

Abstract
Despite years of research, the glycome of the model nematode Caenorhabditis elegans is still not fully understood. Certainly, data over the years have indicated that this organism synthesizes unusual N-glycans with a range of galactose and fucose modifications on the Man2–3GlcNAc2 core region. Previously, up to four fucose residues were detected on its N-glycans, despite these lacking the fucosylated antennae typical of many other eukaryotes; some of these fucose residues are capped with hexose residues as shown by the studies of us and others. There have, though, been contrasting reports regarding the maximal number of fucose substitutions in C. elegans, which in part may be due to different methodological approaches, including use of either peptide:N-glycosidases F and A (PNGase F and A) or anhydrous hydrazine to cleave the N-glycans from glycopeptides. Here we compare the use of hydrazine with that of a new enzyme (rice PNGase Ar) and show that both enable release of glycans with more sugar residues on the proximal GlcNAc than previously resolved. By use of exoglycosidase sequencing, in conjunction with high-performance liquid chromatography (HPLC) and matrix-assisted laser desorption ionization time-of-flight tandem mass spectrometry (MALDI-TOF MS/MS), we now reveal that actually up to five fucose residues modify the core region of C. elegans N-glycans and that the α1,3-fucose on the reducing terminus can be substituted by an α-linked galactose. Thus, traditional PNGase F and A release may be insufficient for release of the more highly core-modified N-glycans, especially those occurring in C. elegans, but novel enzymes can compete against chemical methods in terms of safety, ease of cleanup, and quality of resulting glycomic data.
Caenorhabditis elegans is a widely used genetically tractable experimental model for, e.g., development and aging biology research and was the first multicellular organism whose genome was sequenced.1 As a nonparasitic nematode, it is also suitable for comparisons with parasitic species of the same phylum. However, C. elegans continues to surprise glycobiologists in terms of its glycome; advances in methodology and the use of mutant strains over the past 15 years of N-glycan analyses have resulted in complex data sets and sometimes contradictory interpretations from different research groups.2 Early on, it was clear that C. elegans has an unparalleled fucosylation machinery, and up to four fucose residues were proposed to be present on its N-glycans with compositions such as Hex5–8HexNAc2Fuc3 or Hex5–7HexNAc2Fuc4Me0–1;3−7 furthermore, over 20 possible fucosyltransferase homologues are encoded by its genome, of which only a few are enzymatically characterized and proven to be able to fucosylate N-glycan structures.6,8−12 In terms of N-glycans, the roles of two α1,3-fucosyltransferases (FUT-1 and FUT-6) and one α1,6-fucosyltransferase (FUT-8) have been defined by us on the basis of analyses of the recombinant enzymes and of the glycomes of the corresponding single, double, and triple fut-1, fut-6, and fut-8 knockout mutants.13,14
Nevertheless, there remain open questions. On one hand, the glycomic studies on mutants revealed five positions for fucosylation, even though only maximally four fucoses were present on any verified structure. On the other, the maximal degree of galactosylation of these C. elegans N-glycans is unclear, despite the recent detection of bisecting galactose on the β1,4-mannose13 as well as the more established “GalFuc” 6-linked modification of the reducing-terminal (proximal) GlcNAc15 and a more unusual GalFuc 3-linked to the distal GlcNAc.16 In one study in which hydrazine was employed, the presence of a hexose (assumed to be β1,4-linked galactose) on the proximal 3-linked core fucose was proposed.7
Hydrazinolysis has been used for some decades for release of glycans,17,18 but is also associated with safety issues as well as artifacts. As hydrazine cleaves amide bonds, it not only will destroy peptide and N-glycosidic bonds, but also deacetylates GlcNAc, GalNAc, and neuraminic acids which must be re-N-acetylated (thus affecting the glycan structure in the case of, e.g., N-glycolylneuraminic acid) and may cause some “peeling” of the N-glycan core.19 Other chemical methods, e.g., under alkaline conditions,20 have not gained wide acceptance for N-glycomic studies. On the other hand, enzymatic release can be dependent on glycopeptide length and the specificity of the required peptide:N-glycosidases (PNGases; 3.5.1.52).21,22 However, some newly discovered enzymes have a wider substrate specificity in terms of cleaving core α1,3-fucosylated glycans from even intact proteins.23−25 Here we have compared the use of hydrazine with an enzyme of plant origin (PNGase Ar, a commercially available recombinant form of rice vacuolar PNGase) and show not only that there is an α-linked galactose attached to the core α1,3-fucose on a subset of C. elegans N-glycans but that pentafucosylated structures also exist in this organism with “maximal” compositions of Hex7HexNAc2Fuc4–5Me1–2. Thus, new enzymes have the promise of replacing chemical methods for safe and effective release of N-glycans with complex core modifications.
Methods
Biological Material
Wild-type C. elegans (N2) was obtained from the Caenorhabditis Genetics Centre (CGC), University of Minnesota, U.S.A. The fut-1;fut-8 and fut-6;fut-8 strains (lacking either the proximal core α1,3- and α1,6-fucose residues or the distal α1,3- and proximal α1,6-fucose) were previously prepared as described.14C. elegans were grown in liquid culture with Escherichia coli OP50 in standard S complete medium; mixed stages were harvested after cultivation at 20 °C (160 rpm) for 4–6 days and purified by sucrose density centrifugation.26,27 Harvested worms were boiled, homogenized, and treated with either pepsin (Sigma) overnight at 37 °C or thermolysin (Promega) for 2 h at 70 °C.28 Glycopeptides were purified by cation-exchange chromatography (Dowex 50WX8) followed by gel filtration (Sephadex G25); for further details refer to the Supporting Information.
N-Glycan Release, Purification, and Labeling
Enzymatic release of N-glycans from worm peptic glycopeptides was done using three different peptide:N-glycosidases: (i) recombinant bacterial PNGase F (from Flavobacterium [Elizabethkingia] meningosepticum, Roche; at pH 8.0), (ii) native almond PNGase A (from Prunus amygdalus, Roche; at pH 5.0), and (iii) recombinant rice PNGase Ar (from Oryza sativa expressed in Pichia pastoris and Endo H treated, New England Biolabs; also at pH 5.0). In the first experiments, digestion was either by PNGase F followed by PNGase A, and then PNGase Ar, or PNGase A followed by PNGase Ar of peptic peptides. In another experiment, thermolysin was used for proteolysis prior to two rounds of PNGase Ar. For chemical release, peptic glycopeptides were incubated with anhydrous hydrazine at 100 °C for 5 h. After centrifugal evaporation, the glycans were re-N-acetylated and treated with 5% (v/v) trifluoroacetic acid in order to liberate the reducing end.29 Note that hydrazine is a hazardous reagent and must only be used when applying relevant safety procedures.
After either enzymatic or chemical release, N-glycan pools resulting from the final round of Dowex 50WX8 chromatography were separately purified by solid-phase extraction using nonporous graphitized carbon (nPGC; elution with 40% acetonitrile) and LiChroprep RP-18 (C18; elution with water) prior to pyridylamination as previously described.28,30 In case of glycans released by two rounds of PNGase Ar digestion, solely C18 purification was performed after Dowex chromatography prior to pyridylamination. For a flowchart as well as further details, refer to the scheme and extended methods in the Supporting Information.
Mass Spectrometric Analysis
The N-glycomes of the different glycan preparations were profiled by matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF MS) (Autoflex Speed, Bruker Daltonics, Germany) in positive-ion mode using FlexControl 3.4 software. 2-Aminopyridine (PA)-labeled N-glycans were fractionated by reversed-phase high-performance liquid chromatography (RP-HPLC), also in combination with a hydrophilic interaction column (see below),28 and all collected HPLC peaks were also examined by MALDI-TOF MS, using 6-aza-2-thiothymine (ATT) as matrix; tandem mass spectrometry (MS/MS) to confirm the composition of all proposed structures was performed by laser-induced dissociation (precursor ion selector was generally set to ±0.6%). The detector voltage was generally set at 1977–2260 V for MS and 2133–2714 V for MS/MS; 1000–3000 shots from different regions of the sample spots were summed. Spectra were processed with the manufacturer’s software (Bruker Flexanalysis 3.3.80) using the SNAP algorithm with a signal-to-noise threshold of 6 for MS (unsmoothed) and 3 for MS/MS (4-times smoothed). In total approximately 2500 MS and MS/MS spectra were manually interpreted on the basis of the mass, fragmentation pattern, and results of chemical and enzymatic treatments; isomeric structures present in different RP-HPLC or 2D-HPLC fractions were defined on the basis of comparisons of the aforementioned parameters. At least five MS/MS fragment ions, including Y-type and B-type ions, were used to aid definition of each structure. Three selected HPLC-purified N-glycans were subject to LC–MSn as previously described using a 5 μm porous graphitized carbon column and an LTQ ion trap mass spectrometer (Thermo Scientific) in negative-ion mode14 with the spectral interpretation being performed in comparison to the literature.31
HPLC Purification of N-Glycans
Separation of PA-labeled glycans was carried out on a Shimadzu HPLC system equipped with a fluorescence detector. In case of RP-HPLC, a Hypersil ODS column (C18; Agilent) was used with 100 mM ammonium acetate, pH 4.0, and 30% (v/v) methanol; a gradient of the latter (1% per minute) was programmed. The column was calibrated daily in terms of glucose units (g.u.) with a pyridylaminated partial dextran hydrolysate. For 2D-HPLC, selected fractions were reapplied to a combined hydrophilic-interaction anionic-exchange HPLC (HIAX, Dionex IonPac AS11) with an inverse gradient of acetonitrile in 800 mM ammonium acetate, pH 3.85, as previously described.32 For further details refer to the Supporting Information.
Structural Elucidation Using Exoglycosidases and Chemical Treatment
In general, a 1 μL aliquot of an HPLC fraction was mixed with 0.2 μL of exoglycosidase and 0.8 μL of 100 mM ammonium acetate solution, pH 5.0, and incubated overnight. Exoglycosidases employed were: α-galactosidase from green coffee beans (Sigma), β-galactosidase from either Aspergillus niger(33) (recombinant, produced in-house) or Aspergillus oryzae (native; Sigma), jack bean α-mannosidase (Sigma), recombinant Xanthomonas manihotis α1,2/3-mannosidase34 (New England Biolabs), and α-L-fucosidase from bovine kidney (Sigma). For removal of α1,2/3-linked fucose or methylfucose, glycan samples were dried in a SpeedVac, and then incubated with 3 μL of 48% (w/v) hydrofluoric acid (HF) on ice for 24 h. The HF was immediately removed in a SpeedVac. Chemically or enzymatically treated glycans were generally reanalyzed, unless otherwise stated, by MALDI-TOF MS and MS/MS without further purification. For further details refer to the Supporting Information.
Results and Discussion
Comparison of Chemical and Enzymatic N-Glycan Release
In order to resolve the contrasting data regarding the maximal degree of core modification of N-glycans in C. elegans, especially whether only chemical release is suitable for isolation of structures carrying a 3-linked HexFuc element,7 we compared the results obtained after hydrazinolysis with those with different PNGases, including a new commercially available plant-derived enzyme (here named PNGase Ar). Typically, to release nematode N-glycans, we and others have serially digested with Flavobacterium PNGase F and almond PNGase A or just used PNGase A alone,4,29,35,36 whereas hydrazinolysis has been used relatively rarely for “worm” samples.7,29,37 There are also differences in procedures prior to glycan release, with either trypsin or pepsin being commonly used to prepare glycopeptides; recently, we have begun to adopt thermolysin to proteolyze samples prior to PNGase treatment,28 as the proteolysis step is at neutral pH and is more rapid.
In this study, the following glycan release protocols were performed: (i) PNGase F, PNGase A, and PNGase Ar, (ii) PNGase A and PNGase Ar, (iii) two rounds of PNGase Ar, or (iv) with hydrazine. The N-glycans were then subject to pyridylamination, RP-HPLC (Supplementary Figure 1), and MALDI-TOF MS. The most complex RP-HPLC profiles were those resulting after release with hydrazine or with PNGase Ar alone, while those chromatograms obtained after use of either PNGases F or A are overall apparently simpler; the major peaks in these four cases are of 4.4, 4.7, 7.0–7.2, and 11.2 g.u. On the basis of MALDI-TOF MS of the individual fractions, oligomannosidic (Man8,9GlcNAc2 and Man5GlcNAc2 4.4, 4.7, and 7.0 g.u.) and paucimannosidic glycans (Man3GlcNAc2Fuc0–1; 7.2 and 11.2 g.u., with and without core α1,6-fucose) dominate the profile after use of PNGase F, an enzyme which cannot cleave glycans with a core α1,3-fucose.22 The glycans released by PNGase A or PNGase Ar included, e.g., standard core di-α1,3/α1,6-fucosylated paucimannosidic structures eluting at 6–8 g.u. as well as earlier-eluting galactosylated forms thereof; all pools contained some phosphorylcholine-modified glycans which are not further discussed here, as the focus is on the galactose-containing fucosylated structures, whose bulky 3D conformation probably influences the ability to release them.
Hydrazine and PNGase Ar Released Glycans with a Novel α-Galactose Residue
When considering the hydrazine-released glycans, we focused on those which presented previously unobserved elution and fragmentation properties. In former studies on wild-type nematode glycomes, we have observed Y-fragments (derived from the pyridylaminated core) such as m/z 446, 592, 608, and 754 (Fuc1–2Hex0–1GlcNAc1–PA29,35), whereas MALDI-TOF MS screening of each RP-HPLC fraction of the hydrazine-released pool indicated the presence of fragment ions at m/z 916 for glycans eluting at 4.4 and 4.7 g.u. (Figure 1); this would correspond to a core of Fuc2Hex2GlcNAc1–PA. Typically, the MS/MS spectra with such a fragment also contained ions at m/z 754 (minor), 608 (relatively intense), and 446 (minor) suggesting that each core fucose on the reducing-terminal GlcNAc (both the α1,3- and α1,6-linked) was modified by a hexose, considering also that the core α1,3-fucose is usually more labile in MS/MS. To investigate these structures more exactly, a number of glycosidase and chemical treatments were performed. Three galactosidases (coffee bean α- and two Aspergillus β-specific) as well as bovine α-fucosidase were used in addition to hydrofluoric acid, and shifts in MS and MS/MS profiles were assessed.
Figure 1.

Detection of novel core diagnostic fragments in wild-type C. elegans N-glycans. N-Glycans released by hydrazinolysis from the N2 wild-type C. elegans were PA-labeled and separated on RP-HPLC (A); collected fractions were subject to MALDI-TOF MS and MS/MS analyses in positive-ion mode. In two neighboring fractions, eluting around 4.4–4.7 glucose units (g.u.), in addition to the oligomannosidic Man8B and Man9 structures of m/z 1799 and 1961, five fucosylated N-glycans with compositions of Hex6–7HexNAc2Fuc2–3Me0–1 (m/z 1767, 1913, 1927, 1929, and 2075 as [M + H]+) were identified. (B). Fragmentation of these glycans yielded novel core Y-fragment ions at m/z 916 (Hex2dHex2GlcNAc–PA), indicative of an additional modification of the innermost GlcNAc residue (C). Proposed structures are shown according to the Standard Nomenclature for Glycans (ref (42)) (circles, mannose or galactose; squares, N-acetylglucosamine; triangles, fucose with α1,3- and α1,6-linkages being indicated by the downward or upward orientation). For a comparison with enzymatic release, refer to Supplementary Figures 1 and 2.
Focusing on the example 4.4 g.u. fraction containing Hex6HexNAc2Fuc2 (m/z 1767; Figure 2A), α-galactosidase removed one residue (Figure 2B) and β-galactosidase up to two, i.e., the bisecting β1,4-galactose, thus “unblocking” the α1,3-linked mannose (Figure 2, parts C and E), as well as the β1,4-galactose capping the α1,6-fucose as shown by the subsequent susceptibility of the latter to α-fucosidase treatment (Figure 2, parts C and D); both galactosidases resulted in loss of the m/z 916 MS/MS Y-fragment, but had different effects on that at m/z 608. Hydrofluoric acid treatment, which can cleave α1,3-fucose but not α1,6-fucose, resulted in concomitant loss of a fucose and a hexose fragment and also resulted in loss of the m/z 916 Y-fragment (Figure 2F). Considering the effect of α-galactosidase on the core fragmentation pattern, it was concluded that an α-galactose residue is attached to the core α1,3-fucose. Thereby, our α-galactosidase treatment data is in contrast to the older report in which the substitution of the α1,3-fucose by β1,4-galactose was proposed.7
Figure 2.

Enzymatic and chemical treatments of an RP-HPLC fractionated hydrazine-released wild-type N-glycan. The PA-labeled Hex6HexNAc2Fuc2 structure (m/z 1767; A), on RP-HPLC eluting at 4.4 g.u., was subject to structural characterization using MALDI-TOF MS/MS in combination with glycosidase and hydrofluoric acid treatments. Incubation of this glycan with coffee bean α-galactosidase resulted in a loss of one hexose residue (B), whereas with recombinant A. niger β-galactosidase up to two hexose residues were removed (C), demonstrating the presence of three galactose residues (one α-Gal and two β-Gal) on the Hex6HexNAc2Fuc2 structure. Treatments of the structure with either a mixture of β-galactosidase (β-Gal) and α-fucosidase (α-Fuc) or a mixture of β-galactosidase (β-Gal) and α1,2/3-mannosidase (α3-Man) yielded two different final products, Hex4HexNAc2Fuc1 (m/z 1297, D) and Hex3HexNAc2Fuc2 (m/z 1281, E). The Hex6HexNAc2Fuc2 glycan was also sensitive to hydrofluoric acid (HF) treatment as judged by the loss of a 308 unit concluded to be a Galα-Fucα1,3-linked disaccharide (F). Relevant MS/MS spectra of glycans from parts A–F are shown in the right panel. Especially, the differential changes of the core m/z 608 and 916 fragments upon either β- or α-galactosidase digestion are a clear indication that the two hexoses modifying the difucosylated GlcNAc core are, respectively, β-linked to the α1,6-fucose or α-linked to the α1,3-fucose. Asterisks indicate non-glycan contaminants with m/z 1522 (in part A) or, originating from the α-galactosidase, 1296, 1470, and 1499 (in part B); M signifies coeluting mannosidic N-glycans. [M + H]+ ions are annotated, but sodium adducts (22 mass units larger) dominate in some spectra (E and F); MS/MS was solely performed on protonated parent ions.
On the basis of these findings with the chemically released glycans, we reappraised enzymatic release from peptides prepared using pepsin with three different PNGases. As mentioned above, use of these enzymes in series resulted in distinct RP-HPLC profiles (Supplementary Figure 1). In the 4.4–4.7 g.u. region for the PNGase Ar-released pools (after prior PNGase A treatment), glycans of the same mass as in the hydrazine-released pool were detected, whose MS/MS patterns also displayed the m/z 916 Y-fragment ion (Supplementary Figure 2). In another experiment, we performed two rounds of PNGase Ar digestion of thermolysin-digested material; in this case the m/z 916 cores were present in both rounds but were more obvious after the second treatment with this enzyme (Supplementary Figure 2, parts C and H). As such glycans were not detected when using PNGase A (either alone or after PNGase F treatment) or PNGase F, we conclude that only PNGase Ar can (regardless of the prior proteolysis method) release the same Fuc2Hex2GlcNAc1 cores as hydrazine, while these are not cleaved by either PNGase F or A (for MS of corresponding fractions from each release method, see Supplementary Figure 3).
PNGase Ar Released Glycans from a Mutant Strain
An independent means of proving the presence of α-galactose on the reducing-terminal core α1,3-fucose was to reappraise the glycome of the fut-6;fut-8 double knockout strain, in which only the FUT-1 α1,3-fucosyltransferase is capable of modifying the core chitobiose region, but neither the FUT-6 nor FUT-8 enzymes.14 Thus, any m/z 446 or 608 fragments (Hex0–1HexNAc1Fuc1–PA) can only originate from, respectively, a free or modified core α1,3-fucose on the proximal GlcNAc. Use of PNGase Ar to release glycans from this mutant indeed resulted in detection of glycans with an m/z 608 fragment (Fuc1Hex1GlcNAc1–PA). A set of such glycans, eluting rather early on RP-HPLC (2.3–2.4 g.u.; Hex4–5GlcNAc2Fuc1–2Me0–1–PA), displayed sensitivity to either α-galactosidase or hydrofluoric acid, with both treatments resulting in loss of the m/z 608 MS/MS ion and appearance of ones at m/z 446 or 300 (Figure 3). Treatment with α-galactosidase also resulted in small shifts in retention time so that one product coeluted with a previously defined core α1,3-fucosylated structure from this mutant (Supplementary Figure 4).
Figure 3.

Detection of N-glycans from a mutant strain carrying a GalαFucα1,3 disaccharide unit on the proximal GlcNAc residue. Glycopeptides from the fut-6;fut-8 double knockout (lacking the distal α1,3- and proximal α1,6-fucosyltransferase genes; thus, the only core fucose is that α1,3-linked to the reducing terminus) were treated with the recombinant PNGase Ar, and the released N-glycans separated on a RP-HPLC. In two early eluting fractions (2.3 and 2.4 g.u.), four glycans (Hex4–5HexNAc2Fuc1–2Me0–1; m/z 1297, 1459, 1605, and 1619) were observed by MALDI-TOF MS (A and B). The MS/MS fragment ions of m/z 608 (Hex1Fuc1HexNAc1–PA) and the loss of 308 from their parent ions indicated the presence of a HexFuc unit on the proximal GlcNAc residue (C–F). These glycans were treated with either α-galactosidase (G) or hydrofluoric acid (H), and the resulted products were fragmented (I–L). The sensitivity of these glycans to α-galactosidase and HF treatments verified that the novel GalαFucα1,3 epitope is present on the proximal GlcNAc. Non-glycan contaminants are marked with asterisks, whereas reapplication on RP-HPLC of the two major α-galactosidase products, Hex3HexNAc2Fuc1–PA and Hex4HexNAc2Fuc1–PA (m/z 1135 and 1297, eluting at 2.6 and 2.8 g.u., respectively), confirmed their basic structure (Supplementary Figure 4).
Negative-mode ESI-MS/MS was also employed to examine two isomeric Hex4HexNAc2Fuc1–PA glycans (m/z 1295, [M – H]−) proposed to contain a galactosylated core α1,3-fucose (one found in the fut-6;fut-8 mutant and the other a digestion product of wild-type m/z 1767; Supplementary Figure 5); both show fragment ions at m/z 588 and 606 indicative of galactosylation of the core fucose, but the relative paucity of fragments for the first isomer and differences in intensity for the second isomer correlate, as shown in our previous study,13 with either the presence of a bisecting galactose on the core βMan or with a standard trimannosyl core. The MS3 spectrum for the ions at m/z 588 (Hex1Fuc1GlcNAc1–PA) displays only one cross-ring cleavage (m/z 221), which is either 1,3AFuc or 2,4AFuc; thus, the linkage between the α-galactose (as defined by enzyme digests) and the core fucose cannot be conclusively defined, but is either α1,3 or α1,4. In contrast, analysis of a glycan from the fut-1;fut-8 mutant (incapable of adding α1,3-fucose to the reducing-terminal GlcNAc), has a contrasting fragmentation pattern as galactosylation of the distal fucose is associated with fragmentation ions at m/z 435/465/510, while the m/z 588/606 ions are absent. Previously, the galactose on the α1,3-fucose at the distal GlcNAc was proposed to be α1,2-linked based on GC/MS data of a glycan from a hex-2;hex-3 mutant.16 Thus, the three Gal–Fuc disaccharide units attached to the chitobiose core are present in different conformations.
Maximal Degree of Fucosylation of Wild-Type C. elegans N-Glycans
Previously, five different fucose linkages on the core region of C. elegans N-glycans have been characterized, i.e., “proximal” core α1,3- and α1,6-Fuc, distal α1,3-Fuc, α1,2-Fuc on the bisecting galactose, and α1,2-Fuc on the Galβ1,4Fucα1,6 substitution of the core.14 However, only maximally four fucoses on a single wild-type glycan have been detected previously, i.e., on Hex7HexNAc2Fuc4Me0–1.7,38 A thorough examination of the hydrazine-released N-glycan pool identified glycans of m/z 1897, 1911, 2059, 2073, and 2235 or of 2333 and 2395 putatively corresponding to Hex5–7HexNAc2Fuc4Me0–1 or Hex6–7HexNAc2Fuc5Me2. The first five tetrafucosylated masses were also observed in the glycan pool released by PNGase Ar, whereas the minor pentafucosylated forms were only detected after hydrazine release. MS/MS of m/z 2235, 2233, and 2395 revealed fragments of, e.g., m/z 770 (Fuc1Hex2GlcNAc1–PA; otherwise previously only detected in analyses of fut-1;fut-6 and hex-2;hex-3 mutant strains), 1076 (Fuc3Hex2GlcNAc1Me1–PA), and 1078 (Fuc2Hex3GlcNAc1–PA), which means that some of these glycans contain either an extra hexose or a methylfucose more on the core than those containing the Galα1,3/4Fucα1,3(Galβ1,4Fucα1,6)GlcNAc (m/z 916) element. The extra hexose and methylfucose can be assumed to be a β1,4-galactose or methylated α1,2-fucose on the Galβ1,4Fucα1,6 moiety. Galβ1,4Galβ1,4Fucα1,6 is known from keyhole limpet hemocyanin as well as from previous work on C. elegans,14,16,39 while the methylfucose is probably α1,2-linked as in the fut-1;fut-6 and hex-2;hex-3 mutants.14,16 To corroborate the conclusions as to the numbers of additional galactose and fucose residues, hydrofluoric acid and galactosidase treatments were performed.
In the case of the largest tetrafucosylated glycan eluting at 5.2 g.u. (m/z 2235; Hex7HexNAc2Fuc4Me1), MS/MS suggested that more than one structure was cofragmented, because, in addition to the m/z 916 ion (Fuc2Hex2GlcNAc1–PA), a novel m/z 1078 ion (Fuc2Hex3GlcNAc1–PA) was seen on the spectrum (Figure 4A and Supplementary Figure 6). Therefore, the 5.2 g.u. RP-HPLC fractions of the hydrazine and PNGase Ar pools were reseparated on a HIAX column, which yielded two overlapping peaks eluting at approximately 20 min (Figure 4B), both containing glycans of m/z 2235. This procedure allowed us to demonstrate that two forms of Hex7HexNAc2Fuc4Me1 existed, displaying different sensitivities to β-galactosidase and HF treatments (Supplementary Figure 6; compare panels A–C and H–J). The less abundant form is concluded to possess a second β1,4-galactose on the proximal 6-linked GalFuc, which accounts for the m/z 770/1078 Y1 fragment pair converted to one at m/z 446/754 upon β-galactosidase digestion and finally to a sole fragment of m/z 446 after subsequent hydrofluoric acid treatment (Supplementary Figure 6D–G). The major form of m/z 2235 has an α-galactose attached to a mannose as compared to the m/z 2059 glycan; thereby both display core fragments at m/z 608/916 converted, respectively, to m/z 608/754, m/z 446/754, and m/z 446 or 608 after α-galactosidase, β-galactosidase, and hydrofluoric acid treatments (Figure 4C and Supplementary Figure 6, parts K–N and O–R). Another tetrafucosylated glycan (Hex6HexNAc2Fuc4Me1; m/z 2073) was also shown to have shifts in the Y-fragmentation (conversion of the m/z 916 ion to one at m/z 754) upon α-galactosidase treatment (Figure 4D).
Figure 4.
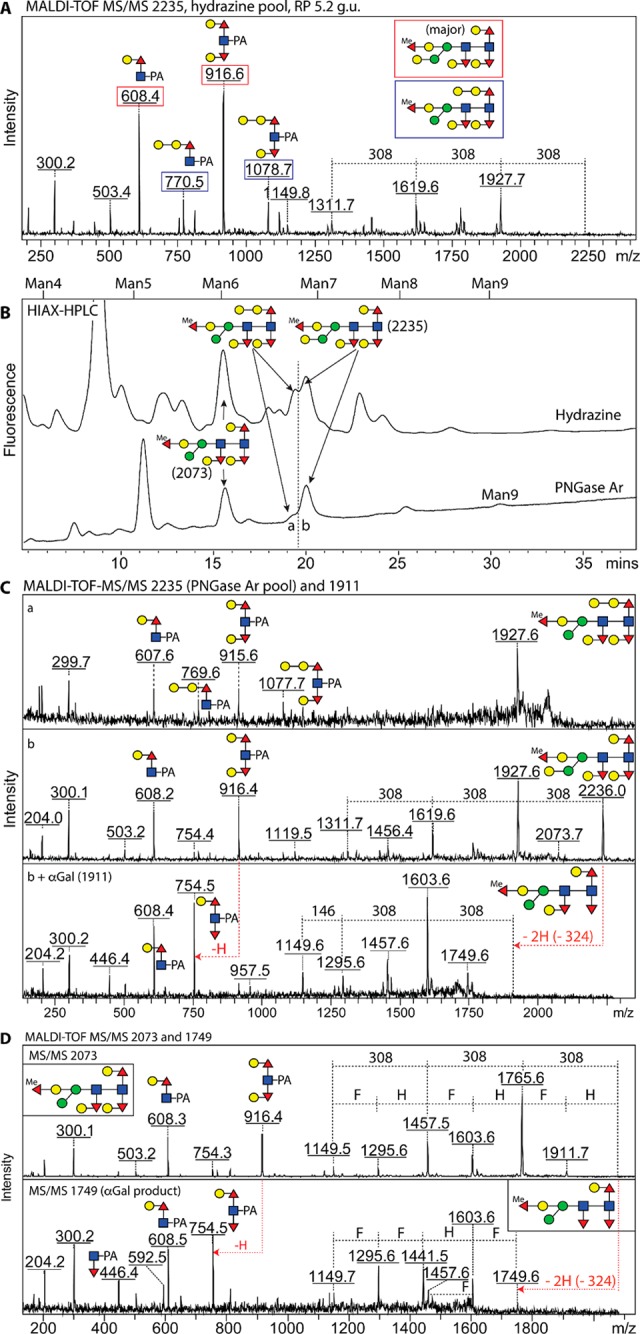
HIAX-HPLC separation of Hex6–7HexNAc2Fuc4Me1 glycans released with hydrazine or PNGase Ar from wild-type C. elegans glycopeptides. (A) The MS/MS spectrum of m/z 2235 in the 5.2 g.u. RP-HPLC fraction displayed two sets of Y-fragment ions of m/z 608/916 and m/z 770/1078, suggestive of cofragmentation of two coeluting isomers. (B) The isomers were separated by applying the 5.2 g.u. fractions to a HIAX column externally calibrated with a mixture of oligomannosidic structures (Man4–9GlcNAc2). (C) MS/MS spectra of the m/z 2235 structures in the HIAX fractions (a and b) confirmed that PNGase Ar released the same forms of Hex7HexNAc2Fuc4Me1 as hydrazine, whereby the m/z 916 fragment of the later-eluting isomer is replaced by one at m/z 754 after α-galactosidase treatment; for other structural analyses and digests on these glycans, see Supplementary Figure 6. (D) 2D-HPLC also purified an m/z 2073 Hex6HexNAc2Fuc4Me1 glycan with a fully modified core with three Gal–Fuc subunits as shown by its MS/MS pattern; the galactose caps on the proximal and distal α1,3-linked fucose residues were sensitive to α-galactosidase resulting in a final product of m/z 1749 after extended incubation.
In addition, we have discovered in a trace amount two glycans at m/z 2233 and 2395 corresponding to pentafucosylated compositions of Hex6–7HexNAc2Fuc5Me2. MS/MS of these two glycans revealed fragments of, e.g., m/z 768, 916, and 1076 (Fuc2–3Hex1–2GlcNAc1Me0–1–PA), which means that these glycans contain an extra methylfucose more on the core than those containing the Galα1,3/4Fucα1,3(Galβ1,4Fucα1,6)GlcNAc element. Hydrofluoric acid treatment resulted in both cases in loss of the m/z 1076 fragment, which would correlate with removal of the hexosylated reducing-terminal core α1,3-fucose in addition to a α1,2-linked methylfucose (Figure 5). On the basis of these data, we conclude that all five known linkages of fucose can be detected on at least two hydrazine-released N-glycans synthesized by the wild-type worm, which might be the maximal degree of fucosylation of C. elegans N-glycans.
Figure 5.
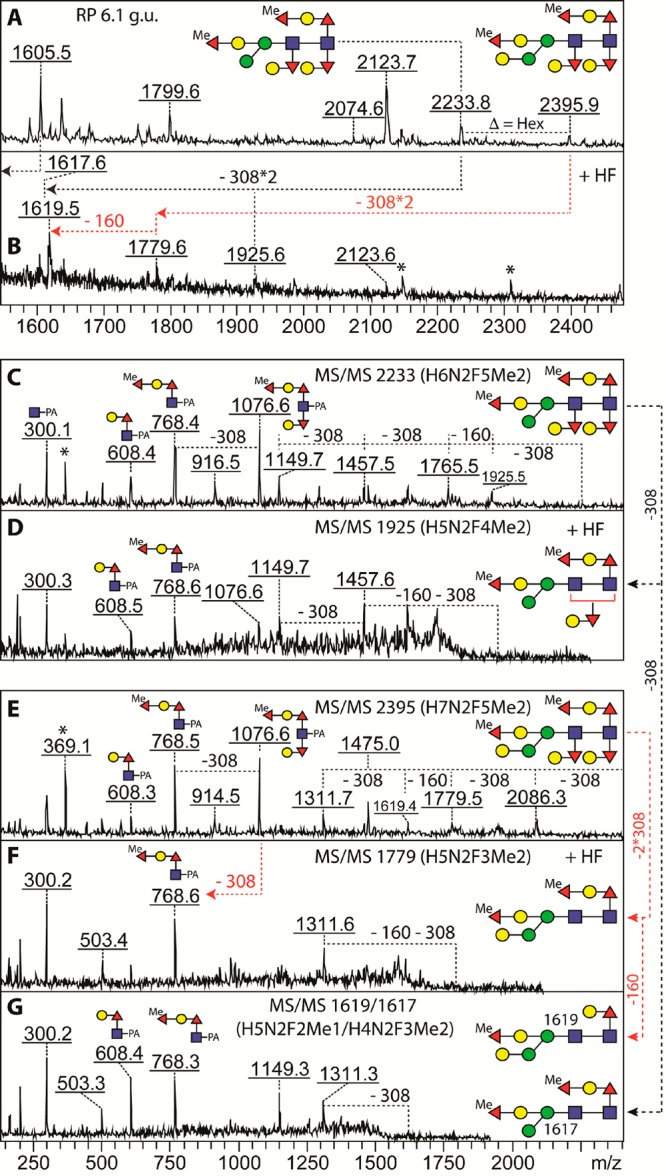
Maximal degree of fucosylation in wild-type C. elegans N-glycans. (A) The 6.1 g.u. fraction contained glycans of Hex6–7HexNAc2Fuc5Me2 (m/z 2233 and 2395) which both displayed a novel MS/MS fragment at m/z 1076 (Hex2GlcNAc1Fuc3Me1–PA; C and E) in addition to one at m/z 768 (Hex1HexNAc1Fuc2Me1–PA). (B) HF treatment resulted in sequential loss of two GalFuc units (308 Da) and, in the case of the m/z 2395 glycan, also a methylfucose (160 Da), leading to products with m/z 1925, 1779, and 1617/1619 whose MS/MS indicated loss of the m/z 1076 fragment (D, F–G). The two HF products at m/z 1617 (from m/z 2233, black-dashed arrows) and 1619 (from m/z 2395, red dashed arrows) were cofragmented, displaying two diagnostic ions (m/z 608 and 768) with equal intensities; the methylated fucose at the bisecting position was more resistant to HF treatment than the one on the core Galβ1,4Fucα1,6 motif. Asterisks in part B indicate non-glycan contaminants, whereas those in parts C and E indicate a PC-containing fragment (m/z 369; PC–HexNAc) originating from coeluting glycans.
Conclusion
From this study, we conclude that the recently introduced PNGase Ar enzyme can release the same GalαFucα1,3 reducing-terminal core modification as hydrazine, but without the complications (safety, artifacts, and cleanup) as the chemical method. Indeed, peeling of core residues from plant N-glycans,19 deacylation (requiring subsequent re-N-acetylation), and partial demethylation of phosphorylcholine (our own unpublished observations) are problems associated with the use of hydrazine. Due to subsequent labeling of the reducing end, the Y1 fragment ions are highly diagnostic of the various core modifications we have found in this and other studies, the key example in this study being the core fragment at m/z 916 (Gal2Fuc2GlcNAc1–PA). The combination of off-line LC–MS and chemical/enzymatic treatments leads us to conclude that the presence of both GalαFucα1,3 and modified GalβFucα1,6 moieties can only be detected after use of either hydrazine or PNGase Ar.
Although the PNGase A and Ar enzymes are both of plant origin, only the latter is capable of releasing glycans with core m/z 916 fragments; apparently, PNGase Ar can also deglycosylate intact glycoproteins,40 which is an indication that it is less fastidious regarding its substrates. Thereby, it is interesting to note that substitutions of the 3-OH of the Asn-bound GlcNAc have different effects on the ability to cleave N-glycans; thus, while an unsubstituted core α1,3-fucose can be cleaved by both PNGase A and PNGase Ar, a core β1,3-mannose motif as found in a marine snail is also sensitive to PNGase F.41 Obviously, the exact modifications of the reducing terminus do have an impact on what methods can be used for N-glycan release. Certainly, PNGase Ar should prove a convenient tool in order to efficiently cleave a wider range of glycans than other PNGase enzymes and so generally increase our knowledge about glycomic complexities in invertebrate species.
Our data also expand the range of C. elegans glycans to pentafucosylated forms, whereby it is to be noted that pentafucosylated structures with an additional fucose residue on the 6-linked GalFuc element were only released by hydrazine. On the other hand, having performed both enzymatic (PNGase F and A, but not Ar) and chemical release of the N-glycans from another nematode, Oesophagostomum dentatum, and not finding the GalαFucα1,3-element,29 it can only be speculated whether this modification is to be found in any parasitic nematode or whether it is specific to C. elegans. Certainly, the model nematode still represents a source of glycomic surprises, which must then be combined with other studies to reveal why a simple organism needs so much variation in its oligosaccharide structures.
Acknowledgments
This work was funded in part by the Austrian Fonds zur Förderung der wissenschaftlichen Forschung (FWF; Grants P21946 and P25058 to K.P. and P23922 and P29466 to I.B.H.W.) and the GlycoPar Marie-Curie Initial Training Network (European Union; PITN-GA-2013-608295). K.P. is an FWF Fellow; J.V. was an experienced postdoctoral researcher within the GlycoPar network. We also thank the staff of Ludger Ltd. for introduction to hydrazinolysis, New England Biolabs for providing the PNGase Ar and assistance in our initial studies, Dr. Martin Dragosits for the recombinant β-galactosidase, Daniel Malzl for help with figure preparation and some mass spectrometric measurements, and Dr. Niclas Karlsson for access to the LTQ mass spectrometer.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.analchem.7b03898.
Scheme summarizing the workflow, an extended version of the methods, Supplementary Figures 1–6 (HPLC, MS, and MS/MS data), and further information regarding the glycomic analyses (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Blaxter M. PLoS Biol. 2011, 9, e1001050. 10.1371/journal.pbio.1001050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschinger K.; Gutternigg M.; Rendić D.; Wilson I. B. H. Carbohydr. Res. 2008, 343, 2041–2049. 10.1016/j.carres.2007.12.018. [DOI] [PubMed] [Google Scholar]
- Altmann F.; Fabini G.; Ahorn H.; Wilson I. B. H. Biochimie 2001, 83, 703–712. 10.1016/S0300-9084(01)01297-4. [DOI] [PubMed] [Google Scholar]
- Haslam S. M.; Gems D.; Morris H. R.; Dell A. Biochem. Soc. Symp. 2002, 69, 117–134. 10.1042/bss0690117. [DOI] [PubMed] [Google Scholar]
- Hirabayashi J.; Hayama K.; Kaji H.; Isobe T.; Kasai K. J. Biochem. 2002, 132, 103–114. 10.1093/oxfordjournals.jbchem.a003186. [DOI] [PubMed] [Google Scholar]
- Paschinger K.; Rendić D.; Lochnit G.; Jantsch V.; Wilson I. B. H. J. Biol. Chem. 2004, 279, 49588–49598. 10.1074/jbc.M408978200. [DOI] [PubMed] [Google Scholar]
- Hanneman A. J.; Rosa J. C.; Ashline D.; Reinhold V. Glycobiology 2006, 16, 874–890. 10.1093/glycob/cwl011. [DOI] [PubMed] [Google Scholar]
- Paschinger K.; Staudacher E.; Stemmer U.; Fabini G.; Wilson I. B. H. Glycobiology 2005, 15, 463–474. 10.1093/glycob/cwi028. [DOI] [PubMed] [Google Scholar]
- Nguyen K.; van Die I.; Grundahl K. M.; Kawar Z. S.; Cummings R. D. Glycobiology 2007, 17, 586–599. 10.1093/glycob/cwm023. [DOI] [PubMed] [Google Scholar]
- Zheng Q.; van Die I.; Cummings R. D. J. Biol. Chem. 2002, 277, 39823–39832. 10.1074/jbc.M207487200. [DOI] [PubMed] [Google Scholar]
- Zheng Q.; Van Die I.; Cummings R. D. Glycobiology 2008, 18, 290–302. 10.1093/glycob/cwn007. [DOI] [PubMed] [Google Scholar]
- Yan S.; Serna S.; Reichardt N. C.; Paschinger K.; Wilson I. B. H. J. Biol. Chem. 2013, 288, 21015–21028. 10.1074/jbc.M113.479147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S.; Brecker L.; Jin C.; Titz A.; Dragosits M.; Karlsson N.; Jantsch V.; Wilson I. B. H.; Paschinger K. Mol. Cell. Proteomics 2015, 14, 2111–2125. 10.1074/mcp.M115.049817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S.; Jin C.; Wilson I. B. H.; Paschinger K. J. Proteome Res. 2015, 14, 5291–5305. 10.1021/acs.jproteome.5b00746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butschi A.; Titz A.; Wälti M.; Olieric V.; Paschinger K.; Nöbauer K.; Guo X.; Seeberger P. H.; Wilson I. B. H.; Aebi M.; Hengartner M.; Künzler M. PLoS Pathog. 2010, 6, e1000717. 10.1371/journal.ppat.1000717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan S.; Bleuler-Martinez S.; Plaza D. F.; Künzler M.; Aebi M.; Joachim A.; Razzazi-Fazeli E.; Jantsch V.; Geyer R.; Wilson I. B. H.; Paschinger K. J. Biol. Chem. 2012, 287, 28276–28290. 10.1074/jbc.M112.353128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yosizawa Z.; Sato T.; Schmid K. Biochim. Biophys. Acta, Gen. Subj. 1966, 121, 417–420. 10.1016/0304-4165(66)90134-6. [DOI] [PubMed] [Google Scholar]
- Mizuochi T. Methods Mol. Biol. 1993, 14, 55–68. 10.1385/0-89603-226-4:55. [DOI] [PubMed] [Google Scholar]
- Ashford D.; Dwek R. A.; Welply J. K.; Amatayakul S.; Homans S. W.; Lis H.; Taylor G. N.; Sharon N.; Rademacher T. W. Eur. J. Biochem. 1987, 166, 311–320. 10.1111/j.1432-1033.1987.tb13516.x. [DOI] [PubMed] [Google Scholar]
- Likhosherstov L. M.; Novikova O. S.; Derevitskaya V. A.; Kochetkov N. K. Carbohydr. Res. 1990, 199, 67–76. 10.1016/0008-6215(90)84093-A. [DOI] [PubMed] [Google Scholar]
- Tarentino A. L.; Gomez C. M.; Plummer T. H. Jr. Biochemistry 1985, 24, 4665–4671. 10.1021/bi00338a028. [DOI] [PubMed] [Google Scholar]
- Tretter V.; Altmann F.; März L. Eur. J. Biochem. 1991, 199, 647–652. 10.1111/j.1432-1033.1991.tb16166.x. [DOI] [PubMed] [Google Scholar]
- Lee K. J.; Gil J. Y.; Kim S. Y.; Kwon O.; Ko K.; Kim D. I.; Kim D. K.; Kim H. H.; Oh D. B. J. Biochem. 2015, 157, 35–43. 10.1093/jb/mvu051. [DOI] [PubMed] [Google Scholar]
- Wang T.; Cai Z. P.; Gu X. Q.; Ma H. Y.; Du Y. M.; Huang K.; Voglmeir J.; Liu L. Biosci. Rep. 2014, 34, e00149. 10.1042/BSR20140148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G.; Yu X.; Bao C.; Wang L.; Li M.; Gan J.; Qu D.; Ma J.; Chen L. J. Biol. Chem. 2015, 290, 7452–7462. 10.1074/jbc.M114.605493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brenner S. Genetics 1974, 77, 71–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis J. A.; Fleming J. T. Methods Cell Biol. 1995, 48, 3–29. 10.1016/S0091-679X(08)61381-3. [DOI] [PubMed] [Google Scholar]
- Hykollari A.; Paschinger K.; Eckmair B.; Wilson I. B. H. Methods Mol. Biol. 2017, 1503, 167–184. 10.1007/978-1-4939-6493-2_13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez-Castells C.; Vanbeselaere J.; Kohlhuber S.; Ruttkowski B.; Joachim A.; Paschinger K. Biochim. Biophys. Acta, Gen. Subj. 2017, 1861, 418–430. 10.1016/j.bbagen.2016.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paschinger K.; Hykollari A.; Razzazi-Fazeli E.; Greenwell P.; Leitsch D.; Walochnik J.; Wilson I. B. H. Glycobiology 2012, 22, 300–313. 10.1093/glycob/cwr149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Everest-Dass A. V.; Abrahams J. L.; Kolarich D.; Packer N. H.; Campbell M. P. J. Am. Soc. Mass Spectrom. 2013, 24, 895–906. 10.1007/s13361-013-0610-4. [DOI] [PubMed] [Google Scholar]
- Hykollari A.; Balog C. I.; Rendić D.; Braulke T.; Wilson I. B. H.; Paschinger K. J. Proteome Res. 2013, 12, 1173–1187. 10.1021/pr300806b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dragosits M.; Pflugl S.; Kurz S.; Razzazi-Fazeli E.; Wilson I. B. H.; Rendić D. Appl. Microbiol. Biotechnol. 2014, 98, 3553–3567. 10.1007/s00253-013-5192-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wong-Madden S. T.; Landry D. Glycobiology 1995, 5, 19–28. 10.1093/glycob/5.1.19. [DOI] [PubMed] [Google Scholar]
- Yan S.; Wilson I. B. H.; Paschinger K. Electrophoresis 2015, 36, 1314–1329. 10.1002/elps.201400528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haslam S. M.; Coles G. C.; Reason A. J.; Morris H. R.; Dell A. Mol. Biochem. Parasitol. 1998, 93, 143–147. 10.1016/S0166-6851(98)00020-6. [DOI] [PubMed] [Google Scholar]
- Natsuka S.; Adachi J.; Kawaguchi M.; Nakakita S.; Hase S.; Ichikawa A.; Ikura K. J. Biochem. 2002, 131, 807–813. 10.1093/oxfordjournals.jbchem.a003169. [DOI] [PubMed] [Google Scholar]
- Struwe W. B.; Reinhold V. N. Glycobiology 2012, 22, 863–875. 10.1093/glycob/cws053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wuhrer M.; Robijn M. L.; Koeleman C. A.; Balog C. I.; Geyer R.; Deelder A. M.; Hokke C. H. Biochem. J. 2004, 378, 625–632. 10.1042/bj20031380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magnelli P.; Guthrie E.; Taron C. H.; Xu M.-Q.; Buswell J.. WO2015184325A2, 2015.
- Eckmair B.; Jin C.; Abed-Navandi D.; Paschinger K. Mol. Cell. Proteomics 2016, 15, 573–597. 10.1074/mcp.M115.051573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varki A.; Cummings R. D.; Aebi M.; Packer N. H.; Seeberger P. H.; Esko J. D.; Stanley P.; Hart G.; Darvill A.; Kinoshita T.; Prestegard J. J.; Schnaar R. L.; Freeze H. H.; Marth J. D.; Bertozzi C. R.; Etzler M. E.; Frank M.; Vliegenthart J. F.; Lutteke T.; Perez S.; et al. Glycobiology 2015, 25, 1323–1324. 10.1093/glycob/cwv091. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.