

Abstract
Shigella deploys a unique mechanism to manipulate macrophage pyroptosis by delivering the IpaH7.8 E3 ubiquitin ligase via its type III secretion system. IpaH7.8 ubiquitinates glomulin (GLMN) and elicits its degradation, thereby inducing inflammasome activation and pyroptotic cell death of macrophages. Here, we show that GLMN specifically binds cellular inhibitor of apoptosis proteins 1 and 2 (cIAP1 and cIAP2), members of the inhibitor of apoptosis (IAP) family of RING‐E3 ligases, which results in reduced E3 ligase activity, and consequently inflammasome‐mediated death of macrophages. Importantly, reducing the levels of GLMN in macrophages via IpaH7.8, or siRNA‐mediated knockdown, enhances inflammasome activation in response to infection by Shigella, Salmonella, or Pseudomonas, stimulation with NLRP3 inflammasome activators (including SiO2, alum, or MSU), or stimulation of the AIM2 inflammasome by poly dA:dT. GLMN binds specifically to the RING domain of both cIAPs, which inhibits their self‐ubiquitination activity. These findings suggest that GLMN is a negative regulator of cIAP‐mediated inflammasome activation, and highlight a unique Shigella stratagem to kill macrophages, promoting severe inflammation.
Keywords: cIAPs, GLMN, inflammasome activation, Shigella
Subject Categories: Immunology; Microbiology, Virology & Host Pathogen Interaction; Post-translational Modifications, Proteolysis & Proteomics
Introduction
Inflammasome activation acts as an important defense mechanism against microbial infection, since it leads to innate immune responses including caspase‐1 activation. Upon bacterial infection, for example, members of the nucleotide‐binding oligomerization domain‐like receptor (NLR) family induce the formation of inflammasomes and initiate innate defense responses. Aberrant inflammasome activation or its failure to shut down causes tissue damage and chronic inflammatory disorders 1, 2, 3, 4, suggesting that inflammasome activation is under tight regulation. Several bacteria including Shigella, Salmonella, Legionella, and Pseudomonas trigger activation of caspase‐1 via the NLRC4 inflammasome in infected macrophages 3, 5. NLRC4 recognizes the bacterial flagellin 6, 7, inner rod protein 8, 9, and needle protein 10 of the bacterial type III secretion system (T3SS). Following recognition of these bacterial pathogen‐associated molecular patterns (PAMPs), NLRC4 is assembled into the inflammasome complex, which induces proteolytic maturation of caspase‐1 and pro‐inflammatory cytokines such as IL‐1β and IL‐18, resulting in a form of cell death called pyroptosis 3, 5. In recent studies, gasdermin D, a member of the gasdermin protein family, was identified as the key caspase‐1 substrate leading to pyroptosis 11, 12, 13. Caspase‐1 (or caspase‐11) cleaves gasdermin D to separate its N‐terminal pore‐forming domain from its C‐terminal repressor domain 11, 12. The oligomerized N‐terminal domain of gasdermin D permeabilizes the plasma membrane by forming large pores in the membrane that consequently cause membrane rupture and destruction of the cell (i.e., pyroptosis) 13.
Shigella stimulates NLR inflammasomes by delivering various effectors and T3SS components into host cells via the T3SS 14, 15, 16, 17. We previously reported that Shigella induces rapid macrophage pyroptotic cell death, a prerequisite for bacterial egress from macrophages, by delivering the IpaH7.8 E3 ubiquitin ligase effector via the T3SS, thereby activating NLRP3 and NLRC4 inflammasomes and caspase‐1 18. In that study, we identified glomulin (GLMN) as an IpaH7.8 target involved in IpaH7.8 E3 ligase‐dependent inflammasome activation. Indeed, upregulation or downregulation of GLMN levels leads to reduced or augmented inflammasome activation, respectively. Macrophages stimulated with lipopolysaccharide/ATP induce GLMN puncta that colocalize with the active form of caspase‐1 18. Although the mechanism of how GLMN is involved in inflammasome activation during Shigella infection remains unknown, the study suggested Shigella possess some as yet uncharacterized mechanisms to manipulate inflammasome activity, via the interaction of IpaH7.8 and GLMN 18. Importantly, GLMN was originally identified through its association with glomuvenous malformations 19, and later, it was identified as acting as an inhibitor of Cullin‐RBX1 E3 ligases 20, 21. Structural and biochemical analyses showed that GLMN can associate with RBX1‐containing Cullin‐RING E3 ligase (CRL) by targeting the RING domain and masking its E2‐binding surface, thereby inhibiting CRL E3 ligase activity 20, 21. Moreover, there is increasing evidence that ubiquitination modification of inflammasome component proteins is important for the regulation of inflammasome activity 22, 23, 24, 25, 26, 27. Given the interplay between GLMN and inflammasomes, we hypothesized that GLMN could have targets involved in modulating NLR inflammasome activation, for example, by interacting with and inhibiting certain putative RING‐E3 ligases.
Results
GLMN targets cIAP1 and cIAP2
We screened a subset of host proteins, previously identified proteins capable of interacting with IpaH7.8 by GST pulldown with GST‐IpaH7.8‐coated beads 18, in‐gel digestion and LC‐TOF/TOF‐MS analyses, for the ability to bind GLMN. Among the 18 proteins, we found cellular inhibitor of apoptosis protein (cIAP) 1 [also known as baculoviral IAP repeat‐containing (BIRC) 2] bound GLMN in the yeast two‐hybrid system using pPC86‐GLMN and pDBleu‐cIAP1 (positive control: pDBleu‐IpaH7.8; negative control: pDBleu‐empty; Fig 1A). cIAP1 belongs to the IAP family (also known as the BIRC family), whose members act as E3 ligases 28 and are critical regulators of multiple cellular pathways that control cell death, proliferation, and differentiation 28, 29. Because GLMN acts as an inhibitor of E3 ligases 20, 21, and cIAP1 and cIAP2 (cIAPs) are required for efficient caspase‐1 activation by NLR inflammasomes 30, we investigated whether cIAPs interact with GLMN in immunoprecipitation assays. 293T cells were cotransfected with 6myc‐cIAPs (or empty vector) and FLAG‐tagged GLMN (or empty vector), and the cell lysates were precipitated with anti‐FLAG M2‐conjugated beads. As shown in Fig 1B, 6myc‐cIAP1 and 6myc‐cIAP2 precipitated with FLAG‐GLMN (Fig 1B), and coprecipitation was also observed when cell lysates containing 6myc‐cIAP1 (and 6myc‐cIAP2) and GFP‐IpaH7.8CA (in which Cys residue 357 in IpaH7.8 is replaced with Ala, resulting in loss of E3 ligase activity) were pulled down with anti‐GFP‐conjugated beads (Fig 1C). As shown in Fig 1C, cIAPs precipitated with GLMN as well as IpaH7.8CA, suggesting that GLMN can interact with cIAPs in a complex with IpaH7.8. Consistent with this, immunohistochemical image analysis revealed colocalization of endogenous GLMN and cIAP2 in bone marrow‐derived macrophages (BMDMs) 1 h after infection with Shigella WT (Figs 1D and EV1). Note that no binding between GLMN and X‐linked inhibitor of apoptosis protein (XIAP) was detected by yeast two‐hybrid assay (Fig 1A), suggesting that GLMN interacts specifically with cIAP1 and cIAP2.
Figure 1. GLMN binds to cIAPs and forms a complex with Shigella IpaH7.8.
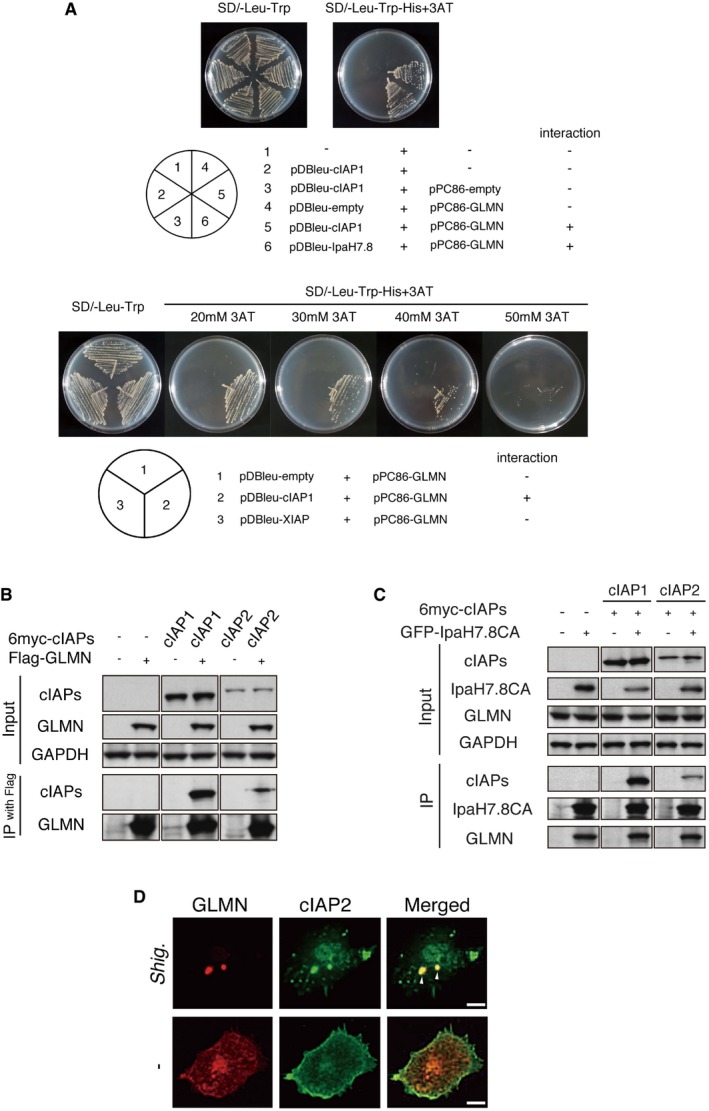
- Yeast two‐hybrid assay revealed direct binding between cIAP1 and GLMN. Saccharomyces cerevisiae strain MaV203 was transformed with the plasmid combinations indicated in the lower panel. The chart in the lower right summarizes the interaction results.
- Immunoprecipitation assay demonstrating the interaction between cIAPs and GLMN.
- cIAPs and GLMN co‐immunoprecipitated with Shigella IpaH7.8.
- Colocalization of endogenous GLMN (red) and cIAP2 (green) in BMDMs, visualized by confocal microscopy. WT BMDMs were infected with Shigella WT for 1 h and then subjected to immunohistochemical analysis using anti‐cIAP2 and anti‐GLMN antibodies. Scale bars, 10 μm. See also Fig EV1.
Source data are available online for this figure.
Figure EV1. GLMN colocalizes with cIAPs in macrophages.
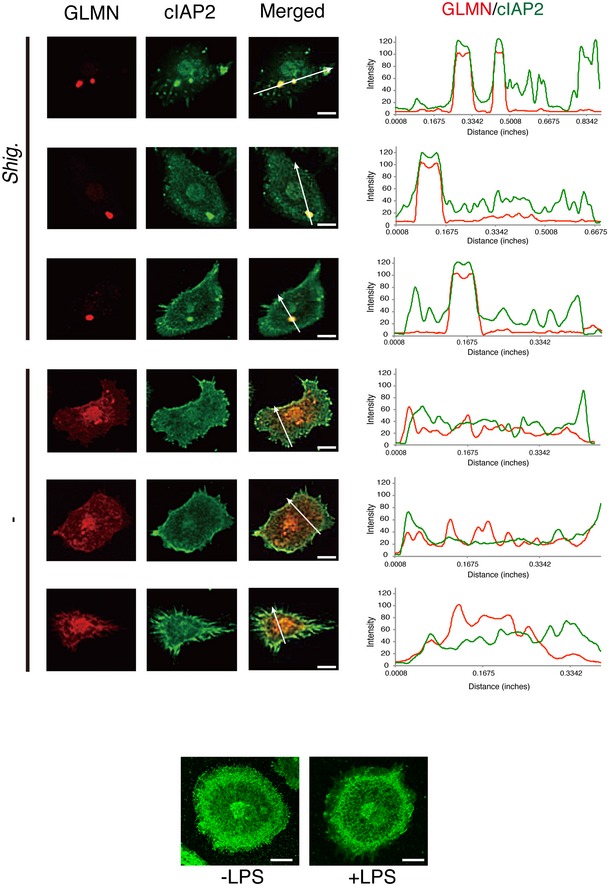
Upper panel: Immunohistochemical analysis reveals the colocalization of endogenous GLMN (red) and cIAP2 (green) in Shigella WT‐infected BMDMs (see also Fig 1D). The intensities of the fluorescence signals along with the white arrows were measured and are shown in the graph. Scale bars, 10 μm. Data represent one of three similar independent experiments. Bottom panel: Intracellular localization of endogenous cIAP2 is not affected by LPS priming. Scale bar, 10 μm.
cIAP1 and cIAP2 are involved in activation of NLRC4, AIM2, and NLRP3 inflammasomes
Two recent studies reported that cIAP1, cIAP2, and XIAP play important roles in regulating inflammasome activation, although there were significant discrepancies in their results 30, 31; Labbe et al reported that cIAP1 and cIAP2 are required for efficient caspase‐1 and NLR inflammasome activation, whereas Vince et al reported that cIAP1, cIAP2, and XIAP inhibit NLR inflammasome activation. Therefore, we knocked down cIAP1, cIAP2, or XIAP in BMDMs using siRNA (Fig EV2A and Table EV1) and then infected each knockdown with Shigella, Salmonella (Salmonella enterica serovar. Typhimurium), or Pseudomonas aeruginosa (Figs 2A and B, and EV2C). The levels of IL‐1β, IL‐18, and active caspase‐1, but not CXCL2 or IL‐6, were decreased by knockdown of cIAP1 or cIAP2, but not XIAP (Figs 2A and B, and EV2C). The same was also true in cIAP1/2‐ or XIAP‐knockdown cells treated with stimulators of AIM2 inflammasomes (poly dA:dT, plasmid DNA; Figs 2C and EV2D) or NLRP3 inflammasomes [silicon dioxide nanoparticles (SiO2), aluminum hydroxide (alum), monosodium urate (MSU)] (Figs 2D and EV2D). To confirm these findings, we generated cIAP1‐knockout (KO), cIAP2‐KO, cIAP1/2‐double KO, and XIAP‐KO macrophages using the CRISPR–Cas9 system. The knockout cells were infected with Shigella or treated with stimulators of AIM2 or NLRP3 inflammasomes such as SiO2 or Alum. The levels of IL‐1β were decreased in cIAP1‐KO, cIAP2‐KO, and cIAP1/2‐double KO cells, but not in XIAP‐KO cells, in response to these stimulations (Fig 2E). To further confirm this, we also measured the levels of IL‐1β and active caspase‐1 in BMDMs overexpressing cIAP1, cIAP2, or XIAP (Fig EV2B) infected with Shigella WT or the T3SS‐deficient mutant S325 (mxiA::Tn5; Fig 2F and G). The overexpression of cIAP1 and cIAP2, but not XIAP, augmented the production of active caspase‐1 (Fig 2F) and IL‐1β (Fig 2G). Based on these results, we concluded that cIAP1 and cIAP2, but not XIAP, functionally participate in activation of NLR inflammasomes.
Figure EV2. cIAP1, cIAP2, XIAP, and GLMN influence inflammasome activation.
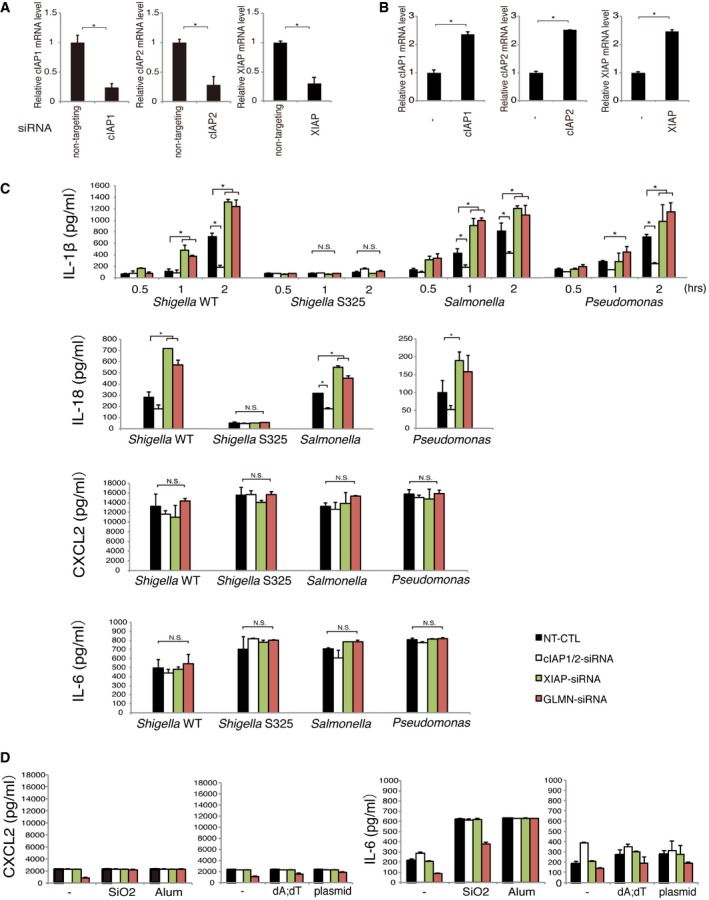
- Knockdown efficiencies of cIAP1, cIAP2, and XIAP in the cells used in this study were assessed by RT–PCR (see also Fig 2A–D). Actin mRNA levels were used for normalization. *P < 0.01.
- Expression levels of cIAP1, cIAP2, and XIAP in the cells used in this study (see also Fig 2F and G). *P < 0.01.
- Influence of cIAP1, cIAP2, XIAP, and GLMN on inflammasome activation caused by infection with Shigella, Salmonella, or Pseudomonas aeruginosa. WT BMDMs were subjected to siRNA‐mediated knockdown of cIAP1/cIAP2, XIAP, or GLMN. Non‐targeting siRNA (NT‐CTL) was used as a negative control. Two days after siRNA nucleofection, cells were infected with each type of bacteria at an MOI of 10 (Shigella) or 1 (Salmonella and P. aeruginosa). IL‐1β levels were measured at the indicated times post‐infection. IL‐18, CXCL2, and IL‐6 levels were measured at 2 h post‐infection (see also Figs 2A and B, and 3A and D). Knockdown of cIAP1/cIAP2 decreased IL‐1β and IL‐18 levels, indicating downregulation of inflammasomes. On the contrary, knockdown of XIAP or GLMN increased IL‐1β and IL‐18 levels. CXCL2 and IL‐6 cytokine levels were not meaningfully affected by the presence or absence of cIAP1/2, XIAP, and GLMN. *P < 0.01.
- CXCL2 and IL‐6 cytokine levels in cIAP1/2‐, XIAP‐, and GLMN‐knockdown cells treated with stimulators of AIM2 or NLRP3 inflammasomes. cIAP1/2‐, XIAP‐, or GLMN‐knockdown cells were stimulated with stimulators of AIM2 or NLRP3 inflammasomes, and supernatants were collected and subjected to CXCL2 and IL‐6 ELISA as described for IL‐1β (see also Figs 2C and D, and 3B and C).
Figure 2. cIAP1 and cIAP2 are important for inflammasome activation.
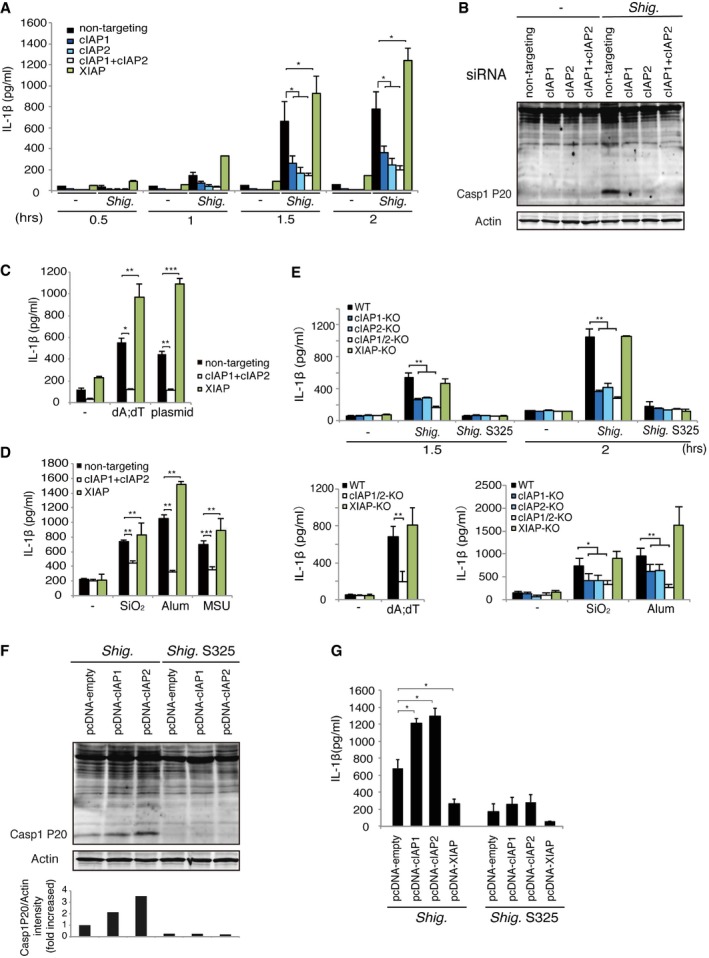
-
A, BWT BMDMs were subjected to siRNA‐mediated knockdown of cIAP1, cIAP2, and XIAP. Two days after siRNA nucleofection, cells were infected with Shigella for 30 min to 2 h, and then, IL‐1β production (A) and caspase‐1 activation (B) were measured. (A) IL‐1β ELISA. n = 3, *P < 0.01. (B) Immunoblot of active mature form (P20) of caspase‐1.
-
C, DcIAP1/2‐ or XIAP‐knockdown cells were treated with stimulators of AIM2 inflammasomes (C) or NLRP3 inflammasomes (D). After stimulation, supernatants were collected and subjected to IL‐1β ELISA. n = 3, *P < 0.01, **P < 0.005, ***P < 0.0005.
-
EcIAP1‐KO, cIAP2‐KO, cIAP1/2‐double KO, or XIAP‐KO macrophages were generated using the CRISPR–Cas9 system. KO cells were infected with Shigella or treated with stimulators of AIM2 inflammasomes or NLRP3 inflammasomes, and then, IL‐1β production was measured. n = 3, *P < 0.01, **P < 0.005.
-
F, GOverexpression of cIAPs promotes inflammasome activation induced by Shigella infection. WT macrophages overexpressing cIAP1, cIAP2, or XIAP were infected with Shigella for 1 h, and then, the levels of caspase‐1 activation were determined by immunoblotting using anti‐caspase‐1 P20 antibody (F) and IL‐1β levels were measured (G). n = 3. *P < 0.01.
GLMN inhibits self‐ubiquitination of cIAPs
Based on the results described above and those of our previous study 18, we hypothesized that GLMN acts as an inhibitor of cIAPs and that Shigella IpaH7.8 E3 ligase‐mediated degradation of GLMN leads to activation of NLR inflammasomes. Therefore, we investigated how GLMN affects cIAP activity. To this end, we knocked down endogenous GLMN in BMDMs (Fig EV3A and Table EV2), infected the knockdown cells with Shigella WT or S325, Salmonella WT [or Salmonella mutant lacking flagella (ΔfliA)], or Pseudomonas, and measured the levels of IL‐1β. GLMN‐knockdown cells infected with Shigella WT, Salmonella WT, or Pseudomonas produced more IL‐1β than those treated with non‐targeting siRNA controls (Figs 3A and EV2C). Similarly, treatment of GLMN‐knockdown cells with AIM2 (poly dA:dT) or NLRP3 (SiO2, alum, MSU) stimulators increased IL‐1β levels compared with controls (Figs 3B and C, and EV2D). The same was true for the mature cleaved form of caspase‐1, which was present at higher levels in GLMN‐knockdown cells than in controls (Fig 3D). These results supported our hypothesis that GLMN acts as a negative regulator of the NLR inflammasome.
Figure EV3. Efficiency of siRNA‐mediated knockdown of GLMN .
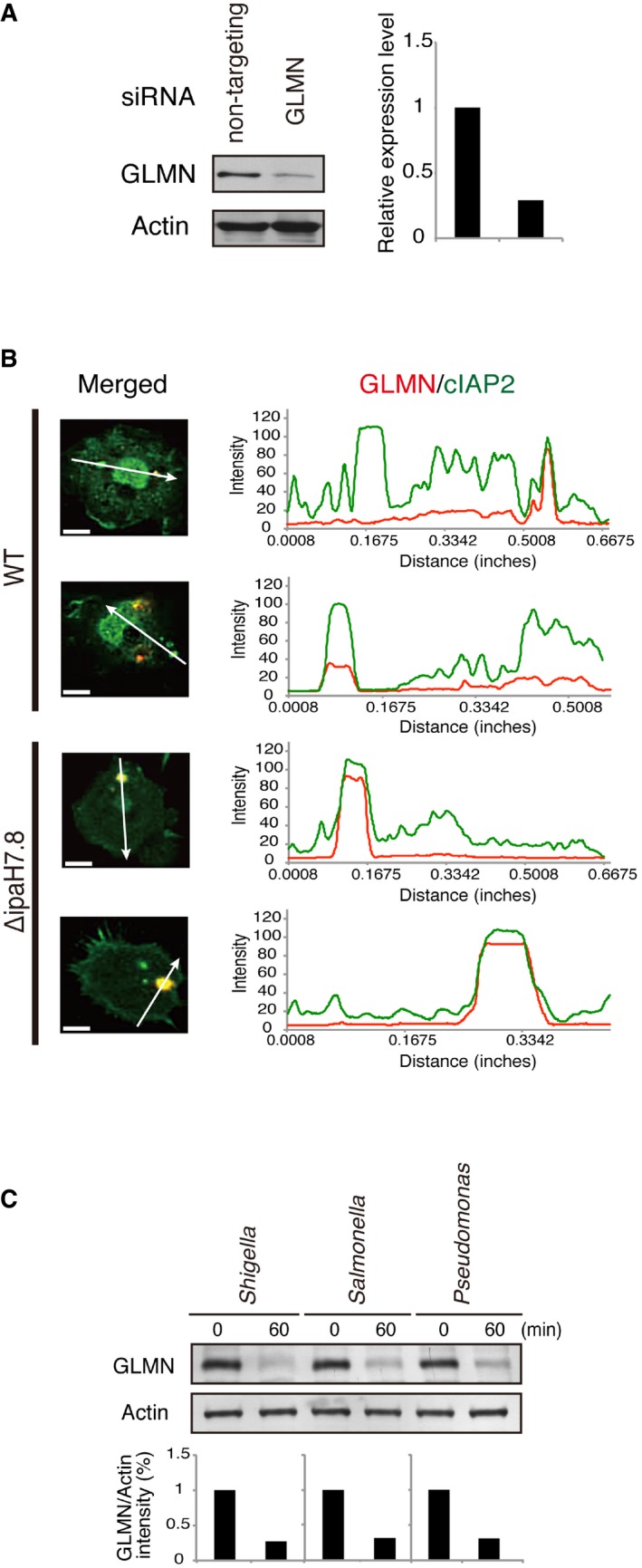
- Macrophages were subjected to siRNA‐mediated knockdown of GLMN (or non‐targeting siRNA as the control), and then, GLMN expression levels were assessed by immunoblotting with anti‐GLMN antibody (see also Fig 3).
- Colocalization analysis. Graph shows the intensities of the fluorescence signals along the white arrows. Scale bars: 10 μm.
- BMDMs were infected with indicated bacterial strains. Cells were harvested 1 h post‐infection, followed by immunoblotting to measure the level of endogenous GLMN. Data represent one of three similar experiments.
Source data are available online for this figure.
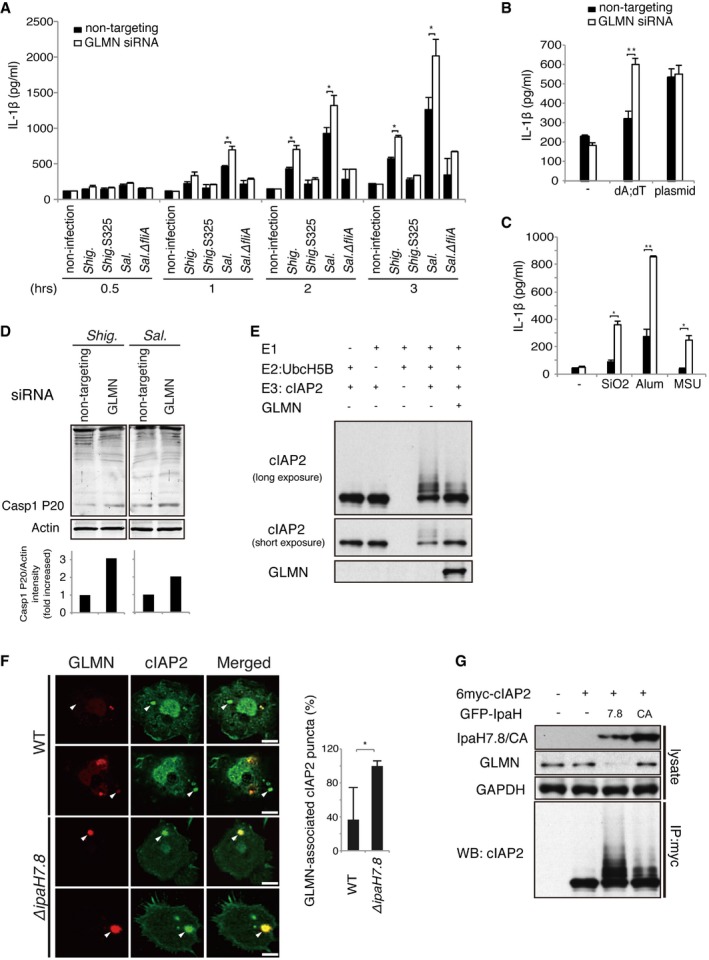
Figure 3. GLMN inhibits self‐ubiquitination by cIAPs, which results in negative regulation of inflammasomes.
-
AGLMN‐knockdown macrophages were infected with Shigella at an MOI of 5 or Salmonella at an MOI of 1, and IL‐1β levels were measured at the indicated time points. Non‐targeting siRNA was used as a control. n = 3, *P < 0.01.
-
B, CGLMN‐knockdown cells were treated with stimulators of AIM2 (B) or NLRP3 (C) inflammasomes for 16 h, and then, levels of IL‐1β were measured. n = 3, *P < 0.01, **P < 0.005.
-
DGLMN‐knockdown cells were infected with Shigella at an MOI of 5 or Salmonella at an MOI of 1, and then, the active mature form (P20) of caspase‐1 was detected by immunoblotting.
-
EIn vitro ubiquitination assay showing that GLMN inhibits self‐ubiquitination by cIAP2. Ubiquitination reactions were performed in the presence of purified ubiquitin, ATP, E1, E2 (UbcH5B), E3 (cIAP2), and GLMN.
-
FConfocal imaging analysis of cIAP2 (green) and GLMN (red) at the late stage of Shigella infection. BMDMs from caspase‐1 KO mice were infected with Shigella WT or ΔipaH7.8 for 3 h, and then, the GLMN/cIAP2 complex was visualized by immunohistochemistry using anti‐cIAP2 and anti‐GLMN antibodies. White arrows indicate the localization of GLMN‐associated cIAP2 puncta. Scale bars, 10 μm. Graph indicates the ratios of GLMN‐associated cIAP2 puncta to total cIAP2 puncta in Shigella‐infected BMDMs, expressed as a percentage (%). n = 20, *P < 0.0001.
-
G293T cells were transfected overnight with or without 6myc‐tagged cIAP2 and GFP‐tagged IpaH7.8 or IpaH7.8CA. 6myc‐tagged cIAP2 in cell lysates was immunoprecipitated with anti‐Myc antibody and subjected to immunoblot with anti‐cIAP2 antibody.
Recent work showed that GLMN acts as an inhibitor of RING E3 ligase by masking its E2‐binding surface 20, 32. We found that GLMN could bind RING‐possessing E3 ubiquitin ligases, including cIAP1 and cIAP2 (Fig 1A–C), suggesting that GLMN modulates the E3 ligase activity of cIAPs in this manner. To verify this, we purified GLMN from Escherichia coli and monitored its ability to inhibit self‐ubiquitination of cIAP2 in vitro. Indeed, GLMN decreased the level of cIAP2 self‐ubiquitination (Fig 3E).
To confirm the physical linkage between GLMN and cIAPs in macrophages, we investigated the behavior of GLMN and cIAP2 in BMDMs infected with Shigella. As shown in Fig 1D, in infected cells the GLMN signal (red) was visible as puncta that colocalized with cIAP2 foci (green) 1 h post‐infection, whereas in uninfected cells both signals were dispersed throughout the cytoplasm (Fig 1D, bottom panels). We subsequently investigated GLMN and cIAP2 localization in caspase‐1 KO BMDMs infected with Shigella WT or ∆ipaH7.8 (Figs 3F and EV3B). Although the GLMN puncta were still observed in cells infected with Shigella WT, the number of GLMN‐associated cIAP2 puncta was much smaller than in cells infected with ∆ipaH7.8 (Figs 3F and EV3B). In the cotransfection of 6myc‐cIAP2 and GFP‐IpaH7.8 (or GFP‐IpaH7.8CA) into 293T cells, the level of GLMN was much lower in lysate containing 6myc‐cIAP2 and GFP‐IpaH7.8 than that in lysate containing 6myc‐cIAP2 and GFP‐IpaH7.8CA (Fig 3G). Similar GLMN degradation was also observed in BMDMs infected with Shigella, Salmonella, or Pseudomonas (Fig EV3C). More importantly, when lysates containing 6myc‐cIAP2 and GFP‐IpaH7.8 (or GFP‐IpaH7.8CA) were pulled down by anti‐Myc antibody, the rate of self‐ubiquitination of precipitated cIAP2 was higher than that in lysate containing 6myc‐cIAP2 and GFP‐IpaH7.8CA (Fig 3G). These data further support the premise that GLMN undergoes proteasomal degradation due to ubiquitination by IpaH7.8 E3 ligase, resulting in upregulation of cIAP2 E3 ligase activity and activation of NLR inflammasomes.
A serine residue in the RING domain of cIAPs is critical for interacting with GLMN
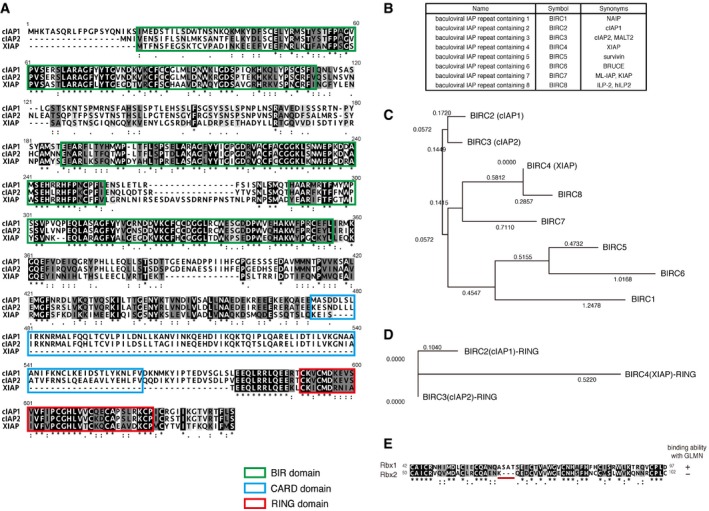
cIAP1 and cIAP2 share several domains: three baculovirus IAP repeat (BIR) domains; the ubiquitin‐association (UBA) domain, which allows interaction with ubiquitin chains; the caspase‐recruitment domain (CARD), which regulates E3 activity; and the RING (really interesting new gene) domain, which acts as the E2 recruitment domain (Figs 4A and EV4A) 28, 29, 33. Importantly, XIAP shares many of these domains with cIAP1 and cIAP2, but lacks the CARD domain (Figs 4A and EV4A). These domains are highly conserved and exhibit remarkably high amino acid sequence similarity (94.8%) between cIAP1 and cIAP2 (Table EV3). In contrast, XIAP is less similar to cIAP1 and cIAP2, with 55–57% nucleotide identity and 73–74% amino acid similarity with cIAP1 and cIAP2 (Table EV3). The amino acid sequences of the RING domains are identical (100% similarity) between cIAP1 and cIAP2 (Table EV3). The human IAP family consists of eight members (Fig EV4B), and phylogenetic analysis revealed that cIAP1 and cIAP2 are closely related to each other, whereas XIAP is on another branch (Fig EV4C and D).
Figure 4. The serine residue on the cIAP RING domain is important for the interaction with GLMN.
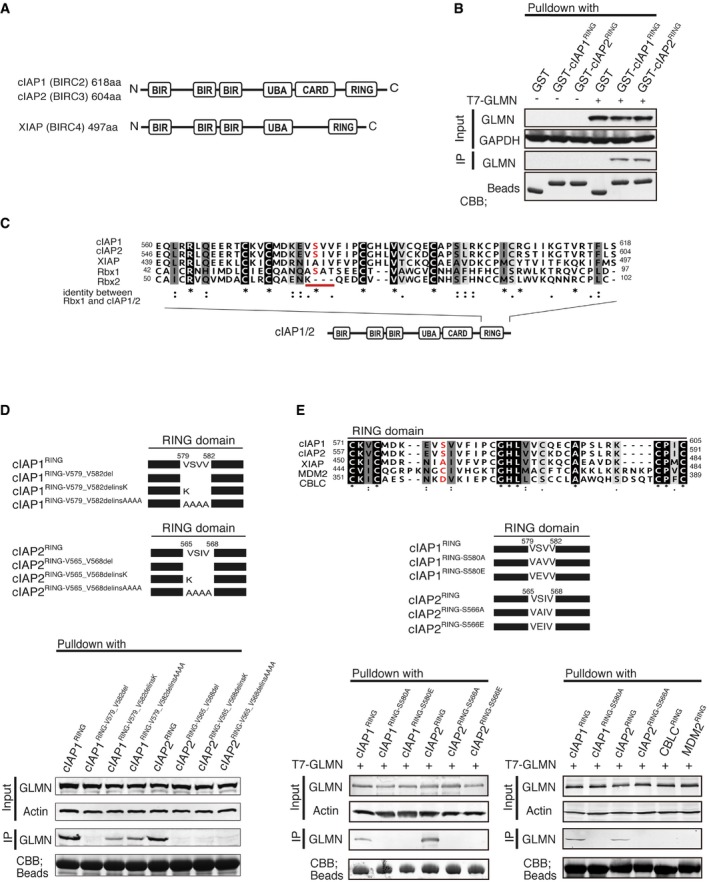
- A schematic representation of cIAP1 (BIRC2, 618 aa) and cIAP2 (BIRC3, 604 aa), and XIAP (BIRC4, 497 aa).
- GST‐pulldown assays revealed that GLMN binds to the RING domain of cIAPs.
- Comparison of the RING‐domain amino acids of Rbx1, Rbx2, cIAP1, cIAP2, and XIAP. Amino acids that are identical (black) or similar amino acids (gray) among the five sequences are shaded. cIAP1 and cIAP2 have a serine residue in the red‐underlined motif that is present in Rbx1 but not Rbx2 or XIAP.
- Point‐mutation analysis indicated that the VSV(I)V motif of the cIAP RING domain is important for the interaction with GLMN. VSV(I)V motifs were deleted or mutated as indicated in the upper panel, and the mutants were subjected to pulldown assays.
- Upper panel: RING domain sequences were compared with those of two other E3 ligases, MDM2 and CBLC. An amino acid sequence alignment of the RING domains of cIAP1, cIAP2, XIAP, MDM2, and CBLC is shown. MDM2 and CBLC do not have a serine residue in the motif corresponding to the VSV(I)V motif in the cIAPs‐RING domain. Middle panel: schematic representation of point mutations of the serine residue in the VSV(I)V motifs of the cIAPs‐RING domains. Lower panel: MDM2−RING, CBLC−RING, and point‐mutated RING domains were expressed, purified, and subjected to GST‐pulldown assays.
Figure EV4. Sequence alignment and phylogenetic tree building for cIAP, cIAP2, and XIAP.
-
AComparison of amino acid sequences of cIAP, cIAP2, and XIAP, aligned using ClustalW. Green, blue, and red boxes indicate the BIRs, CARD, and RING, respectively. The three BIRs and RING are highly conserved among all three proteins. XIAP lacks the CARD, which is well conserved between cIAP1 and cIAP2.
-
BA list of the BIRC family members. The human BIRC family consists of eight members. A common feature of all BIRCs is the presence of BIR domain(s) in one to three copies. BIRC2 (cIAP1), BIRC3 (cIAP2), BIRC4 (XIAP), BIRC7, and BIRC8 possess RING zinc‐finger domains that can act as E3 ligases.
-
C, DPhylogenetic analysis of BIRC family members. The neighbor‐joining (NJ) method was used to create a phylogenetic tree using the Genetyx‐Mac software. The amino acid sequences of full‐length of BIRC family members (C) or RING domains (D) were analyzed. The phylogenetic tree shows that cIAP1 and cIAP2 are closely related to each other, whereas XIAP is located on another branch.
-
EAmino acid sequence alignment of the RING domains of Rbx1 and Rbx2. Identical amino acids (black) and similar amino acids (gray) are shaded. The RING domain is highly conserved between Rbx1 and Rbx2, except for the motif underlined in red.
A recent study showed that GLMN inhibits CRL1 activity by masking the RBX1 RING finger that is essential for E2 binding 20, 32, suggesting that GLMN interacts with cIAP1 and cIAP2 via their RING domains. In agreement with this hypothesis, GST‐pulldown assays revealed that GLMN binds to the RING domains of both cIAP1 and cIAP2 (Fig 4B). Because GLMN binds to Rbx1 but not Rbx2 21, we carefully examined the aligned sequences of these two proteins. The RING domains are highly conserved between Rbx1 and Rbx2, except for four amino acids (A61 to T64, –A–S–A–T–, in Rbx1; these residues are underlined with red in Fig EV4E). Therefore, we compared the RING domain amino acids between Rbx1, Rbx2, cIAP1, cIAP2, and XIAP and noted that cIAP1, cIAP2, and Rbx1 (which interact with GLMN) share a serine residue (underlined red in Fig 4C) that is not present in Rbx2 (which does not interact with GLMN) or XIAP (Fig 4C). Hence, we hypothesized that the RING‐domain amino acids common to cIAP1, cIAP2, and Rbx1 might play an important role in the interaction with GLMN.
To test this hypothesis, we constructed a series of point mutants in which the serine residue of interest was replaced with other amino acids (Fig 4D, upper panel). Each of the RING‐domain mutants was investigated for its ability to interact with GLMN in a GST‐pulldown assay. Deletion of –V579–S580–V581–V582– of cIAP1 or –V565–S566–I567–V568– of cIAP2 RING domains abolished the interaction with GLMN (Fig 4D). Similarly, interaction between GLMN and the cIAP RING domains was greatly diminished by the amino acid replacement(s) indicated in the upper panel (Fig 4D), implicating that the VSV(I)V motif of cIAP RING domains is important for interaction with GLMN. Therefore, we checked the amino acid sequences of other E3 ligases possessing RING domains (MDM2 and CBLC) and found that they do not have the serine residue of interest (Fig 4E, upper panel). To reinforce our findings, we compared the interaction of GLMN to the RING domains of MDM2, CBLC, and cIAPs. We also constructed cIAP1/2‐RING point‐mutated clones in which the serine residue was replaced with alanine (A) or glutamic acid (E). These RING domains were expressed in E. coli, purified, and subjected to GST‐pulldown assays. The results revealed that the RING domains of MDM2 and CBLC did not interact with GLMN (Fig 4E, bottom panel). Moreover, the cIAP1RING‐S580A, cIAP1RING‐S580E, cIAP2RING‐S566A, and cIAP2RING‐S566E point mutants also lacked the ability to interact with GLMN (Fig 4E, bottom panels). Finally, we created point mutants in full‐length cIAP1 (cIAP1S580A, cIAP1S580E) and cIAP2 (cIAP2S566A, cIAP2S566E) in which the key RING domain serine residues were replaced with alanine or glutamic acid residues. The resulting mutants were expressed at high levels in macrophages, which were subsequently infected with Shigella (Fig 5A), or stimulated with poly dA:dT (Fig 5B), SiO2, or alum (Fig 5C). The serine‐to‐alanine/glutamic acid mutant cIAPs failed to activate inflammasomes (Fig 5A–C). The results of the series of experiments suggest that the serine residue of the RING domain is critical for facilitating interaction with GLMN as well as for inflammasome activation.
Figure 5. Mutation of the serine residue in the cIAP VSV(I)V motif exerts a dominant‐negative effect on inflammasome activation.
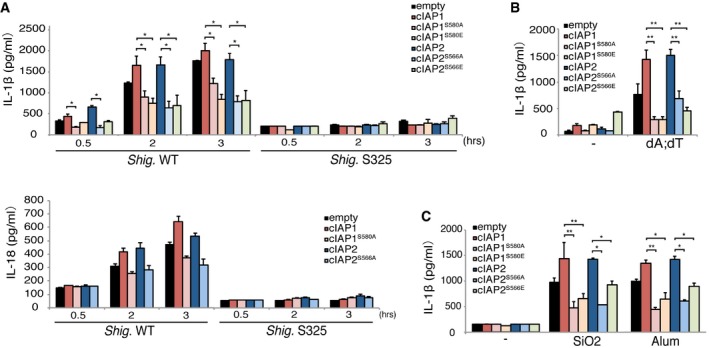
-
A–CWT macrophages overexpressing cIAP1, cIAP1S580A, cIAP1S580E, cIAP2, cIAP2S566A, or cIAP2S566E were infected with Shigella for 0.5–3 h (A), stimulated with poly dA:dT (B), or treated with NLRP3 stimulators (SiO2, Alum) (C), and then, IL‐1β levels were measured. n = 3, *P < 0.05, **P < 0.005. Error bars represent the SD of the measurements. Statistical analyses were performed using the Mann–Whitney U‐test. The results are representative of three independent experiments.
Discussion
In this study, we discovered that the GLMN–cIAPs axis controls inflammasome activation. Our data strongly suggest that the IpaH7.8–GLMN–cIAPs axis (Fig 6) acts as a unique mechanism by which Shigella can accelerate macrophage pyroptosis causing severe inflammation. GLMN has recently attracted much attention due to the discovery of its capacity to directly bind the RING domain of RBX1, thereby inhibiting the E3 ubiquitin ligase activity 20, 21. Structural and biochemical analyses indicated that GLMN adopts a HEAT‐like repeat fold that tightly binds the E2‐interacting surface of RBX1, inhibiting CRL‐mediated ubiquitin chain formation by the E2 20. Meanwhile, cIAPs have also been shown to play pivotal roles in gut homeostasis 34, immunity 35, inflammation, cell death, and cancer development. Notably, cIAP1 and cIAP2 are known modulators of the inflammasome and involved in efficient caspase‐1 activation. Together with the adaptor protein TRAF2, cIAPs interact with caspase‐1‐containing complex and mediate the K63‐linked non‐degradative ubiquitination of caspase‐1 30. Deficiency in either cIAP1 or cIAP2, therefore, severely impairs caspase‐1 activation, blunting the inflammatory reaction to various inflammasome agonists 30. As shown in this study, binding of cIAP1 and cIAP2 by GLMN suppresses inflammasome activation. Upon overexpression of cIAP1 or cIAP2 in macrophages, the levels of IL‐1β and pyroptotic cell death increased in response to bacterial infection or stimulation with NLR inflammasome agonists. In addition, the levels of IL‐1β and cell death were greatly reduced in cells subjected to siRNA‐mediated knockdown of cIAP1 and cIAP2. Based on these results, together with those of previous studies 20, 21, 30, we conclude that the GLMN–cIAPs axis is involved in regulating inflammasome activation. In the context of Shigella‐directed inflammasome activation, where the level of GLMN in macrophages can be modified by the IpaH7.8 E3 ligase, leading to the upregulation of the E3 ligase activities of cIAPs, it is likely that the GLMN–cIAPs axis may act as a suppression circuit for modulating inflammasome activation. In our study, other pathogenic bacteria such as Salmonella and Pseudomonas also showed cIAP‐mediated inflammasome activation on infection of macrophages (Fig EV2C). Some pathogenic bacteria are also known to possess bacterial effectors that are similar to Shigella IpaHs. For example, the Salmonella effector, SspH2, is homologous to Shigella IpaH7.8 and shares a similar molecular structure with IpaH7.8. SspH2 may also function in controlling the GLMN–cIAPs–casp1 axis, and this is a subject for future analysis.
Figure 6. A proposed model for the role of GLMN and IAPs in regulation of inflammasomes.
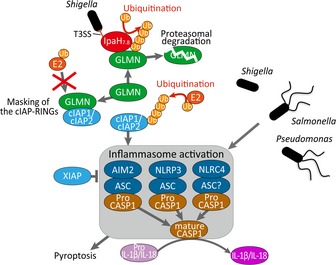
A schematic representation shows the machinery for the regulation of inflammasome activation caused by bacterial infection. In this study, we demonstrated that cIAP1 and cIAP2 are important for efficient inflammasome activation, whereas XIAP acts as a negative regulator of inflammasomes. Recent work showed that GLMN binds to small RING‐finger protein RBX1 (RING‐box1, an component of the Rbx1‐Cul1 E3 ligase), inhibiting its E3 ligase activity by masking the E2‐binding surface. In accordance with these observations, GLMN binds to the RING domains of cIAP1 and cIAP2, thereby inhibiting their functions. Shigella delivers the T3SS effector IpaH7.8, which targets GLMN at a specific stage of infection, thereby conferring an advantage on the bacterial invader. In our model, IpaH7.8 removes the masking GLMN from cIAPs via E3 ligase‐dependent proteolysis, leading to aberrant upregulation of inflammasomes.
Our studies strongly suggest that the GLMN–cIAPs axis plays a pivotal role in modulating inflammasome activity, governing the fate of pathogen colonization and host defense, and thus determining the outcome and severity of disease. Although further studies are needed to determine what kind of cellular or physiological conditions are necessary for regulation of inflammasome activity by the GLMN–cIAPs axis, since GLMN is ubiquitously produced in various cell types, our findings regarding the GLMN–cIAPs–inflammasome pathway reveal a previously unidentified mechanism of NLR inflammasomes to be negatively controlled. Although whether the GLMN–cIAPs axis could directly take part in terminating inflammasome activation or in preventing excess inflammation has yet to be determined, it must be an important mechanism for sustaining tissue homeostasis. cIAPs act as caspase inhibitors to modulate NF‐κB and inflammation, are associated with many types of cancer, and are involved in chemoresistance, disease progression, and malignancy (reviewed in 29, 33, 36). In light of these diverse activities of cIAPs, GLMN or the GLMN–cIAPs axis may become a promising therapeutic target for the cIAP‐related diseases.
Materials and Methods
Macrophages and cells
Primary bone marrow‐derived macrophages (BMDMs) obtained from wild‐type (WT) C57BL/6 mice were used in this study. Mice were housed in a specific pathogen‐free facility. All animal studies followed protocols approved by the Animal Care and Use Committee of the Institute of Medical Science, The University of Tokyo (Tokyo, Japan). BMDMs were prepared from femurs and tibias of mice and cultured for 5 days in RPMI (Gibco) supplemented with 30% L‐cell supernatant and 10% FCS. For siRNA/DNA transfection, macrophage cells immortalized by J2 retrovirus carrying the v‐Myc and v‐Raf oncogenes were used because such cells have a very high transfection efficiency. Retroviral supernatant was harvested from AMJ2‐C11 macrophage cell lines (ATCC CRL‐2456) 37. Immortalized WT macrophages were generated by infecting primary BMDMs with J2 recombinant retrovirus. 293T cells were cultured in Dulbecco's modified Eagle's medium (Sigma) containing 10% FCS.
Bacteria strains and cDNA clones
Shigella flexneri strain YSH6000 38 was used as the WT strain, and S325 (mxiA::Tn5) 39 was used as the type III secretion system‐deficient negative control. The Ipa7.8‐deficient Shigella strain (ΔipaH7.8) was constructed as previously described 15, 40. The WT S. enterica serovar Typhimurium SR‐11 v3181 and isogenic FliA‐deficient strain (fliA::Tn10; ΔfliA) were kindly provided by Dr. Hidenori Matsui (Kitasato Institute for Life Science, Tokyo, Japan) 41. 6Myc‐tagged cDNA clones of full‐length cIAP1, cIAP2, and XIAP in vector pcDNA3, used in IAP overexpression experiments in macrophages and immunoprecipitation studies, were kindly provided by Dr. Naohiro Inohara (University of Michigan, Ann Arbor, USA).
Antibodies
Rabbit polyclonal antibodies against GLMN and cleaved forms of caspase‐1 (P20) were described previously 18. Mouse monoclonal cIAP2 antibody (R&D, MAB817), mouse monoclonal anti‐actin antibody (clone C4, Chemicon, MAB1501), rabbit polyclonal anti‐GAPDH antibody (Sigma), rabbit polyclonal anti‐Myc antibody (A‐14, Santa Cruz Biotechnology, sc‐789), mouse T7•Tag® monoclonal antibody (Merck Millipore, 69522), mouse monoclonal anti‐FLAG antibody (M2, Sigma, F3165), and rabbit polyclonal anti‐GFP antibody (MBL, 598) were purchased from the indicated suppliers. HRP‐ or AP‐conjugated goat anti‐rabbit (Santa Cruz Biotechnology, sc‐2030, sc‐2007) and anti‐mouse (Santa Cruz Biotechnology, sc‐2005, sc‐2008) IgGs were used as secondary antibodies for immunoblotting. FITC‐ or TRITC‐conjugated goat anti‐rabbit and anti‐mouse IgGs (Sigma) were used as secondary antibodies for immunostaining.
Yeast two‐hybrid assay
To identify GLMN target protein(s), yeast two‐hybrid screening was performed using the ProQuest Two‐Hybrid System (Invitrogen) and a mouse brain cDNA library (cloned into pPC86 plasmid, Invitrogen). Full‐length GLMN was cloned into pDBleu plasmid and used as bait. Saccharomyces cerevisiae strain MaV203 was transformed with the bait and library plasmids. Activation of the His3 reporter was assessed by growing yeast in histidine‐deficient minimal medium in the presence of 20–50 mM tri‐amino‐triazole (3‐AT).
Immunoprecipitation
Mouse monoclonal anti‐FLAG M2 affinity gel (Sigma), rat monoclonal anti‐GFP‐agarose beads (MBL) or rabbit polyclonal anti‐Myc antibody A‐14 (Santa Cruz Biotechnology) were used for immunoprecipitation. All cell lysates for immunoprecipitation were prepared using CelLytic M buffer (Sigma) and complete protease inhibitor cocktail (Roche). For anti‐FLAG immunoprecipitation, 293T cells were cotransfected with 6Myc‐tagged cIAPs (or an empty vector control) and FLAG‐tagged GLMN (or an empty vector control) for 20 h, and then, the cell lysates were prepared and mixed with anti‐FLAG M2‐conjugated beads. For anti‐GFP immunoprecipitation, 293T cells were cotransfected with 6myc‐tagged cIAPs and GFP‐tagged IpaH7.8CA, and then, cell lysates were mixed with anti‐GFP‐conjugated agarose beads. For anti‐Myc immunoprecipitation, 293T transfectants expressing 6myc‐tagged cIAPs and GFP‐tagged IpaH7.8 (or IpaH7.8CA) were lysed in CelLytic M buffer and reacted with anti‐Myc antibody for 1 h and then mixed with protein G Sepharose (GE Healthcare). After overnight incubation with gentle rotation, beads were washed with CelLytic M buffer, mixed with SDS sample buffer (10% glycerol, 0.1% bromophenol blue, 62.5 mM Tris–HCl, pH 6.8, 5% 2‐mercaptoethanol, and 2% SDS) and boiled at 100°C for 6 min. The samples were subjected to immunoblot analysis with anti‐Myc, anti‐GFP, anti‐GLMN, and anti‐cIAP2 antibodies.
Immunofluorescence staining
For immunofluorescence studies, infected cells were fixed and immunostained as previously described 18, and images were captured in a single confocal plane with a confocal laser‐scanning microscope (Carl Zeiss).
Bacterial infection
The procedure for Shigella infection was described previously 9, 18. Shigella strains were pre‐cultured overnight in Mueller–Hinton broth (Difco) at 30°C. Bacterial cultures were inoculated into brain–heart infusion broth (Difco) and incubated for 2 h at 37°C prior to infection. Macrophages were stimulated with 0.1 μg/ml LPS (from E. coli O55:B5, Sigma) for 2 h and then infected with Shigella at an MOI of 5–10. The plates were centrifuged at 700 g for 5 min to synchronize infection, and gentamicin (100 μg/ml) and kanamycin (60 μg/ml) were added after 20 min. At the indicated times after infection, cytokine levels in culture supernatants were measured using specific ELISA kits (R&D Systems, eBioscience). Active forms of caspase‐1 (P20) were assessed by immunoblotting. To compare protein levels, bands were quantitated by densitometry, analyzed using the ImageJ software, and normalized against actin or GAPDH protein levels. Error bars represent standard deviations.
Stimulation of Nlrp3 and AIM2 inflammasomes
SiO2 (Sigma), alum (Sigma), and monosodium urate (MSU, Adipogen) were used to stimulate the Nlrp3 inflammasome. Macrophages were seeded overnight in a CO2 incubator at 37°C. They were pre‐stimulated with 0.1 μg/ml LPS for 2 h to upregulate IL‐1β mRNA levels and then treated with SiO2, alum, and MSU. To stimulate the AIM2 inflammasome, macrophages were transfected with poly (dA:dT; Sigma) or plasmid DNA using Lipofectamine 2000 (Invitrogen). After stimulation for 24 h, supernatants were collected, and cytokine levels were measured by ELISA.
siRNA and DNA transfection
siRNA/DNA transfection into macrophages was performed on an Amaxa Nucleofector system (Lonza) using Kit V and program D‐032. siRNAs specific for mouse cIAP1, cIAP2, XIAP, and GLMN were prepared by RNAi Inc. (Tokyo, Japan). Non‐targeting siRNAs were purchased from Dharmacon. For siRNA transfection, two to four siRNAs per targeting gene were constructed, mixed, and transfected into cells to increase the knockdown efficiency. Two days after siRNA nucleofection, cells were infected with bacteria or treated with stimulators. For overexpression of cIAP1, cIAP2, and XIAP, macrophages were nucleofected with pcDNA vectors carrying cDNAs of cIAP1, cIAP2, and XIAP. One day after nucleofection, cells were subjected to the experiments. For transfection into 293T cells, FuGENE 6 (Roche) or Lipofectamine 2000 (Invitrogen) was used. The standard error was calculated based on at least three independent experiments.
RT–PCR
Total RNA was prepared from cells using the RNeasy mini kit (Qiagen) and RNAse‐free DNase (Qiagen). To prepare templates for PCRs, total RNA was converted to cDNA using the SuperScript™ III First‐Strand Synthesis System (Invitrogen) with random hexamers. cDNA templates (100 ng) were used for PCRs with Ex Taq DNA polymerase (TaKaRa) and specific primer sets. Bands were scanned, and their intensities were quantitated using the ImageJ software. mRNA levels were normalized against the corresponding level of actin mRNA.
Generation of KO cells using the CRISPR–Cas9 system
cIAP1‐KO, cIAP2‐KO, cIAP1/2‐double KO, and XIAP‐KO macrophages were generated using the IDT Alt‐R® CRISPR–Cas9 System (Integrated DNA Technologies). CRISPR–Cas9 ribonucleoprotein (RNP) complex, containing Alt‐R® CRISPR–Cas9 crRNA:tracrRNA and a Cas9 enzyme, was prepared following the manufacturer's instructions. Two RNA oligos (crRNA and tracrRNA) were mixed in equimolar concentrations in a tube to a final duplex concentration of 100 μM. The tube was heated at 95°C for 5 min and then cooled to room temperature to allow for the formation of the crRNA:tracrRNA duplex. The duplex was diluted in PBS and incubated with purified recombinant Streptococcus pyogenes Cas9 nuclease (Integrated DNA Technologies) at room temperature for 10–20 min to form the RNP complex. Subsequently, the CRISPR–Cas9 RNP complex was delivered into immortalized WT macrophages by electroporation using the Amaxa® Nucleofector® system (Lonza). The same molar concentration of Alt‐R Cas9 electroporation enhancer (Integrated DNA Technologies) was added to improve electroporation efficiency. After incubation for 2 days, individual cells were isolated by limiting dilution. Isolated cells were expanded, and Cas9‐mediated mutagenesis of the target genes was confirmed by a T7E1 assay following the manufacturer's protocol (Integrated DNA Technologies).
In vitro ubiquitination assays
In vitro ubiquitination assays were performed as previously described 18. Samples were analyzed by immunoblotting using anti‐cIAP2 and anti‐GLMN antibodies.
Expression and purification of recombinant proteins
Point‐mutated strains of IAPs‐RING domains (amino acid residues composing the VSV(I)V motif were replaced with another amino acids) were created with a QuikChange site‐directed mutagenesis kit (Agilent Technologies). To express GST‐fusion proteins, E. coli BL21 Rosetta (DE3) strain harboring the pGEX‐6P‐1 derivatives was cultured for 8 h at 25°C in L‐broth supplemented with ampicillin (50 g/ml). Protein expression was induced by adding 1 mM IPTG and incubating the cells for 20 h at 25°C. Bacteria were disrupted by sonication and lysozyme treatment. GST‐fusion proteins were purified by affinity chromatography with Glutathione Sepharose 4B (GE Healthcare). The GST tag was removed by treating the beads that had bound the GST‐fusion protein with PreScission protease (GE Healthcare).
GST‐pulldown assay
Cell lysates of transfectants were prepared using CelLytic M buffer (Sigma) containing complete protease inhibitor cocktail (Roche). Glutathione Sepharose 4B beads (GE Healthcare) bound to GST‐fusion proteins or control GST were mixed with lysates and incubated overnight at 4°C. The beads were then washed with CelLytic M buffer and analyzed by immunoblotting.
Statistical analyses
The error bars represent the standard deviation (SD) of the measurements. Statistical analyses were performed using the Mann–Whitney U‐test. Differences were considered significant at P < 0.05. All results shown in figures are representative of at least three independent experiments.
Author contributions
SS conceived the original hypothesis and designed the study. CS and TS supervised the project. SS performed experiments. HM provided materials and bacterial strains. SS, CS, HM and TM analyzed results. SS and CS wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Acknowledgements
We thank N. Inohara for providing cDNA clones of full‐length cIAP1, cIAP2, and XIAP, and H. Matsui for providing Salmonella strains. This work was supported by a Grant‐in‐Aid for Specially Promoted Research 23000012 (C.S.), a Grant‐in‐Aid for Scientific Research (C) 16K08772 (S.S.), a Grant‐in‐Aid for Young Scientists (B) 19790315 (S.S.), the Research Grant of Takeda Science Foundation (S.S.), the Research Grant of Kato Memorial Bioscience Foundation (S.S.), a grant for Joint Research Project of the Institute of Medical Science, The University of Tokyo (T.S.), the Research Grant of Astellas Foundation for Research on Metabolic Disorders (S.S.), the Uehara Memorial Foundation Research Fellowship (S.S.), a Grant‐in‐Aid for JSPS Fellows 21·10053 (S.S.), the JSPS Excellent Young Researchers Overseas Visit Program 21·10053 (S.S.), and the Japan Initiative for Global Research Network on Infectious Diseases (C.S.). Part of this work was supported by Grants from the Yakult Central Institute (C.S.).
EMBO Reports (2018) 19: 89–101
Contributor Information
Shiho Suzuki, Email: ss.bact@tmd.ac.jp.
Chihiro Sasakawa, Email: sasakawa@ims.u-tokyo.ac.jp.
References
- 1. Cohen TS, Prince AS (2013) Activation of inflammasome signaling mediates pathology of acute P. aeruginosa pneumonia. J Clin Invest 123: 1630–1637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Faure E, Mear JB, Faure K, Normand S, Couturier‐Maillard A, Grandjean T, Balloy V, Ryffel B, Dessein R, Chignard M et al (2014) Pseudomonas aeruginosa type‐3 secretion system dampens host defense by exploiting the NLRC4‐coupled inflammasome. Am J Respir Crit Care Med 189: 799–811 [DOI] [PubMed] [Google Scholar]
- 3. Man SM, Kanneganti TD (2015) Regulation of inflammasome activation. Immunol Rev 265: 6–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Pedraza‐Alva G, Perez‐Martinez L, Valdez‐Hernandez L, Meza‐Sosa K, Ando‐Kuri M (2015) Negative regulation of the inflammasome: keeping inflammation under control. Immunol Rev 265: 231–257 [DOI] [PubMed] [Google Scholar]
- 5. Franchi L, Munoz‐Planillo R, Nunez G (2012) Sensing and reacting to microbes through the inflammasomes. Nat Immunol 13: 325–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Franchi L, Amer A, Body‐Malapel M, Kanneganti TD, Ozoren N, Jagirdar R, Inohara N, Vandenabeele P, Bertin J, Coyle A et al (2006) Cytosolic flagellin requires Ipaf for activation of caspase‐1 and interleukin 1beta in salmonella‐infected macrophages. Nat Immunol 7: 576–582 [DOI] [PubMed] [Google Scholar]
- 7. Miao EA, Alpuche‐Aranda CM, Dors M, Clark AE, Bader MW, Miller SI, Aderem A (2006) Cytoplasmic flagellin activates caspase‐1 and secretion of interleukin 1beta via Ipaf. Nat Immunol 7: 569–575 [DOI] [PubMed] [Google Scholar]
- 8. Miao EA, Mao DP, Yudkovsky N, Bonneau R, Lorang CG, Warren SE, Leaf IA, Aderem A (2010) Innate immune detection of the type III secretion apparatus through the NLRC4 inflammasome. Proc Natl Acad Sci USA 107: 3076–3080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Suzuki S, Franchi L, He Y, Munoz‐Planillo R, Mimuro H, Suzuki T, Sasakawa C, Nunez G (2014a) Shigella type III secretion protein MxiI is recognized by Naip2 to induce Nlrc4 inflammasome activation independently of Pkcdelta. PLoS Pathog 10: e1003926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Yang J, Zhao Y, Shi J, Shao F (2013) Human NAIP and mouse NAIP1 recognize bacterial type III secretion needle protein for inflammasome activation. Proc Natl Acad Sci USA 110: 14408–14413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F (2015) Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 526: 660–665 [DOI] [PubMed] [Google Scholar]
- 12. Kayagaki N, Stowe IB, Lee BL, O'Rourke K, Anderson K, Warming S, Cuellar T, Haley B, Roose‐Girma M, Phung QT et al (2015) Caspase‐11 cleaves gasdermin D for non‐canonical inflammasome signalling. Nature 526: 666–671 [DOI] [PubMed] [Google Scholar]
- 13. Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J (2016) Inflammasome‐activated gasdermin D causes pyroptosis by forming membrane pores. Nature 535: 153–158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ashida H, Ogawa M, Kim M, Suzuki S, Sanada T, Punginelli C, Mimuro H, Sasakawa C (2011a) Shigella deploy multiple countermeasures against host innate immune responses. Curr Opin Microbiol 14: 16–23 [DOI] [PubMed] [Google Scholar]
- 15. Ashida H, Ogawa M, Mimuro H, Kobayashi T, Sanada T, Sasakawa C (2011b) Shigella are versatile mucosal pathogens that circumvent the host innate immune system. Curr Opin Immunol 23: 448–455 [DOI] [PubMed] [Google Scholar]
- 16. Ashida H, Kim M, Sasakawa C (2014) Exploitation of the host ubiquitin system by human bacterial pathogens. Nat Rev Microbiol 12: 399–413 [DOI] [PubMed] [Google Scholar]
- 17. Ashida H, Mimuro H, Sasakawa C (2015) Shigella manipulates host immune responses by delivering effector proteins with specific roles. Front Immunol 6: 219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Suzuki S, Mimuro H, Kim M, Ogawa M, Ashida H, Toyotome T, Franchi L, Suzuki M, Sanada T, Suzuki T et al (2014b) Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc Natl Acad Sci USA 111: E4254–E4263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brouillard P, Boon LM, Mulliken JB, Enjolras O, Ghassibe M, Warman ML, Tan OT, Olsen BR, Vikkula M (2002) Mutations in a novel factor, glomulin, are responsible for glomuvenous malformations (“glomangiomas”). Am J Hum Genet 70: 866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Duda DM, Olszewski JL, Tron AE, Hammel M, Lambert LJ, Waddell MB, Mittag T, DeCaprio JA, Schulman BA (2012) Structure of a glomulin‐RBX1‐CUL1 complex: inhibition of a RING E3 ligase through masking of its E2‐binding surface. Mol Cell 47: 371–382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Tron AE, Arai T, Duda DM, Kuwabara H, Olszewski JL, Fujiwara Y, Bahamon BN, Signoretti S, Schulman BA, DeCaprio JA (2012) The glomuvenous malformation protein Glomulin binds Rbx1 and regulates cullin RING ligase‐mediated turnover of Fbw7. Mol Cell 46: 67–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Py BF, Kim MS, Vakifahmetoglu‐Norberg H, Yuan J (2013) Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 49: 331–338 [DOI] [PubMed] [Google Scholar]
- 23. Juliana C, Fernandes‐Alnemri T, Kang S, Farias A, Qin F, Alnemri ES (2012) Non‐transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 287: 36617–36622 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. López‐Castejón G, Luheshi NM, Compan V, High S, Whitehead RC, Flitsch S, Kirov A, Prudovsky I, Swanton E, Brough D (2013) Deubiquitinases regulate the activity of caspase‐1 and interleukin‐1beta secretion via assembly of the inflammasome. J Biol Chem 288: 2721–2733 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. López‐Castejón G (2014) Regulation of NLRP3 activation by the ubiquitin system. Inflammasome 1: 15–19 [Google Scholar]
- 26. Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q, Amatya R, Kelly TJ, Iwai K, Ting J et al (2014) The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med 211: 1333–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Minton K (2014) Inflammasomes: ubiquitin lines up for inflammasome activity. Nat Rev Immunol 14: 580–581 [DOI] [PubMed] [Google Scholar]
- 28. Beug ST, Cheung HH, LaCasse EC, Korneluk RG (2012) Modulation of immune signalling by inhibitors of apoptosis. Trends Immunol 33: 535–545 [DOI] [PubMed] [Google Scholar]
- 29. Gyrd‐Hansen M, Meier P (2010) IAPs: from caspase inhibitors to modulators of NF‐κB, inflammation and cancer. Nat Rev Cancer 10: 561–574 [DOI] [PubMed] [Google Scholar]
- 30. Labbe K, McIntire CR, Doiron K, Leblanc PM, Saleh M (2011) Cellular inhibitors of apoptosis proteins cIAP1 and cIAP2 are required for efficient caspase‐1 activation by the inflammasome. Immunity 35: 897–907 [DOI] [PubMed] [Google Scholar]
- 31. Vince JE, Wong WW, Gentle I, Lawlor KE, Allam R, O'Reilly L, Mason K, Gross O, Ma S, Guarda G et al (2012) Inhibitor of apoptosis proteins limit RIP3 kinase‐dependent interleukin‐1 activation. Immunity 36: 215–227 [DOI] [PubMed] [Google Scholar]
- 32. Hristova VA, Stringer DK, Weissman AM (2012) Cullin RING ligases: glommed by glomulin. Mol Cell 47: 331–332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vandenabeele P, Bertrand MJ (2012) The role of the IAP E3 ubiquitin ligases in regulating pattern‐recognition receptor signalling. Nat Rev Immunol 12: 833–844 [DOI] [PubMed] [Google Scholar]
- 34. Dagenais M, Dupaul‐Chicoine J, Champagne C, Skeldon A, Morizot A, Saleh M (2016) A critical role for cellular inhibitor of protein 2 (cIAP2) in colitis‐associated colorectal cancer and intestinal homeostasis mediated by the inflammasome and survival pathways. Mucosal Immunol 9: 146–158 [DOI] [PubMed] [Google Scholar]
- 35. Reardon C, Mak TW (2009) cIAP proteins: keystones in NOD receptor signal transduction. Immunity 30: 755–756 [DOI] [PubMed] [Google Scholar]
- 36. Budhidarmo R, Day CL (2015) IAPs: modular regulators of cell signalling. Semin Cell Dev Biol 39: 80–90 [DOI] [PubMed] [Google Scholar]
- 37. Palleroni AV, Varesio L, Wright RB, Brunda MJ (1991) Tumoricidal alveolar macrophage and tumor infiltrating macrophage cell lines. Int J Cancer 49: 296–302 [DOI] [PubMed] [Google Scholar]
- 38. Sasakawa C, Kamata K, Sakai T, Murayama SY, Makino S, Yoshikawa M (1986) Molecular alteration of the 140‐megadalton plasmid associated with loss of virulence and congo red binding activity in Shigella flexneri . Infect Immun 51: 470–475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Watarai M, Tobe T, Yoshikawa M, Sasakawa C (1995) Contact of Shigella with host cells triggers release of Ipa invasins and is an essential function of invasiveness. EMBO J 14: 2461–2470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Datsenko KA, Wanner BL (2000) One‐step inactivation of chromosomal genes in Escherichia coli K‐12 using PCR products. Proc Natl Acad Sci USA 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kodama C, Matsui H (2004) Salmonella flagellin is not a dominant protective antigen in oral immunization with attenuated live vaccine strains. Infect Immun 72: 2449–2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Expanded View Figures PDF
Table EV1
Table EV2
Table EV3
Source Data for Expanded View
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4