the diagnostic criteria currently mandated by the ISHLT (56). These morphological features are suggestive of AMR but are not pathognomonic, and can occur in other disease processes involving the lung such as infection, organising pneumonia, and diffuse alveolar damage (DAD) secondary to other causes, as well as ACR (grade > A3) (56,61,98).
As in the case of DSAs, the absence of these features does not exclude AMR, and once again the clinical context needs to be taken into consideration when contemplating the diagnosis, and an effort needs to be made to exclude other causes such as infection although the two may co-exist (56,99) although the two may co-exist.
C4d immunohistochemistry
The identification of C4d staining on a biopsy sample as a surrogate marker for AMR due to activation of the complement cascade is controversial and its role in the diagnosis of pulmonary AMR may have been overestimated (56,61). It is rarely seen in patients with a diagnosis of pulmonary AMR based on other criteria (26,100), and there is poor inter-pathologist agreement when it comes to recognising a positive stain on trans-bronchial biopsy (101). Notably, C4d is not specific to pulmonary AMR, can be found in any process that is associated with complement activation such as reperfusion injury and infection (86), and studies have shown no little or correlation between positive staining and the presence of DSA (100,102).
Clinical features of AMR
The clinical presentation of pulmonary AMR can be variable depending on the time post-transplant. Hyperacute rejection can occur as soon as the vascular anastomosis is completed and recipient immune cells come into contact with the donor recipient endothelial cells. Pre-formed antibodies already present at the time of transplant are solely responsible for the devastating effects of this type of rejection. Graft failure occurs within minutes to hours and is usually fatal as a result of acute severe refractory hypoxaemia with respiratory failure. Diffuse pulmonary infiltrates are seen on radiological examination, and histopathology reveals neutrophilic margination, vasculitis, fibrinoid necrosis, vascular thrombosis, and pulmonary infarction (61). With the introduction of highly sensitive assays that can detect DSA the incidence of hyperacute rejection has been significantly reduced (103), however antibody responses to donor HLA antigens are not well controlled with current conventional immune-suppression and therefore acute AMR can occur at any time after transplantation (104), with either pre-transplant HLA, de novo HLA, or non-HLA DSA such as autoantibodies to Kα1T and Col-V being responsible in mediating allograft injury. A more chronic and indolent form for AMR is thought to contribute to the development of CLAD due to BOS or even r-CLAD, but there are insufficient data to confirm this association (97).
A high index of suspicion is necessary when considering pulmonary AMR as a possible diagnosis as its clinical presentation is often nonspecific. Patients may often present with an asymptomatic drop in lung function and non-specific symptoms such as cough, dyspnoea, fever, lethargy, which can rapidly progress to profound dyspnoea with progressive respiratory failure requiring increasing oxygen supplementation, mechanical ventilation and in some cases ECMO as an attempt to buy time for rescue therapy to work (105).
Diagnosis of AMR
The diagnosis of AMR is challenging and a high level of clinical vigilance is paramount. The current classification system requires evidence of lung allograft dysfunction, histological changes consistent with AMR, positive C4d staining, and the presence of DSA. A multidisciplinary approach that addresses the need to consider the clinical presentation as well as immunological and pathological information obtained during the diagnostic process is therefore recommended (56,98) and includes:
Measurement of allograft function;
Histopathological assessment including C4d staining;
Evaluation for the presence of DSA.
The diagnosis of AMR is usually made during the process of investigating a patient for a drop in lung function of greater than 10% from their baseline. This usually involves radiological investigations including a computerised tomography scan of the chest, nasopharyngeal swabs for respiratory viruses, bronchoscopy and bronchoalveolar lavage to look for infectious aetiologies as well as any airway abnormalities that may be contributing to the loss of graft function. If safe to do so, a trans-bronchial biopsy is also undertaken as the ‘gold standard’ in diagnosing ACR, and allows assessment for any features consistent with AMR, which includes staining for C4d (54) (Figure 3). Serology for the detection of DSA using SAB is also requested.
Figure 3.
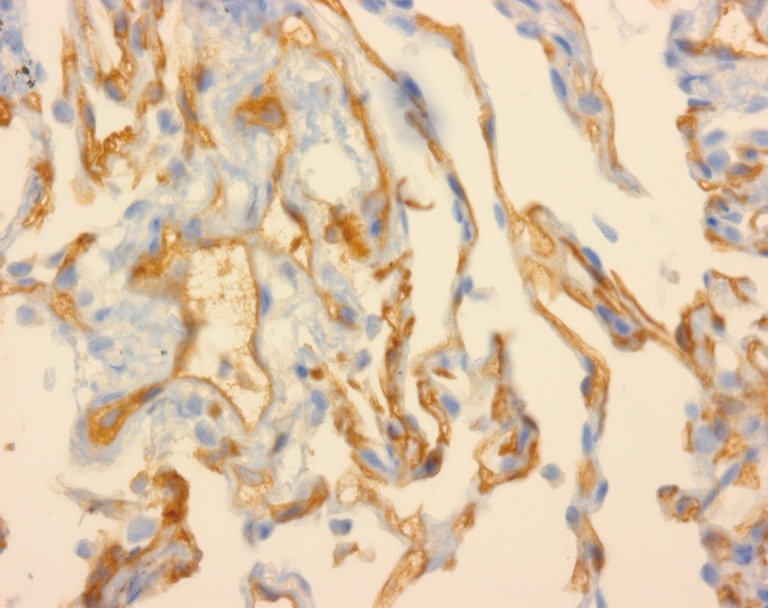
Trans-bronchial biopsy at high power magnification showing C4d capillary staining.
The identification of DSA has evolved over time from the provision of a rather crude measure of the presence of DSA by mixing donor lymphocytes with recipient serum with the finding of recipient cell lysis indicating the presence of DSA, the so called complement-dependent cytotoxic (CDC) assay (106). A modified version of this test relied on the use of a panel of HLA-phenotyped cells to express the result as the percentage of cells in the panel giving a positive result, the “panel reactive antibody” (PRA) percentage, with higher percentage PRA indicating a higher probability of positive crossmatch and vice versa (107).
The development of the solid phase SAB technology has allowed for very sensitive and accurate identification of DSA. The assay involves incubation of the patient’s serum with beads coated with two fluorochromes, which result in a unique signal for each bead. A specific HLA molecule produced by recombinant technology is also attached to each bead. When the recipient’s serum is incubated with the beads, any HLA-antibody present will react with the bead expressing the corresponding antigen molecule. The beads are then washed against a fluoro-labelled anti-human IgG antibody, which provides a mean fluorescence intensity (MFI) representing the amount of fluoro-labelled anti-IgG in complex with anti-HLA antibody. The MFI is not an accurate quantitative measurement of the amount of antibody concentration which requires measurement of the antibody titre (108-110).
Given the above, it is perhaps not surprising that there is ongoing controversy regarding what cut off level of MFI should be considered a clinically significant positive result. Whereas titres are more informative and accurate, random arbitrary selection of MFI thresholds to determine positive SAB results have led to conflicting reports on the clinical significance of DSA and their relationship to clinical outcomes post transplantation, with some studies using low MFI thresholds [100–1,200] showing no negative effect on transplant outcome with pre-existing DSA (62,111), whereas other studies that have chosen a slightly higher MFI (>3,000) showing an association with increased AMR risk following lung transplantation (75). Notably this study reported a lack of effect of DSA with MFI values of 1,000–3,000, clearly demonstrating the importance of selecting MFI threshold values for identifying anti-HLA antibodies. The MFI threshold may explain suggested differences between pre-transplant DSA against Class I and II HLA associated with increased risk for BOS and decreased survival as suggested by the study conducted by Brugiere et al. where they showed that DSA against class II HLA, but not class I, was associated with increased risk for BOS and decreased survival, though there was significant difference in the MFI values for anti-class I (1,048±256) and anti-class II (2,175±597) DSA, precluding definitive assessment of differences in biological impacts between antibodies against class I or class II HLA (76).
Longitudinal variability in the MFI levels for particular antibodies are also seen which is thought to be due to the deposition of antibodies in the graft leading to levels in the circulation, the so called ‘sponge effect’ as described by Girnita and colleagues who noted the detection of DSA in the serum only after removal of the graft suspected to have AMR (73).
It is also unclear as to whether DSA to any HLA locus or DSA with any MFI would have the same clinical outcome (111). Some studies have shown that HLA class II antibodies may be more significant than HLA Class I antibodies (73,112), and that patients who develop class II DSA or have persistent DSA despite antibody-depleting therapy have worse long term outcomes after transplantation (67,113,114).
C1q binding
The C1q binding assay C1q screen™ (One Lambda Inc., Canoga Park, California, USA), was developed in an attempt to determine which patients with detectable DSA are at higher risk of graft injury based on the presence of complement fixing DSA. As much as it is informative it only recognizes binding and not physiological complement activation (115-117). However, its clinical utility has been demonstrated in renal (117), heart (118), and lung transplants (67).
Complement is not always required to cause antibody mediated graft injury since DSA have the ability to induce damage via non-complement dependant pathways, and this is a significant limitation to using C1q binding to predict the likelihood of an a antibody binding complement resulting in allograft damage (119).
Classification of AMR
AMR is classified into clinical and sub-clinical AMR. Clinical AMR indicates that there is allograft dysfunction whereas in sub-clinical AMR the lung function is normal. Clinical AMR is then sub-classified into definite, probable and possible AMR. There needs to be allograft dysfunction for it to be considered clinical AMR. In addition 4 other criteria are considered which include exclusion of other causes, histological changes consistent with AMR, the presence of C4d staining on biopsy, and the presence of DSA. When all criteria are met it is definite AMR. When 3 out of 4 criteria are met it is classified as probable AMR, whereas the presence of 2 out of 4 criteria classifies it as possible AMR. Subclinical AMR is also sub-classified into definite, probable and possible, however there is no allograft dysfunction, and definite subclinical AMR is when there is histology consistent with AMR, positive C4d staining and presence of DSA. Probable subclinical AMR is consistent with the presence of 2 of 3 criteria and possible AMR 1 of 3 criteria (Table 2, Figure 4) (56).
Table 2. Definition and diagnostic certainty of clinical pulmonary AMR depending on criteria present*.
| AMR Grade | Allograft dysfunction | Other causes excluded | Lung histology | Lung biopsy C4d | DSA |
|---|---|---|---|---|---|
| Definite | X | X | X | X | X |
| Probable | X | X | X | O | X |
| Probable | X | X | X | X | O |
| Probable | X | X | O | X | X |
| Probable | X | O | X | X | X |
| Possible | X | X | X | O | O |
| Possible | X | X | O | O | X |
| Possible | X | X | O | X | O |
| Possible | X | O | X | X | O |
| Possible | X | O | X | O | X |
| Possible | X | O | O | X | X |
*, adapted from the 2016 pulmonary AMR consensus document of the ISHLT. X, absent; O, present. AMR, antibody-mediated rejection; DSA, donor-specific antibodies.
Figure 4.

Classification of AMR according to the presence or absence of diagnostic certainty and presence (Clinical) or absence (Sub-clinical) of allograft dysfunction*. *Adapted from the 2016 pulmonary AMR consensus document of the ISHLT. AMR, antibody-mediated rejection.
Hyperacute rejection can occur peri-operatively, acute rejection as discrete clinically-symptomatic episodes and chronic rejection may manifest as persistent allograft dysfunction resulting from cumulative and on-going pathologic events. Rates of hyperacute rejection have significantly been reduced as a result of the introduction of highly sensitive assays used at the time of cross-matching (103).
Treatment of AMR
The management of AMR poses an ongoing dilemma for physicians caring for patients in whom a diagnosis of AMR has been made since there is no clear evidence to determine the best management option for AMR post lung transplantation. There are no randomised controlled or head to head studies and current data supporting various treatment modalities arise from retrospective studies, case reports, or case series. Current treatment strategies aim to deplete and modulate the production of circulating DSA in an attempt to arrest graft dysfunction. As yet, there are no clear guidelines or consensus on the management of AMR post lung transplantation. Current treatment regimens are based on those employed in other solid organ transplants and include the use of plasmapheresis (PP), intravenous human immunoglobulin (IVIG), rituximab, and bortezomib (120), but results are suboptimal (121). PP removes antibodies from the circulation and has been reported to ameliorate lung function in some studies. IVIG causes B-cell apoptosis, reduces B-cell numbers, blocks binding of donor reactive antibodies and may inhibit complement activation (122). Rituximab is a chimeric human/murine monoclonal antibody directed at the CD-20 molecule that is found on mature B cells but not on pro-B cells or plasma cells and therefore it leads to elimination of peripheral B cells in the circulation but it has no effect on mature plasma cells or B cells in lymphoid tissue (123). Bortezomib is a proteasome inhibitor that reduces DSA by depleting plasma cells by causing plasma cell apoptosis. Its clinical use has been supported within the renal (124-128), and lung transplant literature (67,129), and has been effectively used to treat AMR that has been refractory to other treatments. The administration of bortezomib subcutaneously is associated with less side effects such as leucopoenia, neutropenia, thrombocytopenia, and peripheral neuropathy (130). Other monoclonal antibodies which have been used as salvage treatment for AMR include eculizumab and alemtuzumab. Eculizumab is an anti-C5 monoclonal antibody that blocks terminal complement activation. Alemtuzumab is a monoclonal antibody that binds to CD52, a protein present on the surface of mature lymphocytes (131,132).
Once treated, pulmonary AMR may stabilise, progress, or improve. A complete response to treatment is defined as a return to baseline function, abolition of DSA and reversal of pathologic changes. Partial response is improvement in lung function but not all parameters return to baseline. Stabilisation is no further clinical deterioration. No response is defined as on-going clinical deterioration and continued pathology (56).
Conclusions
Lung transplantation is unique to other solid organs in that it is open to the environment and undergoes continuous stimulus from infectious and non-infectious stimuli, which may play a part in up-regulating the immune system and therefore higher immune suppression is necessary. Long-term survival remains disappointing, with CLAD being the main cause of death. Current immunosuppression strategies targeting the T-cell responses do not seem to control CLAD, which may suggest an alternative pathway mechanically causing on-going graft damage. AMR has taken centre stage recently as a possible important mediator of chronic allograft injury and with improved knowledge on its mechanism of injury we may be able to identify targeted treatment that will enable us to gain good control and overall improve our patient survival post transplantation.
Acknowledgements
None.
Footnotes
Conflicts of Interest: The authors have no conflicts of interest to declare.
References
- 1.Yusen RD, Edwards LB, Dipchand AI, et al. The Registry of the International Society for Heart and Lung Transplantation: Thirty-third Adult Lung and Heart-Lung Transplant Report-2016; Focus Theme: Primary Diagnostic Indications for Transplant. J Heart Lung Transplant 2016;35:1170-84. 10.1016/j.healun.2016.09.001 [DOI] [PubMed] [Google Scholar]
- 2.Hopkins PM, Aboyoun CL, Chhajed PN, et al. Prospective analysis of 1,235 transbronchial lung biopsies in lung transplant recipients. J Heart Lung Transplant 2002;21:1062-7. 10.1016/S1053-2498(02)00442-4 [DOI] [PubMed] [Google Scholar]
- 3.Quantz MA, Bennett LE, Meyer DM, et al. Does human leukocyte antigen matching influence the outcome of lung transplantation? an analysis of 3,549 lung transplantations. J Heart Lung Transplant 2000;19:473-9. 10.1016/S1053-2498(00)00081-4 [DOI] [PubMed] [Google Scholar]
- 4.Colobran R, Casamitjana N, Roman A, et al. Copy number variation in the CCL4L gene is associated with susceptibility to acute rejection in lung transplantation. Genes Immun 2009;10:254-9. 10.1038/gene.2008.96 [DOI] [PubMed] [Google Scholar]
- 5.Girnita DM, Webber SA, Zeevi A. Clinical impact of cytokine and growth factor genetic polymorphisms in thoracic organ transplantation. Clin Lab Med 2008;28:423-40, vi. 10.1016/j.cll.2008.08.002 [DOI] [PubMed] [Google Scholar]
- 6.Penninga L, Moller CH, Penninga EI, et al. Antibody induction therapy for lung transplant recipients. Cochrane Database Syst Rev 2013:CD008927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hachem RR, Yusen RD, Chakinala MM, et al. A randomized controlled trial of tacrolimus versus cyclosporine after lung transplantation. J Heart Lung Transplant 2007;26:1012-8. 10.1016/j.healun.2007.07.027 [DOI] [PubMed] [Google Scholar]
- 8.Penninga L, Penninga EI, Moller CH, et al. Tacrolimus versus cyclosporin as primary immunosuppression for lung transplant recipients. Cochrane Database Syst Rev 2013:CD008817. [DOI] [PubMed] [Google Scholar]
- 9.Lafferty KJ, Bootes A, Dart G, et al. Effect of organ culture on the survival of thyroid allografts in mice. Transplantation 1976;22:138-49. 10.1097/00007890-197608000-00009 [DOI] [PubMed] [Google Scholar]
- 10.Ingulli E. Mechanism of cellular rejection in transplantation. Pediatr Nephrol 2010;25:61-74. 10.1007/s00467-008-1020-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.De Vito Dabbs A, Hoffman LA, Iacono AT, et al. Are symptom reports useful for differentiating between acute rejection and pulmonary infection after lung transplantation? Heart Lung 2004;33:372-80. 10.1016/j.hrtlng.2004.05.001 [DOI] [PubMed] [Google Scholar]
- 12.Trull A, Steel L, Cornelissen J, et al. Association between blood eosinophil counts and acute cardiac and pulmonary allograft rejection. J Heart Lung Transplant 1998;17:517-24. [PubMed] [Google Scholar]
- 13.Speck NE, Schuurmans MM, Murer C, et al. Diagnostic value of plasma and bronchoalveolar lavage samples in acute lung allograft rejection: differential cytology. Respir Res 2016;17:74. 10.1186/s12931-016-0391-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Bjortuft O, Johansen B, Boe J, et al. Daily home spirometry facilitates early detection of rejection in single lung transplant recipients with emphysema. Eur Respir J 1993;6:705-8. [PubMed] [Google Scholar]
- 15.Otulana BA, Higenbottam T, Ferrari L, et al. The use of home spirometry in detecting acute lung rejection and infection following heart-lung transplantation. Chest 1990;97:353-7. 10.1378/chest.97.2.353 [DOI] [PubMed] [Google Scholar]
- 16.Becker FS, Martinez FJ, Brunsting LA, et al. Limitations of spirometry in detecting rejection after single-lung transplantation. Am J Respir Crit Care Med 1994;150:159-66. 10.1164/ajrccm.150.1.8025743 [DOI] [PubMed] [Google Scholar]
- 17.Van Muylem A, Melot C, Antoine M, et al. Role of pulmonary function in the detection of allograft dysfunction after heart-lung transplantation. Thorax 1997;52:643-7. 10.1136/thx.52.7.643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Martinu T, Chen DF, Palmer SM. Acute rejection and humoral sensitization in lung transplant recipients. Proc Am Thorac Soc 2009;6:54-65. 10.1513/pats.200808-080GO [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Millet B, Higenbottam TW, Flower CD, et al. The radiographic appearances of infection and acute rejection of the lung after heart-lung transplantation. Am Rev Respir Dis 1989;140:62-7. 10.1164/ajrccm/140.1.62 [DOI] [PubMed] [Google Scholar]
- 20.Gotway MB, Dawn SK, Sellami D, et al. Acute rejection following lung transplantation: limitations in accuracy of thin-section CT for diagnosis. Radiology 2001;221:207-12. 10.1148/radiol.2211010380 [DOI] [PubMed] [Google Scholar]
- 21.Park CH, Paik HC, Haam SJ, et al. HRCT Features of Acute Rejection in Patients With Bilateral Lung Transplantation: The Usefulness of Lesion Distribution. Transplant Proc 2014;46:1511-6. 10.1016/j.transproceed.2013.12.060 [DOI] [PubMed] [Google Scholar]
- 22.Hasegawa T, Iacono AT, Yousem SA. The anatomic distribution of acute cellular rejection in the allograft lung. Ann Thorac Surg 2000;69:1529-31. 10.1016/S0003-4975(00)01226-1 [DOI] [PubMed] [Google Scholar]
- 23.Stewart S, Fishbein MC, Snell GI, et al. Revision of the 1996 working formulation for the standardization of nomenclature in the diagnosis of lung rejection. J Heart Lung Transplant 2007;26:1229-42. 10.1016/j.healun.2007.10.017 [DOI] [PubMed] [Google Scholar]
- 24.Chan CC, Abi-Saleh WJ, Arroliga AC, et al. Diagnostic yield and therapeutic impact of flexible bronchoscopy in lung transplant recipients. J Heart Lung Transplant 1996;15:196-205. [PubMed] [Google Scholar]
- 25.McWilliams TJ, Williams TJ, Whitford HM, et al. Surveillance bronchoscopy in lung transplant recipients: risk versus benefit. J Heart Lung Transplant 2008;27:1203-9. 10.1016/j.healun.2008.08.004 [DOI] [PubMed] [Google Scholar]
- 26.Roden AC, Aisner DL, Allen TC, et al. Diagnosis of Acute Cellular Rejection and Antibody-Mediated Rejection on Lung Transplant Biopsies: A Perspective From Members of the Pulmonary Pathology Society. Arch Pathol Lab Med 2017;141:437-44. 10.5858/arpa.2016-0459-SA [DOI] [PubMed] [Google Scholar]
- 27.Yousem SA, Martin T, Paradis IL, et al. Can immunohistological analysis of transbronchial biopsy specimens predict responder status in early acute rejection of lung allografts? Hum Pathol 1994;25:525-9. 10.1016/0046-8177(94)90126-0 [DOI] [PubMed] [Google Scholar]
- 28.Chakinala MM, Ritter J, Gage BF, et al. Reliability for grading acute rejection and airway inflammation after lung transplantation. J Heart Lung Transplant 2005;24:652-7. 10.1016/j.healun.2004.04.002 [DOI] [PubMed] [Google Scholar]
- 29.Colombat M, Groussard O, Lautrette A, et al. Analysis of the different histologic lesions observed in transbronchial biopsy for the diagnosis of acute rejection. Clinicopathologic correlations during the first 6 months after lung transplantation. Hum Pathol 2005;36:387-94. 10.1016/j.humpath.2005.01.022 [DOI] [PubMed] [Google Scholar]
- 30.Trulock EP, Ettinger NA, Brunt EM, et al. The role of transbronchial lung biopsy in the treatment of lung transplant recipients. An analysis of 200 consecutive procedures. Chest 1992;102:1049-54. 10.1378/chest.102.4.1049 [DOI] [PubMed] [Google Scholar]
- 31.Arcasoy SM, Berry G, Marboe CC, et al. Pathologic interpretation of transbronchial biopsy for acute rejection of lung allograft is highly variable. Am J Transplant 2011;11:320-8. 10.1111/j.1600-6143.2010.03382.x [DOI] [PubMed] [Google Scholar]
- 32.Poletti V, Casoni GL, Gurioli C, et al. Lung cryobiopsies: a paradigm shift in diagnostic bronchoscopy? Respirology 2014;19:645-54. 10.1111/resp.12309 [DOI] [PubMed] [Google Scholar]
- 33.Roden AC, Kern RM, Aubry MC, et al. Transbronchial Cryobiopsies in the Evaluation of Lung Allografts: Do the Benefits Outweigh the Risks? Arch Pathol Lab Med 2016;140:303-11. 10.5858/arpa.2015-0294-OA [DOI] [PubMed] [Google Scholar]
- 34.Glanville AR, Aboyoun CL, Havryk A, et al. Severity of lymphocytic bronchiolitis predicts long-term outcome after lung transplantation. Am J Respir Crit Care Med 2008;177:1033-40. 10.1164/rccm.200706-951OC [DOI] [PubMed] [Google Scholar]
- 35.Valentine VG, Gupta MR, Weill D, et al. Single-institution study evaluating the utility of surveillance bronchoscopy after lung transplantation. J Heart Lung Transplant 2009;28:14-20. 10.1016/j.healun.2008.10.010 [DOI] [PubMed] [Google Scholar]
- 36.Ahya VN, Douglas LP, Andreadis C, et al. Association between elevated whole blood Epstein-Barr virus (EBV)-encoded RNA EBV polymerase chain reaction and reduced incidence of acute lung allograft rejection. J Heart Lung Transplant 2007;26:839-44. 10.1016/j.healun.2007.05.009 [DOI] [PubMed] [Google Scholar]
- 37.Silkoff PE, Caramori M, Tremblay L, et al. Exhaled nitric oxide in human lung transplantation. A noninvasive marker of acute rejection. Am J Respir Crit Care Med 1998;157:1822-8. 10.1164/ajrccm.157.6.9707159 [DOI] [PubMed] [Google Scholar]
- 38.Antus B, Csiszer E, Czebe K, et al. Pulmonary infections increase exhaled nitric oxide in lung transplant recipients: a longitudinal study. Clin Transplant 2005;19:377-82. 10.1111/j.1399-0012.2005.00354.x [DOI] [PubMed] [Google Scholar]
- 39.Deborah HY, Anu K, Sharron C, et al. Non-invasive assessment of exhaled biomarkers in lung transplantation. J Breath Res 2011;5:024001. 10.1088/1752-7155/5/2/024001 [DOI] [PubMed] [Google Scholar]
- 40.De Vlaminck I, Martin L, Kertesz M, et al. Noninvasive monitoring of infection and rejection after lung transplantation. Proc Natl Acad Sci U S A 2015;112:13336-41. 10.1073/pnas.1517494112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Shino MY, Weigt SS, Saggar R, et al. Usefulness of immune monitoring in lung transplantation using adenosine triphosphate production in activated lymphocytes. J Heart Lung Transplant 2012;31:996-1002. 10.1016/j.healun.2012.05.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Greenland JR, Wong CM, Ahuja R, et al. Donor-Reactive Regulatory T Cell Frequency Increases During Acute Cellular Rejection of Lung Allografts. Transplantation 2016;100:2090-8. 10.1097/TP.0000000000001191 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duong Van Huyen JP, Tible M, Gay A, et al. MicroRNAs as non-invasive biomarkers of heart transplant rejection. Eur Heart J 2014;35:3194-202. 10.1093/eurheartj/ehu346 [DOI] [PubMed] [Google Scholar]
- 44.Wilflingseder J, Regele H, Perco P, et al. miRNA profiling discriminates types of rejection and injury in human renal allografts. Transplantation 2013;95:835-41. 10.1097/TP.0b013e318280b385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gharib SA, Edelman JD, Ge L, et al. Acute Cellular Rejection Elicits Distinct MicroRNA Signatures in Airway Epithelium of Lung Transplant Patients. Transplant Direct 2015;1. pii: e44. 10.1097/TXD.0000000000000551 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khalifah AP, Hachem RR, Chakinala MM, et al. Minimal acute rejection after lung transplantation: a risk for bronchiolitis obliterans syndrome. Am J Transplant 2005;5:2022-30. 10.1111/j.1600-6143.2005.00953.x [DOI] [PubMed] [Google Scholar]
- 47.Aboyoun CL, Tamm M, Chhajed PN, et al. Diagnostic value of follow-up transbronchial lung biopsy after lung rejection. Am J Respir Crit Care Med 2001;164:460-3. 10.1164/ajrccm.164.3.2011152 [DOI] [PubMed] [Google Scholar]
- 48.Guilinger RA, Paradis IL, Dauber JH, et al. The importance of bronchoscopy with transbronchial biopsy and bronchoalveolar lavage in the management of lung transplant recipients. Am J Respir Crit Care Med 1995;152:2037-43. 10.1164/ajrccm.152.6.8520773 [DOI] [PubMed] [Google Scholar]
- 49.Sarahrudi K, Estenne M, Corris P, et al. International experience with conversion from cyclosporine to tacrolimus for acute and chronic lung allograft rejection. J Thorac Cardiovasc Surg 2004;127:1126-32. 10.1016/j.jtcvs.2003.11.009 [DOI] [PubMed] [Google Scholar]
- 50.Vitulo P, Oggionni T, Cascina A, et al. Efficacy of tacrolimus rescue therapy in refractory acute rejection after lung transplantation. J Heart Lung Transplant 2002;21:435-9. 10.1016/S1053-2498(01)00379-5 [DOI] [PubMed] [Google Scholar]
- 51.Reams BD, Musselwhite LW, Zaas DW, et al. Alemtuzumab in the treatment of refractory acute rejection and bronchiolitis obliterans syndrome after human lung transplantation. Am J Transplant 2007;7:2802-8. 10.1111/j.1600-6143.2007.02000.x [DOI] [PubMed] [Google Scholar]
- 52.Benden C, Speich R, Hofbauer GF, et al. Extracorporeal photopheresis after lung transplantation: a 10-year single-center experience. Transplantation 2008;86:1625-7. 10.1097/TP.0b013e31818bc024 [DOI] [PubMed] [Google Scholar]
- 53.Hachem R. Antibody-Mediated Lung Transplant Rejection. Curr Respir Care Rep 2012;1:157-61. 10.1007/s13665-012-0019-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Westall GP, Snell GI. Antibody-mediated rejection in lung transplantation: fable, spin, or fact? Transplantation 2014;98:927-30. 10.1097/TP.0000000000000392 [DOI] [PubMed] [Google Scholar]
- 55.Glanville AR. Antibody-mediated rejection in lung transplantation: myth or reality? J Heart Lung Transplant 2010;29:395-400. 10.1016/j.healun.2010.01.012 [DOI] [PubMed] [Google Scholar]
- 56.Levine DJ, Glanville AR, Aboyoun C, et al. Antibody-mediated rejection of the lung: A consensus report of the International Society for Heart and Lung Transplantation. J Heart Lung Transplant 2016;35:397-406. 10.1016/j.healun.2016.01.1223 [DOI] [PubMed] [Google Scholar]
- 57.Stewart S, Winters GL, Fishbein MC, et al. Revision of the 1990 working formulation for the standardization of nomenclature in the diagnosis of heart rejection. J Heart Lung Transplant 2005;24:1710-20. 10.1016/j.healun.2005.03.019 [DOI] [PubMed] [Google Scholar]
- 58.Haas M, Sis B, Racusen LC, et al. Banff 2013 meeting report: inclusion of c4d-negative antibody-mediated rejection and antibody-associated arterial lesions. Am J Transplant 2014;14:272-83. 10.1111/ajt.12590 [DOI] [PubMed] [Google Scholar]
- 59.Felix NJ, Allen PM. Specificity of T-cell alloreactivity. Nat Rev Immunol 2007;7:942-53. 10.1038/nri2200 [DOI] [PubMed] [Google Scholar]
- 60.Lefaucheur C, Suberbielle-Boissel C, Hill GS, et al. Clinical Relevance of Preformed HLA Donor-Specific Antibodies in Kidney Transplantation. Am J Transplant 2008;8:324-31. 10.1111/j.1600-6143.2007.02072.x [DOI] [PubMed] [Google Scholar]
- 61.Wallace WD, Weigt SS, Farver CF. Update on pathology of antibody-mediated rejection in the lung allograft. Curr Opin Organ Transplant 2014;19:303-8. 10.1097/MOT.0000000000000079 [DOI] [PubMed] [Google Scholar]
- 62.Tikkanen JM, Singer LG, Kim SJ, et al. De Novo DQ Donor-Specific Antibodies Are Associated with Chronic Lung Allograft Dysfunction after Lung Transplantation. Am J Respir Crit Care Med 2016;194:596-606. 10.1164/rccm.201509-1857OC [DOI] [PubMed] [Google Scholar]
- 63.Snyder LD, Wang Z, Chen DF, et al. Implications for Human Leukocyte Antigen Antibodies after Lung Transplantation: A 10 year experience in 441 patients. Chest 2013;144:226-33. 10.1378/chest.12-0587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ius F, Sommer W, Tudorache I, et al. Early donor-specific antibodies in lung transplantation: risk factors and impact on survival. J Heart Lung Transplant 2014;33:1255-63. 10.1016/j.healun.2014.06.015 [DOI] [PubMed] [Google Scholar]
- 65.Morrell MR, Pilewski JM, Gries CJ, et al. De novo donor-specific HLA antibodies are associated with early and high-grade bronchiolitis obliterans syndrome and death after lung transplantation. J Heart Lung Transplant 2014;33:1288-94. 10.1016/j.healun.2014.07.018 [DOI] [PubMed] [Google Scholar]
- 66.Le Pavec J, Suberbielle C, Lamrani L, et al. De-novo donor-specific anti-HLA antibodies 30 days after lung transplantation are associated with a worse outcome. J Heart Lung Transplant 2016;35:1067-77. 10.1016/j.healun.2016.05.020 [DOI] [PubMed] [Google Scholar]
- 67.Witt CA, Gaut JP, Yusen RD, et al. Acute antibody-mediated rejection after lung transplantation. J Heart Lung Transplant 2013;32:1034-40. 10.1016/j.healun.2013.07.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Reinsmoen NL, Mirocha J, Ensor CR, et al. A 3 Center Study Reveals New Insights into the Impact of NonHLA Antibodies On Lung Transplantation Outcome. Transplantation 2017;101:1215-21. 10.1097/TP.0000000000001389 [DOI] [PubMed] [Google Scholar]
- 69.Roux A, Bendib Le Lan I, Holifanjaniaina S, et al. Antibody-Mediated Rejection in Lung Transplantation: Clinical Outcomes and Donor-Specific Antibody Characteristics. Am J Transplant 2016;16:1216-28. 10.1111/ajt.13589 [DOI] [PubMed] [Google Scholar]
- 70.Kauke T, Kneidinger N, Martin B, et al. Bronchiolitis obliterans syndrome due to donor-specific HLA-antibodies. Tissue Antigens 2015;86:178-85. 10.1111/tan.12626 [DOI] [PubMed] [Google Scholar]
- 71.Safavi S, Robinson DR, Soresi S, et al. De novo donor HLA-specific antibodies predict development of bronchiolitis obliterans syndrome after lung transplantation. J Heart Lung Transplant 2014;33:1273-81. 10.1016/j.healun.2014.07.012 [DOI] [PubMed] [Google Scholar]
- 72.Bosanquet JP, Witt CA, Bemiss BC, et al. The impact of pre-transplant allosensitization on outcomes after lung transplantation. J Heart Lung Transplant 2015;34:1415-22. 10.1016/j.healun.2015.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Girnita AL, McCurry KR, Iacono AT, et al. HLA-specific antibodies are associated with high-grade and persistent-recurrent lung allograft acute rejection. J Heart Lung Transplant 2004;23:1135-41. 10.1016/j.healun.2003.08.030 [DOI] [PubMed] [Google Scholar]
- 74.Girnita AL, Duquesnoy R, Yousem SA, et al. HLA-specific antibodies are risk factors for lymphocytic bronchiolitis and chronic lung allograft dysfunction. Am J Transplant 2005;5:131-8. 10.1111/j.1600-6143.2004.00650.x [DOI] [PubMed] [Google Scholar]
- 75.Kim M, Townsend KR, Wood IG, et al. Impact of pretransplant anti-HLA antibodies on outcomes in lung transplant candidates. Am J Respir Crit Care Med 2014;189:1234-9. 10.1164/rccm.201312-2160OC [DOI] [PubMed] [Google Scholar]
- 76.Brugiere O, Suberbielle C, Thabut G, et al. Lung transplantation in patients with pretransplantation donor-specific antibodies detected by Luminex assay. Transplantation 2013;95:761-5. 10.1097/TP.0b013e31827afb0f [DOI] [PubMed] [Google Scholar]
- 77.Smith JD, Ibrahim MW, Newell H, et al. Pre-transplant donor HLA-specific antibodies: characteristics causing detrimental effects on survival after lung transplantation. J Heart Lung Transplant 2014;33:1074-82. 10.1016/j.healun.2014.02.033 [DOI] [PubMed] [Google Scholar]
- 78.Smith JD, Banner NR, Hamour IM, et al. De novo donor HLA-specific antibodies after heart transplantation are an independent predictor of poor patient survival. Am J Transplant 2011;11:312-9. 10.1111/j.1600-6143.2010.03383.x [DOI] [PubMed] [Google Scholar]
- 79.Lobo LJ, Aris RM, Schmitz J, et al. Donor-specific antibodies are associated with antibody-mediated rejection, acute cellular rejection, bronchiolitis obliterans syndrome, and cystic fibrosis after lung transplantation. J Heart Lung Transplant 2013;32:70-7. 10.1016/j.healun.2012.10.007 [DOI] [PubMed] [Google Scholar]
- 80.Westall GP, Paraskeva MA, Snell GI. Antibody-mediated rejection. Curr Opin Organ Transplant 2015;20:492-7. 10.1097/MOT.0000000000000235 [DOI] [PubMed] [Google Scholar]
- 81.Thomas KA, Valenzuela NM, Reed EF. The perfect storm: HLA antibodies, complement, FcgammaRs, and endothelium in transplant rejection. Trends Mol Med 2015;21:319-29. 10.1016/j.molmed.2015.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mannam VK, Lewis RE, Cruse JM. The fate of renal allografts hinges on responses of the microvascular endothelium. Exp Mol Pathol 2013;94:398-411. 10.1016/j.yexmp.2012.06.002 [DOI] [PubMed] [Google Scholar]
- 83.AlMahri A, Holgersson J, Alheim M. Detection of complement-fixing and non-fixing antibodies specific for endothelial precursor cells and lymphocytes using flow cytometry. Tissue Antigens 2012;80:404-15. 10.1111/j.1399-0039.2012.01954.x [DOI] [PubMed] [Google Scholar]
- 84.Hachem RR, Tiriveedhi V, Patterson GA, et al. Antibodies to K-alpha 1 tubulin and collagen V are associated with chronic rejection after lung transplantation. Am J Transplant 2012;12:2164-71. 10.1111/j.1600-6143.2012.04079.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tait BD, Susal C, Gebel HM, et al. Consensus guidelines on the testing and clinical management issues associated with HLA and non-HLA antibodies in transplantation. Transplantation 2013;95:19-47. 10.1097/TP.0b013e31827a19cc [DOI] [PubMed] [Google Scholar]
- 86.Bharat A, Saini D, Steward N, et al. Antibodies to self-antigens predispose to primary lung allograft dysfunction and chronic rejection. Ann Thorac Surg 2010;90:1094-101. 10.1016/j.athoracsur.2010.06.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Iwata T, Philipovskiy A, Fisher AJ, et al. Anti-type V collagen humoral immunity in lung transplant primary graft dysfunction. J Immunol 2008;181:5738-47. 10.4049/jimmunol.181.8.5738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Burlingham W, Wilkes DS, Sullivan JA. Why is the patient out of breath? Collagen V(alpha1) and K-alpha1-tubulin take center stage in lung transplantation. Am J Transplant 2014;14:2201-3. 10.1111/ajt.12910 [DOI] [PubMed] [Google Scholar]
- 89.Tiriveedhi V, Gautam B, Sarma NJ, et al. Pre-transplant antibodies to Kalpha1 tubulin and collagen-V in lung transplantation: clinical correlations. J Heart Lung Transplant 2013;32:807-14. 10.1016/j.healun.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Nickerson PW, Rush DN. Antibodies beyond HLA. Am J Transplant 2013;13:831-2. 10.1111/ajt.12160 [DOI] [PubMed] [Google Scholar]
- 91.Bharat A, Mohanakumar T. Immune Responses to Tissue-Restricted Nonmajor Histocompatibility Complex Antigens in Allograft Rejection. J Immunol Res 2017;2017:6312514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fukami N, Ramachandran S, Saini D, et al. Antibodies to MHC class I induce autoimmunity: role in the pathogenesis of chronic rejection. J Immunol 2009;182:309-18. 10.4049/jimmunol.182.1.309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Adeegbe D, Matsutani T, Yang J, et al. CD4(+) CD25(+) Foxp3(+) T regulatory cells with limited TCR diversity in control of autoimmunity. J Immunol 2010;184:56-66. 10.4049/jimmunol.0902379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shah PD, McDyer JF. Viral infections in lung transplant recipients. Semin Respir Crit Care Med 2010;31:243-54. 10.1055/s-0030-1249120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Uhlin M, Mattsson J, Maeurer M. Update on viral infections in lung transplantation. Curr Opin Pulm Med 2012;18:264-70. 10.1097/MCP.0b013e3283521066 [DOI] [PubMed] [Google Scholar]
- 96.Papakonstantinou E, Karakiulakis G, Batzios S, et al. Acute exacerbations of COPD are associated with significant activation of matrix metalloproteinase 9 irrespectively of airway obstruction, emphysema and infection. Respir Res 2015;16:78. 10.1186/s12931-015-0240-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Morris GP. Anti-HLA Antibodies in Lung Transplantation. OBM Transplantation 2017;1:002.
- 98.Berry G, Burke M, Anderson C, et al. The pathology of pulmonary antibody mediated rejection: 2012 update from the Pathology Council of ISHLT. J Heart Lung Transplant 2013;32:14-21. 10.1016/j.healun.2012.11.005 [DOI] [PubMed] [Google Scholar]
- 99.Wallace WD, Li N, Andersen CB, et al. Banff study of pathologic changes in lung allograft biopsy specimens with donor-specific antibodies. J Heart Lung Transplant 2016;35:40-8. 10.1016/j.healun.2015.08.021 [DOI] [PubMed] [Google Scholar]
- 100.Roberts JA, Barrios R, Cagle PT, et al. The presence of anti-HLA donor-specific antibodies in lung allograft recipients does not correlate with C4d immunofluorescence in transbronchial biopsy specimens. Arch Pathol Lab Med 2014;138:1053-8. 10.5858/arpa.2013-0539-OA [DOI] [PubMed] [Google Scholar]
- 101.Roden AC, Maleszewski JJ, Yi ES, et al. Reproducibility of Complement 4d deposition by immunofluorescence and immunohistochemistry in lung allograft biopsies. J Heart Lung Transplant 2014;33:1223-32. 10.1016/j.healun.2014.06.006 [DOI] [PubMed] [Google Scholar]
- 102.Westall GP, Snell GI, McLean C, et al. C3d and C4d deposition early after lung transplantation. J Heart Lung Transplant 2008;27:722-8. 10.1016/j.healun.2008.03.018 [DOI] [PubMed] [Google Scholar]
- 103.Appel JZ, 3rd, Hartwig MG, Cantu E, 3rd, et al. Role of flow cytometry to define unacceptable HLA antigens in lung transplant recipients with HLA-specific antibodies. Transplantation 2006;81:1049-57. 10.1097/01.tp.0000204046.89396.c5 [DOI] [PubMed] [Google Scholar]
- 104.Gupta G, Abu Jawdeh BG, Racusen LC, et al. Late antibody-mediated rejection in renal allografts: outcome after conventional and novel therapies. Transplantation 2014;97:1240-6. 10.1097/01.TP.0000442503.85766.91 [DOI] [PubMed] [Google Scholar]
- 105.Marasco SF, Lukas G, McDonald M, et al. Review of ECMO (extra corporeal membrane oxygenation) support in critically ill adult patients. Heart Lung Circ 2008;17 Suppl 4:S41-7. 10.1016/j.hlc.2008.08.009 [DOI] [PubMed] [Google Scholar]
- 106.Pena JR, Fitzpatrick D, Saidman SL. Complement-dependent cytotoxicity crossmatch. Methods Mol Biol 2013;1034:257-83. 10.1007/978-1-62703-493-7_13 [DOI] [PubMed] [Google Scholar]
- 107.Pei R, Lee JH, Shih NJ, et al. Single human leukocyte antigen flow cytometry beads for accurate identification of human leukocyte antigen antibody specificities. Transplantation 2003;75:43-9. 10.1097/00007890-200301150-00008 [DOI] [PubMed] [Google Scholar]
- 108.Susal C, Opelz G, Morath C. Role and Value of Luminex((R))-Detected HLA Antibodies before and after Kidney Transplantation. Transfus Med Hemother 2013;40:190-5. 10.1159/000351314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Liu C, Wetter L, Pang S, et al. Cutoff values and data handling for solid-phase testing for antibodies to HLA: effects on listing unacceptable antigens for thoracic organ transplantation. Hum Immunol 2012;73:597-604. 10.1016/j.humimm.2012.04.016 [DOI] [PubMed] [Google Scholar]
- 110.Tambur AR, Herrera ND, Haarberg KM, et al. Assessing Antibody Strength: Comparison of MFI, C1q, and Titer Information. Am J Transplant 2015;15:2421-30. 10.1111/ajt.13295 [DOI] [PubMed] [Google Scholar]
- 111.Hachem RR, Reinsmoen NL. What is the definition of a clinically relevant donor HLA-specific antibody (DSA)? Am J Transplant 2015;15:299-300. 10.1111/ajt.13079 [DOI] [PubMed] [Google Scholar]
- 112.Palmer SM, Davis RD, Hadjiliadis D, et al. Development of an antibody specific to major histocompatibility antigens detectable by flow cytometry after lung transplant is associated with bronchiolitis obliterans syndrome. Transplantation 2002;74:799-804. 10.1097/00007890-200209270-00011 [DOI] [PubMed] [Google Scholar]
- 113.Hachem RR, Yusen RD, Meyers BF, et al. Anti-human leukocyte antigen antibodies and preemptive antibody-directed therapy after lung transplantation. J Heart Lung Transplant 2010;29:973-80. 10.1016/j.healun.2010.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kulkarni HS, Bemiss BC, Hachem RR. Antibody-mediated Rejection in Lung Transplantation. Curr Transplant Rep 2015;2:316-23. 10.1007/s40472-015-0074-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Piazza A, Poggi E, Ozzella G, et al. Post-transplant development of C1q-positive HLA antibodies and kidney graft survival. Clin Transpl 2013:367-75. [PubMed] [Google Scholar]
- 116.Chen G, Sequeira F, Tyan DB. Novel C1q assay reveals a clinically relevant subset of human leukocyte antigen antibodies independent of immunoglobulin G strength on single antigen beads. Hum Immunol 2011;72:849-58. 10.1016/j.humimm.2011.07.001 [DOI] [PubMed] [Google Scholar]
- 117.Loupy A, Lefaucheur C, Vernerey D, et al. Complement-binding anti-HLA antibodies and kidney-allograft survival. N Engl J Med 2013;369:1215-26. 10.1056/NEJMoa1302506 [DOI] [PubMed] [Google Scholar]
- 118.Chin C, Chen G, Sequeria F, et al. Clinical usefulness of a novel C1q assay to detect immunoglobulin G antibodies capable of fixing complement in sensitized pediatric heart transplant patients. J Heart Lung Transplant 2011;30:158-63. 10.1016/j.healun.2010.08.020 [DOI] [PubMed] [Google Scholar]
- 119.Hirohashi T, Uehara S, Chase CM, et al. Complement independent antibody-mediated endarteritis and transplant arteriopathy in mice. Am J Transplant 2010;10:510-7. 10.1111/j.1600-6143.2009.02958.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kidney Disease: Improving Global Outcomes Transplant Work G. KDIGO clinical practice guideline for the care of kidney transplant recipients. Am J Transplant 2009;9 Suppl 3:S1-155. 10.1111/j.1600-6143.2009.02834.x [DOI] [PubMed] [Google Scholar]
- 121.Djamali A, Kaufman DB, Ellis TM, et al. Diagnosis and management of antibody-mediated rejection: current status and novel approaches. Am J Transplant 2014;14:255-71. 10.1111/ajt.12589 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McManigle W, Pavlisko EN, Martinu T. Acute cellular and antibody-mediated allograft rejection. Semin Respir Crit Care Med 2013;34:320-35. 10.1055/s-0033-1348471 [DOI] [PubMed] [Google Scholar]
- 123.Bansal SB. Rituximab use in late antibody-mediated rejection. Indian J Nephrol 2016;26:315-6. 10.4103/0971-4065.179305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Roberts DM, Jiang SH, Chadban SJ. The treatment of acute antibody-mediated rejection in kidney transplant recipients-a systematic review. Transplantation 2012;94:775-83. 10.1097/TP.0b013e31825d1587 [DOI] [PubMed] [Google Scholar]
- 125.Ejaz NS, Alloway RR, Halleck F, et al. Review of bortezomib treatment of antibody-mediated rejection in renal transplantation. Antioxid Redox Signal 2014;21:2401-18. 10.1089/ars.2014.5892 [DOI] [PubMed] [Google Scholar]
- 126.Walsh RC, Alloway RR, Girnita AL, et al. Proteasome inhibitor-based therapy for antibody-mediated rejection. Kidney Int 2012;81:1067-74. 10.1038/ki.2011.502 [DOI] [PubMed] [Google Scholar]
- 127.Walsh RC, Brailey P, Girnita A, et al. Early and late acute antibody-mediated rejection differ immunologically and in response to proteasome inhibition. Transplantation 2011;91:1218-26. 10.1097/TP.0b013e318218e901 [DOI] [PubMed] [Google Scholar]
- 128.Yang KS, Jeon H, Park Y, et al. Use of bortezomib as anti-humoral therapy in kidney transplantation. J Korean Med Sci 2014;29:648-51. 10.3346/jkms.2014.29.5.648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Flechner SM, Fatica R, Askar M, et al. The role of proteasome inhibition with bortezomib in the treatment of antibody-mediated rejection after kidney-only or kidney-combined organ transplantation. Transplantation 2010;90:1486-92. 10.1097/TP.0b013e3181fdd9b0 [DOI] [PubMed] [Google Scholar]
- 130.Moreau P, Pylypenko H, Grosicki S, et al. Subcutaneous versus intravenous administration of bortezomib in patients with relapsed multiple myeloma: a randomised, phase 3, non-inferiority study. Lancet Oncol 2011;12:431-40. 10.1016/S1470-2045(11)70081-X [DOI] [PubMed] [Google Scholar]
- 131.Locke JE, Magro CM, Singer AL, et al. The use of antibody to complement protein C5 for salvage treatment of severe antibody-mediated rejection. Am J Transplant 2009;9:231-5. 10.1111/j.1600-6143.2008.02451.x [DOI] [PubMed] [Google Scholar]
- 132.Yeung MY, Gabardi S, Sayegh MH. Use of polyclonal/monoclonal antibody therapies in transplantation. Expert Opin Biol Ther 2017;17:339-52. 10.1080/14712598.2017.1283400 [DOI] [PubMed] [Google Scholar]