

Abstract
This report describes the metabolic glycoengineering (MGE) of intracellular esterase activity in human colon cancer (LS174T) and Chinese hamster ovary (CHO) cells. In silico analysis of the carboxylesterases CES1 and CES2 suggested that these enzymes are modified with sialylated N-glycans, which are proposed to stabilize the active multimeric forms of these enzymes. This premise was supported by treating cells with butanolylated ManNAc to increase sialylation, which in turn increased esterase activity. By contrast, hexosamine analogues not targeted to sialic acid biosynthesis (e.g., butanoylated GlcNAc or GalNAc) had minimal impact. Measurement of mRNA and protein confirmed that esterase activity was controlled through glycosylation and not through transcription or translation. Azide-modified ManNAc analogues widely used in MGE also enhanced esterase activity and provided a way to enrich targeted “glycoengineered” proteins (such as CES2), thereby providing unambiguous evidence that the compounds were converted to sialosides and installed into the glycan structures of esterases as intended. Overall, this study provides a pioneering example of the modulation of intracellular enzyme activity through MGE, which expands the value of this technology from its current status as a labeling strategy and modulator of cell surface biological events.
Introduction
Manipulation of glycosylation – especially in living cells – remains difficult with the notable exception of metabolic glycoengineering (MGE).[1,2] This methodology, also referred to as metabolic oligosaccharide engineering (MOE[1,3]), relies on the substrate promiscuity of mammalian biosynthetic pathways that process hexosamines and fucose to incorporate chemically modified versions of these monosaccharides into larger glycans. In vivo applications of this methodology were largely pioneered by Reutter's research group, colleagues, and collaborators who demonstrated 25 years ago that exogenously-supplied, non-natural sugars – exemplified by conversion of ManNAc analogues bearing extended N-acyl alkyl into the corresponding sialosides – could be introduced into biosynthetic pathways to modulate glycosylation in living cells and animals.[4]
The MGE field was taken in a new direction when the Bertozzi lab demonstrated that bioorthogonal chemical functional goups could be installed into the glycocalyx by targeting sialic acid biosynthesis with “ManNLev,” a ketone-derivatized ManNAc analogue. Subsequently, MGE has been used to label glycans with a bevy of chemically-tagged monosaccharides including the canonical click chemistry coupling partners (azides or alkynes, [5-7]), thiols,[8] or photo-activated functional groups (diazirines or phenylarylazides[9,10]). The unstated assumption in many of these studies has been that MGE labeling has negligible impact on biological activity; one example of this premise is provided by the visualization of glycans in zebrafish embryos,[11] which had no reported impact on the subsequent development. In other cases, it is explicitly assumed – or at least desired – that MGE not alter the natural functions and interactions of modified glycans; for example, when using this strategy to capture and identify binding partners of glycoconjugates in situ.[9,10,12]
The Reutter group, however, showed that given the correct circumstances MGE can modulate biological activity. In some cases, exemplified by the inhibition of viral infection,[13-16] the mechanism is straightforward. In particular, the N-acyl group of sialic acid is in a spatially confined region of the hemagglutinin binding pocket of the influenza virus.[17] As a consequence, the extra bulk found at the N-acyl position of most MGE analogues sterically reduces viral binding to an analogue-treated host cell. In other cases, for example changes to the developmental fate of embryonic neural cells,[18] human stem cells, [8] or the behavior of cancer cells[19,20] remain unexplained at a detailed mechanistic level. (A reasonable assumption, based on the role of glycosylation in modulating signal pathway activity, is that MGE perturbs receptor ligand interactions.)
Despite consolidating (although often circumstantial) evidence that MGE can modulate multiple types of biological receptor ligand interactions, this approach has not yet been shown to directly govern intracellular enzyme activity in living cells. We were interested whether this possibility – which would extend Reutter's legacy of manipulating biological activity via MGE in a new direction – was feasible. In this report we use esterases, a diverse and versatile group of enzymes that play many important roles maintaining healthy cell physiology, to demonstrate the ability of MGE to modulate enzyme activity. Esterases provide an appropriate test platform for this purpose based on two decades of evidence that the biosynthesis, enzymatic activity, and pharmacological properties of two secreted esterases – acetylcholinesterase and butyrylcholinesterase – are modulated by glycosylation.[21-26] Subsequent reports that intracellular esterases – in particular carboxylesterases 1 and 2 (CES1 and 2) – also are glycosylated[27-30] raised the intriguing possibility that the activity of these enzymes, and by extension cellular functions that depend on esterase activity, could be modulated by manipulating their glycosylation.
Specifically, we focused on reports that an N-linked oligosaccharide is located at the interface between CES1 monomers[28,31] to propose that MGE analogues that increase sialylation could modulate intracellular esterase activity. This premise – as described in detail later in this manuscript – is based on the hypothesis that sialic acid stabilizes the multimeric assembly of CES1 and thereby increases its activity; in particular, CES is not active in monomeric form but instead requires trimeric (and/or hexameric) assembly.[27,28,31,32] Based on this information, we proposed that cells treated with “high flux” ManNAc analogues that increase sialic acid production would promote and stabilize the formation of active multimeric forms of these esterases and increase their activity. Our results, which are consistent with this hypothesis, establish a new paradigm by which MGE can be used to manipulate enzyme activity, and ultimately, provide cells with novel and enhanced properties.
Materials and Methods
Prediction of N-glycan site occupancy using NetNGlyc 1.0
To provide a perspective on the glycosylation status of common esterases found in human cells, we used an in silico tool (NetNGlyc 1.0, see the Supporting Information) to predict N-glycan site occupancy.
Modeling of N-Glycosylation of CES1 with Discovery Studio
The “Build Fragment” and molecule sketching capabilities of Accelrys Discovery Studio 3.5 Visualizer were used to generate models of insect and human glycans into the crystal structure from PDB ID 1YA8 after water and other small molecules were removed. Initial models were minimized with a Dreiding-like forcefield to optimize geometries.
Cell culture and incubation with sugar analogues
LS174T (ATCC® CL-188) cells were grown in Minimal Essential Medium Eagle supplemented with heat-inactivated 10 % fetal bovine serum (FBS), 100 units/mL penicillin and 100 μg/mL streptomycin (pen/strep), 1x (100x dilution of 100x stock) non-essential amino acids, and 110 mg/mL sodium pyruvate (Invitrogen, Carlsbad, CA). Chinese hamster ovary (CHO) cells - wild-type (WT, gift from the Betenbaugh laboratory, JHU), K1 (ATCC® CCL-61), Pro-5 (ATCC® CRL-1781), and Lec2 (gift from the Betenbaugh laboratory, JHU) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with heat-inactivated 10% FBS, penicillin/streptomycin (Pen/Strep) solution, and 2.0 mM L-glutamine. SCFA hexosamine analogues were synthesized and characterized using published methods (Bu4ManNAc,[33] 1,3,4-O-Bu3ManNAc and 3,4,6-O-Bu3ManNAc,[34,35] and 1,3,4-O-Bu3ManNAz and 3,4,6-O-Bu3ManNAz,[36] For treatment of cells, analogues (from a 100 mM stock solution in ethanol (EtOH)) were added to 6-well tissue culture plates and the EtOH was allowed to evaporate. Cells were suspended in 2.0 mL of culture media and then added to each well and incubated with the sugar analogues for time periods up to 48 h.
Intracellular esterase activity
Analogue-treated cells were detached using 1.0 mL of enzyme free cell dissociation buffer (Millipore) and washed with D-PBS (Mediatech). Cells (200,000 cells) were then incubated with 600 μL of 1.0 μM carboxy fluorescein diacetate (CFDA; Marker Gene Technologies) or 400 μM resorufin acetate (Marker Gene Technologies) in the dark at room temperature under gentle vibration for 40 min or 10 min respectively. An aliquot (150 μL) of each sample was then loaded into a black 96 well plate and the fluorescence level was read using the Synergy™ 2 Multi-Mode Microplate Reader (Biotek). For CFDA, the reader was set to excite 475 nm and read at 530 nm for CFDA whereas for resorufin acetate, the reader was set to excite at 540 nm and read at 590 nm.
Tunicamycin treatment
LS174T cells were incubated with 0.125 μg/mL of tunicamycin (Sigma-Aldrich) for 24 h. The appropriate volume of sugar analogue was then added to each well to give concentrations ranging from 0 to 100 μM and the cells were then further incubated for 48 h at which time intracellular esterase activity was measured as described above.
Reverse transcription-polymerase chain reaction (rt-PCR)
Total RNA was isolated using Trisol® reagents (Gibco BRL) and reversed transcribed using the high capacity RNA-to-cDNA kit (Applied Biosystems). PCR amplifications were performed using the following primer pairs: GAPDH, 5′-AGGTCATCCCTGAGCTGAACGG-3′ (sense) and 5′-CGCCTGCTTCACCACCTTCTTG-3′ (antisense); acetylcholinesterase (ACHE), 5′-TTCGCCCAGCGACTGATGCG-3′ (sense) and 5′-GCCATTGTGGGGCCTTGGGG-3′ (antisense); butrylcholinesterase (BCHE), 5′-AGATCCATAGTGAAACGGTGGGCA-3′ (sense) and 5′-TGAAGACAGGCCAGCTTGTGCT-3′ (antisense); carboxyl ester lipase (CEL), 5′-GTGGGTTCGTGGAAGGCGTCAA-3′ (sense) and 5′-GGAACCCAAGGGGGCCGACA-3′ (antisense); carboxylesterase 1 (CES1), 5′- AGCGCAGGGCGGTAACTCTGG-3′ (sense) and 5′-CGAGGACGGATGCCCTGCCCA-3′; carboxylesterase 3 (CES3), 5′-GGGCGTGAAGGGCACAGACC-3′ (sense) and 5′- CAGTGCTGGCATCCCGCACA-3′ (antisense); carboxylesterase 7 (CES7), 5′-GCTCGATATCACAGAGAAGGAGCCA-3′ (sense) and 5′-CCGGTTCGAGCAAAGGTAGCCC-3′ (antisense). qRT-PCR was performed using the Step-One Plus Real-Time PCR system (Applied Biosystems) with the thermocycling conditions of 50 °C for 30 min, 95 °C for 10 min followed by 40 cycles of 95 °C for 15 s and 60 °C for 1.0 min. PCR amplification for carboxyesterase 2 (CES2) was performed using TaqMan® Gene Expression Assay (cat. No. 4331182, Applied Biosystems).
Western blot analysis
Proteins were immunodetected using the following commercial antibodies: anti-CES1 (AV41877, Sigma-Aldrich), anti-CES2 (514-621, Novus Bio.) and HRP-linked anti-rabbit antibody (Cell Signaling).
Intracellular immunofluorescence staining and flow cytometry
Analogue treated cells were detached using 1.0 mL of enzyme free cell dissociation buffer and washed with D-PBS. The cells were then washed once with permeation buffer (D-PBS, 2.0 % heat inactivated FBS, 0.2 % sodium azide, 0.5 % saponin), washed once with super-permeation buffer (3 parts permeation buffer to 1 part FBS) and fixed using 3.75 % formaldehyde in DPBS for 15 min. Cells were then washed twice with staining buffer (PBS, 2.0 % heat inactivated FBS, 0.2 % sodium azide) and incubated with human-specific anti-CES1 (Sigma-Aldrich) primary antibody in permeation buffer for 1.0 h at room temperature. Cells were then washed with permeation buffer, incubated with FITC-linked anti-rabbit antibody (Sigma-Aldrich) secondary antibody for 1.0 h and washed again with permeation buffer. The cells were then re-suspended in PBS and analyzed using the C6 Flow Cytometry system (Accuri Cytometers).
Determination of sialic acid levels in analogue-treated cells
Cells were incubated for 48 h and then lysed by subjecting them to three freeze-thaw cycles. Lysates were analyzed using an adaptation of the periodate-resorcinol assay[37] to a 96 well plate format[38] to quantify total sialic acid. For each experiment, test samples were compared to a standard curve created using known concentrations of N-acetylneuraminic acid (Invitrogen).
Click chemistry enrichment of azide-labeled proteins in ManNAz-analogue treated cells
Cells (5.0 × 107) were incubated for 48 h with 0 μM or 100 μM 1,3,4-O-Bu3ManNAz after which the cells were collected using 3.0 mL enzyme free cell dissociation buffer and washed once with 3.0 mL D-PBS. Cells were then lysed in 1.0 mL lysis buffer (Click Chemistry Tools from the Bioconjugate Technology Company). Lysates were pelleted by centrifugation at 10,000 g for 5.0 min and the supernatant was collected. Copper catalyst solution (1.0 mL of a 2x solution, Bioconjugate Technology Company) and 0.2 mL of washed alkyne agarose resin (Bioconjugate Technology Company) were added and mixed on an end-over-end rotary shaker for 48 h at 4 °C. The resin was then washed 10 times with agarose wash buffer (Click Chemistry Tools) using a spin column and centrifuging at 1,000 g for 1.0 min, the column then was washed 5 times with 8 M urea/100 mM Tris pH-8 and finally washed 5 times with 20% acetonitrile. The washed resin was then resuspended in 0.2 mL elution buffer (100 mM sodium phosphate with 2% (v/v) hydrazine) and incubated for 120 min at room temperature: during this step the hydrazine cleaves a Dde linker thus releasing the bound proteins from the agarose resin. The eluted solution was collected by centrifugation and analyzed by western blotting.
Statistical analysis
Data was expressed as means ± standard error (SEM). Statistical significance was determined using one way ANOVA to compare means of different samples with control. The null hypothesis was rejected in cases where p-values were < 0.05.
Results
In silico analyses of hCES glycosylation
N-Linked glycosylation of human carboxylesterases has been established for over a decade[27,28] (and our in silico [see the Supporting Information] analysis provides conceptual support for the idea that almost all human esterases bear this modification) but even recent publications[29,30] leave many facets of CES1 and CES2 glycosylation unexplained. For example, only one or at most, two GlcNAc residues of the glycan located proximal to the peptide chain, and an undefined sialic acid, were observed in crystal structures of human CES1 (hCES1) produced in Spodoptera frugiperda insect cells. We assumed that the sialic acid was a legitimate part of an N-glycan chain attached to hCES1 because the free monosaccharide form of this sugar is only present in very low concentrations in biological milieu and was particularly unexpected in insect cells, where sialylation remains controversial. Our team has employed MGE techniques to verify sialic acid production in insect cells[39]) giving us confidence that hCES1 could legitimately be sialylated. Levels of sialic acid in insect cells nevertheless were unlikely to high enough to allow the monosaccharide form of this sugar to co-crystallize with the protein (especially after its enrichment); based on this reasoning, we discounted the possibility that the observed sialic acid was due to co-crystallization artifacts. We instead pursued the hypothesis that undefined sialic acid residue was the terminal residue of an N-glycan structure attached to ASN79, which is the one known N-glycosylation site of hCES1, by conducting a series of in silico modeling simulations to establish or discount the feasibility of various scenarios.
We first asked whether a glycan attached to trimeric hCES1 that comprises one crystal unit could reach an adjacent hCES1. As shown in Figure 1A, the distance between a proximal GlcNAc and the closest sialic acid (as observed in the crystal structures) on an adjacent crystal unit is ∼45 Å. This distance, combined with the length of the longest known S. frugiperda N-glycan (as well as the majority of human N-glycans[40]) of < 25 Å when fully extended, convinced us that sialic acid observed in crystal structures could only be appended to a N-glycan chain located within the same trimeric unit of CES1. Accordingly, we focused on a single CES1 trimer (Figure 1B) and showed (i) the location of the proximal GlcNAc attached to ASN79 at the interface of two CES1 peptide chains along with the nearby sialic acid observed in the crystal structures. We then (ii) modeled a typical N-glycan structure at the interface between the two CES1 subunits, which showed that the glycan is not large enough for its terminal sialic acid(s) to extend to either of the two more distant sites where this sugar is seen in the crystal structure (e.g., to the sialic acid shown in (ii)). Finally, the glycan is not large enough – and is sufficiently far removed from the enzyme's active site (as indicated in (iii)) – to interfere with substrate binding. Therefore, any impact that the glycan has on catalysis – as described subsequently in this report – must occur via allosteric effects or stabilization of the trimeric form of the enzyme.
Figure 1. In silico evaluation of CES glycosylation.
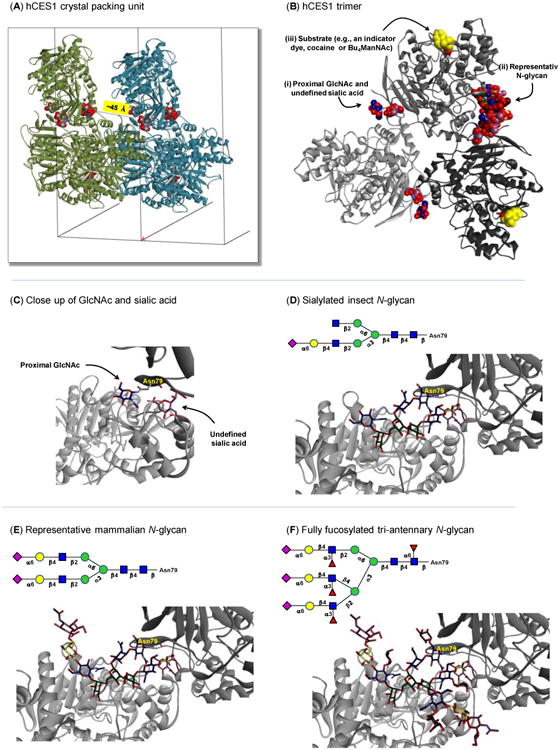
(A) The hCES1 trimer and its closest crystal partner in the periodic image along the a axis. The a unit cell vector length is 55.78 Å. Glycan residues are shown as space-filling models and colored by element. The distance from a GlcNAc residue to the closest sialic acid residue is ∼45 Å. (B) The hCES1 trimer with each monomer shown in a different shade of gray. Non-peptide residues and molecules are shown as space-filling models and colored by element. Carbon atoms for saccharide residues are shown in standard CFG colors. Carbon atoms for the substrate analogues are shown in yellow and the GlcNAc of this glycan that is proximal to the peptide chain as well as the undefined sialic acid are shown at higher resolution in Panel C. (C) Protein subunits A and B of hCES1 are shown using dark gray and light gray ribbons, respectively, from the published crystal structure,[28] with Asn79 (labeled) along with the proximal GlcNAc and undefined sialic acid shown in the position observed in the published crystal structure. (D–F) Protein subunits A and B of hCES1 are shown along with appended glycans representative of (D) an low-abundance complex-type insect N-glycan w/ sialic acid, (E) a representative mammalian bis-sialylated complex N-glycan, and (F) a tri-antennary fully fucosylated mammalian N-glycan (All carbohydrate atoms in C–F are shown with sticks and colored by element with representations of glycans relevant to hCES activity determined using the CFG standard symbol notation generated using GlycoWorkbench.[74] Models in A–F were created with Accelrys Discovery Studio Visualizer 3.5.)
Based on the results presented in Figure 1A and B, the sialic acid in the crystal structure of hCES1 could only have been part of an N-glycan attached to spatially closest ASN97. The portion of the N-glycan chain ostensibly connecting the proximal GlcNAc and terminal sialic acid was not observed in the crystal form of hCES1; the “missing” monosaccharide residues can be explained by the motion or heterogeneity of the glycan chain (glycans are often removed from proteins before crystallization to avoid this type of ambiguity). In the absence of definite structural data, we modeled various representative N-glycans to determine if the proximal GlcNAc reasonably could be connected to the nearby sialic acid as part of the same oligosaccharide chain. We conducted these simulations by “pinning” the GlcNAc located at the reducing end of representative N-glycan structures to ASN79 of the protein and the sialic acid to the position observed in the hCES1 crystal structures.
To select representative glycans, we noted that Spodoptera frugiperda cells (the source of crystallized CES1 in published studies) preferentially produce high mannose N-glycans that lack sialic acid; these glycans are incompatible with the presence of sialic acid in crystal structures. Therefore we turned to Drosophila (another type of insect) cells that are now known to sialylate a small fraction of glycan structures (< 10%) and can process ManNAc analogues into sialosides.[39] Based on this capacity for insect cells to produce at least a limited amount of sialic acid, we appended a complex-type arthropod N-glycan[41] with a single sialic acid on the α3 antennary branch to ASN79 of hCES1 and modeled whether this structure could fill the missing density between the observed peptide-attached GlcNAc and the ambiguous sialic acid. The glycan was minimized to correct for deviations in idealized geometries, and there were no geometric or steric difficulties in closing a loop from GlcNAc to the terminal sialic acid (Figure 1D). This result was not surprising because of ample “open space” extending away from the multimeric hCES1 structure, allowing the glycan to adopt a configuration that positioned the sialic acid as observed in the published crystal structures.
Next, the α6 antennary branch of the singly sialylated arthropod glycan was extended by a β-Gal-6α-Neu5Ac unit to represent complex-type biantennary N-glycans found in mammalian cells. This glycan ensured the relevance of the modeling efforts to the experiments subsequently reported in this paper conducted in human and Chinese hamster ovary (CHO) cells. This result showed that there are no constraints on extending the second sialylated arm of the N-glycan away from the surface of the CES1 complex while positioning the other terminal sialic acid at as observed in the crystal structure (Figure 1E). Finally, a fully fucosylated, triantennary N-glycan was modeled; although this particular glycan structure is not abundant we used it to represent an extreme case to ask whether the extra steric bulk from added branching introduced by the third arm or by the fucose residues could prevent correct positioning of the sialic acid. The results of this simulation (Figure 1F) showed that the terminal sialic acid again could be properly positioned. Together, the modeling results illustrated that the hCES1 trimer can accommodate a broad range of N-glycans (including numerous examples not shown here) that properly position both the peptide-attached GlcNAc and the sialic acid whose source was undefined in the published crystal structures.
These in silico results were significant from several perspectives. First, they confirmed that the sialic acid observed in hCES crystal structures legitimately could be a part of an appended N-glycan (as compared to a serendipitously co-crystallized soluble sialic acid monosaccharide). Second, to be observed in the crystal structure, the sialic acid is necessarily confined in space and relatively motionless, unlike the connecting monosaccharide residues that were not observed in the crystal structure presumably because of a combination of rapid unconfined motion and the heterogeneity of intervening N-glycan chains. Third, once the minimal requirement of having a sialylated α3 antennary branch is met (as evidenced by the arthropod glycan shown in Figure 1D), substitution with various additional N-glycans including those found in mammals is not detrimental for hCES1 subunit assembly because the “extra” oligosaccharide chains can be oriented away from the globular protein into open space. Finally, the ability to observe sialic acid in the crystal structures implied that sialylated N-glycans were over-represented in crystallized CES1. In particular, because only a relatively small fraction insect glycans are sialylated, this sugar would not be observed in the hCES1 structures if the crystallized material was representative of the overall glycan profile produced in S. frugiperda cells; instead, sialylated species are enriched in the crystal structures leading us to speculate that sialylation stabilizes the trimeric assembly of hCES1.
In brief, the in silico modeling results presented above allowed us to formulate a hypothesis wherein sialic acid plays a stabilizing role in hCES1 trimer formation. To experimentally evaluate this idea, we increased esterase sialylation using MGE. Importantly, we had at our disposal ManNAc analogues that were expected to increase esterase activity via sialylation as well as hexosamines (e.g., GlcNAc and GalNAc analogues) that have minimal impact on sialic acid. The control analogues instead modulate other aspects of glycosylation (e.g., N-glycan branching by increasing flux through the hexosamine biosynthetic pathway) that our modeling suggested was not important for esterase activity.
Increased esterase activity was observed in ManNAc analogue treated cells
The hypothesis that sialic acid-bearing species become enriched during crystal formation of CES1 was interesting to us because multimeric assembly of this enzyme enhances catalytic activity.[27,28] Accordingly, we predicted that a ManNAc analogue-based MGE approach that enhances flux through the sialic acid pathway[33,34] and in turn increases protein sialylation,[36,42] would increase esterase activity (Figure 2). As a caveat, because these analogues lack plasma membrane transporters, a common strategy to increase cellular uptake has been to add ester-linked short chain fatty acids (SCFA) such as acetate[43,44] and more recently n-butyrate[33,45] to the core sugar. The SCFA groups increase the lipophilicity of the sugar and facilitate passive diffusion across the plasma membrane thereby bypassing the need for transporter-mediated intake. Once inside a cell, esterases – presumably CES1 and CE2 that detoxify a wide range of both narcotics and pharmaceuticals – remove the SCFAs over a period of a few hours allowing the sugar to intercept the targeted glycosylation pathway;[46] at the outset of our experiments we were concerned that this “added” burden on esterase activity might offset any MGE-mediated increase when monitored by colorimetric (or other) assays (the esterase processing of SCFA-hexosamine analogues is discussed extensively in our previous publications.[34,44-46] Therefore, as a precaution in case a substantial portion of intracellular esterase activity was diverted for hexosamine analogue processing a high basal level of activity was desired, leading us to select the human colon adenocarcinoma LS174T line for testing because of the high level of esterase activity reported for these cells.[47]
Figure 2. Overview of esterase processing of SCFA-derivatized ManNAc analogues and the subsequent impact on esterase activity.
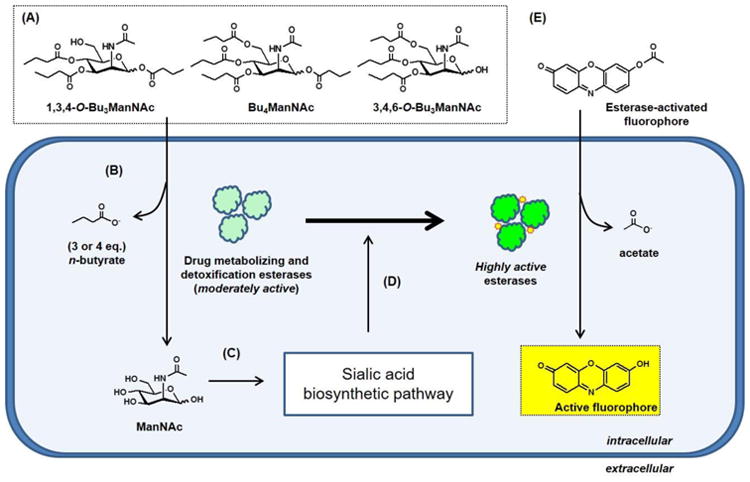
(A) Structures of butanoylated ManNAc analogues that readily enter cells through membrane diffusion where (B) drug metabolizing esterases remove their ester-linked n-butyrate groups to generate unmodified ManNAc, which (C) acts as a feedstock that increased metabolic flux through the sialic acid biosynthetic pathway. (D) Increased sialylation is proposed to increase the activity of intracellular esterases as measured by (E) resorufin acetate (shown) or carboxy fluorescein diacetate (CFDA) assays; experimental results for both of these ester-activated fluorophores are shown in Figure 3.
To test the prediction that increased esterase activity could be achieved through MGE, indicator dye assays that rely on esterase activation of fluorophores were used to evaluate the impact of “high flux” butanoylated ManNAc analogues that increase sialylation[34,36,42] The fluorescent dyes used to measure esterase activity in our assays are widely employed in live/dead cell assays. Therefore a second possible pitfall was that, if the cells being tested experienced reduced viability due to analogue cytotoxicity,[33] the ensuing decrease in esterase activity could offset the predicted sialic acid-driven increase. To avoid this possibility, two steps were taken. First, the analogues were used at sub-growth inhibitory doses (i.e., < 150 μM for Bu4ManNAc and 1,3,4-O-Bu3ManNAc and < 25 μM for 3,4,6-O-Bu3ManNAc) and further, these three analogues have different toxicities[34] allowing esterase activity in cells treated with the more toxic analogues (Bu4ManNAc and 3,4,6-O-Bu3ManNAc) and the less toxic analogue 1,3,4-O-Bu3ManNAc to be deconvoluted by comparing these three compounds if necessary. These concerns, however, were rendered moot when independent assays using different indicator dyes (carboxy fluorescein diacetate, CFDA; Figure 3A or resorufin acetate; Figure 3B) both showed unambiguous increases in esterase-generated fluorescent signal. These results confirmed our prediction that increased sialylation enhances esterase activity, and furthermore, they demonstrated that the increased activity was sufficiently robust to outweigh secondary effects that theoretically could diminish esterase activity (in other words, the observed results – if anything – understate the magnitude of increased esterase activity achieved through increased sialylation).
Figure 3. Intracellular esterase activity activities measured by activation of fluorescent dyes.
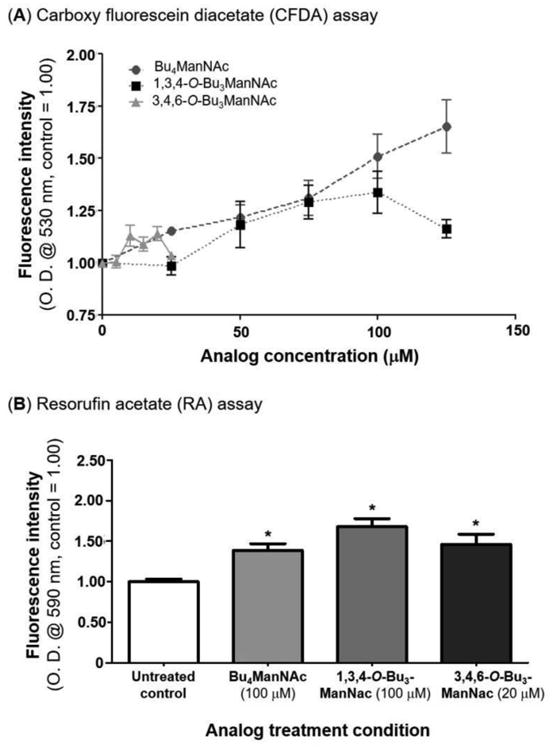
Fluorescence levels are shown for LS174T cells treated with Bu4ManNAc, 1,3,4-O-Bu3ManNAc, and 3,4,6-O-Bu3ManNAc analyzed by two assays (A) carboxy fluorescein diacetate and (B) resorufin acetate. * - indicates a P value of < 0.05 in a two-tailed t test.
Increased esterase activity is not correlated with gene expression or protein levels
After confirming the prediction that MGE could be exploited to increase esterase activity through sialylation, we conducted a several experiments to gain evidence for the mechanism proposed in Figure 1 and the accompanying discussion. Our first concern was that the butanoylated analogues, that can function as epigenetic modulators from released butyrate that acts as an histone deacetylase inhibitor,[8] was changing the transcription and subsequent translation of one or more esterases. At the outset of these experiments, the most likely esterase targeted by MGE in the LS174T cell line was thought to be CES2 because, although both CES1 and CES2 are implicated in drug detoxification and metabolism,[48] robust CES1 expression is restricted to liver- and monocyte-derived cell lineages[49,50] However, because of conservation of important catalytic and structural residues[51] and similar glycosylation patterns, we expected that MGE would affect both carboxylesterases in a similar manner and initially focused on testing CES2.
We found that neither gene expression or protein levels could explain in the increased esterase activity observed in ManNAc analogue-treated cells by evaluating CES2 transcription by qRT-PCR (Figure 4A) and protein levels by western blot assays (Figure 4B), respectively. Although Bu4ManNAc increased transcription roughly equivalent to the magnitude of increased esterase activity (e.g., to 150 to 200 % of controls, Figure 3), the two tributanoylated analogues 1,3,4-O-Bu3ManNAc, and 3,4,6-O-Bu3ManNAc, did not increase transcription or translation of CES2 despite enhancing esterase activity as effectively as Bu4ManNAc (Figure 3). Therefore we concluded that the Bu4ManNAc-driven increase in carboxylesterase mRNA levels was either unrelated, unnecessary, or at most only a partial explanation for the observed increase in esterase activity.
Figure 4. Transcriptional and translational control of esterases in LS174T cells.
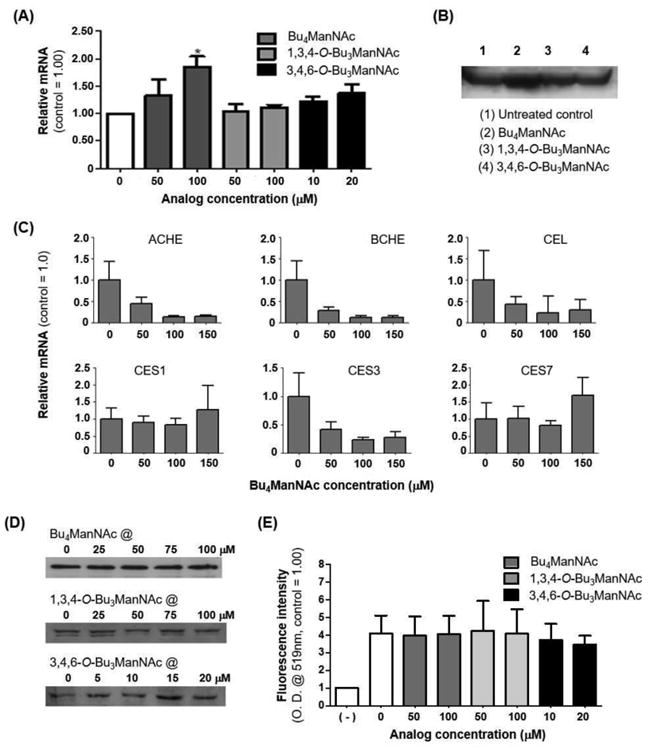
Quantification of CES2 protein and RNA levels from LS174T cells via (A) qRT-PCR and (B) western blot respectively showed that overall levels of this esterase remained unchanged upon sugar analogue treatment, with the exception of Bu4ManNAc treatment where the signal increased. (C) qRT-PCR showed that mRNA levels for ACHE, BCHE, CEL, and CES3 in LS174T cells treated with Bu4ManNAc were significantly down-regulated at higher concentrations while CES1 and CES7 were not affected. Quantification of CES1 from LS174T cells via (D) western blots and (E) immunofluorescence assays using flow cytometry showed that the overall levels of this esterase remained the same upon sugar analogue treatment. * - indicates a P value of < 0.05 in a two-tailed t test.
An alternate explanation for the discrepancy between the inconsistent changes in transcription and translation of CES2 and the consistent overall increase in overall cellular esterase activity was that esterases other than CES2 played an unexpectedly large role in responses to ManNAc analogues in LS174T cells. Accordingly, we expanded analysis to additional esterases expressed in human cells and found that mRNA levels for several of these enzymes were either measurably down-regulated (e.g., for AChE, BChE, CEL, and CES3) or not affected (e.g., for CES1 and CES7) by Bu4ManNAc (Figure 4C). Although the down-regulation of several of these enzymes ran counter to the overall increase in esterase activity observed in ManNAc analogue-treated cells, the specific esterases that experienced reduced mRNA levels have limited relevance to drug detoxification and were not expected to process SCFA-hexosamine analogues or indicator dyes and therefore were largely irrelevant to the activity results presented in Figure 3.
An unanticipated finding of the mRNA results was that CES1 was transcribed in the LS174T cell line; this finding was not expected because, as mentioned above, CES1 is primarily associated with liver and monocyte-derived cell lineages; however, epigenetic control of CES1 in cancer[52] provides a potential explanation for the expression of CES1 in the prostate LS174T line. To further probe the significance of CES1 transcription in these cells, western blot (Figure 4D) and immunofluorescence (Figure 4E) assays confirmed expression at the protein level. Therefore, instead of, or in addition to CES2, which started out as the most likely candidate for SCFA-hexosamine processing in LS174T cells, CES1 also likely plays a major role in the results presented in this paper and indeed, shows no change in transcription or translation upon treatment with any of the three analogues tested.
Increased esterase activity was experimentally confirmed to be linked to glycosylation
Having discounted transcription or translation as the controlling mechanism for esterase activity in ManNAc analogue-treated cells, we next conducted a series of experiments to confirm that the glycosylation of these enzymes contributed to the observed increased activity in cells. First, cells were treated with tunicamycin, which blocks N-glycosylation and is known to inhibit liver carboxylesterase activity;[53] a similar result was obtained in the LS174T prostate cell line where tunicamycin prevented the ManNAc analogue-driven increases in esterase activity. Second, evaluation of an expanded panel of hexosamine analogues revealed that compounds that did not alter sialylation had minimal if any impact on esterase activity. Specifically, GalNAc and GlcNAc analogues (Bu4GalNAc and Bu4GlcNAc, Figure 5B) had only a negligible effect on esterase activity compared to Bu4ManNAc (Figure 5C) even though these analogues increase the production of UDP-GlcNAc, which increases N-glycan branching.[54] However, as is indicated in Figure 1, highly branched N-glycans can be oriented away from trimeric CES1 and therefore increased branching is not necessarily expected to modulated catalytic activity. Together, the tunicamycin and Gal/GlcNAc analogue experiments demonstrated that N-glycans were necessary but not sufficient to bolster esterase activity in metabolically-glycoengineered cells.
Figure 5. Expanded analysis of the effects of glycosylation on esterase activity.
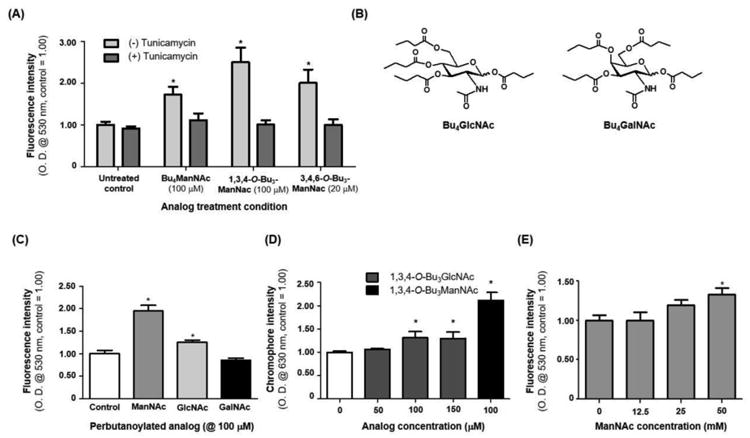
(A) Tunicamycin treated LS174T cells that were also treated with ManNAc analogues did not experience an increase in esterase activity. (B) Structures of Bu4GlcNAc and Bu4GalNAc analogues, which (C) elicited a small or no increase in esterase activity (respectively) in LS174T cells (Bu4ManNAc was included in this experiment as a positive control). (D) The periodate resorcinol assay was used to measure sialic acid content in 1,3,4-O-Bu3GlcNAc-treated cells, with 1,3,4-O-Bu3ManNAc again used as the positive control (no increase in sialic acid levels was observed for GalNAc analogue treated cells, data not shown). (E) Intracellular esterase activity in cells treated with increasing concentrations of unmodified ManNAc. * - indicates a P value of < 0.05 in a two-tailed t test.
A minimal, but measurable, increase in esterase activity was detected in cells treated with butanoylated GlcNAc, which was commensurate with a small increase in sialic acid detected in these cells (Figure 5D); the increased sialic acid is likely a consequence of GlcNAc epimerization to ManNAc, the committed feedstock for sialic acid biosynthesis.[55] A final piece of evidence implicating sialic acid as the critical factor responsible for enhanced esterase activity was provided by media supplementation with unmodified ManNAc (Figure 5E). High levels of ManNAc (e.g., 30 to 75 mM.[56] which preclude routine use of the unmodified form of this sugar in MGE experiments) were able to recapitulation the activity-enhancing effects of the butanoylated ManNAc analogues. This result is significant because the exclusive metabolic role of ManNAc is to supply flux into the sialic acid pathway. Therefore, unlike the butanoylated analogues that can also have epigenetic effects[35,45]), this experiment unambiguously implicated sialylation as a critical and necessary determinant of enhanced esterase activity.
Increased sialylation also modulates esterase activity in CHO cells
Esterase activity next was tested in Chinese hamster ovary (CHO) cells because we were interested whether ManNAc analogue-mediated enhancement of esterase activity held across cell lines and species. First, we tested wild-type (WT) CHO cells and found an increase in activity (e.g., roughly two-fold, Figure 6A) similar in magnitude to human cells (Figure 3). Importantly, CHO mutant lines were available that allowed us to confirm that N-glycosylation (in general) and sialic acid (more specifically) was required to enhance esterase activity. Specifically, a comparison of three mutant CHO lines (summarized in Table 1) yielded results consistent with the human LS174T cells. First, the K1 line, which has mutations that ablate both N-glycan production and sialylation, did not respond to ManNAc analogue treatment (Figure 6B). Second, the CHO5 line, which has a reduced (but measurable) capacity for sialylation of N-glycans, showed an attenuated but still measurably increased level of esterase activity upon analogue supplementation. Finally, the Lec-2 line, which has defective Golgi transport of CMP-Neu5Ac that can be rescued by increased flux through the sialic acid pathway (e.g., via ManNAc analogue supplementation in the current experiments), had a robust increase in esterase activity similar in magnitude to the WT cells. An interesting facet of the CHO cell experiments was that although all ManNAc analogues solicited qualitatively similar responses, the two analogues with “whole molecule” activity (e.g., Bu4ManNAc and 3,4,6-O-Bu3ManNAc as we discuss elsewhere [34,35,57]) enhanced esterase activity to a greater extent than the “high flux only” 1,3,4-O-Bu3ManNAc. [36] Therefore, it is noteworthy that while ManNAc – and by extension sialylation – is a critical component of esterase modulation in the CHO cells, other factors (beyond the scope of this report) may also contribute to this phenomenon.
Figure 6. Analysis of esterase activity in various analogue-treated CHO cell lines.
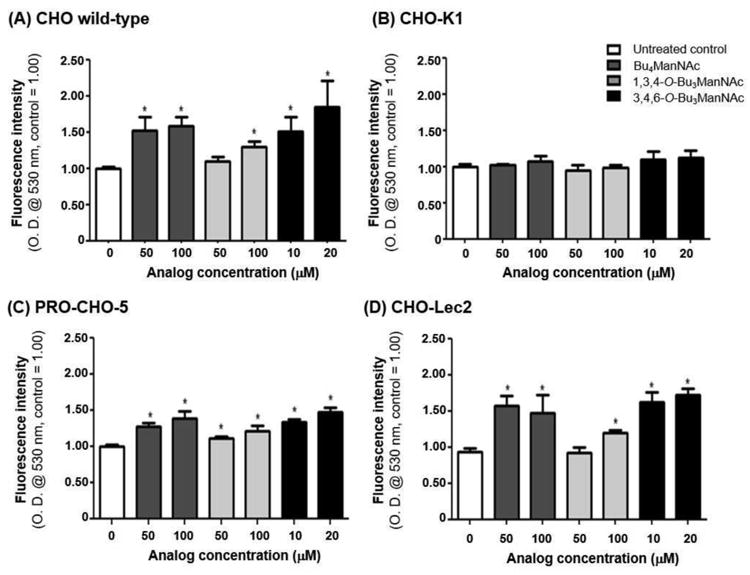
(A) Wild-type cells that treated with ManNAc analogues experienced an increase in esterase activity while (B) CHO-K1 cells did not experience a change in esterase activity under any condition; (C) Pro-CHO-5 cells demonstrated a less dramatic increase; and (D) CHO-Lec2 cells showed an increase in esterase activity comparable to the wild-type cells. * - indicates a P value of < 0.05 in a two-tailed t test.
Table 1. Synopsis of CHO cell line experiments.
| Cell line | Deficiency | Esterase activity | Possible explanation |
|---|---|---|---|
| K1 | - Lack asparagine-linked glycosylation homolog ALG13 which heterodimerizes with ALG14 to catalyze the second step in N-linked glycosylation[75] - Lack ST6Gal sialyltransferase responsible for formation of α-2,6-linked sialic acid terminal bonds [76] |
No increase | - Absence of ALG 13 could lead to N-glycosylation deficiencies in some of the esterases - Formation of α-2,6 sialic acid terminal bonds plays an important role in the changes in esterase activity |
| CHO-5 | - Lack β4-Galactosyl transferase-6 essential for galactosylation of N-glycans[77] - This may reduce N-glycan terminal sialic acid as they are attached to galactose |
Small increase | - Reduced N-glycan capacity for addition of terminal sialic acid reduces the increase in esterase activity |
| Lec2 | - Defective CMP-sialic acid transporters required to transport sialic acid into the Golgi complex [78] | Increase | - Increase in cytoplasm sialic acid concentration makes up for the defective transporter |
Azido-modified sialic acids modulate esterase activity and are incorporated into CES2
Moving beyond the use of ManNAc analogues to increase expression of natural sialic acid, as has been reported so far in this paper, many examples of MGE employ non-natural analogues modified with abiotic chemical functionalities. A common modification of this type involves substitution of the natural N-acetyl group of ManNAc with an N-azido group (e.g., Bu4ManNAz or 1,3,4-O-Bu3ManNAz, Figure 7A); these analogues install the corresponding azido-modified sialic acid into cell surface glycans. We tested Bu4ManNAz and 1,3,4-O-Bu3ManNAz and found that both compounds increased esterase activity; furthermore the magnitude of the increase of up to ∼2-fold was similar to natural ManNAc (Figure 7B). Moreover, the kinetics of the increase in esterase activity (e.g., over a ∼ 2 day period) was consistent with the incorporation of various (e.g,. ketone-[56] or azide-derivatized[36]) ManNAc analogues into glycoproteins, which are distinctively slower than treatment with 1,3,4-O-Bu3ManNAc, which more quickly introduces flux of the natural core sugar into the pathway (i.e., increased esterase activity is observed within an hour (Figure 7C), consistent with previously-observed kinetics for sialylation with the similar analogue Ac4ManNAc[58]).
Figure 7. Time course of ManNAc analogue induced changes and the effect of azido-labeled analogues on esterase activity.
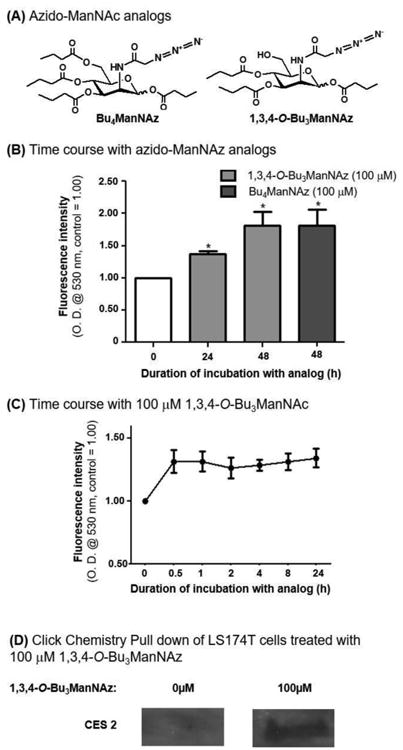
(A) Structures of Bu4ManNAz and 1,3,4-O-Bu3ManNAz, which (B) elicited an increase in esterase activity after 24 and 48 h of incubation of LS174T cells with the respective analogues. (C) Time course of esterase activity upon treatment with 1,3,4-O-Bu3ManNAc. (D)Western blotting of proteins pulled down by click chemistry shows incorporation of Sia5Az into CES2 in 1,3,4-O-Bu3ManNAz treated LS174T cells. * - indicates a P value of < 0.05 in a two-tailed t test.
The ability of azide-modified ManNAc analogues to increase esterase activity was significant because MGE labeling strategies, including the use of 1,3,4-O-Bu3ManNAc and 1,3,4-O-Bu3ManNAz used in this study, broadly impact large numbers (e.g., dozens to hundreds) of cellular sialoglycoconjugates.[42,59] Therefore 1,3,4-O-Bu3ManNAc treatment, an analogue that increases the natural Neu5Ac form of sialic acid, makes it difficult to verify that CES1, CES2, or any another esterase, had been directly affected by MGE. By contrast 1,3,4-O-Bu3ManNAz installs the non-natural azido-form of sialic acid into glycoconjugates making it unambiguous that they have been altered by MGE, moreover the modified species can by enriched in click chemistry-based “pull-down” experiments.[59] This strategy allows identification of specific glycoproteins labeled with exogenously-supplied MGE sugar analogues. By using this approach, analogue incorporation into CES2 was confirmed by the western blot results shown in Figure 7D, where signal was only detected in ManNAz analogue-treated samples. This experiment unambiguously establishes that ManNAc analogues were incorporated into the N-glycans of CES2; furthermore the “1,3,4” pattern of analogue butanoylation avoids off-target effects[34,35,57] (e.g., changes to esterase transcription or translation as shown for 1,3,4-O-Bu3ManNAc in Figure 4A, B, D, & E) while increasing sialylation.[36] Consequently, the results obtained with 1,3,4-O-Bu3ManNAz further support the hypothesis outlined in Figure 1 where sialic acid – presumably including non-natural forms such as “Sia5Az” – play an important role in stabilizing the highly active, trimeric form of this esterase.
Discussion
Humans express a diverse and versatile group of esterases that play many important roles ranging from maintaining healthy cell physiology in the human body, facilitating therapeutic drug efficacy, to biotechnology applications. The current study focuses on situations where increased intracellular esterase activity in living cells is desired, including the intracellular activation of short chain fatty acid-modified hexosamine analogues the facilitate MGE. Interestingly, when ManNAc analogues that enhance sialylation are used, esterase activity required to activate these compounds is also enhanced, in essence providing a simple feed-forward metabolic network. As of now, it is not clear that esterase processing is a rate limiting step in MGE (later steps in the conversion of ManNAc to Neu5Ac or to CMP-Neu5Ac are more likely candidates[60,61]) but this result is important because it rules out esterase depletion or hijacking as a mechanism for the almost ubiquitous (albeit often mild) cytotoxicity of fully acylated hexosamines used in MGE.
A potentially more significant outcome of MGE-enhanced esterase activity is treatment of pathologies characterized by deficient esterase activity ranging from congenital mutations that lower esterase activity[62] to neurological disorders such as Alzheimer's disease.[63] Another example is cancer where reduced esterase activity hinders monocyte-mediated killing of tumor cells[64] and down-regulation of esterases in certain types of cancers hinders treatment with esterase-activated prodrugs[65] such as CPT-11, a semisynthetic derivative of camptothecin, which is activated to SN-38 by carboxylesterases.[47,66] Esterase-activated drugs include acetyl salicylic acid (aspirin).[67] Esterase activity also provides critical protection against intoxicants and narcotics such as cocaine; the intracellular degradation/detoxification of these compounds is primarily carried out by CES1 and CES2.[68,69]
Because of their impact on cell physiology, and ultimately human health, it is not surprising that many efforts to modulate esterase activity have been reported; in particular, multiple esterase inhibitors have been developed.[70] By comparison esterase activators are virtually unknown despite the potential utility of such compounds to ameliorate diseases caused by insufficient esterase activity, to augment prodrug processing, or to protect against narcotic overdoses. To begin to fill this void, we demonstrated MGE approaches that use monosaccharide analogues to increase sialic acid production and thereby elicit a concomitant increase in intracellular esterase activity due to enhanced sialylation of the N-glycans found on CES1 and CES2, the two enzymes most strongly tied to drug metabolism and detoxification in humans.
Another scenario where the correlation between sialic acid and esterase activity may prove important is provided by nascent efforts to produce recombinant versions of these enzymes (e.g., carboxylesterase CES2[71]), which could exploit ManNAc analogue supplementation to maximize activity. Finally, the ability of butanoylated ManNAc analogues to down-regulate transcription of AChE, BChE, CEL, and CES3 (as shown in Figure 4) may be valuable in several disease contexts; one example is attenuation of increased BChE activity associated with Alzheimer's disease.[72,73] Overall, this paper demonstrates that hexosamine analogues that manipulate glycosylation can be used as small molecule, pharmacologically relevant agents to fill the current void consisting of a lack of activators for intracellular drug processing and detoxification esterases.
Supplementary Material
Acknowledgments
Funding for this project was provided by the National Institutes of Health (NCI R01CA112314, NCI 1F32 CA189246, and the Willowcroft Foundation).
References
- 1.Campbell CT, Sampathkumar SG, Weier C, Yarema KJ. Mol Biosys. 2007;3:187–194. doi: 10.1039/b614939c. [DOI] [PubMed] [Google Scholar]
- 2.Du J, Meledeo MA, Wang Z, Khanna HS, Paruchuri VDP, Yarema KJ. Glycobiology. 2009;19:1382–1401. doi: 10.1093/glycob/cwp115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dube DH, Bertozzi CR. Curr Opin Chem Biol. 2003;7:616–625. doi: 10.1016/j.cbpa.2003.08.006. [DOI] [PubMed] [Google Scholar]
- 4.Kayser H, Zeitler R, Kannicht C, Grunow D, Nuck R, Reutter W. J Biol Chem. 1992;267:16934–16938. [PubMed] [Google Scholar]
- 5.Saxon E, Bertozzi CR. Cell surface engineering by a modified Staudinger reaction. Science. 2000;287:2007–2010. doi: 10.1126/science.287.5460.2007. [DOI] [PubMed] [Google Scholar]
- 6.Hsu TL, Hanson SR, Kishikawa K, Wang SK, Sawa M, Wong CH. Proc Natl Acad Sci U S A. 2007;104:2614–2619. doi: 10.1073/pnas.0611307104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hong V, Steinmetz NF, Manchester M, Finn MG. Bioconjug Chem. 2010;21:1912–1916. doi: 10.1021/bc100272z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sampathkumar SG, Li AV, Jones MB, Sun Z, Yarema KJ. Nat Chem Biol. 2006;2:149–152. doi: 10.1038/nchembio770. [DOI] [PubMed] [Google Scholar]
- 9.Han S, Collins BE, Bengtson P, Paulson JC. Nat Chem Biol. 2005;1:93–97. doi: 10.1038/nchembio713. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka Y, Kohler JJ. J Am Chem Soc. 2008;130:3278–3279. doi: 10.1021/ja7109772. [DOI] [PubMed] [Google Scholar]
- 11.Laughlin ST, Baskin JM, Amacher SL, Bertozzi CR. Science. 2008;320:664–667. doi: 10.1126/science.1155106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bond MR, Zhang H, Vu PD, Kohler JJ. Nat Protoc. 2009;4:1044–1063. doi: 10.1038/nprot.2009.85. [DOI] [PubMed] [Google Scholar]
- 13.Herrler G, Gross HJ, Milks G, Paulson JC, Klenk HD, Brossmer R. Acta Histochem Suppl. 1990;40:39–41. [PubMed] [Google Scholar]
- 14.Kelm S, Paulson JC, Rose U, Brossmer R, Schmid W, Bandgar BP, Schreiner E, Hartmann M, Zbiral E. Eur J Biochem. 1992;205:147–153. doi: 10.1111/j.1432-1033.1992.tb16762.x. [DOI] [PubMed] [Google Scholar]
- 15.Keppler OT, Herrmann M, von der Lieth CW, Stehling P, Reutter W, Pawlita M. Biochem Biophys Res Commun. 1998;253:437–442. doi: 10.1006/bbrc.1998.9650. [DOI] [PubMed] [Google Scholar]
- 16.Keppler OT, Horstkorte R, Pawlita M, Schmidt C, Reutter W. Glycobiology. 2010;11:11R–18R. doi: 10.1093/glycob/11.2.11r. [DOI] [PubMed] [Google Scholar]
- 17.Watowich SJ, Skehel JJ, Wiley DC. Structure. 1994;2:719–731. doi: 10.1016/s0969-2126(00)00073-3. [DOI] [PubMed] [Google Scholar]
- 18.Schmidt C, Stehling P, Schnitzer J, Reutter W, Horstkorte R. J Biol Chem. 1998;i:19146–19152. doi: 10.1074/jbc.273.30.19146. [DOI] [PubMed] [Google Scholar]
- 19.Du J, Che PL, Aich U, Tan E, Kim HJ, Sampathkumar SG, Yarema KJ. Bioorg Med Chem Lett. 2011;i:4980–4984. doi: 10.1016/j.bmcl.2011.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Du J, Che PL, Wang ZY, Aich U, Yarema KJ. Biomaterials. 2011;32:5427–5437. doi: 10.1016/j.biomaterials.2011.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Weikert T, Ebert C, Rasched I, Layer PG. J Neurochem. 1994;63:318–325. doi: 10.1046/j.1471-4159.1994.63010318.x. [DOI] [PubMed] [Google Scholar]
- 22.Saez-Valero J, Barquero M, Marcos A, McLean CA, Small DH. J Neurol Neurosurg Psych. 2000;69:664–667. doi: 10.1136/jnnp.69.5.664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Kolarich D, Weber A, Pabst M, Stadlmann J, Teschner W, Ehrlich H, Schwarz HP, Altmann F. Proteomics. 2008;8:254–263. doi: 10.1002/pmic.200700720. [DOI] [PubMed] [Google Scholar]
- 24.Schneider JD, Castilho A, Neumann L, Altmann F, Loos A, Kannan L, Mor TS, Steinkellner H. Biotechnol J. 2013;9:501–510. doi: 10.1002/biot.201300229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schneider JD, Marillonnet S, Castilho A, Gruber C, Werner S, Mach L, Klimyuk V, Mor TS, Steinkellner H. Plant Biotechnol J. 2014;12:832–839. doi: 10.1111/pbi.12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xu ML, Luk WK, Lau KM, Bi CW, Cheng AW, Gong AG, Lin H, Tsim KW. J Mol Neurosci. 2015;57:486–491. doi: 10.1007/s12031-015-0629-z. [DOI] [PubMed] [Google Scholar]
- 27.Bencharit S, Morton CL, Hyatt JL, Kuhn P, Danks MK, Potter PM, Redinbo MR. Chem Biol. 2003;10:341–349. doi: 10.1016/s1074-5521(03)00071-1. [DOI] [PubMed] [Google Scholar]
- 28.Fleming CD, Bencharit S, Edwards CC, Hyatt JL, Tsurkan L, Bai F, Fraga C, Morton CL, Howard-Williams EL, Potter PM, Redinbo MR. J Mol Biol. 2005;352:165–177. doi: 10.1016/j.jmb.2005.07.016. [DOI] [PubMed] [Google Scholar]
- 29.Arena de Souza V, Scott DJ, Nettleship JE, Rahman N, Charlton MH, Walsh MA, Owens RJ. PLoS One. 2015;10:e0143919. doi: 10.1371/journal.pone.0143919. 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alves M, Lamego J, Bandeiras T, Castro R, Tomás H, Coroadinha AS, Costa J, Simplícioa AL. Biochem Biophys Rep. 2016;5:105–110. doi: 10.1016/j.bbrep.2015.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bencharit S, Morton CL, Xue Y, Potter PM, Redinbo MR. Nat Struct Biol. 2003;i:349–356. doi: 10.1038/nsb919. [DOI] [PubMed] [Google Scholar]
- 32.Fleming CD, Edwards CC, Kirby SD, Maxwell DM, Potter PM, Cerasoli DM, Redinbo MR. Biochemistry. 2003;46:5063–5071. doi: 10.1021/bi700246n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim EJ, Sampathkumar SG, Jones MB, Rhee JK, Baskaran G, Yarema KJ. J Biol Chem. 2004;279:18342–18352. doi: 10.1074/jbc.M400205200. [DOI] [PubMed] [Google Scholar]
- 34.Aich U, Campbell CT, Elmouelhi N, Weier CA, Sampathkumar SG, Choi SS, Yarema KJ. ACS Chem Biol. 2008;3:230–240. doi: 10.1021/cb7002708. [DOI] [PubMed] [Google Scholar]
- 35.Elmouelhi N, Aich U, Paruchuri VDP, Meledeo MA, Campbell CT, Wang JJ, Srinivas R, Khanna HS, Yarema KJ. J Med Chem. 2009;52:2515–2530. doi: 10.1021/jm801661m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Almaraz RT, Aich U, Khanna HS, Tan E, Bhattacharya R, Shah S, Yarema KJ. Biotechnol Bioeng. 2012;109:992–1006. doi: 10.1002/bit.24363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jourdian GW, Dean L, Roseman S. J Biol Chem. 1971;246:430–435. [PubMed] [Google Scholar]
- 38.Yarema KJ, Goon S, Bertozzi CR. Nat Biotechnol. 2001;19:553–558. doi: 10.1038/89305. [DOI] [PubMed] [Google Scholar]
- 39.Granell AE, Palter KB, Akan I, Aich U, Yarema KJ, Betenbaugh MJ, Thornhill WB, Recio-Pinto E. ACS Chem Biol. 2011;6:1287–1295. doi: 10.1021/cb200238k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Werz DB, Ranzinger R, Herget S, Adibekian A, Von der Lieth CW, Seeberger PH. ACS Chem Biol. 2007;2:685–691. doi: 10.1021/cb700178s. [DOI] [PubMed] [Google Scholar]
- 41.Tiemeyer M, Selleck SB, Esko JD. In: Varki A, Cummings RD, Esko JD, et al., editors. Cold Spring Harbor Laboratory Press; Cold Spring Harbor (NY): 2009. [PubMed] [Google Scholar]
- 42.Almaraz RT, Tian Y, Bhattarcharya R, Tan E, Chen SH, Dallas MR, Chen L, Zhang Z, Zhang H, Konstantopoulos K, Yarema KJ. Mol Cell Proteomics. 2012 doi: 10.1074/mcp.M112.017558. 10.1074/mcp.M1112.017558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sarkar AK, Fritz TA, Taylor WH, Esko JD. Proc Natl Acad Sci U S A. 1995;92:3323–3327. doi: 10.1073/pnas.92.8.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jones MB, Teng H, Rhee JK, Baskaran G, Lahar N, Yarema KJ. Biotechnol Bioeng. 2004;85:394–405. doi: 10.1002/bit.10901. [DOI] [PubMed] [Google Scholar]
- 45.Sampathkumar SG, Jones MB, Meledeo MA, Campbell CT, Choi SS, Hida K, Gomutputra P, Sheh A, Gilmartin T, Head SR, Yarema KJ. Chem Biol. 2006;13:1265–1275. doi: 10.1016/j.chembiol.2006.09.016. [DOI] [PubMed] [Google Scholar]
- 46.Mathew MP, Tan E, Shah S, Bhattacharya R, Meledeo MA, Huang J, Espinoza FA, Yarema KJ. Bioorg Med Chem Lett. 2012;22:6929–6933. doi: 10.1016/j.bmcl.2012.09.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jansen WJ, Zwart B, Hulscher ST, Giaccone G, Pinedo HM, Boven E. Int J Cancer. 1997;70:335–340. doi: 10.1002/(sici)1097-0215(19970127)70:3<335::aid-ijc15>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 48.Fukami T, Yokoi T. Drug Metab Pharmacokinet. 2012;27:466–477. doi: 10.2133/dmpk.dmpk-12-rv-042. [DOI] [PubMed] [Google Scholar]
- 49.Imai T. Human carboxylesterase isozymes: catalytic properties and rational drug design. Drug Metab Pharmacokinet. 2006;21:173–185. doi: 10.2133/dmpk.21.173. [DOI] [PubMed] [Google Scholar]
- 50.Markey GM. J Clin Pathol. 2011;64:107–109. doi: 10.1136/jcp.2010.084657. [DOI] [PubMed] [Google Scholar]
- 51.Sanghani SP, Sanghani PC, Schiel MA, Bosron WF. Protein Pept Lett. 2009;16:1207–1214. doi: 10.2174/092986609789071324. [DOI] [PubMed] [Google Scholar]
- 52.Hori T, Hosokawa M. Xenobiotica. 2010;40:119–128. doi: 10.3109/00498250903431794. [DOI] [PubMed] [Google Scholar]
- 53.Kroetz DL, McBride OW, Gonzalez FJ. Biochemistry. 1993;32:11606–11617. doi: 10.1021/bi00094a018. [DOI] [PubMed] [Google Scholar]
- 54.Dennis JW, Nabi IR, Demetriou M. Cell. 2009;139:1229–1241. doi: 10.1016/j.cell.2009.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Luchansky SJ, Yarema KJ, Takahashi S, Bertozzi CR. J Biol Chem. 2003;278:8036–8042. doi: 10.1074/jbc.M212127200. [DOI] [PubMed] [Google Scholar]
- 56.Yarema KJ, Mahal LK, Bruehl RE, Rodriguez EC, Bertozzi CR. J Biol Chem. 1998;273:31168–31179. doi: 10.1074/jbc.273.47.31168. [DOI] [PubMed] [Google Scholar]
- 57.Wang Z, Du J, Che PL, Meledeo MA, Yarema KJ. Curr Opin Chem Biol. 2009;13:565–572. doi: 10.1016/j.cbpa.2009.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yarema KJ. in Cell Engineering 3. In: Al-Rubeai M, editor. Glycosylation. Vol. 3. Kluwer Academic Publishers; Dordrecht, The Netherlands: 2002. pp. 171–196. [Google Scholar]
- 59.Tian Y, Almaraz RT, Choi CH, Li QK, Saeui C, Li D, Shah P, Bhattacharya R, Yarema KJ, Zhang H. Clin Proteomics. 2015;12 doi: 10.1186/s12014-12015-19083-12018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Jacobs CL, Goon S, Yarema KJ, Hinderlich S, Hang HC, Chai DH, Bertozzi CR. Biochemistry. 2001;40:12864–12874. doi: 10.1021/bi010862s. [DOI] [PubMed] [Google Scholar]
- 61.Viswanathan K, Lawrence S, Hinderlich S, Yarema KJ, Lee YC, Betenbaugh M. Biochemistry. 2003;42:15215–15225. doi: 10.1021/bi034994s. [DOI] [PubMed] [Google Scholar]
- 62.Zhu HJ, Patrick KS, Yuan HJ, Wang JS, Donovan JL, DeVane CL, Malcolm R, Johnson JA, Youngblood GL, Sweet DH, Langaee TY, Markowitz JS. Am J Hum Genet. 2008;82:1241–1248. doi: 10.1016/j.ajhg.2008.04.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schedin-Weiss S, Winblad B, Tjernberg LOT. FEBS J. 2014;281:46–62. doi: 10.1111/febs.12590. [DOI] [PubMed] [Google Scholar]
- 64.Oertel J, Hagner G, Kastner M, Huhn D. Br J Heamatol. 1985;61:171–726. doi: 10.1111/j.1365-2141.1985.tb02886.x. [DOI] [PubMed] [Google Scholar]
- 65.Burkhart DJ, Barthel BL, Post GC, Kalet BT, Nafie JW, Shoemaker RK, Koch TH. J Med Chem. 2006;49:7002–7012. doi: 10.1021/jm060597e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tsuji T, Kaneda N, Kado K, Yokokura T, Yoshimoto T, Tsuru D. J Pharmacobiodyn. 1991;14:341–349. doi: 10.1248/bpb1978.14.341. [DOI] [PubMed] [Google Scholar]
- 67.Lavis LD. Ester bonds in prodrugs. ACS Chem Biol. 2008;3:203–206. doi: 10.1021/cb800065s. [DOI] [PubMed] [Google Scholar]
- 68.Pindel EV, Kedishvili NY, Abraham TL, Brzezinski MR, Zhang J, Dean RA, Bosron WF. J Biol Chem. 1997;272:14769–14775. doi: 10.1074/jbc.272.23.14769. [DOI] [PubMed] [Google Scholar]
- 69.Gao D, Narasimhan DL, Macdonald J, Brim R, Ko MC, Landry DW, Woods JH, Sunahara RK, Zhan CG. Mol Pharmacol. 2009;75:318–323. doi: 10.1124/mol.108.049486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Hatfield MJ, Potter PM. Expert Opin Ther Pat. 2011;21:1159–1171. doi: 10.1517/13543776.2011.586339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lamego J, Cunha B, Peixoto C, Sousa MF, Alves PM, Simplício AL, Coroadinha AS. Appl Microbiol Biotechnol. 2013;97:1161–1173. doi: 10.1007/s00253-012-3994-3. [DOI] [PubMed] [Google Scholar]
- 72.Greig NH, Lahiri DK, Sambamurti K. Int Psychogeriatr. 2002;14(1):77–91. doi: 10.1017/s1041610203008676. [DOI] [PubMed] [Google Scholar]
- 73.Nordberg A, Ballard C, Bullock R, Darreh-Shori T, Somogyi M. Prim Care Companion CNS Disord. 2013;15 doi: 10.4088/PCC.12r01412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Ceroni A, Maass K, Geyer H, Geyer R, Dell A, Haslam SM. J Proteome Res. 2008;7:1650–1659. doi: 10.1021/pr7008252. [DOI] [PubMed] [Google Scholar]
- 75.Xu X, Nagarajan H, Lewis NE, Pan S, Cai Z, Liu X, Chen W, Xie M, Wang W, Hammond S. Nat Biotechnol. 2011;29:735–741. doi: 10.1038/nbt.1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Gao XD, Tachikawa H, Sato T, Jigami Y, Dean NA. J Biol Chem. 2005;280:36254–36362. doi: 10.1074/jbc.M507569200. [DOI] [PubMed] [Google Scholar]
- 77.Lee JH, Sundaram S, Shaper NL, Raju S, Stanley P. J Biol Chem. 2001;276:2001. doi: 10.1074/jbc.M010046200. [DOI] [PubMed] [Google Scholar]
- 78.Eckhardt M, Gotza B, Gerardy-Schahn R. J Biol Chem. 1998;274:20189–20195. doi: 10.1074/jbc.273.32.20189. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.