

Abstract
T‐box transcription factors play essential roles in multiple aspects of vertebrate development. Here, we show that cooperative function of BRACHYURY (T) with histone‐modifying enzymes is essential for mouse embryogenesis. A single point mutation (TY88A) results in decreased histone 3 lysine 27 acetylation (H3K27ac) at T target sites, including the T locus, suggesting that T autoregulates the maintenance of its expression and functions by recruiting permissive chromatin modifications to putative enhancers during mesoderm specification. Our data indicate that T mediates H3K27ac recruitment through a physical interaction with p300. In addition, we determine that T plays a prominent role in the specification of hematopoietic and endothelial cell types. Hematopoietic and endothelial gene expression programs are disrupted in T Y88A mutant embryos, leading to a defect in the differentiation of hematopoietic progenitors. We show that this role of T is mediated, at least in part, through activation of a distal Lmo2 enhancer.
Keywords: autoregulation, Brachyury, H3K27 acetylation, Lmo2, T‐box factors
Subject Categories: Chromatin, Epigenetics, Genomics & Functional Genomics; Development & Differentiation
Introduction
The Brachyury gene (symbol T) was discovered by phenotypic analysis of a spontaneously occurring mouse mutant characterized by a short tail 1. Positional cloning of the T gene and biochemical analysis of its protein product led to its classification as a DNA‐binding transcription factor 2, 3, 4, 5. Examination of the DNA‐binding domain, termed the T‐box, and comparison to other genes resulted in the discovery of a large family of related T‐box transcription factors, all of which have since been shown to play roles during vertebrate development 6. Furthermore, analysis of the primary protein sequence and structural analysis of several T‐box factors identified additional conserved domains within the T‐box that are not involved in DNA binding, but instead contain important regions for binding to histone‐modifying enzymes 7.
T is transiently expressed in the nascent mesoderm, which originates during gastrulation, and mesodermal cells lose T expression after ingression through the primitive streak and subsequent migration to lateral parts of the embryo. During later embryonic development, T expression is restricted to the caudal end of the embryo, the site of new mesodermal cell generation, and the notochord. Null mutants of T (T 2J/2J) display phenotypes which include a severe posterior axis truncation and loss of notochord tissue, resulting in the formation of a kinked neural tube and 7–9 abnormally formed anterior somites 8, 9, 10, 11. In these embryos, failure of the allantois to properly connect to the chorion results in embryonic lethality at embryonic day 10.0 (E10.0). The activation of T expression by the WNT and FGF signaling pathways, followed by subsequent activation of Wnt and Fgf genes by T, has been shown to maintain the pool of mesodermal progenitor cells during axis elongation 8, 12, 13, 14, 15, 16. Loss of these mesodermal progenitors results in the severe posterior truncation seen in T null embryos and is further corroborated by similar axis truncation phenotypes in Wnt3a and Fgf4/8 mutant mouse embryos 15, 17, 18.
T has also been shown to be expressed in mesodermal progenitors that will give rise to hematopoietic and endothelial cell lineages 19, 20, and there have been reports implicating T in the process of hematopoietic and endothelial cell development. For example, vascular patterning and Flk1 expression in the yolk sac and allantois of T c/T c mutants were found to be disrupted during early mouse development 21. Additionally, siRNA knockdown of T in a model of human embryonic stem cell (ESC) differentiation resulted in a significant decrease in CD45+ definitive hematopoietic cells 22. Despite the preliminary evidence that T plays a role in the development of hematopoietic and endothelial lineages, the mechanism as to how this occurs has not yet been explored in detail.
Members of the T‐box family of transcription factors, including T, have been shown to physically interact with histone‐modifying enzymes, suggesting a role for these transcription factors in regulating genome activity through modulation of the chromatin landscape. Studies performed in T‐lymphocytes using the T‐box factor T‐BET (TBX21) demonstrated that amino acid residues in conserved binding pockets, which reside within the T‐box but face away from the DNA‐binding domain, are important for binding to RBBP5 (retinoblastoma‐binding protein‐5), a protein within the TrxG/MLL (Trithorax group/MLL protein complexes) H3K4 methyltransferase complex, and for association with H3K27 demethylase activity, which is carried out by either KDM6A (UTX) or KDM6B (JMJD3) in vertebrates 7. This interaction was found to be necessary for activation of endogenous target gene expression through deposition of active H3K4me2 and removal of repressive H3K27me3 at target promoters.
We show here that a conserved tyrosine residue previously demonstrated to be important for interaction of T‐box factors with histone‐modifying enzymes is essential for BRACHYURY/T function in vivo and is important for the modulation of chromatin modification at a subset of T occupancy sites in vitro. Furthermore, our data suggest that T plays a role in the deposition of H3K27ac at target genes during mesodermal development, through a physical interaction with the p300 histone acetyltransferase enzyme. Through this mechanism, we show that T regulates the expression of Lmo2 (LIM domain only 2), a key gene essential for hematopoietic and endothelial development, in the early embryo.
Results
Maintenance of T expression requires the conserved Y88 residue
The exchange of the conserved tyrosine residue 88 to an alanine (Y88A) in T was previously shown in vitro to disrupt the interaction of T with multiple components of the histone‐modifying machinery, including RBBP5 and H3K27 demethylase activity, mediated by KDM6A and KDM6B 7. A similarly mutated T‐BET (Y182A) was able to activate reporter expression in transiently transfected cells (a non‐chromatin environment), arguing for the intact DNA binding and transactivation properties of T‐BETY182A. However, activation of endogenous target genes in a chromatin context was impaired due to the loss of a permissive chromatin environment at T‐BET target genes 7. In order to identify whether there is a histone‐specific function of T in early mesoderm development in vivo, we mutated the endogenous T allele in mouse ESCs (mESCs) containing a single wild‐type allele (T 2J/+) to generate the TY88A mutant (T 2J/Y88A) (Fig 1A–C). We directly generated embryos via tetraploid morula aggregation 23 of either T 2J/+ (T WT), T 2J/2J (T null), or T 2J/Y88A (T Y88A) mESCs. During early mesodermal specification (E7.0–E7.5), the expression of T mRNA in T WT and T Y88A mutant embryos by whole‐mount in situ hybridization (WISH) was comparable. However, at E8.0, the expression of T was markedly decreased in T Y88A compared to T WT embryos (Fig 1D). RT‐qPCR analysis of T expression in whole embryos verified the reduction of T in T Y88A mutants compared to T WT (Fig 1D). Hence, while the initiation of T expression is unaffected in T Y88A mutants, expression cannot be maintained, leading to a concordant decrease in T protein levels (Fig 1E). This disruption of T maintenance in T Y88A mutant embryos resulted in a phenotype similar to T null embryos (Fig 1F), with embryos exhibiting a failure in allantoic development, a complete truncation of posterior mesodermal tissue, and lethality between E9.5 and E10.0 (Figs 1F and EV1A).
Figure 1. Y88 is essential for T function.
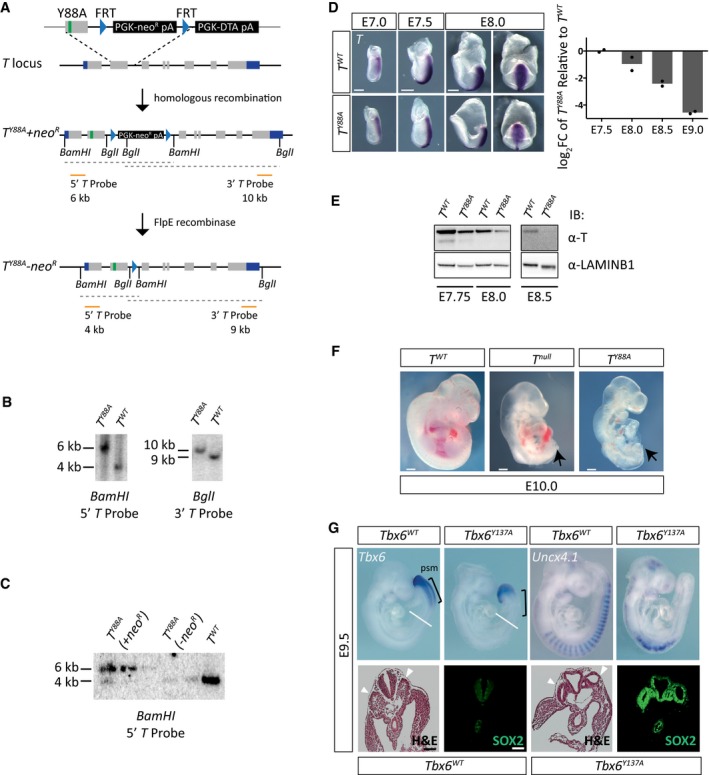
- Schematic illustrating the homologous recombination strategy to insert the Y88A point mutation in the endogenous T locus. The neomycin selection cassette was removed by transient expression of FlpE recombinase. Orange lines depict probes used for Southern blotting to confirm integration of the mutation. Restriction enzyme sites used for Southern blotting are depicted.
- Southern blot of genomic DNA from mESCs to detect homologous recombination of the Y88A mutation using T probes depicted in (A). A 4‐kb and 9‐kb band correspond to the wild‐type T allele using the 5′ and 3′ T probe, respectively, while a 6‐kb band and 10‐kb band correspond to the mutated T Y88A allele containing the neomycin selection cassette.
- Southern blot of genomic DNA from mESCs to detect removal of the neomycin selection cassette after expression of FlpE recombinase using the 5′ T probe.
- WISH analysis of T Y88A mutants shows T expression is normal at early stages, E7.0–7.5, and begins to decrease at E8.0. At E8.0, the left panels show a lateral view of the embryo, while the right panels show a dorsal view. The graph depicts RT‐qPCR analysis of whole embryos to confirm loss of T maintenance in T Y88A mutants compared to T WT controls. The mean of n = 2 biological replicates of pools of three to four embryos is depicted. Scale bar: 200 μm.
- Immunoblot from whole embryo lysates using an α‐T antibody shows a progressive loss of T protein during development in the T Y88A mutant. α‐LAMIN‐B1 was used as a loading control.
- T Y88A mutants resemble T null embryos on the gross morphological level, displaying a complete loss of posterior mesoderm (arrow). Scale bar: 200 μm.
- Tbx6 and Uncx4.1 expression in E9.5 Tbx6 Y137A embryos by WISH. Black brackets indicate the PSM, and white lines indicate the plane of section shown in the lower panels. Hematoxylin and eosin staining of sections through the trunk of E9.5 Tbx6 Y137A embryos revealed the presence of two ectopic neural tubes (arrowheads) at the expense of somite tissue, which was confirmed by immunofluorescent staining for the neural tube marker SOX2. Scale bar: 180 μm.
Figure EV1. Y to A mutations in T and Tbx6 .
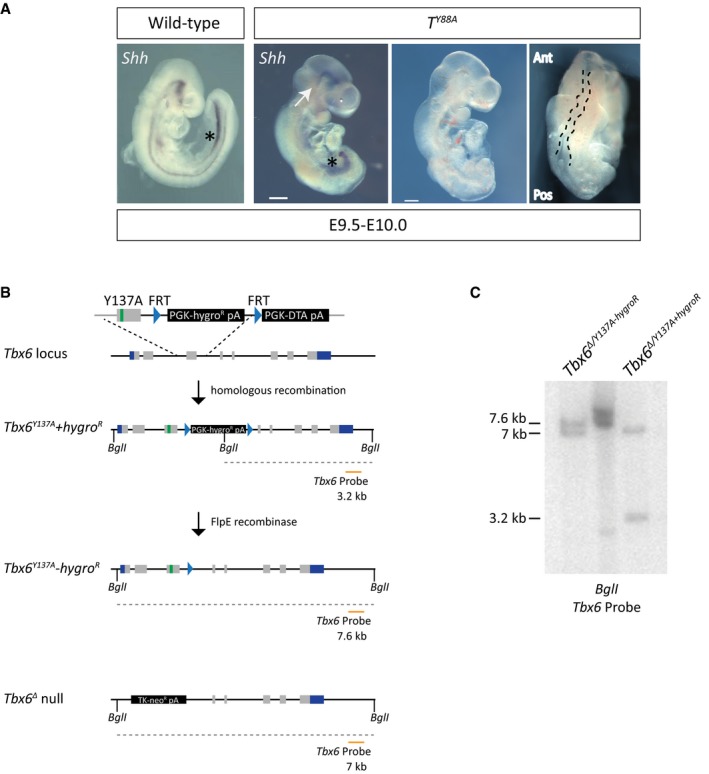
- Whole‐mount in situ hybridization for Shh expression reveals the loss of notochordal structures in T Y88A mutant embryos at E9.5–10.0. White arrows point to the remnants of notochord at the rostral end of the embryo. Asterisks depict Shh staining in the foregut. The panels on the right display the kinked neural tube (outlined with a black dashed line) and truncated posterior mesoderm in the T Y88A mutant embryos at E9.5. The T Y88A mutant embryo is the same as that in Fig 1F. Scale bar: 200 μm. Ant, anterior; Pos, posterior.
- Schematic illustrating the homologous recombination strategy to insert the Y137A point mutation in the endogenous Tbx6 locus. The hygromycin selection was removed by expression of FlpE recombinase. The orange line depicts the probe used for Southern blotting to confirm integration of the mutation and removal of the hygromycin resistance cassette. BglI restriction enzyme sites used for Southern blotting are depicted.
- Southern blot of mESC genomic DNA to detect homologous recombination of the Y137A mutation and removal of the hygromycin cassette using the probe depicted in (B). The 3.2‐kb band corresponds to the Tbx6 Y137A mutant allele containing the hygromycin selection cassette, while the 7.6‐kb band corresponds to the Tbx6 Y137A allele with the selection cassette removed. The 7.0‐kb band corresponds to the Tbx6 Δ null allele.
Considering that the tyrosine residue is conserved among multiple T‐box family members, we hypothesized that the analogous Y to A mutation in another T‐box factor would lead to corresponding phenotypes to those observed in the T Y88A mutant. We chose to focus on the T‐box factor family member TBX6, which is also involved in mesodermal development. TBX6 has previously been shown to play a role in the specification of presomitic mesoderm (PSM) in early embryonic development 24. We generated the analogous Y137A mutation by homologous recombination in Tbx6 +/Δ heterozygous mESCs (Fig EV1B and C) and generated embryos via tetraploid aggregation. Tbx6 Y137A mutant embryos display phenotypes comparable to the Tbx6 null mutant 24, with a similar loss in the maintenance of Tbx6 expression. WISH analysis of Tbx6 expression indicated that while Tbx6 expression is initiated in the caudal end of the embryo, its expression is not maintained in the PSM (Fig 1G). WISH analysis of Uncx4.1, expressed in the caudal half of the developing somite, revealed a profound defect in somitogenesis in the Tbx6 Y137A mutant (Fig 1G). We further confirmed this defect by histological staining and SOX2 immunofluorescence of embryo sections and found that Tbx6 Y137A mutants display ectopic neural tubes at the expense of somite tissue, similar to the Tbx6 null phenotype (Fig 1G). This observation argues that the conserved tyrosine residue in the T‐box family of transcription factors, and presumably interaction with histone‐modifying enzymes, is essential for maintenance of expression and for their function in vivo.
It was previously suggested that Brachyury regulates its expression in a positive autoregulatory loop, in combination with FGF and WNT signaling molecules, to maintain the pool of mesodermal progenitors 16, 25, 26, 27. To test whether the defective maintenance of T expression in T Y88A mutants is a consequence of disruption of this autoregulatory loop, we used a method to differentiate mESCs into the mesodermal cell lineage in vitro, in the presence of defined growth factors 28. The presence of WNT and FGF ligands throughout the in vitro differentiation protocol, both of which are known to activate T expression, was not sufficient to maintain the expression of T in T Y88A mutant mESCs (Fig 2A). In order to determine whether the decrease in T level was a consequence of reduced generation of mesodermal cells, we generated a T reporter BAC. We utilized the RP24‐530D23 BAC, containing the T gene and approximately 160 kb of surrounding genomic sequence. First, to determine whether the BAC contains sufficient regulatory elements to drive normal T expression, we performed a rescue experiment using the wild‐type BAC in the T Y88A mutant background. Genomic integration of the wild‐type T BAC into the T Y88A mutant (T Y88A ‐Tg(RP24‐530D23, T WT neo)) was able to rescue both expression of T and the T Y88A mutant phenotype, suggesting that the BAC contains all of the regulatory elements required for wild‐type T expression (Fig EV2A–D). We then generated a reporter by inserting the mCherry open reading frame into the start ATG of the T gene in RP24‐520D23 and integrated the T mCherry reporter BAC (Tg(RP24‐530D23, T mCherry)1Bgh, hereafter referred to as Tg(T mcherry)) into T 2J/+ (T WT), T 2J/2J (T null), and T 2J/Y88A (T Y88A) mESCs to monitor mesoderm formation (Figs 2B and EV2E). The T mCherry BAC was able to faithfully recapitulate the T expression pattern, as evidenced by WISH of mCherry expression in T WT ‐ and T Y88A ‐Tg(T mCherry) embryos (Fig 2C). The mCHERRY protein was localized in a broader domain than the T mRNA expression domain, likely reflecting the stability of mCHERRY protein (Fig 2C). RT‐qPCR analysis of T WT ‐ and T Y88A ‐Tg(T mCherry) mESCs differentiated into mesoderm revealed that neither T nor mCherry expression was maintained in T Y88A cells to the same degree as in T WT cells during the differentiation time course (Fig 2D). However, due to the stability of the mCHERRY protein, FACS analysis revealed that there was no difference in the percentage of T‐mCHERRY‐positive cells in T WT compared to T Y88A and T null (Fig 2E), suggesting that while initiation of T expression occurs normally in the T Y88A mutant, expression cannot be maintained. Furthermore, maintenance of T expression appears to be independent of WNT and FGF ligands, as these ligands are provided continuously throughout the differentiation time course. However, we cannot rule out that the Y88A mutation affects a component of the WNT/FGF pathway that alters the ability of these cells to respond to WNT/FGF ligand. Altogether, our results suggest that the conserved tyrosine residue in T is essential for the maintenance of T expression during mesodermal development.
Figure 2. Y88 is necessary for T maintenance.
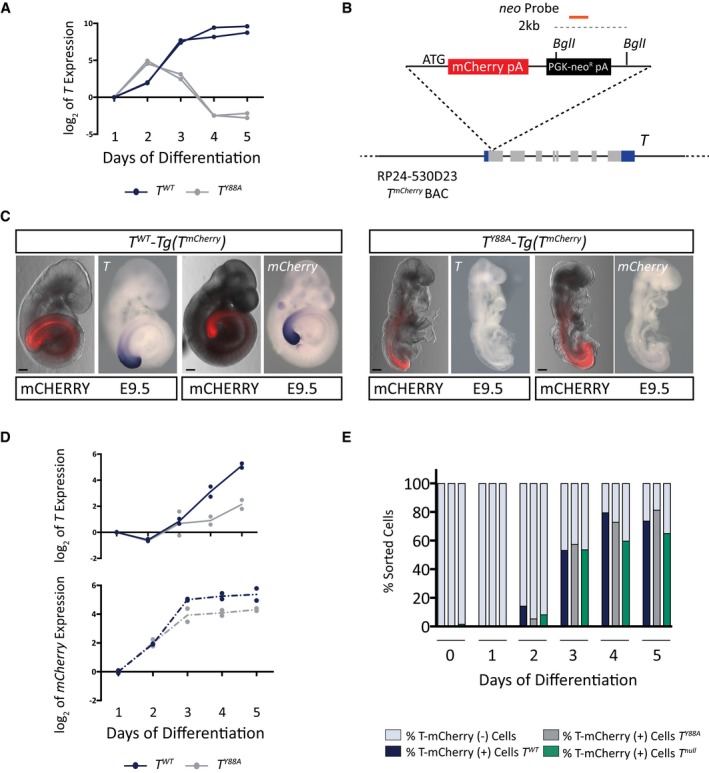
- T expression in in vitro‐differentiated mesodermal cells from control T WT and T Y88A during the 5‐day differentiation protocol as measured by RT‐qPCR. n = 2 biological replicates.
- Schematic illustrating the RP24‐530D23 T mCherry reporter BAC. The orange line depicts the probe used to confirm genomic integration of the reporter BAC by Southern blot. Restriction enzyme sites used for Southern blotting are depicted.
- Fluorescence images and WISH analysis of T WT‐ and T Y88A ‐Tg(T mCherry) with T and mCherry probes at E9.5 reveals the co‐localization of T and mCherry expression from the T mCherry reporter BAC. mCHERRY protein is more stable than the mCherry RNA and is localized to a broader domain. Scale bars: 200 μm.
- T and mCherry expression in T WT ‐ and T Y88A ‐Tg(T mCherry) cells differentiated into mesoderm in vitro during the 5‐day differentiation protocol, as measured by RT‐qPCR. The mean of n = 2 biological replicates is depicted.
- Bar graph depicting the percentage of T‐mCHERRY(+) mesodermal cells and T‐mCHERRY(−) cells, as assayed by FACS, in T WT ‐, T Y88A ‐, and T null ‐Tg(T mCherry) cells during the 5‐day in vitro differentiation protocol.
Figure EV2. The T BAC is able to rescue the TY 88A mutant phenotype.
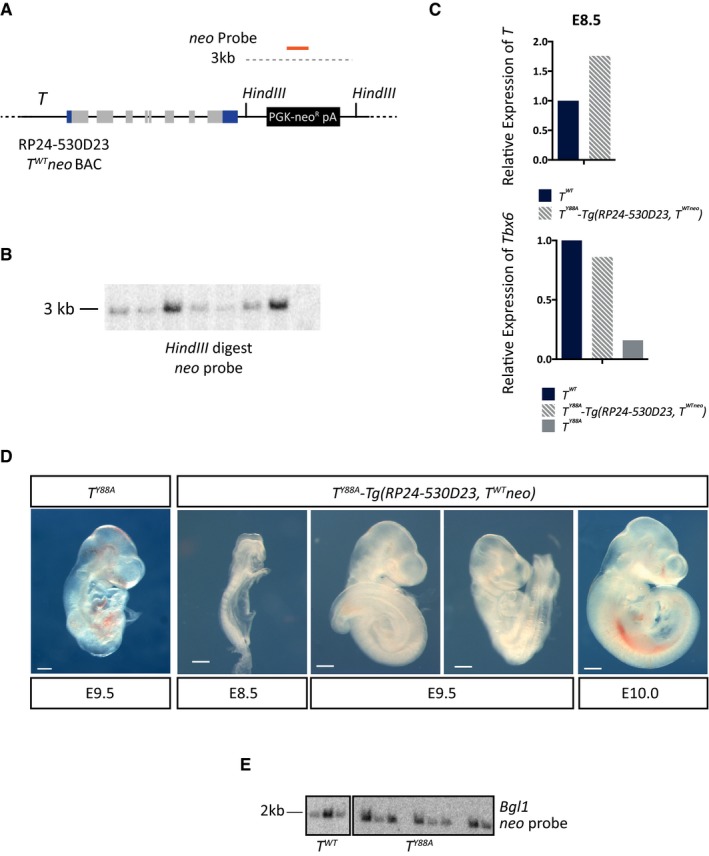
- Schematic illustrating the wild‐type RP24‐530D23‐T WT neo BAC, containing a neomycin selection cassette recombined into the BAC backbone. The orange line depicts the probe used to confirm genomic integration of the T WT neo BAC by Southern blot. Restriction enzyme sites used for Southern blotting are also depicted.
- Southern blot of genomic DNA from T Y88A mESC clones in which the T WT neo BAC was randomly integrated. Positive clones display a band at 3 kb when using a probe against the neomycin resistance gene.
- RT–qPCR analysis of T and Tbx6 expression in whole embryos at E8.5 reveals that T expression and Tbx6 expression are rescued in T Y88A ‐Tg(RP24‐530D23, T WT neo) embryos.
- Bright field images of T Y88A ‐Tg(RP24‐530D23, T WT neo) embryos from E8.5 to E10.0 reveals rescue of gross morphological phenotypes when compared to the T Y88A mutant. Scale bar: 200 μm.
- Southern blot of genomic DNA from T WT and T Y88A mESC clones in which the T mCherry reporter BAC was randomly integrated. Positive clones display a band at 2 kb when using a probe against the neomycin resistance gene.
RNA‐Seq analysis reveals a role for T in the regulation of hematopoietic and endothelial gene expression programs
To investigate the T Y88A mutant on the molecular level, we analyzed the transcriptome in mesodermal cells of the mutant using RNA‐Seq. To this end, T‐mCHERRY‐positive cells were isolated by FACS from T WT and T Y88A embryos harboring the T mCherry reporter at E8.25 and E8.5 (embryonic stages where gross morphological phenotypes are not yet visible) and transcriptomic analysis was performed (Figs 3A and EV3A). Pairwise scatter plots of FPKM values from E8.25 versus E8.5 revealed that gene expression at the two developmental timepoints was similar (R 2 = 0.989, Fig EV3B). Due to the high correlation, we merged the two expression data sets and compared gene expression to a T WT control data set (n = 1). This comparison revealed a number of genes that were dysregulated in T Y88A mesodermal cells (log2 fold change ≥ 1.3, Figs 3B and EV3C). 190 genes were downregulated at both developmental timepoints and 129 were upregulated (log2 fold change ≥ 1.3, Fig EV3D), consistent with previous reports that T contains transcriptional activator and repressor domains 4. Gene ontology (GO) term enrichment analysis of these 190 downregulated genes revealed a significant enrichment of genes involved in somitogenesis and mesoderm development, including known T targets such as Tbx6, Msgn1, Dkk1, and Foxc1 (Fig 3C) 29, 30, 31. There was also a significant enrichment of genes involved in erythrocyte development and differentiation, myeloid cell development, angiogenesis and blood vessel development, and cardiovascular system development (Fig 3C). Interestingly, both the hematopoietic and endothelial cell lineages arise from a common mesodermal progenitor and there is evidence that T regulates aspects of hematopoiesis and blood vessel development 19, 20, 21, 22. We found and verified the downregulation in expression of a number of important regulators of hematopoietic and endothelial cell development in T Y88A mutants in vivo by RT‐qPCR (Fig 3D and E), including Lmo2, Kdr, Tal1, Gata1, Klf1, Cdh5, and Tie1. Downregulated expression of two of these genes in T Y88A caudal end mesoderm was also validated using WISH. Lmo2 expression in the caudal end of T Y88A mutants was nearly absent when compared to T WT controls (Fig 3F), while Cdh5 expression in the developing dorsal aorta was significantly downregulated, and at later stages of development, Cdh5 expression was barely detectable (Fig 3F).
Figure 3. RNA‐Seq analysis reveals a role for T in hematopoietic and endothelial cell development.
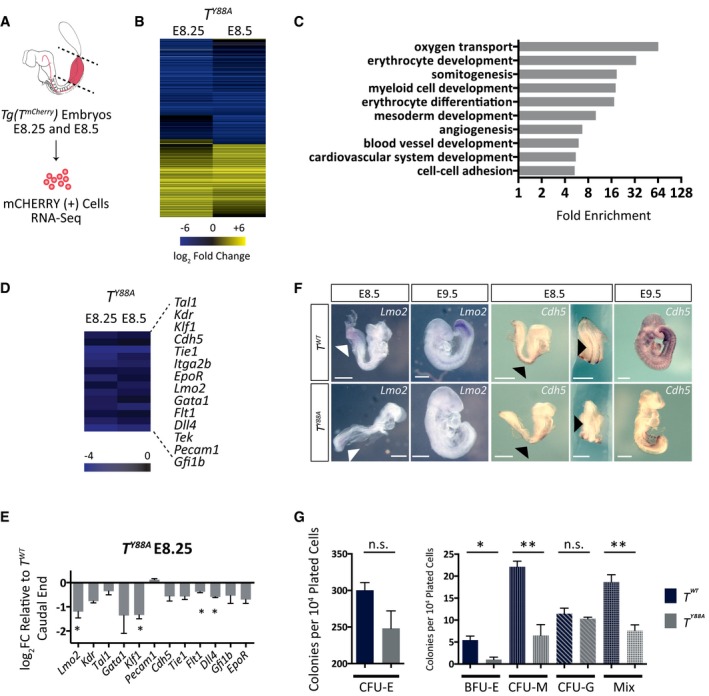
- Schematic illustrating microdissection and FACS of T‐mCHERRY(+) mesodermal cells from caudal ends of Tg(T mCherry) embryos for RNA‐Seq analysis. Dotted lines indicate microdissection planes before FACS.
- Heat map illustrating dysregulated gene expression (log2 fold change ≥ 1.3 when compared to T WT) in T Y88A mutant mesodermal cells at E8.25 and E8.5.
- Statistical overrepresentation test (PANTHER) of GO terms from dysregulated genes in T Y88A mutant mesodermal cells at E8.25 and E8.5 (log2 fold change ≥ 1.3 when compared to T WT). Gray bars indicate fold enrichment over the reference Mus musculus gene list.
- Heat map displaying downregulation of multiple genes important for hematopoietic and endothelial cell development in T Y88A mutants at E8.25 and E8.5 from the RNA‐Seq.
- RT‐qPCR validation of gene expression in microdissected caudal ends of E8.25 T Y88A mutants compared to T WT control. n = 3 biological replicates of pools of five microdissected caudal ends. Error bars = SEM. *P ≤ 0.05 using a one‐sample t‐test.
- WISH analysis of Lmo2 expression in T Y88A compared to T WT control embryos from E8.5 to E9.5. White arrowheads indicate Lmo2 expression in the caudal end of embryos. WISH analysis of Cdh5 expression in T Y88A compared to T WT control embryos from E8.5 to E9.5. Black arrowheads indicate expression of Cdh5 in the developing dorsal aorta. Scale bar: 500 μm.
- CFC assay using cells derived from embryoid bodies of T WT and T Y88A mESCs. The graph represents the number of colonies per 104 plated cells. n = 3 biological replicates, with each replicate comprised of three technical replicates. Error bars = SEM. *P < 0.05, **P < 0.01 using the unpaired t‐test. n.s. not significant. CFU‐E, colony‐forming unit erythroid; BFU‐E, burst‐forming unit erythroid; CFU‐M, colony‐forming unit macrophage; CFU‐G, colony‐forming unit granulocyte; Mix, presence of two or more myeloid progenitor cell types (macrophage, granulocyte, or erythroid).
Figure EV3. Transcriptome analysis in TY 88A mutants in vivo .
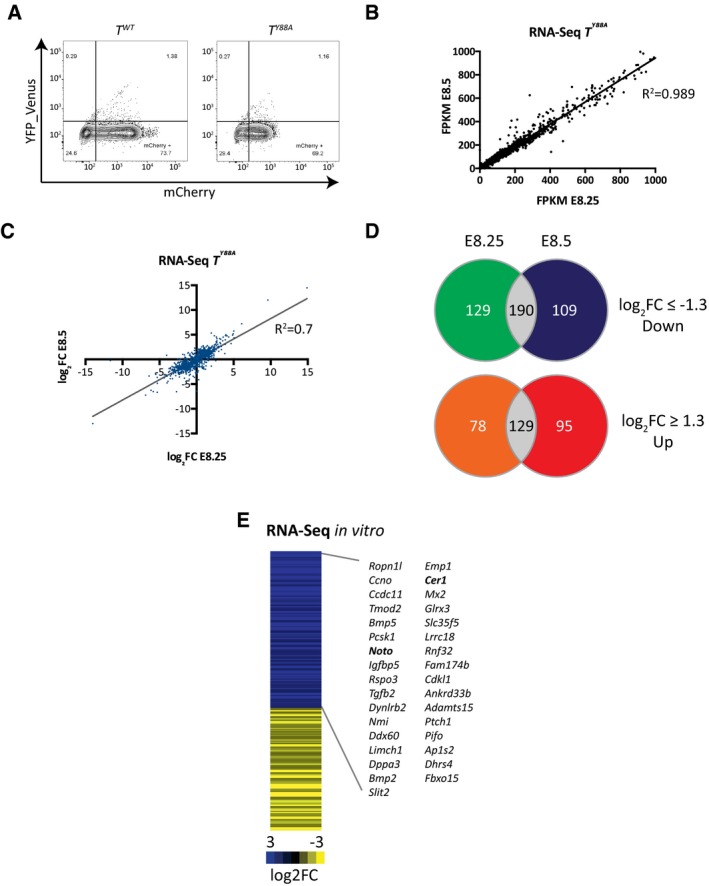
- FACS plots from the sorting of T‐mCHERRY‐positive cells from T WT and T Y88A embryos for RNA‐seq.
- Scatter plot of FPKM values from RNA‐Seq using mesodermal cells of T Y88A embryos at E8.25 and E8.5 (one biological replicate for each timepoint).
- Scatter plot of log2 fold change values comparing T Y88A mutant mesodermal cells to T WT at E8.25 and E8.5 (log2 fold change ≥ 1.3).
- Venn diagrams illustrating genes that are dysregulated both at E8.25 and E8.5 in T Y88A mutant mesodermal cells compared to T WT (log2 fold change ≥ 1.3).
- Heat map depicting dysregulated gene expression (log2 fold change ≥ 1.3) in T Y88A mesodermal cells differentiated in vitro compared to T WT from RNA‐Seq of one biological replicate. Gene names correspond to genes that are downregulated and contain a nearby TBS with reduced H3K27ac (fold change > 1.4). Bold gene names indicate known T targets.
In order to determine whether the downregulation of hematopoietic gene expression in T Y88A mutants resulted in a defect in the ability to differentiate into hematopoietic progenitors, we assayed the colony‐forming potential of these cells using a colony‐forming cell (CFC) assay. Embryoid bodies were generated from T WT and T Y88A mESCs, and single cells were dissociated and plated in defined methylcellulose medium containing cytokines to support the growth and differentiation of hematopoietic progenitors 32. In line with the RNA‐Seq data, there was a significant decrease in the ability of T Y88A mutant cells to differentiate into erythroid progenitors (BFU‐E), macrophages (CFU‐M), and myeloid progenitors (Mix, consisting of 2 or more myeloid cell types, that is, granulocyte, macrophage, or erythroid), while the number of CFU‐E erythroid progenitors and granulocytes (CFU‐G) were comparable to T WT (Fig 3G). The strong downregulation of a set of genes known to play important roles in both hematopoietic and endothelial development in T Y88A mutant mesodermal cells in vivo, combined with the defect in the ability of mutant cells to differentiate into some hematopoietic lineages in vitro, indicates that T plays a prominent role in the early specification of certain hematopoietic cell lineages.
The Y88A mutation in T affects H3K27 acetylation at T binding sites
In order to evaluate whether the disruption of T interaction with histone‐modifying enzymes contributes to the morphological and molecular phenotypes described above, we performed genome‐wide ChIP‐Seq analysis to screen for differences in the histone modification landscape in mesodermal cells derived from in vitro differentiation of mESC in T WT and the T Y88A mutant. We chose the in vitro differentiation model due to the limiting amounts of material for ChIP‐Seq from mesodermal cells in vivo. T‐mCHERRY‐positive mesodermal cells from each genotype were sorted by FACS and subjected to ChIP using antibodies specific for H3K27me3 and H3K27ac, as T was previously shown to interact with proteins involved in H3K27 demethylation 7. We included genome‐wide analysis of H3K27ac, as H3K27 methylation and acetylation are thought to be mutually exclusive 33, 34. We compared the levels of H3K27me3 and H3K27ac at regions (±1 kb) around T binding sites (TBS) identified by T ChIP‐Seq in in vitro‐differentiated mesodermal cells. A number of TBS (29.8%) exhibited an increase in H3K27me3 levels (fold change > 1.4), in accordance with previously published data that show that the Y88 residue is important for binding of T‐box factors to H3K27 demethylase proteins (Fig EV4A) 7. We also found a proportion of TBS (18.9%) which exhibited a decrease in H3K27ac levels (fold change > 1.4) (Fig EV4B). We chose to focus our analyses on H3K27ac, as an interaction between T and histone acetyltransferase enzymes has not yet been reported. Furthermore, when we examined the overlap between TBS with higher H3K27me3 and lower H3K27ac (fold change > 1.4), we found that only 30% of TBS with lower H3K27ac also displayed increased H3K27me3 (Fig EV4C). This observation is suggestive that the majority of TBS with lower H3K27ac cannot simply be explained by the failure of H3K27me3 removal at target sites. However, as these data stem from a single experiment, further experiments need to be performed to draw firm conclusions. Our data suggest that T is necessary for the recruitment of H3K27ac to target regulatory regions, which to our knowledge has not yet been shown.
Figure EV4. Disruption of H3K27ac at TBS in the TY 88A mutant.
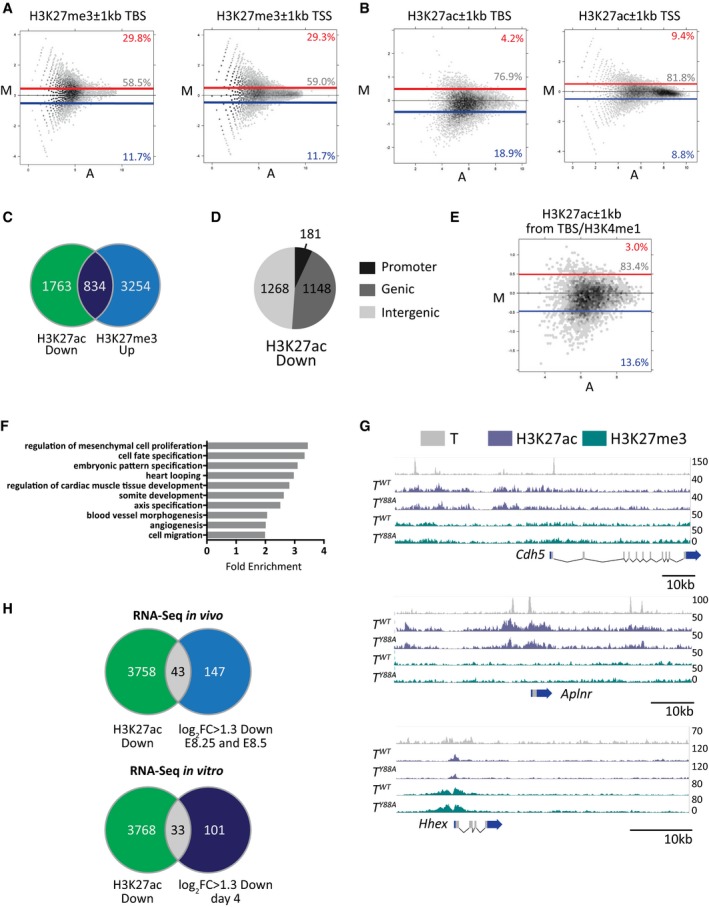
- MA plots of H3K27me3 ChIP‐Seq (one biological replicate) read density ±1 kb from transcriptional start sites (TSS, with TBS removed) and TBS. Blue and red lines depict fold change values of −1.4 and 1.4, respectively. The percentage of regions above and below this threshold (red and blue, respectively) are indicated on the plots.
- MA plot of H3K27ac ChIP‐seq (one biological replicate) read density ±1 kb from T binding sites and TSS (with TBS removed) in T WT versus T Y88A mutant mesodermal cells differentiated in vitro. Blue and red lines depict fold change values of −1.4 and 1.4, respectively. The percentage of regions above and below this threshold (red and blue, respectively) are indicated on the plots.
- Overlap of TBS with higher H3K27me3 and lower H3K27ac (fold change > 1.4) from one biological ChIP‐Seq replicate each.
- Genomic distribution of TBS with lower H3K27ac (fold change > 1.4).
- MA plots of H3K27ac read density ±1 kb from TBS regions that overlap K4me1 peaks. Blue and red lines depict fold change values of −1.4 and 1.4, respectively. The percentage of regions above and below this threshold (red and blue, respectively) are indicated on the plots.
- Statistical overrepresentation test (PANTHER) of GO terms from genes nearby regions of lower H3K27ac in T Y88A mutant mesodermal cells differentiated in vitro (fold change > 1.4 when compared to T WT). Gray bars indicate fold enrichment over the reference Mus musculus gene list.
- Genome browser tracks depicting T binding in mesodermal cells differentiated in vitro, and H3K27ac and H3K27me3 at TBS of target genes.
- Venn diagram illustrating the overlap between downregulated gene expression from RNA‐Seq in vivo (log2 fold change ≥ 1.3) and the neighboring two to three genes from each TBS with reduced H3K27ac (fold change > 1.4) from one biological ChIP‐Seq replicate. The lower Venn diagram illustrates the overlap between downregulated gene expression from RNA‐Seq from in vitro‐differentiated mesodermal cell (log2 fold change ≥ 1.3) and the neighboring two to three genes from each TBS with reduced H3K27ac (fold change > 1.4).
T controls H3K27ac levels at distal regulatory regions of target genes
We further analyzed TBS with lower H3K27ac in T Y88A mutant mesodermal cells and determined the genomic distribution of these regions. We found that only 7% (181/2,597) were found within promoter regions of genes (−5 kb to +2 kb of TSS), while 49% (1,268/2,597) were found in intergenic regions and 44% (1,148/2,597) were found within genic regions (Fig EV4D). We overlapped TBS with genome‐wide H3K4me1 peaks from T WT mesodermal cells and found the same trend of decreased H3K27ac within ±1 kb of TBS/H3K4me1 sites (Fig EV4E), suggesting that T functions to coordinate H3K27ac at putative enhancer regions throughout the genome, and may regulate the switch between poised H3K4me1(+) enhancers and active H3K27ac/H3K4me1(+) enhancer regions 35. We then associated each TBS containing lower H3K27ac in T Y88A mutants to the neighboring two to three genes and performed GO term analysis to determine whether these T targets were enriched for specific biological functions. Interestingly, the nearest genes associated with TBS exhibiting lower H3K27ac were enriched for GO terms such as heart looping, regulation of cardiac muscle tissue development, angiogenesis, and blood vessel morphogenesis (Fig EV4F), suggesting that T may play a role in regulating the chromatin environment at regulatory regions of genes important for cardiovascular development. Genome browser tracks of H3K27ac at TBS nearby Cdh5, Aplnr, and Hhex, genes that are important for vascular development, depict the decreased H3K27ac at TBS in T Y88A mutant mesodermal cells (Fig EV4G). These observations, in addition to those from the RNA‐Seq in T Y88A mutants in vivo, indicate a role for T in regulating putative enhancer activity in mesodermal precursors that will give rise to hematopoietic and endothelial cell types.
However, when we tested whether the neighboring two to three genes of TBS regions with reduced H3K27ac were dysregulated in the RNA‐Seq in vivo, we found little overlap (43/190, 23%) (Fig EV4H). As this observation may reflect differences in comparing in vivo and in vitro models, we analyzed the transcriptome of in vitro‐differentiated mesodermal cells by RNA‐Seq, using the same conditions as those for the H3K27ac ChIP‐Seq (Fig EV3E). Similarly, there was little overlap of dysregulated gene expression in the neighboring two to three genes of TBS regions with reduced H3K27ac (33/104, 25%) (Fig EV4H). This lack of overlap is in accordance with previously published data, which reported a 13% overlap between downregulated genes in an Xbra/Xbra3 knockdown compared to Xbra binding by ChIP‐Seq in Xenopus 29. Another study reported a 40% overlap between downregulated genes in a BRACHYURY knockdown model of human ESC differentiation compared to BRACHYURY binding by ChIP‐Seq 31. This relative lack of overlap between downregulated gene expression in T Y88A mutants and lower H3K27ac at TBS may point to a broader function for T than simply direct activation or repression of transcription. Rather, T likely cooperates with histone‐modifying enzymes to create a permissive chromatin environment during early mesodermal development for later differentiation of various cell types derived from the mesoderm, including hematopoietic and endothelial cells.
T controls H3K27ac at the T locus and interacts with p300
Strikingly, loss of H3K27ac was found at TBS upstream of the T genomic locus itself by ChIP‐Seq (Fig 4A). We validated this decrease in H3K27ac at the T genomic locus by ChIP‐qPCR in T Y88A and T WT mesodermal cells differentiated in vitro (Fig 4B). This observation suggests that T regulates the maintenance of its own expression through recruitment of chromatin‐influencing factors to create a permissive chromatin environment for continuous T activation. Furthermore, this autoregulation occurs independently and downstream of regulation by WNT/FGF ligands (Fig 2A), as providing exogenous WNT/FGF ligands is not able to rescue T expression in T Y88A mutant mesodermal cells.
Figure 4. H3K27ac is decreased at T binding sites in the TY 88A mutant.
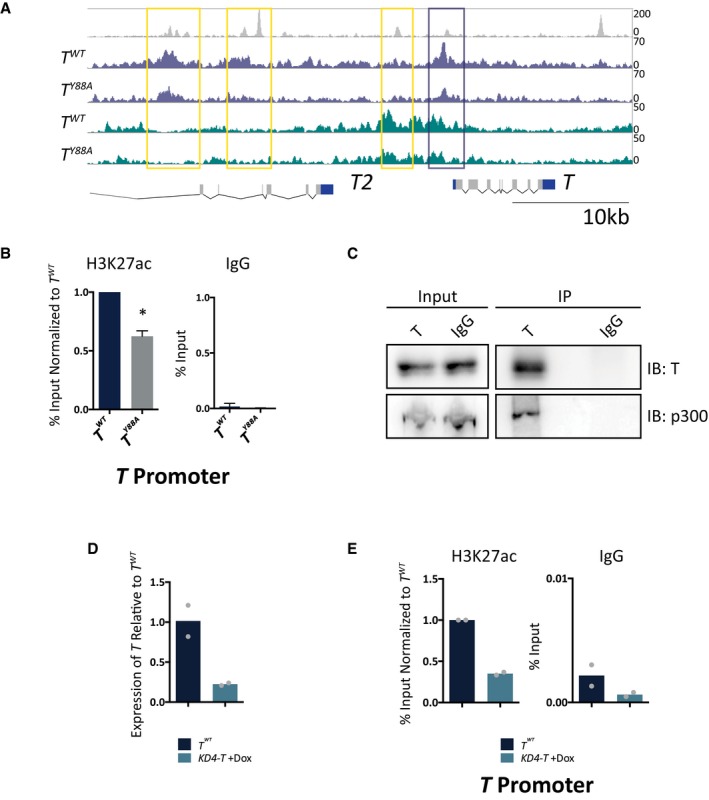
- Genome browser tracks depicting H3K27ac/me3 and T binding at the T locus in mesodermal cells differentiated in vitro. Yellow and purple boxes indicate T binding sites with decreased H3K27ac.
- ChIP‐qPCR analysis of H3K27ac at the TBS of the T promoter, corresponding to the purple box, in T WT and T Y88A in vitro‐differentiated mesodermal cells at day 4. IgG ChIP was used as a negative control. Error bars depict SD of n = 3–4 biological replicates. *P = 0.005 using a one‐sample t‐test.
- Immunoprecipitation of endogenous T from in vitro‐differentiated mesodermal cells at day 4 revealed an interaction with endogenous p300, as assayed by immunoblotting. IgG was used as a negative control. Blot is representative of n = 3 biological replicates.
- RT‐qPCR analysis of T expression in KD4‐T mESCs compared to T WT at day 4 of the in vitro differentiation protocol. Doxycycline was administered at day 2 of the protocol to induce expression of shRNA hairpins directed against T 39. The mean of n = 2 biological replicates is depicted.
- ChIP‐qPCR analysis of H3K27ac at the T promoter in KD4‐T in vitro‐differentiated mesodermal cells at day 4. The mean of n = 2 biological replicates is depicted.
There are two main histone H3K27 acetyltransferases, p300 and CBP, that are known to deposit H3K27ac on target histones 33, 36, 37, 38. We performed a co‐immunoprecipitation analysis from in vitro‐differentiated mesodermal cells and found an interaction between endogenous T and p300 (Fig 4C). This observation, in combination with our observation that H3K27ac levels are downregulated at TBS in T Y88A mutants, suggests that T, together with p300, plays a role in depositing H3K27ac at target loci and regulating the histone modification landscape in mesodermal cells.
Loss of T also results in lower H3K27ac at TBS
In order to distinguish whether it is the TY88A amino acid substitution itself, or the loss of T maintenance in T Y88A mutants, that causes the decrease in H3K27ac at TBS, we used a mESC model to knockdown T expression and survey H3K27ac when T expression is not maintained 39. The KD4‐T mESC line contains doxycycline‐inducible expression of shRNAs directed against the T mRNA. We administered doxycycline at day 2 of the in vitro mesodermal differentiation protocol to knockdown T expression at a similar timepoint when T expression decreases in the T Y88A mutant. RT‐qPCR analysis of T expression at day 4 in KD4‐T knockdown mesodermal cells revealed a loss of T expression when compared to the T WT control at day 4 (Fig 4D). Further, ChIP‐qPCR analysis revealed that the loss of T resulted in a subsequent loss of H3K27ac enrichment at TBS within the T promoter (Fig 4E). This observation suggests that it is the loss of T expression in the T Y88A mutants that causes the loss of H3K27ac enrichment at TBS. However, it is likely that both the Y88A point mutation in T and the loss of T maintenance are responsible for the reduced H3K27ac at TBS, as the initial loss of T maintenance in T Y88A mutants occurs due to the lack of recruitment of H3K27ac to the T genomic locus itself.
T controls the expression of Lmo2, a master regulator of hematopoietic and endothelial development
In order to further elucidate the mechanism by which T regulates endothelial and hematopoietic development, we took a closer look at genes that were dysregulated in T Y88A mutant embryos. One of these genes, Lmo2, is known to be an early upstream regulator of blood and endothelial development 40, 41. T binds to a region corresponding to the previously described −70 enhancer of Lmo2 (−70 kb from the TSS), which was shown by chromosome conformation capture assays to be in close proximity to the TSS of Lmo2 42, 43. Both the −70 enhancer and the TSS of Lmo2 display a decrease in H3K27ac in T Y88A mutant mesodermal cells, in the ChIP‐Seq data and confirmed by ChIP‐qPCR (Figs 5A and B, and EV5A). We reasoned that T binding to the −70 enhancer of Lmo2 may recruit permissive chromatin marks to the TSS to regulate expression, and that this regulation is disrupted in T Y88A mutant mesodermal cells.
Figure 5. T regulates Lmo2 expression through binding to a distal enhancer.
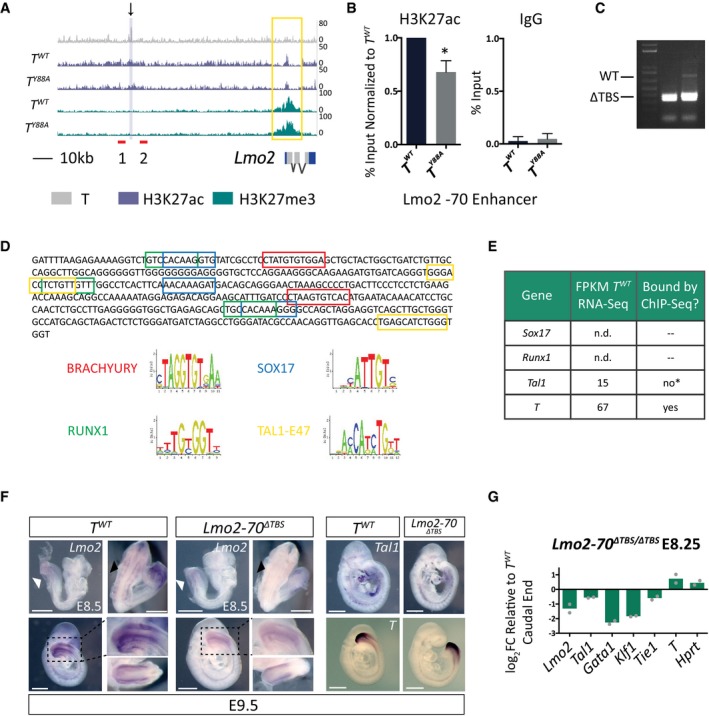
- T binding and H3K27ac/me3 tracks from T WT and T Y88A mutant mesodermal cells differentiated in vitro at the Lmo2 locus. The black arrow indicates the T binding region that was removed using CRISPR/Cas9 nickase technology. Red lines indicate primers used to genotype the CRISPR/Cas9‐mediated deletion. The yellow box indicates altered H3K27ac at the TSS. The purple‐shaded box indicates the region amplified in the ChIP‐qPCR analysis.
- ChIP‐qPCR analysis of H3K27ac at the −70 enhancer of Lmo2. IgG ChIP was used as a negative control. Error bars depict SD of n = 3–4 biological replicates. *P = 0.009 using a one‐sample t‐test.
- Genotyping PCR from genomic DNA of Lmo2‐70 ΔTBS/ΔTBS mESC clones, using primers depicted in (A), revealed homozygous mutant deletion clones.
- Motif analysis of the 400‐bp deleted region within the −70 enhancer of Lmo2. Colored boxes represent motifs pictured below, as predicted by ConSite 60.
- Table illustrating the lack of Sox17 and Runx1 expression in RNA‐Seq of T WT embryos in vivo at E8.25. While Tal1 is expressed, previous reports have shown that it binds to the Lmo2 −75 enhancer in mesodermal cells, and not the Lmo2 −70 enhancer. *ChIP‐Seq tracks from 45 can be found in Fig EV5C. The only factor that is expressed and bound at the Lmo2‐70 enhancer by ChIP‐Seq is T.
- WISH analysis of Lmo2, Tal1, and T expression in control T WT compared to Lmo2‐70 ΔTBS/ΔTBS embryos from E8.5 to E9.5. Black dotted lines indicate the magnification of a lateral view of the caudal end of the embryo, shown in the inset. The second inset pictured below is a dorsocaudal view of the tail. White arrowheads indicate Lmo2 expression in the caudal end mesoderm. Black arrowheads indicate Lmo2 expression in the intersomitic vessels. Scale bar: 500 μm.
- RT‐qPCR analysis of gene expression in the caudal ends of E8.25 Lmo2‐70 ΔTBS/ΔTBS embryos compared to T WT control. The mean of n = 2 biological replicates of pools of five microdissected caudal ends is depicted.
Figure EV5. T regulation of the Lmo2 locus.
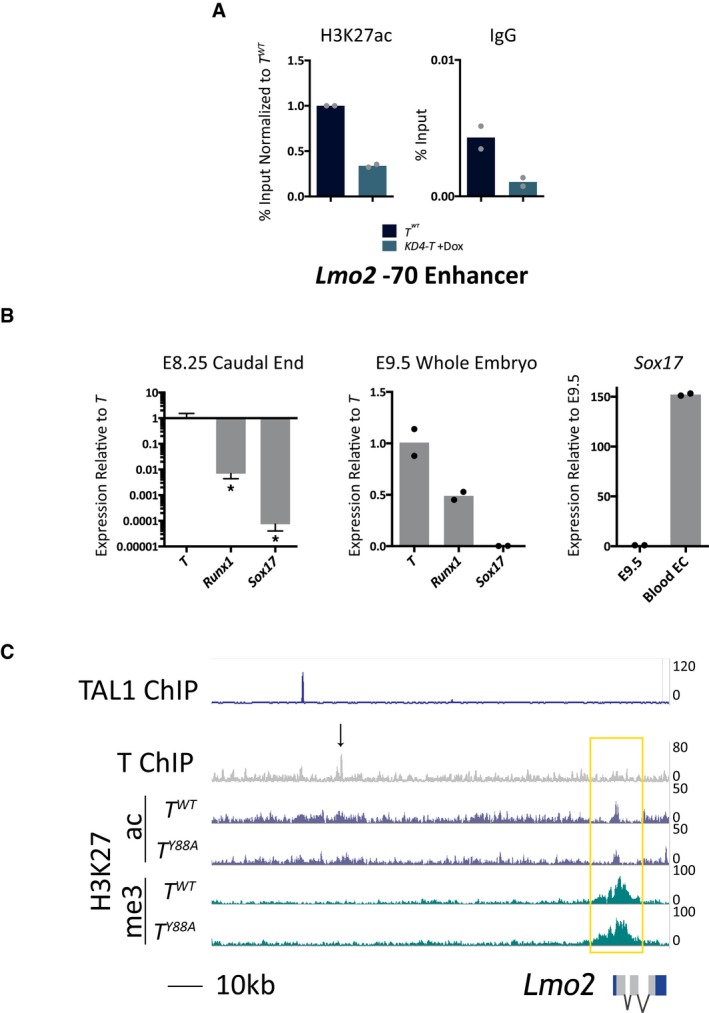
- ChIP‐qPCR analysis of H3K27ac at the Lmo2 −70 enhancer in KD4‐T in vitro‐differentiated mesodermal cells at day 4. The mean of n = 2 biological replicates is depicted.
- RT‐qPCR analysis of T, Runx1, and Sox17 expression in E8.25 caudal ends of T WT embryos. Levels of Runx1 and Sox17 are significantly lower than that of T in E8.25 caudal ends. Error bars depict SD of n = 3 biological replicates. *P < 0.02 using an unpaired t‐test. Middle graph depicts T, Runx1, and Sox17 levels in whole E9.5 embryos as a positive control for the qPCR primers. The mean of n = 2 biological replicates is depicted. Sox17 expression is low in E9.5 embryos, and the graph on the right depicts high Sox17 expression in CD31+ blood endothelial cells isolated from embryonic skin at E15.5. The mean of n = 2 biological replicates is depicted.
- Genome browser tracks of the Lmo2 genomic locus with T, H3K27ac, and H3K27me3 tracks from the ChIP‐Seq analysis in this study. TAL1 ChIP‐Seq tracks were depicted using data from 45, and illustrate TAL1 binding in mesodermal cells at the Lmo2 −75 enhancer, approximately 7 kb upstream of T binding at the −70 enhancer. The black arrow indicates the region removed by CRISPR/Cas9‐mediated genome editing at the TBS of the Lmo2 −70 enhancer.
In order to test this, we removed a 400‐bp genomic region within the Lmo2 −70 enhancer containing the TBS in wild‐type mESCs by genome engineering using CRISPR/Cas9 nickase technology 44 to generate homozygous Lmo2‐70 ΔTBS/ΔTBS mESCs (Fig 5A and C). Inspection of this 400‐bp region for transcription factor binding motifs revealed the presence of BRACHYURY motifs, in addition to SOX17, RUNX1, and TAL1 (Fig 5D). We were not able to detect expression of Runx1 or Sox17 in either in vivo or in vitro mesodermal cell RNA‐Seq analyses (Fig 5E). Furthermore, RT‐qPCR analysis of Runx1 and Sox17 in the caudal end of E8.25 embryos confirmed that Runx1 and Sox17 were expressed at levels 100× and 10,000× fold less, respectively, relative to T (Fig EV5B). Hence, it is unlikely that these two factors are binding the Lmo2 −70 enhancer region at E8.25. While Tal1 is expressed in E8.25 caudal ends, previously published ChIP‐Seq analysis of in vitro‐differentiated FLK1‐positive mesodermal cells revealed TAL1 binding at the Lmo2 −75 enhancer, but not at the Lmo2 −70 enhancer 45 (Figs 5E and EV5C). These observations suggest that regulation of H3K27ac at these 400 bp of the Lmo2 −70 enhancer most likely stem from T binding.
We generated embryos from the homozygous Lmo2‐70 ΔTBS/ΔTBS mutant mESC line by tetraploid morula aggregation and compared the expression levels of a set of hematopoietic and endothelial marker genes from the caudal end of E8.25 embryos. The expression levels of these marker genes were reduced in Lmo2‐70 ΔTBS/ΔTBS mutants, similar to the T Y88A mutants, when compared to T WT (Fig 5G). Expression of Lmo2 itself, along with Tal1, Gata1, Klf1, and Tie1, were downregulated when compared to T WT control embryos, while the expression of T and the housekeeping gene Hprt were unchanged. WISH analysis of Lmo2 expression in Lmo2‐70 ΔTBS/ΔTBS mutant embryos revealed a decrease in expression in the caudal end of E8.5 embryos compared to T WT control. At later stages (E9.5), Lmo2 expression was decreased in the intersomitic vessels and the caudal end of mutant embryos (Fig 5F). Furthermore, we confirmed by WISH the decrease in Tal1 expression, while T expression remained unchanged in Lmo2‐70 ΔTBS/ΔTBS mutant embryos at E9.5 (Fig 5F). Altogether, our data indicate that T binding at the −70 enhancer of Lmo2 is important for Lmo2 expression. The gene expression changes in hematopoietic and endothelial genes seen in T Y88A mutants are most likely due to the dysregulation of an upstream player, Lmo2, in the cascade of hematopoietic and endothelial development.
Discussion
While a few reports have implicated T in hematopoietic and endothelial development, we show here that T regulates Lmo2 expression through recruitment of histone acetylation to a distal enhancer. Lmo2 was previously shown to be a major regulator of both primitive and definitive hematopoiesis and vascular remodeling during angiogenesis 40, 41, 46, and its dysregulation is likely responsible for the defects observed in hematopoietic and endothelial gene expression in T Y88A mutants. T‐mediated recruitment of H3K27 acetylation to the −70 enhancer of Lmo2 during early mesoderm development potentially creates a permissive chromatin environment for downstream transcription factors to fully activate Lmo2. We identified consensus binding site sequences for RUNX1, SOX17, and TAL1, flanking the T‐bound −70 enhancer; all of which are important regulators for hematopoietic and endothelial development (Fig 5D). T‐mediated recruitment of H3K27 acetylation seemingly occurs at distal regions of other genes important for cardiovascular and hematopoietic development, including Cdh5, Aplnr, and Hhex (Fig EV4G). This observation, in combination with our data showing that the vast majority of T‐mediated recruitment of H3K27 acetylation (93%) occurs outside of promoter regions (Fig EV4D), suggests that the major function of T may not be in direct transcriptional activation. Rather, T may play a crucial role in setting up a permissive chromatin environment within nascent mesoderm, in order to permit downstream factors to fully activate cell‐type specification programs in cardiovascular and hematopoietic cells (Fig 6A).
Figure 6. Model of T function in early mesodermal development.
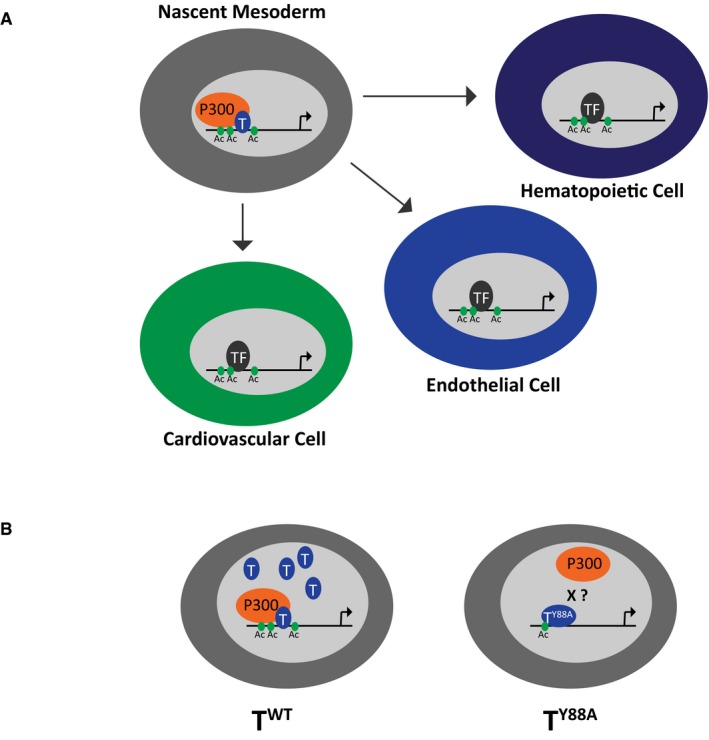
- Schematic illustrating T‐mediated recruitment of p300 and H3K27ac in nascent mesoderm to target loci involved in development of cell types derived from the mesoderm, including hematopoietic, endothelial, and cardiovascular cells.
- Schematic illustrating our proposed model showing that in wild‐type T mesodermal cells, recruitment of H3K27ac occurs at target loci. In the T Y88A mutant mesodermal cells, less H3K27ac is recruited to T target loci and there is less TY88A protein present. The X represents speculation that the Y88A mutation disrupts the interaction between T and p300.
We found a role for T in the maintenance of T expression, and this phenomenon likely extends to other members of the T‐box family of transcription factors, including TBX6 (Fig 1). We show that while T expression is initiated normally in T Y88A mutant mesodermal cells, expression of both T and mCherry from a T mCherry reporter BAC is not maintained throughout an in vitro differentiation protocol (Fig 2). The observation that mCherry expression is also not maintained suggests that lack of T maintenance in T Y88A mutants is not due to defects in T mRNA stability or mRNA elongation. Instead, we show that this lack of T maintenance likely occurs through defective recruitment of permissive chromatin modifications to the T genomic locus. Further, while WNT/FGF signaling molecules are sufficient to initiate T expression, we propose that maintenance of expression requires T‐mediated recruitment of permissive chromatin modifications, including H3K27ac, to the T locus. Interestingly, this tyrosine residue is conserved throughout all members of the T‐box family and is mutated in many human patients with congenital diseases 47, 48, 49. Our findings suggest that mutation of this conserved tyrosine residue may disrupt autoregulation and maintenance of expression, resulting in haploinsufficiency of the T‐box factor gene, and may help to explain mechanisms of congenital disease etiology.
We were unable to definitively determine whether the defects seen in the T Y88A mutant arise from the point mutation itself, and potential lack of binding to p300, or whether the phenotype arises due to lack of T maintenance. However, we believe these phenomena go hand‐in‐hand, as it is the initial defect in recruitment of permissive chromatin modifications that result in the lack of T maintenance. We propose a model (Fig 6B) in which wild‐type T, through an interaction with p300, is able to mediate the deposition of permissive H3K27ac to target loci during mesodermal development and specification. In T Y88A mutant mesodermal cells, we speculate that mutant TY88A cannot bind p300, leading to a decrease in H3K27ac at T target sites genome‐wide. We were unable to determine whether TY88A mutant protein lacks binding to p300 due to technical limitations arising from the severely decreased levels of T protein in the T Y88A mutant. However, biochemical or in vitro studies of TY88A mutant protein and its interaction with p300 would perhaps provide conclusive evidence.
Previous reports have shown that T‐box factors, including T, recruit H3K27 demethylase activity to target genomic regions to remove repressive H3K27 methylation through the conserved tyrosine residue described here 7. We observe a similar phenomenon in the T Y88A mutant, although the increase in H3K27 methylation was not entirely specific to TBS (Fig EV4A). This difference likely arises due to the restricted set of target loci that were analyzed for H3K27 methylation in previous studies, whereas our genome‐wide analyses allowed for an unbiased survey of the H3K27 methylation landscape. Instead, we found a more specific role for T in the recruitment of H3K27 acetylation to target genomic regions. Furthermore, we show that T physically interacts with p300 to mediate H3K27ac recruitment to target loci (Fig 4C). Members of the T‐box family of transcription factors have emerged as regulators of the histone modification landscape through their previously described interactions with multiple histone‐modifying enzymes. For example, T‐box factors including T‐BET and T were shown to physically interact with RBBP5, a member of the H3K4 methyltransferase complex and to control H3K4me2 at target genes 7. The same authors were able to show that multiple T‐box factors interact with proteins that facilitate H3K27 demethylation, including KDM6A and KDM6B, and, further, that this interaction was able to mediate an interaction between T‐box factors and members of the BRG1‐containing SWI/SNF chromatin remodeling complex 7, 50. Furthermore, a physical interaction between T‐BET and p300 has previously been shown 51, although unlike in our study, the authors did not show a functional requirement for T‐BET in the recruitment of H3K27ac to T‐BET target sites. TBX5, a T‐box factor involved in cardiac development, has been shown to physically interact with KDM6A, a histone H3K27 demethylase enzyme, and promote enhancer activation of cardiac‐specific genes including ANF 52. Recently, TBX1 was reported to physically interact with histone methyltransferase enzymes, including MLL3, and Tbx1 knockdown resulted in a decrease in H3K4me1 levels at a subset of TBX1 binding sites 53. The results from our study, in addition to previously published studies, support a general role for T‐box factors in regulating the histone modification landscape through interaction with multiple histone‐ and chromatin‐modifying enzymes during embryonic development. Furthermore, regulation of the histone modification landscape by T‐box factors appears to be complex, in that the same T‐box factor interacts with multiple histone‐modifying enzymes and controls the recruitment of different histone modifications within the same cell type. This complexity likely contributes to the severe developmental mutant phenotypes that arise in T‐box factor knockout models and to congenital disease when T‐box factors are mutated in humans.
Materials and Methods
Culture of mESCs
mESCs were maintained on feeder cells (mouse embryonic fibroblasts) in ESC medium [Dulbecco's Modified Eagle's Medium (DMEM) containing 4,500 mg/ml glucose and without sodium pyruvate (Gibco #41965), 15% ES cell‐grade fetal bovine serum (Pan‐Biotech GmbH), 2 mM l‐glutamine (Lonza), 50 U/ml penicillin, 50 μg/ml streptomycin (Lonza), 1% 100× non‐essential amino acids (Gibco), 0.1 mM β‐mercaptoethanol (Sigma‐Aldrich), 1% 100× nucleosides (Specialty media), and 1,000 U/ml murine leukemia inhibitory factor (LIF; Chemicon)]. Medium was changed daily.
Generation of mutant mESCs
T 2J/Y88A mESCs were generated by homologous recombination of heterozygous T 2J/+ F1G4 cells using a donor plasmid containing the Y88A mutation in exon 2 of T, an FRT‐flanked neomycin resistance cassette in the neighboring intron, and a diphtheria toxin A gene expression cassette (PGK‐DTA) within the plasmid backbone to prevent integration of the entire plasmid. Positive clones, as detected by neomycin resistance and Southern blot, were transfected with a mammalian FlpE expression vector to remove the neomycin resistance cassette using Lipofectamine 2000 (Invitrogen). The targeting strategy for Tbx6 Y137A/Δ mutant mESCs was similar to the strategy for T 2J/Y88A mESC generation, with the exception of the use of an FRT‐flanked hygromycin resistance cassette instead of neomycin. The remaining Tbx6 wild‐type allele in a heterozygous Tbx6 Δ/+ mESC line, generated from Tbx6 heterozygous blastocysts 24, was targeted by homologous recombination. Lmo2‐70 ΔTBS/ΔTBS mESCs were generated using a double‐nicking CRISPR/Cas9(D10A) approach with four guide RNAs (gRNAs, sequences in Table 1) surrounding the T binding site at the −70 enhancer of Lmo2. Wild‐type F1G4 cells 23 were transfected with a modified pX335 plasmid (a kind gift from Dr. Boris Greber) containing the gRNAs and a GFP/Puromycin selection cassette 54, and transiently selected for transfection with 1 μg/ml puromycin (Gibco, #10130127) for 2 days. Single mESC clones were picked 7–9 days after transfection and plated onto 96‐well plates and screened for genomic DNA deletion by PCR using primers outside of the deletion region, listed in Table 1.
Table 1.
Primer sequences
| Primer name | Sequence | Purpose |
|---|---|---|
|
Lmo2‐70del.for Lmo2‐70del.rev |
CGGAGACCTGTCTGAGACCTCAG CGTCAGAATGTGATTGATGTGAG |
Genotyping Lmo2‐70 ΔTBS/ΔTBS mESC |
|
T_5′ Southern.for T_5′ Southern.rev |
GTCGCTTCTAGCCCGGAAAC GAAGGAAGCCAGAGCTCGCG |
5′ T Southern blot probe |
|
T_3′ Southern.for T_3′ Southern.rev |
CACTACACAGTCCAGAGCTCAAGG CACAGACCAGAGACTGGGATACTGG |
3′ T Southern blot probe |
|
Tbx6_Southern.for Tbx6_Southern.rev |
GACGTGTCCTACAGTGAGGC AGTTTCCTCTTCACACGGGC |
Tbx6 Southern blot probe |
|
Lmo2‐70 gRNA#1 Lmo2‐70 gRNA#2 Lmo2‐70 gRNA#3 Lmo2‐70 gRNA#4 |
CCTACACCACAGTCGTTGT ATCATAGATAAGAAAGTTA GCACCTGAGCATCTGGGTGG CAACCTGTTGGCGTATCCC |
Sequences of gRNAs used for generating Lmo2‐70 ΔTBS/ΔTBS mESCs |
Generation of mouse embryos from mESCs
All animal procedures were conducted as approved by local authorities (LaGeSo Berlin) under the license numbers G0368/08, G0253/10 and G0247/13. Embryos were generated by tetraploid morula aggregation of ESCs as described in 23. SWISS or CD1 strains of mice were used for either wild‐type donor (to generate tetraploid morula) or transgenic recipient host (as foster mothers for transgenic mutant embryos). All transgenic embryos and mESC lines were on a hybrid F1 (C57Bl6/129S6) background.
Directed differentiation of mESCs to mesoderm
Feeder cells were removed from mESCs by serial plating, and mESCs were aggregated by the hanging drop method overnight in ESC medium (40 cells/μl, 5 μl/drop). The next day, embryoid bodies were washed thoroughly with Advanced DMEM (Gibco #12491015) and plated at a density of approximately 45 drops/cm2 onto tissue culture dishes coated with fibronectin (1:100 dilution in 1× PBS, Calbiochem #341631) in serum‐free medium [SFM, 1:1 Neurobasal:F12/DMEM (Gibco), 2 mM l‐glutamine (Gibco), 50 U/ml penicillin, 50 μg/ml streptomycin (Lonza), 0.5× B‐27 supplement, serum‐free (Gibco), 0.5× N‐2 supplement (Gibco), 0.05% Albumin, bovine Fraction V (7.5%; Sigma‐Aldrich), 0.001% 1‐thioglycerol (Sigma‐Aldrich)] containing recombinant BMP4 (20 ng/ml, R&D Systems) for 18 h. After 18 h, SFM + rBMP4 was removed and replaced with SFM containing 40 ng/ml rWNT3A (R&D Systems) and 40 ng/ml rFGF8 (R&D Systems) for the remainder of the experiment.
KD4‐T mESCs 39 were used to knockdown T expression during the in vitro differentiation to mesodermal cells. Doxycycline (2 μg/ml) was added to SFM on days 2–4 of the differentiation protocol to induce expression of shRNA hairpins against T.
Colony‐forming cell assays
Colony‐forming cell assays using dissociated embryoid bodies from T WT and T Y88A mESCs were performed as previously described 32, with embryoid bodies generated in SFM. Embryoid bodies were treated from day 2.5 to day 5 with 5 ng/ml of ACTIVIN A, VEGF, and bFGF (R&D Systems), as described in 32. At day 5 of embryoid body formation, cells were dissociated, counted, and 104 cells (per ml of methylcellulose medium) were plated in MethoCult GF M3434 (STEMCELL Technologies) according to manufacturer's instructions. CFU‐E colonies were counted at day 5 after plating in M3434 medium, while BFU‐E, CFU‐M, CFU‐G, and mixed colonies were counted at day 8. Scoring of colonies was performed blinded, with the genotype of the plate's contents unknown to the counter.
RNA‐Seq
The caudal ends of E8.25–E8.5 mouse embryos were microdissected (n = 10–20 caudal ends per genotype and timepoint), pooled, and T‐mCHERRY(+) mesodermal cells from T WT and T Y88A embryos were sorted by FACS (FACS Aria II SORP, BD Bioscience). The RNA from 20,000 cells was purified using the Qiagen RNeasy Micro kit (Qiagen), according to manufacturer's instructions. Ribosomal RNA was depleted and libraries were prepared using the ScriptSeq v.2 Complete (Human/Mouse/Rat) Low Input kit (epicenter), according to manufacturer's instructions. Libraries (one biological replicate per genotype and timepoint) were sequenced with the Illumina Hi‐Seq 2500 platform, with paired‐end (2 × 51 bp) and multiplexed sequencing. Sequences were mapped using the RefSeq mm10 annotation from UCSC using TopHat 2.0.8b, and only uniquely mapped reads were chosen. Normalization and FPKM values were calculated using Cufflinks 2.1.1 with the RefSeq mm10 annotation from UCSC. RNA‐Seq from in vitro‐differentiated mesodermal cells was performed using 50,000 T‐mCHERRY‐positive cells sorted by FACS on day 4 of the in vitro differentiation protocol. RNA was isolated using the Qiagen RNeasy Micro kit (Qiagen), according to manufacturer's instructions. RNA‐Seq libraries (one biological replicate per genotype) were generated and sequenced the same as described above. Normalized FPKM values from all RNA‐Seq experiments can be found in Dataset EV1.
ChIP‐Seq
Histone ChIP was performed using in vitro‐differentiated T‐mCHERRY(+) mesodermal cells sorted by FACS on day 4 of the differentiation protocol. 100,000 cells were used per ChIP with the iDeal ChIP‐Seq kit (Diagenode), according to manufacturer's instructions. Antibodies used were α‐H3K27me3 (Diagenode, A1811‐001P), α‐H3K27ac (Abcam, ab4729), and α‐H3K4me1 (Diagenode, A1883‐0010). T‐ChIP was performed from 2.5 × 107 mesodermal cells differentiated in vitro at day 4 of the differentiation protocol using an α‐T antibody (5 μg, Santa Cruz, sc‐17743). The ChIP protocol was performed according to 55. ChIP DNA libraries (one biological replicate per genotype) were generated using the TruSeq ChIP Sample Preparation Kit (Illumina), according to manufacturer's instructions, with minor adjustments. 18× cycles of amplification were used but with 5× cycles after adaptor ligation and the remaining 13× cycles after purification of ligation products. Sequencing was performed using the Illumina Hi‐Seq 2500 platform, and sequences were mapped to the 10‐mm genome using Bowtie 1.0, with only uniquely mapped reads chosen for further analysis.
RNA‐Seq and ChIP‐Seq data analysis
Normalized FPKM values from RNA‐Seq were filtered, and only genes with a FPKM > 2 in at least one sample (T WT or T Y88A) were examined; log2 fold change values were calculated using normalized FPKM comparing T Y88A(T 2J/Y88A) to T WT(T 2J/+). Heat maps and hierarchical clustering of dysregulated gene expression data (log2 fold change ≥ 1.3) were performed using Cluster 3.0 software 56. GO term enrichment analysis of dysregulated genes (log2 fold change > 1.3) was performed using the PANTHER classification system (www.pantherdb.org), version 12.0 57.
Uniquely mapped ChIP‐Seq data were analyzed using seqMINER v. 1.3.3 and Galaxy open‐source software 58, 59. H3K27ac and H3K27me3 read density at ±1 kb from TBS and TSS (with TBS removed) genome‐wide was calculated using seqMINER. Fold change values in read density were calculated from T Y88A compared to T WT in vitro‐differentiated mesodermal cells and plotted as an MA plot (M – log ratio, A – mean average). TBS with lower H3K27ac (fold change > 1.4) were associated with the nearest two to three genes for GO term analysis and comparison to dysregulated gene expression from RNA‐Seq. For comparison to the RNA‐Seq, if the TBS with lower H3K27ac (fold change > 1.4) was found within the promoter region (−5 kb to +2 kb of the TSS), it was assigned to that gene. If the TBS with lower H3K27ac was found within a genic region (either intronic or exonic), it was assigned to that gene and the nearest upstream and downstream gene. Lastly, intergenic TBS with lower H3K27ac were assigned to the nearest upstream and downstream gene.
ChIP‐qPCR
Histone ChIP was performed as described above. Immunoprecipitated DNA was subjected to qPCR using the primers listed in Table 3. qPCR was run using GoTaq qPCR Master Mix (Promega), and reactions were performed in a MicroAmp Fast Optical 96‐well Plate (Applied Biosystems) and run in triplicates using a StepOne Real‐Time PCR System. Percentage input was calculated using the ΔΔC T method.
Table 3.
ChIP‐qPCR primers
| Primer name | Sequence | Purpose |
|---|---|---|
| T promoter |
CTCCTTTGGCGAATGTGCAG ACCTTCACTTCTCTCGCCAC |
K27ac ChIP‐qPCR |
| Lmo2‐70 TBS |
GGGTGGGACCTCTGTTGTTT TTGGCCTGCTTTGGTCTTCA |
K27ac ChIP‐qPCR |
Gene expression analysis by RT‐qPCR
RNA from embryo samples and cultured cells was isolated using the RNeasy Mini Plus Kit (Qiagen), according to manufacturer's instructions. Reverse transcription of RNA into cDNA was performed using the SuperScript II First Strand Synthesis Kit (Invitrogen) with Random Hexamer primers (Roche), according to manufacturer's instructions. 500 ng of RNA was used as a template in each cDNA synthesis reaction. Detection and quantification of individual genes of interest were performed using RT‐qPCR with SYBR green. Approximately 2.5 ng of total cDNA was used in a reaction with 5 μM transcript‐specific primers and GoTaq qPCR Master Mix (Promega). Reactions were performed in a MicroAmp Fast Optical 96‐well Plate (Applied Biosystems) and run in triplicate using a StepOne Real‐Time PCR System. Data were analyzed using ΔΔC T method provided by the StepOnePlus Software v2.0.2 (Applied Biosystems). Expression of genes of interest was normalized using Pmm2 as a reference gene. The transcript‐specific primer sequences can be found in Table 2.
Table 2.
Gene expression analysis primers
| Primer name | Sequence | Purpose |
|---|---|---|
| T |
CAATGGAGGGGGACAGATCAT CTGGTGATCATGCGTTGCG |
RT–qPCR |
| Lmo2 |
GACCCGTCTGAGGAACCC AGTAGCGGTCCCCTATGTTCT |
RT–qPCR |
| Kdr |
TTCCTTAGGTGCCTCCCCAT CAGGAGCCCGCATTCTAGTT |
RT–qPCR |
| Tal1 |
GTTGTTTACCGCGCAGGAC GAGAGGGGTCGAGGTAGGAC |
RT–qPCR |
| Gata1 |
ACCACTACAACACTCTGGCG CGGTTCACCTGATGGAGCTT |
RT–qPCR |
| Klf1 |
CTTCCTCAAGTGGTGGCGG GTCCTCCGATTTCAGACTCACG |
RT–qPCR |
| Pecam1 |
TCACCAAGAGAACGGAAGGC CTCTTCTCGGGACATGGACG |
RT–qPCR |
| Cdh5 |
GGCAATCAACTGTGCTCTCC GAGGAGCTGATCTTGTCCGT |
RT–qPCR |
| Tie1 |
TACTGTGGCATGACCTGTGC GCATCAGCTCGTACACTTCG |
RT–qPCR |
| Flt1 |
TACCTCACCGTGCAAGGAAC AAGGAGCCAAAAGAGGGTCG |
RT–qPCR |
| Dll4 |
GCAGCTGTAAGGACCAGGA GCAGGTCAAGGTACTATGCTCA |
RT–qPCR |
| Gfi1b |
GCCCTTTGCCTGTGATGTCT CTTCGCTCCTGTGAGTGGAC |
RT–qPCR |
| EpoR |
TAGGCCCCCTCTGTCTCCT GCAGGGCCGCTTTGCTC |
RT–qPCR |
| Hprt |
GCTTTCCCTGGTTAAGCAGTACA GAGAGGTCCTTTTCACCAGCAA |
RT–qPCR |
| Pmm2 |
AGGGAAAGGCCTCACGTTCT AATACCGCTTATCCCATCCTTCA |
RT–qPCR |
| mCherry |
GCCACTACGACGCTGAGG CAACTTGATGTTGACGTTGTAGGC |
RT–qPCR |
| Lmo2 WISH |
AACGAAGCTTAGATACCCCCATCCCTGCTG AACGTCTAGAGTTAGTGCCCAGGAAGAGTCC |
RNA in situ hybridization probe |
| mCherry WISH |
TGGTGAGCAAGGGCGAGGAGG TAATACGACTCACTATAGGGCGGCGCTTCGTACTGTTCCA |
RNA in situ hybridization probe |
Whole‐mount in situ hybridization
WISH was performed according to the protocol available online from the Molecular Atlas of the Mouse Embryo Project (MAMEP; Berlin, Germany; http://mamep.molgen.mpg.de/), with minor adjustments described below. RNA ISH probes were generated from plasmid templates contained in the MAMEP database according to the online protocol, with the exception of Lmo2 and mCherry, which were generated using a PCR template amplified with the primers shown in Table 2. Clean‐up of the probes was performed using ProbeQuant G50 Sephadex Columns (Pharmacia). After overnight hybridization with the ISH probe, embryos were washed 2× in Solution I for 30 min each at 68°C, followed by 2× washes in Solution 3T for 30 min each at 68°C. Embryos were then washed 1× with 50% Solution 3T:50% 1× MABT (100 mM Maleic acid, 150 mM NaCl, pH 7.5) for 20 min, followed by 3× washes in 1× MABT with 2 mM Levamisole (Sigma‐Aldrich) at room temperature. Blocking was performed for 1 h at room temperature in blocking reagent (2% Blocking Reagent (Roche), 20% fetal bovine serum, 2 mM Levamisole in 1× MABT). Embryos were incubated overnight at 4°C with α‐DIG‐AP antibody (Roche) in blocking reagent. Post‐antibody washes were performed as described in the MAMEP online protocol, and staining was performed as described, using BM Purple AP Substrate (Roche) as a staining solution. The staining was monitored periodically under a binocular microscope, and staining was stopped as described in the online protocol. Images were obtained using a SteREO Discovery.V12 microscope and AxioCam Color Camera (Zeiss). Images of the T and mCherry WISH from T mCherry reporter embryos were obtained using the AxioZoom.V16 and AxioCam Color Camera (Zeiss). Fluorescence images of the T mCherry reporter embryos were obtained using the AxioZoom.V16 in combination with the AxioCam503 (Zeiss). Images were taken as z‐stacks and processed with the “Enhanced Depth of Focus” module of the ZEN software (Zeiss). All WISH images shown are representative of n > 3 embryos at each developmental timepoint, from each genotype, and with each WISH probe.
Histology and immunofluorescent staining
Embryos were fixed overnight in 4% paraformaldehyde, washed in 1× PBS, and washed in an ethanol series and xylenes to dehydrate before embedding in paraffin; 5‐μm transverse sections were cut with a Microm HM355S microtome and positioned on SuperFrost Ultra Plus slides (ROTH). Sections were dewaxed by incubation at 60°C for 30 min, followed by incubation in xylene (ROTH) 3× for 5 min. Ethanol series (100, 96, 90, 80, 70, 50%) followed, for 3 min each, prior to staining with hematoxylin (Sigma‐Aldrich) for 10 min and eosin (Sigma‐Aldrich) for a few seconds. The sections were washed in distilled water, 80% ethanol, and two times in 100% ethanol for 3 min, and were incubated in xylene 3× for 5 min prior to mounting under coverslips (Menzel Gläser) using Entellan new mounting buffer (MERCK). Hematoxylin and eosin‐stained sections were imaged using the Zeiss AXIO Observer Z1 using the AxioVision 40 software (Version 4.5.3.SP1).
Immunofluorescent staining was performed by rehydrating paraffin sections from above, followed by antigen retrieval by boiling for 10 min in 0.01 M Citrate Buffer, pH 6.0. Sections were blocked in 1.5% BSA/10% lamb serum in PBS for 30 min at room temperature and incubated overnight at 4°C with mouse monoclonal anti‐SOX2 antibody (Abcam), diluted 1:400 in blocking solution. Sections were washed 3 × 5 min in PBS and incubated with Alexa Fluor 488 goat anti‐mouse (Invitrogen) secondary antibody, diluted 1:1,000 in blocking solution, for 1 h at room temperature. Sections were rinsed again 3 × 5 min in PBS prior to mounting with Hardset Mounting Medium containing DAPI (Vectashield). Immunostained sections were imaged under the Zeiss AXIO Observer Z1, and images were analyzed by AxioVision 40 software (Version 4.5.3.SP1).
Immunoblotting
Embryo lysates were collected in lysis buffer containing 1% SDS, 10 mM Tris–HCl pH7.5, 2 mM EDTA, 1× Complete protease inhibitor (Roche), and 1× PhosStop phosphatase inhibitor (Roche) for 10 min on ice. Proteins were separated by electrophoresis (Bio‐Rad Mini Trans‐Blot cell) and transferred onto a nitrocellulose membrane. Membranes were incubated with an α‐T antibody (R&D Systems, #AF2085, 1:10,000) and an α‐LaminB1 antibody (Abcam, ab16048, 1:3,500), followed by washes in 1× TBST (50 mM Tris–HCl pH 7.5, 150 mM NaCl, 0.1% Tween‐20). Membranes were then incubated with HRP‐coupled secondary antibodies (Cell Signaling), and proteins were detected using the Amersham ECL kit (GE Healthcare) according to manufacturer's instructions. Chemiluminescent signal was visualized using the Fusion SL Advance Chemiluminescence Detection System.
Co‐immunoprecipitation
Approximately 2.5 × 107 mesodermal cells from day 4 of the in vitro differentiation protocol were used for each immunoprecipitation assay. Cells were lysed in buffer containing 25 mM HEPES, pH 8.0, 25 mM sodium chloride, 2 mM EDTA, 0.5% Tween‐20, and 1× cOmplete EDTA‐free protease inhibitor (Roche) for 15 min and nuclei were extracted by centrifugation at 1,350 g for 5 min. Pelleted nuclei were lysed in 25 mM HEPES, pH 8.0, 500 mM sodium chloride, 2 mM EDTA, 0.5% Tween‐20, and 1× cOmplete EDTA‐free protease inhibitor (Roche) for 15 min on ice. An equal volume of lysis buffer without sodium chloride was added to the nuclear lysate before immunoprecipitation. Nuclear lysates were pre‐cleared by adding 50 μl goat serum and 50 μl of Protein G Dynabeads (Invitrogen) per 1 ml of lysate for 1 h at 4°C. Protein G Dynabeads were removed and replaced with Protein G beads together with α‐T antibody (5 μg, Santa Cruz, sc‐17743) or normal goat IgG (5 μg, R&D Systems). Lysates were incubated on a rotating mixer overnight at 4°C. 5× washes were performed at 4°C using 25 mM HEPES, pH 8.0, 200 mM sodium chloride, 2 mM EDTA, 0.5% Tween‐20, and 1× cOmplete EDTA‐free protease inhibitor (Roche). Immunoprecipitated proteins were eluted by boiling in sample buffer before immunoblotting. Immunoblotting was performed as described above, with protein transfer onto nitrocellulose membrane performed at 4°C overnight (16 hours) at 25V, using α‐T (Santa Cruz, sc‐166962, 1:200) and α‐p300 (Active Motif, Clone NM11, 1:1,000) antibodies.
Sample size, randomization, and inclusion/exclusion criteria
No statistical calculation was performed to pre‐determine sample size for the experiments shown here. There was no randomization or blinding performed when allocating samples or animals to treatments. Blinding was performed in the counting of CFC Assays, as described above. There were no pre‐established inclusion/exclusion criteria in the experiments performed.
Data availability
RNA‐Seq and ChIP‐Seq data were deposited in the Gene Expression Omnibus (GEO) under the accession number GSE94142. Referenced TAL1 ChIP‐Seq data 45 are available at GEO under the accession number GSE47085.
Author contributions
PG and AB conceived the project, designed the experiments, and wrote the manuscript with input from co‐authors. AB, PT, FK, M‐TM, SUS, ADS, and LW performed experiments. FK and AB performed analyses of the RNA‐Seq and ChIP‐Seq data. TP generated the RP24‐530D23‐T WT BAC. KM performed the tetraploid aggregation of mESCs to generate transgenic embryos. BGH supervised the project.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Dataset EV1
Review Process File
Acknowledgements
We would like to thank Dr. Boris Greber for kindly providing the modified pX335‐G2P plasmid for CRISPR/Cas9‐mediated genome editing. We thank Dr. Arnold Schröder and Dr. Eun‐ha Shin for the RP24‐530D23‐T mCherry BAC and for their initial work on the in vitro differentiation protocol, Sandra Währisch, Manuela Scholze, and Gaby Blaess for excellent technical assistance, Judith Fiedler und Mirjam Peetz for excellent animal husbandry and assistance in generating transgenic embryos, and members of the Herrmann laboratory for helpful discussion. This work was supported by funds from the Max Planck Society and the DFG (German Research Foundation) Excellence Cluster Cardio‐Pulmonary System (Exc147‐2).
EMBO Reports (2018) 19: 118–134
References
- 1. Dobrovolskaia‐Zavadskaia N (1927) Sur la mortification sponta‐nee de la queue chez la souris nouveau‐nee et sur l'existence d'un caractere hereditaire “non viable”. Compr Soc Biol 97: 114–116 [Google Scholar]
- 2. Herrmann BG, Labeit S, Poustka A, King TR, Lehrach H (1990) Cloning of the T gene required in mesoderm formation in the mouse. Nature 343: 617–622 [DOI] [PubMed] [Google Scholar]
- 3. Kispert A, Herrmann BG (1993) The Brachyury gene encodes a novel DNA binding protein. EMBO J 12: 3211–3220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kispert A, Koschorz B, Herrmann BG (1995) The T protein encoded by Brachyury is a tissue‐specific transcription factor. EMBO J 14: 4763–4772 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Müller CW, Herrmann BG (1997) Crystallographic structure of the T domain‐DNA complex of the Brachyury transcription factor. Nature 389: 884–888 [DOI] [PubMed] [Google Scholar]
- 6. Bollag RJ, Siegfried Z, Cebra‐Thomas JA, Garvey N, Davison EM, Silver LM (1994) An ancient family of embryonically expressed mouse genes sharing a conserved protein motif with the T locus. Nat Genet 7: 383–389 [DOI] [PubMed] [Google Scholar]
- 7. Miller SA, Huang AC, Miazgowicz MM, Brassil MM, Weinmann AS (2008) Coordinated but physically separable interaction with H3K27‐demethylase and H3K4‐methyltransferase activities are required for T‐box protein‐mediated activation of developmental gene expression. Genes Dev 22: 2980–2993 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Amaya E, Stein PA, Musci TJ, Kirschner MW (1993) FGF signalling in the early specification of mesoderm in Xenopus . Development 118: 477–487 [DOI] [PubMed] [Google Scholar]
- 9. Chesley P, Dunn LC (1936) The inheritance of taillessness (Anury) in the house mouse. Genetics 21: 525–536 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Gluecksohn‐Schoenheimer S (1944) The development of normal and homozygous Brachy (T/T) mouse embryos in the extraembryonic coelom of the chick. Proc Natl Acad Sci USA 30: 134–140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Gruneberg H (1958) Genetical studies on the skeleton of the mouse. XXIII. The development of brachyury and anury. J Embryol Exp Morphol 6: 424–443 [PubMed] [Google Scholar]
- 12. Conlon FL, Sedgwick SG, Weston KM, Smith JC (1996) Inhibition of Xbra transcription activation causes defects in mesodermal patterning and reveals autoregulation of Xbra in dorsal mesoderm. Development 122: 2427–2435 [DOI] [PubMed] [Google Scholar]
- 13. Schulte‐Merker S, Hammerschmidt M, Beuchle D, Cho KW, De Robertis EM, Nüsslein‐Volhard C (1994) Expression of zebrafish goosecoid and no tail gene products in wild‐type and mutant no tail embryos. Development 120: 843–852 [DOI] [PubMed] [Google Scholar]
- 14. Galceran J, Hsu SC, Grosschedl R (2001) Rescue of a Wnt mutation by an activated form of LEF‐1: regulation of maintenance but not initiation of Brachyury expression. Proc Natl Acad Sci USA 98: 8668–8673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yamaguchi TP, Takada S, Yoshikawa Y, Wu N, McMahon AP (1999) T (Brachyury) is a direct target of Wnt3a during paraxial mesoderm specification. Genes Dev 13: 3185–3190 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Martin BL, Kimelman D (2008) Regulation of canonical Wnt signaling by Brachyury is essential for posterior mesoderm formation. Dev Cell 15: 121–133 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Naiche LA, Holder N, Lewandoski M (2011) FGF4 and FGF8 comprise the wavefront activity that controls somitogenesis. Proc Natl Acad Sci USA 108: 4018–4023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Boulet AM, Capecchi MR (2012) Signaling by FGF4 and FGF8 is required for axial elongation of the mouse embryo. Dev Biol 371: 235–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fehling HJ, Lacaud G, Kubo A, Kennedy M, Robertson S, Keller G, Kouskoff V (2003) Tracking mesoderm induction and its specification to the hemangioblast during embryonic stem cell differentiation. Development 130: 4217–4227 [DOI] [PubMed] [Google Scholar]
- 20. Huber TL, Kouskoff V, Fehling HJ, Palis J, Keller G (2004) Haemangioblast commitment is initiated in the primitive streak of the mouse embryo. Nature 432: 625–630 [DOI] [PubMed] [Google Scholar]
- 21. Inman KE, Downs KM (2006) Brachyury is required for elongation and vasculogenesis in the murine allantois. Development 133: 2947–2959 [DOI] [PubMed] [Google Scholar]
- 22. Cerdan C, McIntyre BAS, Mechael R, Levadoux‐Martin M, Yang J, Lee JB, Bhatia M (2012) Activin A promotes hematopoietic fated mesoderm development through upregulation of brachyury in human embryonic stem cells. Stem Cells Dev 21: 2866–2877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. George SHL, Gertsenstein M, Vintersten K, Korets‐Smith E, Murphy J, Stevens ME, Haigh JJ, Nagy A (2007) Developmental and adult phenotyping directly from mutant embryonic stem cells. Proc Natl Acad Sci USA 104: 4455–4460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chapman DL, Papaioannou VE (1998) Three neural tubes in mouse embryos with mutations in the T‐box gene Tbx6. Nature 391: 695–697 [DOI] [PubMed] [Google Scholar]
- 25. Isaacs HV, Pownall ME, Slack JM (1994) eFGF regulates Xbra expression during Xenopus gastrulation. EMBO J 13: 4469–4481 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Schulte‐Merker S, Smith JC (1995) Mesoderm formation in response to Brachyury requires FGF signalling. Curr Biol 5: 62–67 [DOI] [PubMed] [Google Scholar]
- 27. Martin BL, Kimelman D (2010) Brachyury establishes the embryonic mesodermal progenitor niche. Genes Dev 24: 2778–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sudheer S, Liu J, Marks M, Koch F, Anurin A, Scholze M, Senft AD, Wittler L, Macura K, Grote P et al (2016) Different concentrations of FGF ligands, FGF2 or FGF8 determine distinct states of WNT‐induced presomitic mesoderm. Stem Cells 34: 1790–1800 [DOI] [PubMed] [Google Scholar]
- 29. Gentsch GE, Owens NDL, Martin SR, Piccinelli P, Faial T, Trotter MWB, Gilchrist MJ, Smith JC (2013) In vivo T‐box transcription factor profiling reveals joint regulation of embryonic neuromesodermal bipotency. Cell Rep 4: 1185–1196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Lolas M, Valenzuela PDT, Tjian R, Liu Z (2014) Charting Brachyury‐mediated developmental pathways during early mouse embryogenesis. Proc Natl Acad Sci USA 111: 4478–4483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Faial T, Bernardo AS, Mendjan S, Diamanti E, Ortmann D, Gentsch GE, Mascetti VL, Trotter MWB, Smith JC, Pedersen RA (2015) Brachyury and SMAD signalling collaboratively orchestrate distinct mesoderm and endoderm gene regulatory networks in differentiating human embryonic stem cells. Development 142: 2121–2135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Sroczynska P, Lancrin C, Pearson S, Kouskoff V, Lacaud G (2009) In vitro differentiation of mouse embryonic stem cells as a model of early hematopoietic development. Methods Mol Biol 538: 317–334 [DOI] [PubMed] [Google Scholar]
- 33. Tie F, Banerjee R, Stratton CA, Prasad‐Sinha J, Stepanik V, Zlobin A, Diaz MO, Scacheri PC, Harte PJ (2009) CBP‐mediated acetylation of histone H3 lysine 27 antagonizes Drosophila Polycomb silencing. Development 136: 3131–3141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pasini D, Malatesta M, Jung HR, Walfridsson J, Willer A, Olsson L, Skotte J, Wutz A, Porse B, Jensen ON et al (2010) Characterization of an antagonistic switch between histone H3 lysine 27 methylation and acetylation in the transcriptional regulation of Polycomb group target genes. Nucleic Acids Res 38: 4958–4969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Calo E, Wysocka J (2013) Modification of enhancer chromatin: what, how, and why? Mol Cell 49: 825–837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ogryzko VV, Schiltz RL, Russanova V, Howard BH, Nakatani Y (1996) The transcriptional coactivators p300 and CBP are histone acetyltransferases. Cell 87: 953–959 [DOI] [PubMed] [Google Scholar]
- 37. Bannister AJ, Kouzarides T (1996) The CBP co‐activator is a histone acetyltransferase. Nature 384: 641–643 [DOI] [PubMed] [Google Scholar]
- 38. Jin Q, Yu L‐R, Wang L, Zhang Z, Kasper LH, Lee J‐E, Wang C, Brindle PK, Dent SYR, Ge K (2011) Distinct roles of GCN5/PCAF‐mediated H3K9ac and CBP/p300‐mediated H3K18/27ac in nuclear receptor transactivation. EMBO J 30: 249–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Vidigal JA, Morkel M, Wittler L, Brouwer‐Lehmitz A, Grote P, Macura K, Herrmann BG (2010) An inducible RNA interference system for the functional dissection of mouse embryogenesis. Nucleic Acids Res 38: e122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Warren AJ, Colledge WH, Carlton MB, Evans MJ, Smith AJ, Rabbitts TH (1994) The oncogenic cysteine‐rich LIM domain protein rbtn2 is essential for erythroid development. Cell 78: 45–57 [DOI] [PubMed] [Google Scholar]
- 41. Yamada Y, Pannell R, Forster A, Rabbitts TH (2000) The oncogenic LIM‐only transcription factor Lmo2 regulates angiogenesis but not vasculogenesis in mice. Proc Natl Acad Sci USA 97: 320–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Landry J‐R, Bonadies N, Kinston S, Knezevic K, Wilson NK, Oram SH, Janes M, Piltz S, Hammett M, Carter J et al (2009) Expression of the leukemia oncogene Lmo2 is controlled by an array of tissue‐specific elements dispersed over 100 kb and bound by Tal1/Lmo2, Ets, and Gata factors. Blood 113: 5783–5792 [DOI] [PubMed] [Google Scholar]
- 43. Bhattacharya A, Chen C‐Y, Ho S, Mitchell JA (2012) Upstream distal regulatory elements contact the Lmo2 promoter in mouse erythroid cells. PLoS One 7: e52880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ran FA, Hsu PD, Lin C‐Y, Gootenberg JS, Konermann S, Trevino AE, Scott DA, Inoue A, Matoba S, Zhang Y et al (2013) Double nicking by RNA‐guided CRISPR Cas9 for enhanced genome editing specificity. Cell 154: 1380–1389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Org T, Duan D, Ferrari R, Montel‐Hagen A, Van Handel B, Kerényi MA, Sasidharan R, Rubbi L, Fujiwara Y, Pellegrini M et al (2015) Scl binds to primed enhancers in mesoderm to regulate hematopoietic and cardiac fate divergence. EMBO J 34: 759–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yamada Y, Warren AJ, Dobson C, Forster A, Pannell R, Rabbitts TH (1998) The T cell leukemia LIM protein Lmo2 is necessary for adult mouse hematopoiesis. Proc Natl Acad Sci USA 95: 3890–3895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bamshad M, Lin RC, Law DJ, Watkins WC, Krakowiak PA, Moore ME, Franceschini P, Lala R, Holmes LB, Gebuhr TC et al (1997) Mutations in human TBX3 alter limb, apocrine and genital development in ulnar‐mammary syndrome. Nat Genet 16: 311–315 [DOI] [PubMed] [Google Scholar]
- 48. Bamshad M, Le T, Watkins WS, Dixon ME, Kramer BE, Roeder AD, Carey JC, Root S, Schinzel A, Van Maldergem L et al (1999) The spectrum of mutations in TBX3: genotype/phenotype relationship in ulnar‐mammary syndrome. Am J Hum Genet 64: 1550–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Reamon‐Buettner SM, Borlak J (2004) TBX5 mutations in non‐Holt‐Oram syndrome (HOS) malformed hearts. Hum Mutat 24: 104 [DOI] [PubMed] [Google Scholar]
- 50. Miller SA, Mohn SE, Weinmann AS (2010) Jmjd3 and UTX play a demethylase‐independent role in chromatin remodeling to regulate T‐box family member‐dependent gene expression. Mol Cell 40: 594–605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Chen G‐Y, Osada H, Santamaria‐Babi LF, Kannagi R (2006) Interaction of GATA‐3/T‐bet transcription factors regulates expression of sialyl Lewis X homing receptors on Th1/Th2 lymphocytes. Proc Natl Acad Sci USA 103: 16894–16899 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Lee S, Lee JW, Lee S‐K (2012) UTX, a histone H3‐lysine 27 demethylase, acts as a critical switch to activate the cardiac developmental program. Dev Cell 22: 25–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Fulcoli FG, Franzese M, Liu X, Zhang Z, Angelini C, Baldini A (2016) Rebalancing gene haploinsufficiency in vivo by targeting chromatin. Nat Commun 7: 11688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Cong L, Ran FA, Cox D, Lin S, Barretto R, Habib N, Hsu PD, Wu X, Jiang W, Marraffini LA et al (2013) Multiplex genome engineering using CRISPR/Cas systems. Science 339: 819–823 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Koch F, Scholze M, Wittler L, Schifferl D, Sudheer S, Grote P, Timmermann B, Macura K, Herrmann BG (2017) Antagonistic activities of Sox2 and Brachyury control the fate choice of neuro‐mesodermal progenitors. Dev Cell 42: 514–526.e517 [DOI] [PubMed] [Google Scholar]
- 56. de Hoon MJL, Imoto S, Nolan J, Miyano S (2004) Open source clustering software. Bioinformatics 20: 1453–1454 [DOI] [PubMed] [Google Scholar]
- 57. Mi H, Muruganujan A, Casagrande JT, Thomas PD (2013) Large‐scale gene function analysis with the PANTHER classification system. Nat Protoc 8: 1551–1566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Ye T, Krebs AR, Choukrallah M‐A, Keime C, Plewniak F, Davidson I, Tora L (2011) seqMINER: an integrated ChIP‐seq data interpretation platform. Nucleic Acids Res 39: e35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Blankenberg D, Von Kuster G, Coraor N, Ananda G, Lazarus R, Mangan M, Nekrutenko A, Taylor J (2010) Galaxy: a web‐based genome analysis tool for experimentalists. Curr Protoc Mol Biol Chapter 19: Unit19.10.1–Unit19.10.21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Sandelin A, Wasserman WW, Lenhard B (2004) ConSite: web‐based prediction of regulatory elements using cross‐species comparison. Nucleic Acids Res 32: W249–W252 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Dataset EV1
Review Process File
Data Availability Statement
RNA‐Seq and ChIP‐Seq data were deposited in the Gene Expression Omnibus (GEO) under the accession number GSE94142. Referenced TAL1 ChIP‐Seq data 45 are available at GEO under the accession number GSE47085.