

Abstract
Although the incidence of Crohn’s disease (CD) in China is not as high as that in European and American countries, there has been a clear increasing trend in recent years. Little is known about its pathogenesis, cause of deferment, and the range of complications associated with the disease. Local and international scholars have presented many hypotheses about CD pathogenesis based on experimental and clinical studies, including genetic susceptibility, immune function defects, intestinal microflora disorders, delayed hypersensitivity, and food antigen stimulation. However, the specific mechanism leading to this immune imbalance, which causes persistent intestinal mucosal damage, and the source of the inflammatory cascade reaction are still unclear. So far, the results of research studies differ locally and internationally. This paper presents the most current research on immune factors in the pathogenesis of CD.
Keywords: Pathogenesis, Immune, Immunotherapy, Intestinal inflammation, Crohn’s disease, Cytokines, Lymphocytes, T lymphocytes
Core tip: It is now clear that Crohn's disease (CD) is an autoimmune disease that involves at least the intestinal mucosal immune system, when the mucosal immune system is invaded by food or bacterial antigens. However, it is worth mentioning that the mechanism remains unclear. Whether activation of the immune system is the internal defects (constitutive activation or regulation mechanism disorder) or changes in the epithelial mucosal barrier leading to continuous stimulation is still not clear. The mechanism of CD is intensively studied by domestic and foreign scholars on the immune destruction. Here we summarize the latest research status on immune factors in the pathogenesis of CD with regard to the mechanisms about T-lymphocyte immunity, innate lymphocyte immunity, cytokines and immune therapy.
INTRODUCTION
Crohn’s disease (CD) is a chronic granulomatous inflammatory disorder that can involve any part of the gastrointestinal tract, predominantly the terminal ileum and adjacent colon, and presents with segmental, asymmetric distribution of granulomatous inflammation[1]. The main clinical symptoms are abdominal pain, diarrhea, fistula, anal lesions, and systemic symptoms of different severity in the body. The incidence and prevalence rates of CD have been increasing rapidly. Zheng et al[2] analyzed the current status and prevalence changes of CD in mainland China in recent decades, and found that the CD incidence and prevalence rates in the last decade were 1.21 per 100000 persons/year and 2.29 per 100000 persons, respectively. These rates are higher than those during 1950-2002, 0.28 per 100000 persons/years and 1.38 per 100000 persons, respectively[2]. However, the pathogenesis, cause of deferment, and variety of complications are not clear[3].
Scholars have presented many hypotheses about the pathogenesis of CD. Some have suggested that the environment is filled with intestinal pathogens or opportunistic pathogens, such as pathogenic Escherichia coli, which spread through patients’ intestinal epithelial cells, and if innate immune cells such as monocytes, neutrophils, and natural killer (NK) cells cannot kill these translocated bacteria, they function as antigens and keep stimulating intestinal mucosal cells to cause immune responses[4], like abnormal Th1 and Th17 activation. Th17 activation results in the formation of granulomas[5]. Some antigens produced by bacteria can induce CD4+ T cells to differentiate into targeted cytotoxic T lymphocytes (CTLs), and these cells release interleukin (IL)-17 to stimulate Th17 cells to produce transforming growth factor and interferon-alpha (IFN-α), which can cause persistent inflammation and fibrosis[6]. Another opinion presented by Papadakis is that after the early antigen enters the body, associated lymphoid tissues are stimulated. When this occurs, the body becomes sensitive to the antigen, creating a sensitive state and mucosal immunity for the antigen of normal intestinal bacteria, and from then on, any secondary damage to the intestinal mucosa barrier results in the antigen contacting the lymphoid tissue again, stimulating a severe local immune response[7,8]. However, intestinal mucosa barrier damage occurs all the time, yet, most often, there is no immune response. This obviously means that this disease is also related to the body’s genetic susceptibility and environmental factors[9].
These theories have some differences related to gene mutations causing immune dysfunction, delayed hypersensitivity, alterations of intestinal flora, and activation induced by some special food[10] or bacterial antigen. However, each mechanism ultimately involves the immune response and broken immune tolerance. Immunity is now a universally acknowledged cause of CD, and hormone therapy can cause remission, demonstrating that CD is an immune-related disease. At present, the specific processes involved in the breakdown of immune tolerance and the associated sequence of genetic changes are still unclear[11-13]. Also, the specific mechanisms leading to this immune imbalance, which causes persistent intestinal mucosal damage, and the source of the inflammatory cascade reaction remain unclear. So far, the results of research studies differ locally and internationally. Here we present the most current research on immune factors in the pathogenesis of CD with regard to the mechanisms about T cell immunity, innate cell immunity, and cytokines.
CELL IMMUNITY AND INFLAMMATORY FACTORS
T lymphocytes
CD4+/CD8+ T lymphocytes and cytokines (Figures 1 and 2): Some studies have found that CD results mainly from chronic inflammation of T lymphocytes, especially CD4+ T cells. As cells that mediate humoral immunity, B lymphocytes do not participate in the occurrence and development of CD. CD4+ T cells are the main effector lymphocytes in intestinal inflammatory tissue. Eventually, despite the differences in the development of the inflammatory process, the process of inflammation is induced by Th1 or Th2 cells. The two subgroups of cytokines produced by Th1 or Th2 cells are mutual antagonists. Once a group of cytokines are produced earlier or more than the other, they will inhibit the other group of cytokines. Therefore, for the same inflammation, the two kinds of CD4+ T cells can lead to different consequences, presenting two different types of immune response, CD and ulcerative colitis (UC). Lymphocytes play a dominant role in CD patients, mainly secreting IL-12, IFN-α, tumor necrosis factor (TNF)-γ, IL-1, IFN-γ, IL-2, and other cytokines[14-16]. But for patients with UC in the intestinal mucosa, the characteristics of Th2 lymphocytes are atypical infiltration and production of IL-4, IL-5, 1L-13, and transforming growth factor (TGF)-β cytokines. Therefore, more autoantibodies are present in UC patients than in CD patients, mainly Th2 type immune response related antibodies. In CD animal models, intestinal bacteria and some specific antigens, such as certain pathogenic Escherichia coli, can induce intestinal mucosal Th1, which induces a cellular immune response. However, cellular immunity can be induced in a specific susceptible animal model such as IL-10 gene knockout mice, whereas cellular immunity cannot be induced in wild type mice[17-19]. This also suggests that gene deletion and mutation cause the natural immunity defects that induce CD. However, there is growing evidence that the Th1-Th2 classification is too simple; the two paths of mutual exclusion hypothesis has been questioned, and increasing evidence shows that IL-4 and IL-13 from the Th2 cells take part in ileal CD. In CD, there are two simultaneous changes in the initial stage of inflammation: the induction phase and the effector phase. Both Th1 and Th2 may participate in each phase simultaneously or sequentially.
Figure 1.
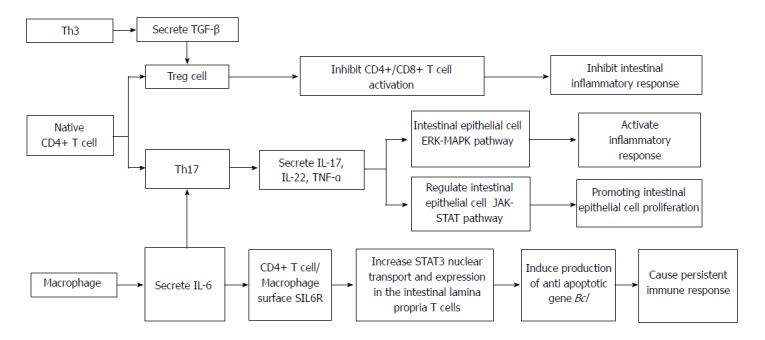
The main immune process of Crohn's disease.
Figure 2.
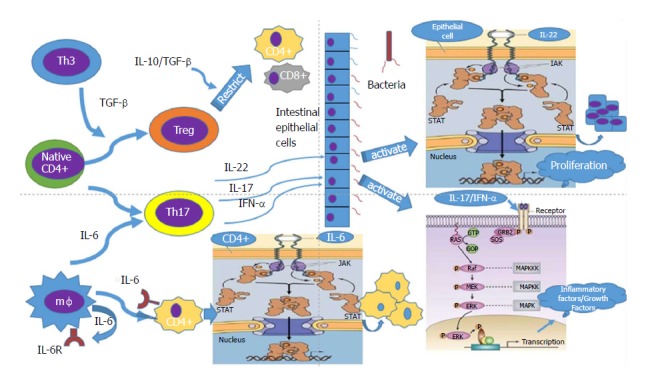
The main immune mechanisms of Crohn's disease.
Studies have found that CD8+ cells also exist in the intestinal mucosa. For example, many CTLs are observed in CD models, indicating a greater effect than in other diseases. When a gene related to the function of CD8+ T cells is deleted, there is no influence on CD inflammation, whereas when genes related to the function of CD4+ T cells are deleted, there is an improvement in intestinal mucosal inflammation. Therefore, the inflammatory response appears to be related to CD4+ T cells, and the role of CD8+ T cells in CD remains unclear. However, the pathological effect of CD8+ cells should not be ruled out completely[19,20]. So far, the role of CTLs in the pathogenesis of CD has been investigated in few studies, but CD8+ cell subsets and T lymphocytes (CTLs, CD8, and CD28+) are believed to play a key role in the recognition and elimination of mutant cells, and inflammation caused by graft-versus-host reactions. Compared with healthy subjects, CTLs in CD patients release enzymes, such as perforin and telomerase, and increase cytotoxic protein activity. Studies have compared the chromosomal changes of CD8+ cells in patients with CD to those of normal patients. Early activation of CD8+ cells may determine subsequent proliferation, cytokine production, and antigen recognition capability. Thus, an intervention causing early activation of CD8 cells is a potential therapeutic option[21]. It has been found that the K+ pathway plays an important role in the activation of T cells in the early stage of CD, and in the expression of Kv1.3 and IKCa1. Additionally, many scholars have reported two kinds of potassium ion channel that play an important role in mouse and human inflammatory bowel disease (IBD) development. The basic function of these ion channels is to maintain intracellular negative potential so that the influx of calcium in the body fluids activates the immune function[22]. These studies have shown that the effects of CD8+ cell toxicity and T cell depletion on CD inflammation are different, but different Th1 mediated inflammatory responses play a major role in the pathogenesis of CD. Therefore, the immune balance is deranged, and persistent inflammatory response occurs.
New T lymphocyte subsets and cytokines (Figures 1 and 2)
In recent years, researchers have found a new subset of T cells, called T regulatory (Treg) lymphocytes. When human or mouse CD4+ T cells were exposed to repeated stimulation with IL-10 in vitro, a new subset of T cells that secrete a high level of IL-10 but a low level of IL-2 was induced. Treg cells have weak proliferation ability[23], but their cytokine secretion pattern is quite stable. They can secrete a small amount of TGF-β, inhibit antigen-specific immune responses, and restrict intestinal inflammation through down-regulation of IL-10[24]. Treg cells are a group of T cell subsets with immunosuppressive effects. They can negatively regulate the immune system by inhibiting the proliferation and activation of CD4+ and CD8+ T cells. Th17 cells are a newly discovered helper T cell subset, named after its secreted cytokine IL-17. Th17 cells play important roles in innate immunity and acquired immunity by secreting IL-17A/F, IL-22, and IL-21. TNF-α is an inflammatory cytokine in host defense against extracellular bacterial infection, in anti-parasite immunity mediated by chronic inflammation, and in organ transplant rejection. Th17 cells have received extensive attention in many important physiological or pathological processes in immune diseases and cancer, especially in the study of the pathogenesis of autoimmune diseases[25]. Researchers have found that Th17 and Treg cells are subsets of CD4+ T cells. Th17 cells promote intestinal inflammation induced by autoimmune diseases, while Treg cells inhibit intestinal mucosal inflammation, which means that they have opposing functions. A recent study found that TGF-β can induce naive T cells into Treg immunosuppression. In the presence of IL-6, TGF-β promotes the differentiation of naive T lymphocytes to Th17 cells, which secrete proinflammatory cytokine IL-17, and the occurrence of autoimmunity and inflammation[26]. Treg cells can repair mucosal inflammation in patients with IBD, but under the influence of IL-6 andor IL-23, Treg cells differentiate into Th17 cells, which secrete large amounts of IL-17[27]. Kinugasa pointed out that IL-17 participates in the regulation of intestinal epithelial barrier function through the extracellular signal regulated (ERK)-mitogen activated protein kinase (MAPK) pathway, which may be a potential cause of intestinal inflammation[28].
Carrier et al[29] reported another kind of T cell that mediates oral tolerance by TGF-β, which is called Th3 cells. The difference between Th3 and Treg cells is mainly that the former secretes high levels of TGF-β, inhibiting immune responses. Due to the lack of an immunomodulatory effect, IL-10 or TGF-α gene knockout mice easily suffer from autoimmune colitis. Therefore, it is speculated that Treg and Th3 cell dysfunction is involved in the pathogenesis of IBD, especially CD. Because previous studies of Th1 lymphocytes cannot fully explain the pathogenesis of CD, new T cell types such as Th17, Treg, and Th3 have become the mainstream of the IBD system, which provides a new direction for the treatment and in-depth study of CD.
Intestinal lamina propria innate lymphocytes and cytokines
NK cells are a lymphoid cell lineage that is considered a part of the natural immune system. Innate immunity is a natural immune defense function formed through the body’s process of evolution and development. However, since the discovery of lymphoid tissue induction (LTi) cells in 1997, the lymphocyte populations in the innate immune system have been continuously expanded[30]. The loss of LTi cells leads to ROR γt expression defects, and the mice could not form lymph nodes and isolated lymphoid follicles, which means that LTi cells are crucial for lymphatic tissues and organs, and the latter is the basis of the intestinal mucosal immune barrier and intestinal immune homeostasis. In recent years, researchers have found some new immune cells in the mouse and human mucosal tissue. These cells belong to the lymph cell lineage in the form and development degree, but do not express specific antigen recognition receptors on the surface of the mature lymphocytes. They are similar to NK cells. Due to the characteristics of LTi cells, they are called innate lymphoid cells (ILCs)[31]. Numerous studies have shown that ILCs participate in the regulation of intestinal mucosa homeostasis and play an important role in the pathogenesis of IBD through multiple pathways. Based on secreted factors and the transcription factor expression pattern of different cells, the family of ILCs is divided into three categories: (1) ILC1, including classic NK cells, tissue residing NK cells, and mucosal ILC1, which express T box transcription factor (T bet) and/or Eomes. Under the stimulus of IL-12 and IL-18, ILC1 produce IFN-γ, which is mainly responsible for antiviral, bacterial, and toxoplasma infections and also plays a role in the immune memory; (2) ILC2, including natural helper (NH) cells, which produce Th2 cytokines, such as IL-5 and IL-13, and play an important role in parasite infection, allergy, and asthma; and (3) ILC3, including all the ILC subsets that produce IL-17 or IL-22, with LTi being the earliest reported ILC subset, which can induce intestinal lamina propria isolated lymphoid follicles and the formation of lymphoid tissue by expressing the ROR-γ transcription factor[32,33]. A study[34] using model mice infected with Citrobacter and Candida albicans showed that ILC3 secrete IL17 and IL22, which can promote Paneth cells to secrete antimicrobial peptides (such as Reg IIIβ and Reg IIIγ). These peptides can block the contact between bacteria and epithelial cells and inhibit intestinal inflammation. Some studies have shown that the reduction of IL-22 + ILC3 in the intestinal mucosa is associated with the occurrence of IBD[35]. IL-22 promotes epithelial cell proliferation through the JAK-STAT pathway, thereby preserving the integrity of the epithelial barrier and hindering intestinal microbial invasion[36]. The deficiency of IL-22 causes damage to the intestinal mucosal barrier, which leads to the exposure of intestinal tissue to many antigens and induces an abnormal immune response in the genetic susceptible host to cause the occurrence of IBD[37]. Meanwhile, macrophages and the intestinal secretion of IL-1β can stimulate ILC3 to produce granulocyte macrophage colony stimulating factor (GM-CSF), which plays a role in T cell proliferation[38]. Inhibition of GM-CSF can lead to a decrease in the number of regulatory T cells and immune tolerance defect, which also plays a role in the occurrence and development of IBD.
Studies have found that increased ILC3 can overexpress major histocompatibility complex (MHC) II[39,40]. ILC3 as antigen-presenting cells can, via MHCII, induce CD4+ T cell apoptosis, and thus avoid the T cell response to produce intolerance to intestinal symbiotic bacteria. Further studies of IBD patients compared with non-IBD patients found that the expression of MHCII by ILC3, which can induce CD4+ T cell apoptosis, was significantly reduced in IBD patients, thus causing an immune reaction against commensal bacteria and damaging the intestinal mucosa. Therefore, the intestinal mucosa barrier damage and immune tolerance defects cause a disorder in intestinal homeostasis, which plays an important role in the pathogenesis of IBD, the maintenance of homeostasis of the body, and the coordination among the different subtypes that work together. Regarding CD, an increasing number of studies have confirmed that the imbalance in the regulation of ILC breaks down the intestinal tolerance to food and bacterial antigens in the gut leading to CD, with ILC3 being the most important[29,34,36,41]. A study found that the deletion of IL-22+ ILC3 caused the spread of Alcaligenes sp. in the intestinal lymph tissue and induced a systemic immune response, which may be closely related to the occurrence of CD[42]. Mast cells stimulate fibroblast proliferation and collagen synthesis, and promote collagen protein aggregation activity and expression of c-kit receptor at the same time. Mast cells also stimulate chemotaxis through stem cell factor receptor (c-kit), and stimulate the proliferation of intestinal cells and fibroblasts[43]. In a previous study, a Salmonella typhi strain was used to induce C57/B16 mice to produce severe and persistent bowel wall fibrosis. In this animal model, chronic infection was caused by intestinal flora; large extracellular matrix (ECM) accumulation was seen in the ileocecal region and colon, and fibrosis and extensive transmural inflammation extended to the colon[44], but the most serious and extensive fibrosis was found in the ileocecal valve[45]. A study showed that ILC3 caused overexpression of IL-22, which activates monocytes, macrophages, and mast cells during inflammatory mucosal repair. Finally, the innate lymphocytes in the tissue result in fibrosis. Many studies on inflammatory factors have revealed that IL-22 arguably secreted by ILC3 can promote Paneth cells to secrete antimicrobial peptides (e.g., Reg IIIβ and Reg IIIγ). These peptides can restrict bacterial contact with epithelial cells and inhibit intestinal inflammation at the same time, and IL-22 promotes epithelial cell proliferation via the JAK-STAT pathway to maintain the integrity of the epithelial barrier and to prevent intestinal microbial invasion. Moreover, deletion of IL-22 causes intestinal mucosa barrier damage, and the intestinal tissue is exposed to many antigens, thus inducing abnormal genetic susceptibility to host immune response and leading to CD[34,36,37]. Scholars believe that Th17 cells, via inflammatory cytokines such as IL-17, IL-22, IL-21, and TNF-α, are involved in the body’s defense of extracellular bacterial infection and parasitic immune resistance, and mediate chronic inflammation, but excessive expression of inflammatory markers leads to CD[25]. Since the results of this study were inconsistent, further studies examining the role of IL-22 in the pathogenesis of CD are needed.
IMMUNOTHERAPY
The traditional view is that CD is due to acquired immune system disorders, and Th1 cytokines are the main factors in the development of CD. However, only some of them can be used in the treatment of CD, including IL-1, 2, 6, 12, 18, IFN-α, and TNF-γ. They can act on intestinal epithelial cells and cause epithelial cell apoptosis by activating lymphocytes. So far, the most successful example is the use of anti-TNF-α antibodies for the treatment of refractory CD with fistula. Mannon et al[46] reported the safety of subcutaneous anti-IL-12 monoclonal antibody for 7 wk, and despite the small number of cases studied, the clinical response was significant when a higher dose of IL-12 monoclonal antibody therapy (3 mg/kg body weight) was given. Long-term safety is a particularly important problem because the Th1 cytokines are important anti-infective factors. For example, long-term inhibition of the host response to tuberculosis (TB) by the treatment of anti-TNF-alpha antibodies may lead to the recurrence of latent TB in the patient; anti-IL-12 treatment should be considered for the possibility of recurrent asthma. Moreover, the etiology of CD is complex, especially in multiple stage disease. In some cases, the cytokines are likely to be harmful rather than beneficial. In some patients, anti-TNF-α antibody (infliximab) treatment does not reduce CD progression, which shows that it is not only due to the Th1 pathway but also due to different mechanisms in the different periods of disease progression. However, the innate immune system is also important, especially in the stage of disease induction; the congenital immune system cells is an important generator of cytokines, such as IL-1, TNF-α, IL-6, and other cytokines, and these factors play an important role in intestinal mucosal inflammation. The CD susceptibility genes encoding intracellular proteins (such as NOD2/CARD15) were found first in the innate immune system, which can activate certain cytokines dependent on NF-kappa B to cause intestinal bacteria to break the immune tolerance of the intestinal mucosa[47]. By using monoclonal antibodies against cell factors, fusion proteins, and receptor antagonists for cytokine blocking, immunomodulation of CD can be done effectively.
CONCLUSION
Research on the relationship between the immune regulation and the pathogenesis of CD has greatly improved the understanding of the role of immune factors in the development of CD. Since the discovery of new types of Th1 cells such as Treg, Th3, and Th17, as well as the application of antibodies to inflammatory factors TNF-α, IL-12, IL-22, and other new cytokines, it is clear that CD is a complex disease, not only due to the Th1 pathway, but also due to different mechanisms in different periods of disease progression, which is why the use of IL-12 or TNF-α antibody treatment in many patients with CD is not so effective. Hereditary susceptibility, the surrounding environment, intestinal flora, and other factors may also be involved. Although there are a variety of cytokine antagonists in clinical application, the results are not satisfactory. So far, we have not found the main immune response pathway involved in CD. So, it is necessary to carry out a large-scale screening of important inflammatory cytokines and inflammatory cells in Asian populations, to find the main cause of the CD immune response pathway.
Footnotes
Manuscript source: Unsolicited manuscript
Specialty type: Gastroenterology and hepatology
Country of origin: China
Peer-review report classification
Grade A (Excellent): 0
Grade B (Very good): B
Grade C (Good): C
Grade D (Fair): 0
Grade E (Poor): 0
Supported by the National Natural Science Foundation of China, No. 81570503.
Conflict-of-interest statement: The authors declare that they have no conflicts of interest to this work.
Peer-review started: November 3, 2017
First decision: November 14, 2017
Article in press: November 27, 2017
P- Reviewer: Gazouli M, Triantafillidis JK S- Editor: Gong ZM L- Editor: Wang TQ E- Editor: Huang Y
Contributor Information
Na Li, Department of Gastroenterology, Zhongda Hospital, Affiliated Hospital of Southeast University, Nanjing 210009, Jiangsu Province, China; Clinical Medical School of Southeast University, Nanjing 210009, Jiangsu Province, China.
Rui-Hua Shi, Department of Gastroenterology, Zhongda Hospital, Affiliated Hospital of Southeast University, Nanjing 210009, Jiangsu Province, China.
References
- 1.Strober W, Fuss I, Mannon P. The fundamental basis of inflammatory bowel disease. J Clin Invest. 2007;117:514–521. doi: 10.1172/JCI30587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zheng JJ, Shi XH, Zhu XS, Huangfu Z, Guo ZR. A comparative study of incidence and prevalence of Crohn 7S disease in mainland China in different periods. Zhonghua Neike Zazhi. 2011;50:597–600. [PubMed] [Google Scholar]
- 3.Chen HZ, Zhong NS. 2013. Internal Medicine. People's Medical Publishing House; pp. 567–569. [Google Scholar]
- 4.Seksik P. [Gut microbiota and IBD] Gastroenterol Clin Biol. 2010;34 Suppl 1:S44–S51. doi: 10.1016/S0399-8320(10)70020-8. [DOI] [PubMed] [Google Scholar]
- 5.Cobrin GM, Abreu MT. Defects in mucosal immunity leading to Crohn’s disease. Immunol Rev. 2005;206:277–295. doi: 10.1111/j.0105-2896.2005.00293.x. [DOI] [PubMed] [Google Scholar]
- 6.Calderón-Gómez E, Bassolas-Molina H, Mora-Buch R, Dotti I, Planell N, Esteller M, Gallego M, Martí M, Garcia-Martín C, Martínez-Torró C, et al. Commensal-Specific CD4(+) Cells From Patients With Crohn’s Disease Have a T-Helper 17 Inflammatory Profile. Gastroenterology. 2016;151:489–500.e3. doi: 10.1053/j.gastro.2016.05.050. [DOI] [PubMed] [Google Scholar]
- 7.Papadakis KA, Targan SR. Current theories on the causes of inflammatory bowel disease. Gastroenterol Clin North Am. 1999;28:283–296. doi: 10.1016/s0889-8553(05)70057-1. [DOI] [PubMed] [Google Scholar]
- 8.Lewis K, Caldwell J, Phan V, Prescott D, Nazli A, Wang A, Soderhölm JD, Perdue MH, Sherman PM, McKay DM. Decreased epithelial barrier function evoked by exposure to metabolic stress and nonpathogenic E. coli is enhanced by TNF-alpha. Am J Physiol Gastrointest Liver Physiol. 2008;294:G669–G678. doi: 10.1152/ajpgi.00382.2007. [DOI] [PubMed] [Google Scholar]
- 9.Smith P, Siddharth J, Pearson R, Holway N, Shaxted M, Butler M, Clark N, Jamontt J, Watson RP, Sanmugalingam D, et al. Host genetics and environmental factors regulate ecological succession of the mouse colon tissue-associated microbiota. PLoS One. 2012;7:e30273. doi: 10.1371/journal.pone.0030273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gray WN, Boyle SL, Graef DM, Janicke DM, Jolley CD, Denson LA, Baldassano RN, Hommel KA. Health-related quality of life in youth with Crohn disease: role of disease activity and parenting stress. J Pediatr Gastroenterol Nutr. 2015;60:749–753. doi: 10.1097/MPG.0000000000000696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lee D, Albenberg L, Compher C, Baldassano R, Piccoli D, Lewis JD, Wu GD. Diet in the pathogenesis and treatment of inflammatory bowel diseases. Gastroenterology. 2015;148:1087–1106. doi: 10.1053/j.gastro.2015.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Powell JJ, Harvey RS, Ashwood P, Wolstencroft R, Gershwin ME, Thompson RP. Immune potentiation of ultrafine dietary particles in normal subjects and patients with inflammatory bowel disease. J Autoimmun. 2000;14:99–105. doi: 10.1006/jaut.1999.0342. [DOI] [PubMed] [Google Scholar]
- 13.Evans SM, Ashwood P, Warley A, Berisha F, Thompson RP, Powell JJ. The role of dietary microparticles and calcium in apoptosis and interleukin-1beta release of intestinal macrophages. Gastroenterology. 2002;123:1543–1553. doi: 10.1053/gast.2002.36554. [DOI] [PubMed] [Google Scholar]
- 14.Bamias G, Sugawara K, Pagnini C, Cominelli F. The Th1 immune pathway as a therapeutic target in Crohn’s disease. Curr Opin Investig Drugs. 2003;4:1279–1286. [PubMed] [Google Scholar]
- 15.Ferretti M, Casini-Raggi V, Pizarro TT, Eisenberg SP, Nast CC, Cominelli F. Neutralization of endogenous IL-1 receptor antagonist exacerbates and prolongs inflammation in rabbit immune colitis. J Clin Invest. 1994;94:449–453. doi: 10.1172/JCI117345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Parronchi P, Romagnani P, Annunziato F, Sampognaro S, Becchio A, Giannarini L, Maggi E, Pupilli C, Tonelli F, Romagnani S. Type 1 T-helper cell predominance and interleukin-12 expression in the gut of patients with Crohn’s disease. Am J Pathol. 1997;150:823–832. [PMC free article] [PubMed] [Google Scholar]
- 17.Fuss IJ, Heller F, Boirivant M, Leon F, Yoshida M, Fichtner-Feigl S, Yang Z, Exley M, Kitani A, Blumberg RS, et al. Nonclassical CD1d-restricted NK T cells that produce IL-13 characterize an atypical Th2 response in ulcerative colitis. J Clin Invest. 2004;113:1490–1497. doi: 10.1172/JCI19836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sartor RB. Microbial influences in inflammatory bowel diseases. Gastroenterology. 2008;134:577–594. doi: 10.1053/j.gastro.2007.11.059. [DOI] [PubMed] [Google Scholar]
- 19.Kim SC, Tonkonogy SL, Karrasch T, Jobin C, Sartor RB. Dual-association of gnotobiotic IL-10-/- mice with 2 nonpathogenic commensal bacteria induces aggressive pancolitis. Inflamm Bowel Dis. 2007;13:1457–1466. doi: 10.1002/ibd.20246. [DOI] [PubMed] [Google Scholar]
- 20.Westendorf AM, Fleissner D, Deppenmeier S, Gruber AD, Bruder D, Hansen W, Liblau R, Buer J. Autoimmune-mediated intestinal inflammation-impact and regulation of antigen-specific CD8+ T cells. Gastroenterology. 2006;131:510–524. doi: 10.1053/j.gastro.2006.05.015. [DOI] [PubMed] [Google Scholar]
- 21.Kappeler A, Mueller C. The role of activated cytotoxic T cells in inflammatory bowel disease. Histol Histopathol. 2000;15:167–172. doi: 10.14670/HH-15.167. [DOI] [PubMed] [Google Scholar]
- 22.Toldi G, Munoz L, Herrmann M, Schett G, Balog A. The effects of Kv1.3 and IKCa1 channel inhibition on cytokine production and calcium influx of T lymphocytes in rheumatoid arthritis and ankylosing spondylitis. Immunol Res. 2016;64:627–631. doi: 10.1007/s12026-015-8683-8. [DOI] [PubMed] [Google Scholar]
- 23.Sydora BC, Macfarlane SM, Walker JW, Dmytrash AL, Churchill TA, Doyle J, Fedorak RN. Epithelial barrier disruption allows nondisease-causing bacteria to initiate and sustain IBD in the IL-10 gene-deficient mouse. Inflamm Bowel Dis. 2007;13:947–954. doi: 10.1002/ibd.20155. [DOI] [PubMed] [Google Scholar]
- 24.Veldhoen M, Moncrieffe H, Hocking RJ, Atkins CJ, Stockinger B. Modulation of dendritic cell function by naive and regulatory CD4+ T cells. J Immunol. 2006;176:6202–6210. doi: 10.4049/jimmunol.176.10.6202. [DOI] [PubMed] [Google Scholar]
- 25.Bettelli E, Carrier Y, Gao W, Korn T, Strom TB, Oukka M, Weiner HL, Kuchroo VK. Reciprocal developmental pathways for the generation of pathogenic effector TH17 and regulatory T cells. Nature. 2006;441:235–238. doi: 10.1038/nature04753. [DOI] [PubMed] [Google Scholar]
- 26.Mucida D, Park Y, Kim G, Turovskaya O, Scott I, Kronenberg M, Cheroutre H. Reciprocal TH17 and regulatory T cell differentiation mediated by retinoic acid. Science. 2007;317:256–260. doi: 10.1126/science.1145697. [DOI] [PubMed] [Google Scholar]
- 27.Kitani A, Xu L. Regulatory T cells and the induction of IL-17. Mucosal Immunol. 2008;1 Suppl 1:S43–S46. doi: 10.1038/mi.2008.51. [DOI] [PubMed] [Google Scholar]
- 28.Kinugasa T, Sakaguchi T, Gu X, Reinecker HC. Claudins regulate the intestinal barrier in response to immune mediators. Gastroenterology. 2000;118:1001–1011. doi: 10.1016/s0016-5085(00)70351-9. [DOI] [PubMed] [Google Scholar]
- 29.Carrier Y, Yuan J, Kuchroo VK, Weiner HL. Th3 cells in peripheral tolerance. II. TGF-beta-transgenic Th3 cells rescue IL-2-deficient mice from autoimmunity. J Immunol. 2007;178:172–178. doi: 10.4049/jimmunol.178.1.172. [DOI] [PubMed] [Google Scholar]
- 30.Mebius RE, Rennert P, Weissman IL. Developing lymph nodes collect CD4+CD3- LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity. 1997;7:493–504. doi: 10.1016/s1074-7613(00)80371-4. [DOI] [PubMed] [Google Scholar]
- 31.Kumar V. Innate lymphoid cells: new paradigm in immunology of inflammation. Immunol Lett. 2014;157:23–37. doi: 10.1016/j.imlet.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 32.Cherrier M, Eberl G. The development of LTi cells. Curr Opin Immunol. 2012;24:178–183. doi: 10.1016/j.coi.2012.02.003. [DOI] [PubMed] [Google Scholar]
- 33.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, et al. Innate lymphoid cells--a proposal for uniform nomenclature. Nat Rev Immunol. 2013;13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 34.Huang XL, Guo XY, Jiang HX. Role of Th22 cells in inflammatory, autoimmune diseases and tumors. Shijie Huaren Xiaohua Zazhi. 2014;22:1812–1819. [Google Scholar]
- 35.Takayama N, Nishimura S, Nakamura S, Shimizu T, Ohnishi R, Endo H, Yamaguchi T, Otsu M, Nishimura K, Nakanishi M, et al. Transient activation of c-MYC expression is critical for efficient platelet generation from human induced pluripotent stem cells. J Exp Med. 2010;207:2817–2830. doi: 10.1084/jem.20100844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo X, Qiu J, Tu T, Yang X, Deng L, Anders RA, Zhou L, Fu YX. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity. 2014;40:25–39. doi: 10.1016/j.immuni.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Guo T. The immunological pathogenesis of inflammatory bowel disease and treatment. Foreign Med Immunol Pathol. 2003;26:190–194. [Google Scholar]
- 38.Matricon J, Barnich N, Ardid D. Immunopathogenesis of inflammatory bowel disease. Self Nonself. 2010;1:299–309. doi: 10.4161/self.1.4.13560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hepworth MR, Fung TC, Masur SH, Kelsen JR, McConnell FM, Dubrot J, Withers DR, Hugues S, Farrar MA, Reith W, et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4+ T cells. Science. 2015;348:1031–1035. doi: 10.1126/science.aaa4812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. Mechanism of IFN-gamma-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell. 2005;16:5040–5052. doi: 10.1091/mbc.E05-03-0193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.De Luca A, Zelante T, D’Angelo C, Zagarella S, Fallarino F, Spreca A, Iannitti RG, Bonifazi P, Renauld JC, Bistoni F, et al. IL-22 defines a novel immune pathway of antifungal resistance. Mucosal Immunol. 2010;3:361–373. doi: 10.1038/mi.2010.22. [DOI] [PubMed] [Google Scholar]
- 42.Sonnenberg GF, Monticelli LA, Alenghat T, Fung TC, Hutnick NA, Kunisawa J, Shibata N, Grunberg S, Sinha R, Zahm AM, et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science. 2012;336:1321–1325. doi: 10.1126/science.1222551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hansen-Wester I, Chakravortty D, Hensel M. Functional transfer of Salmonella pathogenicity island 2 to Salmonella bongori and Escherichia coli. Infect Immun. 2004;72:2879–2888. doi: 10.1128/IAI.72.5.2879-2888.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Di Sabatino A, Pender SL, Jackson CL, Prothero JD, Gordon JN, Picariello L, Rovedatti L, Docena G, Monteleone G, Rampton DS, et al. Functional modulation of Crohn’s disease myofibroblasts by anti-tumor necrosis factor antibodies. Gastroenterology. 2007;133:137–149. doi: 10.1053/j.gastro.2007.04.069. [DOI] [PubMed] [Google Scholar]
- 45.Beddy D, Mulsow J, Watson RW, Fitzpatrick JM, O’Connell PR. Expression and regulation of connective tissue growth factor by transforming growth factor beta and tumour necrosis factor alpha in fibroblasts isolated from strictures in patients with Crohn’s disease. Br J Surg. 2006;93:1290–1296. doi: 10.1002/bjs.5431. [DOI] [PubMed] [Google Scholar]
- 46.Mannon PJ, Fuss IJ, Dill S, Friend J, Groden C, Hornung R, Yang Z, Yi C, Quezado M, Brown M, et al. Excess IL-12 but not IL-23 accompanies the inflammatory bowel disease associated with common variable immunodeficiency. Gastroenterology. 2006;131:748–756. doi: 10.1053/j.gastro.2006.06.022. [DOI] [PubMed] [Google Scholar]
- 47.Lauro ML, Burch JM, Grimes CL. The effect of NOD2 on the microbiota in Crohn’s disease. Curr Opin Biotechnol. 2016;40:97–102. doi: 10.1016/j.copbio.2016.02.028. [DOI] [PMC free article] [PubMed] [Google Scholar]