

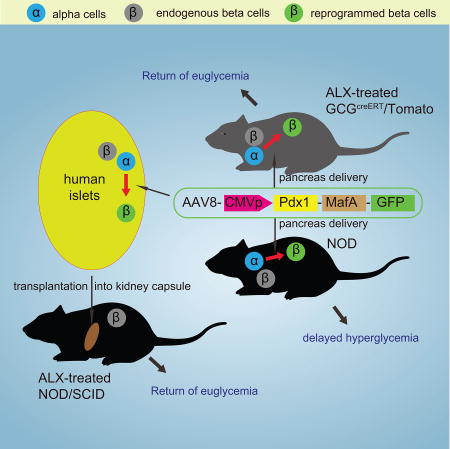
Summary
Successful strategies for treating type 1 diabetes need to restore the function of pancreatic beta cells that are destroyed by the immune system and overcome further destruction of insulin-producing cells. Here, we infused adeno-associated virus carrying Pdx1 and MafA expression cassettes through the pancreatic duct to reprogram alpha-cells into functional beta-cells, and normalized blood glucose in both beta-cell-toxin-induced diabetic mice and in autoimmune non-obese diabetic (NOD) mice. The euglycemia in toxin-induced diabetic mice and new insulin+ cells persisted in the autoimmune NOD mice for 4 months prior to reestablishment of autoimmune diabetes. This gene therapy strategy also induced alpha to beta cell conversion in toxin-treated human islets, which restored blood glucose levels in NOD/SCID mice upon transplantation. Hence, this strategy could represent a new therapeutic approach, perhaps complemented by immunosuppression, to bolster endogenous insulin production. Our study thus provides a potential basis for further investigation in human type 1 diabetes.
Keywords: alpha cells, beta cells, MafA, Pdx1, lineage tracing, intraductal viral infusion, NOD, adoptive transfer, human islets, islet transplantation
Graphical abstract
Introduction
Insulin (INS) is a key regulator of glucose homeostasis, and is produced by pancreatic beta cells. Insufficient INS leads to diabetes mellitus, a metabolic disease that affects over 300 million people worldwide (Ackermann and Gannon, 2007; Pipeleers et al., 2008; Pipeleers et al., 2002; Pipeleers and Ling, 1992; Weir and Bonner-Weir, 1998; Zaret and Grompe, 2008). The fundamental objective of diabetes treatment is to preserve and restore a functional beta cell mass, perhaps through beta-cell replacement therapy. However, beta-cell replacement may fall short in autoimmune type 1 diabetes (T1D) due to persistent, recurrent autoimmunity against the new beta-cells (Ackermann and Gannon, 2007; Pipeleers et al., 2008; Pipeleers et al., 2002; Weir and Bonner-Weir, 1998; Zaret and Grompe, 2008). In fact, this form of renewed autoimmune attack has been found to be particularly aggressive (Purcell and Mottram, 1995). Unfortunately, a clinically applicable strategy leading to a more durable beta-cell mass has yet to be developed for T1D (Campbell et al., 2007).
Although great efforts have been made to identify, isolate and purify beta cell progenitors in the adult pancreas (Kopp et al., 2011a; Kushner et al., 2010), accumulating evidence does not support a substantial contribution of beta-cell neogenesis to a functional beta-cell mass in the adult pancreas (Cavelti-Weder et al., 2013; Chintinne et al., 2012; Desai et al., 2007; Dor et al., 2004; Georgia and Bhushan, 2004; Kopinke et al., 2011; Kopp et al., 2011b; Meier et al., 2008; Pan et al., 2013; Rankin et al., 2013; Solar et al., 2009; Teta et al., 2007; Tonne et al., 2014; Xiao et al., 2013a; Xiao et al., 2013c; Xiao et al., 2013d), except for a few rare situations (Baeyens et al., 2014; Chera et al., 2014; Thorel et al., 2010). Thus, gene therapy may be required in order to generate new beta-cells from other cell types (Lee et al., 2013; Li et al., 2014; Zhou et al., 2008).
Pancreatic and duodenal homeobox 1 (Pdx1) is a transcription factor necessary for pancreatic development, including beta-cell maturation, beta-cell proliferation and function (Gannon et al., 2001). MafA is a transcription factor that binds to the INS promoter to regulate INS expression and beta-cell metabolism (Hang and Stein, 2011). Ectopic expression of a combination of three key pancreatic beta-cell transcription factors [Pdx1, neurogenin 3 (Ngn3) and MafA] has been shown to reprogram adult mouse pancreatic acinar cells into beta-cell-like cells (Akinci et al., 2012; Lee et al., 2013; Zhou et al., 2008). Moreover, co-overexpression of these three genes has been shown to convert Sox9+ liver cells into INS-producing cells (Banga et al., 2012). However, alpha cells may be the ideal source for beta-cell replacement for several reasons. First, as endocrine cells, alpha cells are developmentally similar to beta cells, which may facilitate reprogramming (Bramswig and Kaestner, 2011; Herrera, 2000). Second, alpha cells are already situated within the islet (Bramswig and Kaestner, 2011; Herrera, 2000; Pipeleers et al., 2002) so that a reprogrammed beta cell from an alpha cell would be well-positioned for ideal beta-cell function. Third, alpha cell hyperplasia is commonly seen in diabetic animals and patients, and constitutes a potentially abundant source for reprogramming, and human islets in particular have a large percentage of alpha cells (Zaret and White, 2010). Fourth, according to recent reports, a significant decrease in the number of alpha cells did not appear to harm proper glucose metabolism (Shiota et al., 2013; Thorel et al., 2011). Fifth, glucagon (GCG) signaling appears to be detrimental in diabetes, which suggests that a partial reduction in alpha cell mass due to their conversion to beta cells may be beneficial to blood glucose control (Ackermann and Gannon, 2007; Pipeleers et al., 2008; Pipeleers et al., 2002; Pipeleers and Ling, 1992; Weir and Bonner-Weir, 1998; Zaret and Grompe, 2008). Sixth, alpha-to-beta cell conversion is feasible since it has been reported to occur after an extreme beta cell loss (Thorel et al., 2010), and in an insulinoma model with alpha-cell specific Men1 ablation (Lu et al., 2010). Lastly, a recent ATAC-seq study showed that the alpha cell genome is remarkably accessible, and thus likely to more easily undergo transdifferentiation (Ackermann et al., 2016). Based on these concepts, we were prompted to examine whether forced expression of key beta-cell transcription factors in alpha cells may trigger their reprogramming to generate beta or beta-like cells. We expected that Ngn3 would not be necessary to convert alpha cells to beta cells because Ngn3 is necessary for the formation of endocrine cells, but alpha cells are already endocrine cells.
Non-integrative adeno-associated viral (AAV) vectors can impart long-term expression of transgenes up to 4.5 kb in length. Moreover, AAV vectors have been found to be more efficient than adenoviral and lentiviral vectors in transducing pancreatic cells (Guo et al., 2012a; Guo et al., 2012b; Jimenez et al., 2011). Among AAV vectors, serotype 8 has been shown to have the highest transduction efficiency for mouse islet endocrine cells (Guo et al., 2012a; Guo et al., 2012b; Jimenez et al., 2011; Tonne et al., 2014). Here, we used a transgenic mouse model that allows lineage tracing of alpha cells, and we delivered Pdx1 and MafA expression vector virus to the mouse pancreas through a recently developed pancreatic intraductal infusion system (Xiao et al., 2014a; Xiao et al., 2014b; Xiao et al., 2013c; Xiao et al., 2014c). We saw glucose normalization in beta-cell-toxin-treated mice, and the new INS+ cells were found to derive almost exclusively from alpha cells. We then similarly delivered virus to autoimmune, hyperglycemic non-obese diabetic (NOD) mice, and saw durable euglycemia, typically for 4 months. The mechanisms underlying the prolonged survival of new INS+ cells in the autoimmune environment were further investigated.
Results
Intraductal infusion of PM virus reverses toxin-induced diabetes in mice
Here, we examined the potential reprogramming of alpha cells into beta cells by inducing the expression of Pdx1 and MafA (PM) using AAV (AAV-PM) in mice. We first administered a single dose of either alloxan (ALX) or streptotozocin (STZ) to induce a sustained hyperglycemia in C57BL/6 mice (Supplementary Figure 1A)(Xiao et al., 2013a) due to a significant decrease in beta-cell mass (Supplementary Figure 1B–E, ALX: decreased to 3.8±0.2%, STZ: decreased to 6.5±0.9%). Moreover, both ALX and STZ induced a modest but significant increase in alpha-cell mass (Supplementary Figure 1B–E), similar to that seen in some diabetic patients (Zaret and White, 2010). No significant increase in INS+ cell proliferation was detected after ALX treatment (Supplementary Figure 1F–G). We thus chose to use ALX in further studies to examine alpha-to-beta cell conversion in vivo, due to the higher and more consistent degree of beta-cell ablation at the dosages used.
To allow lineage tracing of alpha cells, we first generated GCG-Cre; R26RTomato reporter mice (Herrera, 2000; Xiao et al., 2013b; Xiao et al., 2013c), in which tomato red fluorescence specifically labels the GCG+ alpha cell lineage in the pancreas (Figure 1A). Quantification showed that the baseline labeling of alpha cells in GCG-Cre; R26RTomato mice was 71.5±5.5%, with absence of detectable off-target labeling (Supplementary Figure 2A). Thus, we then gave ALX to destroy the majority of beta cells in these GCG-Cre; R26RTomato mice. One week later, AAV-PM or control AAV-GFP was directly introduced into the mouse pancreas, using a recently-developed pancreatic intraductal viral infusion technique, in which infusion of 150µl of AAV serotype 8 efficiently transduces endocrine cells (Xiao et al., 2014a; Xiao et al., 2014b; Xiao et al., 2013c; Xiao et al., 2014c). The intrapancreatic ductal delivery of virus did not result in appreciable expression of GFP in the liver. More importantly, no significant levels of INS gene expression were detected in the liver (Supplementary Figure 3A–B). As a quality control, we stained for Pdx1 and MafA in untreated mice (no ALX, no virus), ALX/AAV-GFP-treated mice, and ALX/AAV-PM-treated mice to confirm the expression of these transgenes in the transduced pancreatic cells (Supplementary Figure 3C). We found that ALX-induced hyperglycemia was corrected within 2 weeks by intraductal infusion of AAV-PM, but not with control AAV-GFP mice (Figure 1B). We also saw a significant improvement in the glucose response (intraperitoneal glucose tolerance test, IPGTT) in ALX-treated, AAV-PM-infused mice at 4 weeks after viral infusion (Figure 1C). Moreover, beta cell mass significantly increased in ALX-mice receiving AAV-PM (0.84±0.06mg), compared to mice receiving AAV-GFP (0.09±0.01mg) at 4 weeks after viral infusion, reaching more than 60% of the beta cell mass of untreated mice (UT, no ALX, no virus; 1.35±0.11 mg) (Figure 1D). Alpha cell mass was quantified, showing decreases in ALX+AAV-PM mice, compared to those in ALX+AAV-GFP mice (Supplementary Figure 3D). Transgene (Pdx1 and MafA) expression was then analyzed in purified tomato+ alpha cells 2 days after viral infusion, confirming the expression of these transgenes in alpha cells (Supplementary Figure 3E). GFP could be detected in ALX+AAV-GFP mouse pancreas 5 weeks after viral infusion, suggesting sustained expression of the transgene (black bar, Supplementary Figure 3A). Thus, intraductal infusion of AAV-PM reversed ALX-induced diabetes in mice.
Figure 1. Intraductal infusion of AAV-PM corrects ALX-induced hyperglycemia in GCG-Cre; R26RTomato mice.

(A) Schematic for the generation of GCG-Cre; R26RTomato mice. (B) Hyperglycemia was induced in GCG-Cre; R26RTomato mice by ALX injection. One week after ALX treatment, mice received a pancreatic intraductal infusion of either AAV-PM (red line) or control AAV-GFP (green line). Fasting blood glucose levels were measured. (C) IPGTT was performed in these mice 4 weeks after viral infusion. Untreated mice (no ALX, no virus, in blue) were used as an additional control. (D) Beta cell mass at 4 weeks after virus infusion. The contribution of INS+ cells without tomato red fluorescence is shown by the hatched bar contained within the red bar, compared to the beta cell mass in mice that received AAV-GFP viral infusion (green bar), and the beta cell mass of untreated mice (UT, no ALX, no virus, blue bar). Statistics were analyzed by one-way ANOVA with a Bonferroni correction, followed by Fisher’s Exact Test. Data are presented as mean±S.D. *: p<0.05. **: p<0.01. N=10. Scale bars are 50µm.
Regenerated INS+ cells are mainly derived from reprogrammed alpha cells
We quantified tomato+ cells in the INS+ cell population, based on counting 5000 INS+ cells in each mouse, with 5 mice in each experimental group. In pancreases of GCG-Cre; R26RTomato mice with infusion of control AAV-GFP, no INS+ cells were found to be labeled with tomato (Figure 2A). However, about 78.9±5.6% of the INS+ cells after AAV-PM infusion in ALX-treated mice were labeled with tomato (Figure 1D, 2A–B), suggesting an alpha-cell origin. Very few INS+ cells were not tagged with tomato red (Figure 1D, 2A), presumably representing those cells that either derived from the few surviving beta cells after ALX treatment, or else derived mainly from unlabeled reprogrammed alpha cells. We gave the mice continuous BrdU in the drinking water starting immediately after the viral infusion for 4 weeks. We found that 78.5±6.6% of the tomato+ INS+ cells had incorporated BrdU (Figure 2C). Meanwhile, 13.4±1.6% Ki-67+ INS+ cells were detected (Figure 2D). Islets isolated from the ALX/AAV-PM-treated mice expressed higher levels of the cell-cycle activators cyclinD1 (CCND1) and CDK4, and lower levels of the cell-cycle suppressor p27 (Figure 2E–G). Together, these data suggest that significant proliferation may occur in the neogenic INS+ cells reprogrammed from alpha cells. Moreover, the islets from the ALX/AAV-PM-treated mice showed no difference in glucose-stimulated INS release, compared to normal islets (from untreated mice; Figure 2H).
Figure 2. Neogenic INS+ cells are derived from reprogrammed alpha cells.

(A) Immunostaining for INS after infusion of control AAV-GFP (upper panels) or AAV-PM (lower panels) in ALX-treated GCG-Cre; R26RTomato mice, along with direct fluorescence for tomato (TOM, from GCG-Cre activity) and for green fluorescence (GFP, from viral infection). Both AAV-GFP and AAV-PM viruses carry a GFP cassette. (B) Immunostaining for GCG from a region nearby to (A) after infusion of control AAV-GFP (upper panels) or AAV-PM (lower panels) in ALX-treated GCG-Cre; R26RTomato mice, along with direct fluorescence for tomato (TOM, from GCG-Cre activity) and for green fluorescence (GFP, from viral infection). (C–D) BrdU was continuously provided in the drinking water during the 4 weeks after viral infusion. Immunostaining for BrdU (C) or Ki-67 (D), INS and TOM in ALX-treated, AAV-PM-infused GCG-Cre; R26RTomato mice. (E–G) Western blot for CyclinD1 (CCND1; E), CDK4 (F) and p27 (G) in islets from ALX-treated, AAV-PM-infused mice, compared to control untreated islets (NT, no ALX, no AAV). (F) Perifusion studies of islets from ALX-treated, AAV-PM-infused mice, compared to NT islets. Statistics were analyzed by one-way ANOVA with a Bonferroni correction, followed by Fisher’s Exact Test. Data are presented as mean±S.D. NS: non-significant. N=5. Scale bars are 20µm.
In order to exclude the possibility that pre-existing beta cells may transiently de-differentiate and activate the GCG promoter and thus become TOM-labeled after ALX and AAV-PM, we generated a GCGCreERT knock-in mouse to create an inducible Cre system (GCGCreERT; R26RTomato) to lineage-tag native alpha cells only prior to ALX and AAV-PM treatment (Figure 3A). Quantification showed that the baseline labeling of alpha cells in GCGCreERT; R26RTomato mice was robust at 93.5±6.5% after tamoxifen treatment, with complete absence of off-target labeling (Supplementary Figure 2B), perhaps due to the knock-in strategy. Rare alpha cells were pre-labeled without tamoxifen. Moreover, infusion of AAV-GFP into mice without ALX treatment did not induce tomato labeling of non-alpha cells (Supplementary Figure 2C).
Figure 3. Intraductal infusion of AAV-PM corrects ALX-induced hyperglycemia in GCGCreERT; R26RTomato mice.

(A) Schematic for the generation of GCGCreERT; R26RTomato mice. (B) One week after tamoxifen administration, hyperglycemia was induced in GCGCreERT; R26RTomato mice by ALX injection. One week after ALX treatment, mice received a pancreatic intraductal infusion of either AAV-PM (red line) or control AAV-GFP (green line). Fasting blood glucose levels were measured. (C) IPGTT was performed in these mice 4 weeks after viral infusion. (D) Beta cell mass at 4 weeks and 24 weeks after virus infusion. (E–F) Immunostaining for INS after infusion of AAV-PM in ALX-treated GCGCreERT; R26RTomato mice, along with direct fluorescence for tomato (Cre-activity) and for green fluorescence (GFP, from viral infection) at 4 weeks (E) and 24 weeks (F) after virus infusion. Statistics were analyzed by one-way ANOVA with a Bonferroni correction, followed by Fisher’s Exact Test. Data are presented as mean±S.D. *: p<0.05. **: p<0.01. NS: non-significant. N=5. Scale bars are 50µm.
In the GCGCreERT; R26RTomato mice, we found that ALX-induced hyperglycemia was again corrected within 2 weeks by intraductal infusion with AAV-PM, but not with control AAV-GFP (Figure 3B). The findings on IPGTT and beta cell mass were also reproduced in these mice (Figure 3C–D). Based on counting of 5000 INS+ cells in each mouse, with 5 mice in each experimental group, we found that 95.6±2.3% of INS+ cells after AAV-PM infusion in ALX-treated mice were labeled with tomato (Figure 3E; no tomato+/INS+ cells were found in the control AAV-GFP infused mice), suggesting an alpha-cell origin. Moreover, when these mice were followed for 24 weeks, the mice remained euglycemic and the re-established beta cell mass appeared to be sustained (Figure 3D), and the beta cells remained as tomato-tagged (Figure 3F). Interestingly, when AAV-PM was given to GCGCreERT; R26RTomato mice that did not receive ALX, the conversion rate of alpha cells to beta cells (percentage of tomato+ cells that were INS+) was only 18.5±2.5%, suggesting that the alpha-to-beta conversion may be greatly decreased under normal blood glucose (Supplementary Figure 3D).
In order to determine whether acinar cells may also be reprogrammed into INS+ cells by AAV-PM, we performed the same viral treatment in ALX-treated, Elastase (Ela)-CreERT; R26RTomato mice, in which essentially all acinar cells were lineage tagged with tomato (Xiao et al., 2013c; Xiao et al., 2014c). We did not detect significant numbers of tomato+INS+ cells, suggesting that here acinar cells are not a major contributor to the newly reprogrammed INS+ cells (Supplementary Figure 3E).
In order to determine whether alpha cells in older mice retain the ability to transdifferentiate into beta cells in this model, we treated GCG-Cre; R26RTomato mice at 4 months of age with ALX injection and viral infusion. Our data suggest that alpha cells in older mice seemed to retain the ability to transdifferentiate into beta cells in response to activation of Pdx1 and MafA expression (Supplementary Figure 3H).
Alpha-cell-derived INS+ cells have a similar expression profile to normal beta cells
Next, we examined the differences in gene expression patterns between alpha-cell-derived INS+ cells and normal alpha and beta cells. We thus generated triple transgenic mice (GCG-Cre; R26RTomato; MIP-GFP). In these mice, alpha cells are lineage tagged with tomato and beta cells express GFP. However, the alpha-cell-derived INS+ cells will express both tomato and GFP (and therefore will be yellow), to allow separate isolation by flow cytometry (Figure 4A, Supplementary Figure 2D), similar to our previous study (Xiao et al., 2013a). Tomato+ cells in untreated mice are used as a control for normal alpha cells, while GFP+ cells in untreated mice are used as a control for normal beta cells (Figure 4B–C). Importantly, because of the MIP-GFP, the AAV-PM virus here did not have the GFP sequence. We found that the alpha-cell-derived INS+ cells have a gene expression pattern very close to normal beta cells, but very different from the original alpha cells, by both RNA-seq (Figure 4D–G), and by direct RT-qPCR quantification of some cell-specific transcripts (Supplementary Figure 4). These data suggest that the alpha-cell-derived INS+ cells have undergone a near-complete conversion to beta cells.
Fig 4. Gene expression pattern of alpha-cell-derived INS+ cells, compared to normal alpha and beta cells.

(A) Schematic of GCG-Cre; R26RTomato; MIP-GFP mouse model. (B) Alpha-cell-derived INS+ cells (yellow cells) were isolated 1 month after AAV-PM infusion from ALX-treated GCG-Cre; R26RTomato; MIP-GFP mice, based on expression of tomato red (alpha-cell lineage) and GFP by flow cytometry. Sorted normal GFP+ beta cells and normal TOM+ alpha cells from GCG-Cre; R26RTomato; MIP-GFP mice without any treatment were used as controls. Representative flow cytometry chart for sorting is shown. (C) Immunofluorescence images from the GCG-Cre; R26RTomato; MIP-GFP mouse 4 weeks after ALX/AAV-PM infusion. Note the abundance of the TOM+GFP+ INS+ (yellow) cells. N=5. (D–F) Gene expression analysis of these alpha cell-derived INS+ cells (Neo INS+) was performed by RNA-seq, and compared to purified, normal beta (beta) and alpha (alpha) cells. (D) Pearson correlation plot showing the FPKM values of all genes generated by Cuffnorm for all samples followed by heat map generation using the Pearson correlation R2 values. (E) Heat map shows log2 fold change (adjusted p value <0.05, absolute fold change +/− 2.0). (F) Selected beta-cell-specific and alpha-cell-specific genes show close alignment of the Neo-INS+ cells with beta cells, not alpha cells by RNA-seq. (G) Volcano plot for the group B versus group C comparison, of – log10(p-value) vs. log2(fold change), for differential gene expression analysis using Cuffdiff output. The horizontal dashed line in both plots corresponds to an FDR adjusted p-value of 0.01. All points displayed above this this line on the plot have an adjusted p-value of less than 0.01. N=3. For each sample, purified cells from 4–5 mouse pancreases were pooled together for RNA-seq.
AAV-PM infusion in hyperglycemic NOD mice leads to prolonged normalization of blood glucose
Next, we examined whether the newly formed INS+ cells would be recognized by an autoimmune diabetic immune system. Thus, we gave NOD mice a single ductal infusion of the PM virus early after the onset of hyperglycemia, when the blood glucose of the mice had surpassed 200mg/dl. We found that the glycemia in these mice normalized fairly rapidly, and for about 4 months. The mice receiving control AAV-GFP showed continuously increasing blood glucose and died within 5 weeks (Figure 5A, and Supplementary Figure 5A). Immunohistological analysis showed that the NOD mice that received AAV-PM had significantly greater INS+ cell mass (Figure 5B–C) as a basis for their normalized blood glucose, although the insulitis (based on CD45 staining for immune cells) was still present (Figure 5C). Moreover, we found that early on, some INS+ cells also expressed GCG (Figure 5D), possibly representing in-transit cells. Furthermore, EM images showed single cells with both INS and GCG granules (Figure 5E), suggesting that these INS-producing cells are reprogrammed from alpha cells. When the intraductal infusion with AAV-PM was instead performed later, after the blood glucose had reached 400mg/dl, we found that the blood glucose was normalized in only 3 out of 7 mice, and for about 16 weeks (Supplementary Figure 5B). This variable result may stem from glucose toxicity or from some alpha cell injury. At the 22 week time-point, when the AAV-PM-treated mice were again hyperglycemic, histology showed that in the islet region, there were many infiltrating CD3+ lymphocytes and F4/80+ macrophages, but very few INS+ cells (but were still GFP+), and still many of the surrounding non-islet (acinar) cells were GFP+, confirming persistence of transgene expression (Supplementary Figure 6A–B).
Figure 5. Intraductal infusion of AAV-PM reverses hyperglycemia in NOD mice.

(A) When the blood glucose of female NOD mice surpassed 200mg/dl, the mice received an intraductal infusion of either AAV-PM or control AAV-GFP. Fasting blood glucose levels were measured, showing continuously increasing hyperglycemia in control mice (green line), but rapid stabilization and then, by 2–3 weeks, normalization of hyperglycemia in mice infused with AAV-PM (red line), lasting for about 4 months. (B) Beta cell mass at 5 weeks after viral infusion. (C) Immunostaining for INS (in red) and CD45 (in white) 5 weeks after infusion of control AAV-GFP (upper panels) or AAV-PM (lower panels), along with direct green fluorescence (GFP) from viral infection. HO: Hoechst, nuclear stain. (D) Confocal images for INS (in red) and GCG (in blue) 5 weeks after infusion of AAV-PM, along with direct green fluorescence (GFP) from viral infection, to show presence of double positive cells for both INS and GCG (arrows). (E) EM image showing an islet cell with both INS (red arrow) and GCG (yellow arrow) granules in the left panel. The right panel is the inset of the blue rectangle region in the left panel. Statistics were analyzed by one-way ANOVA with a Bonferroni correction, followed by Fisher’s Exact Test. Data are presented as mean±S.D. **: p<0.01. N=10. Scale bars are 50µm.
Assessment of the status of the NOD immune system following viral therapy
It is well-known that islet transplants into an autoimmune diabetic environment can be quickly destroyed by the “rapid-recurrent” form of immune attack (Ramesh et al., 2013), even if the patient is adequately immunosuppressed for an accompanying kidney transplant (Vendrame et al., 2010). One possible explanation for the resistance of our neogenic INS+ cells to such a rapid-recurrent autoimmune rejection is that the pancreatic ductal infusion with AAV-PM may somehow directly alter the autoimmunity in the NOD mouse, resulting in extended survival of the neogenic INS+ cells. This virus-induced “bystander effect” has been well described in NOD mice (Atkinson and Leiter, 1999). This possibility seemed unlikely since the control AAV-GFP infusion gave no protection to the beta cells. However, to further examine this possibility, we isolated splenocytes from AAV-PM-treated NOD mice 4 weeks after viral infusion and performed an adoptive transfer into NOD/SCID mice. Splenocytes from untreated hyperglycemic NOD mice (UT) and from AAV-GFP-treated NOD mice were used as controls, the latter two showing no significant difference from each other (Figure 6A). We found that the development of diabetes in recipient NOD/SCID mice after delivery of splenocytes from AAV-PM-treated NOD mice was somewhat delayed, but still present (Figure 6A). The timing and kinetics of the hyperglycemia onset mirrored what might be seen if naïve NOD splenocytes (harvested prior to the onset of hyperglycemia) had been used (Christianson et al., 1993). To further test the competence of the NOD autoimmunity, NOD/SCID mouse islets (300) were transplanted under the kidney capsule of AAV-PM-treated and AAV-GFP-treated NOD mice 4 weeks after viral infusion, as well as into undisturbed NOD/SCID mice as controls (Figure 6B). We detected slightly higher graft INS content (Figure 6C) and higher INS+ cell numbers (Figure 6D) in AAV-PM-treated NOD mice compared with AAV-GFP-treated NOD mice (but still much lower than control grafts). This result was further confirmed by INS immunohistochemistry of the graft under the kidney capsule (Figure 6E). Also of importance here is that the presumed reactivation of the autoimmunity in the AAV-PM-treated mice, targeted against the transplanted islets, did not lead to an adjuvant effect with an autoimmune attack on the neogenic INS+ cells (derived from alpha-cells) in the pancreas, as the blood glucose remained normal. These latter data further support that the autoimmunity of the NOD mice is intact, and is behaving as if it were not actively being exposed to beta-cell antigens at the time of transplant, and thus showed a slightly delayed response to the transplanted islets.
Figure 6. Assessment of the status of the NOD autoimmunity following viral therapy.

(A) Splenocytes isolated from untreated diabetic NOD mice (UT), and from AAV-PM-infused and AAV-GFP- infused NOD mice 4 weeks after viral infusion were adoptively transferred into NOD/SCID mice. The development of diabetes in recipient NOD/SCID mice was compared. (B) NOD/SCID mouse islets (300) were transplanted under the kidney capsule of AAV-PM-treated and AAV-GFP-treated NOD mice 4 weeks after viral infusion, and into undisturbed NOD/SCID mice as a control. (C–D) Quantification of graft INS content (C) and INS+ cell number (D) 2 weeks after transplantation. (E) Representative images for INS (in green) and CD45 (in red) in the islet graft under the kidney capsule. Statistics were analyzed by one-way ANOVA with a Bonferroni correction, followed by Fisher’s Exact Test. Data are presented as mean±S.D. *: p<0.05. **: p<0.01. N=5. Scale bars are 50µM.
AAV-PM induces the generation of functional INS+ cells from alpha cells in human islets
Based on data from these mouse studies, we then tested whether human alpha cells may be reprogrammed into functional INS+ cells through a similar strategy. Human islets are resistant to ALX, but high-dose STZ has been reported to ablate a large proportion of human beta cells in vitro (Eizirik et al., 1994). Here, we treated human islets with 20mmol/l STZ for 12 hours to destroy beta cells, after which the islets were treated in vitro with either AAV-PM or AAV-GFP for 24 hours to trigger alpha-to-beta cell conversion, and then transplanted into ALX-treated hyperglycemic NOD/SCID mice (Figure 7A). The beta-cell-toxic effect of STZ was confirmed by examining INS content per islet (Figure 7B). Cells double positive for both INS and GCG were detected 3 days after STZ and AAV-PM treatment in culture (Figure 7C). In addition, the alpha cell mass in human islets was decreased by 35% 3 days after STZ and AAV-PM treatment in culture. We found that within one week after transplantation, the ALX-treated NOD/SCID mice that received human islets treated with STZ and AAV-PM had significantly lower blood glucose levels (Figure 7D), and significantly better glucose tolerance curves, compared to mice transplanted with STZ and AAV-GFP-treated human islets (Figure 7E). The grafts were harvested 4 weeks after transplantation and we found a significantly higher INS content (Figure 7F), greater beta cell mass (Figure 7G) and higher serum human C-peptide (Figure 7H) in the grafts of AAV-PM-treated human islets compared to the graft of AAV-GFP-treated human islets, which was further confirmed by immunohistochemistry (Figure 7I). One week of continuous BrdU labeling was performed after the islet transplantation, only 1.5±0.3% of INS+ cells had incorporated BrdU (Figure 7J). Thus, any contribution of proliferating residual beta cells to the increase in beta cell mass should be minimal. Together, these data suggest that AAV-PM can induce the generation of functional INS+ cells from alpha cells in human islets, although we cannot exclude that human non-alpha/non-beta cells may have contributed to the augmented INS+ cell numbers.
Figure 7. AAV-PM induces generation of functional INS+ cells in human islets.

(A) Human islets were treated with 20 mmol/l STZ for 12 hours, after which the islets were treated with either AAV-PM or AAV-GFP for 24 hours, and then transplanted into ALX-treated hyperglycemic NOD/SCID mice. (B) The beta-cell-destroying effect of STZ was confirmed at 12 hours by examining INS content per islet. (C) Immunostaining for INS and GCG on human islets 2 days after treatment with STZ and AAV-PM. (D–E) The ALX-NOD/SCID mice that received human islets treated with STZ and AAV-PM (in red) had significantly lower fasting blood glucose levels (D), and significantly better glucose tolerance (E), as early as 1 week after transplantation, compared to the ALX-NOD/SCID mice that received human islets treated with STZ and AAV-GFP (in green). (F–H) Graft INS content (F), graft beta cell mass (G) and serum human C-peptide levels (H) were determined 4 weeks after transplantation. (I) Representative images for INS (in green) and GCG (in red) in the graft under the kidney capsule. (J) One week of continuous BrdU labeling was performed after the islet transplantation, only 1.5±0.3% of INS+ cells had incorporated BrdU, shown by a representative image in the left panel. The right panel is an inset of the yellow rectangle region in the left panel. Statistics were analyzed by one-way ANOVA with a Bonferroni correction, followed by Fisher’s Exact Test. Data are presented as mean±S.D. *: p<0.05. **: p<0.01. N=5. White scale bars are 50µM. Yellow scale bars are 20µM.
Discussion
Earlier work has shown that overexpression of the transcription factor Pax4 in pancreatic progenitors (Collombat et al., 2009), or Pdx1 in newly-formed alpha cells (Yang et al., 2011), may result in conversion from an alpha cell phenotype to a beta-like cell phenotype, a phenomenon further supported by epigenetic analysis (Bramswig et al., 2013). However, this phenomenon seems to not occur in mature alpha cells in adults (Yang et al., 2011), except perhaps in the event of severe beta-cell loss (Thorel et al., 2010).
Here, we showed that PM overexpression in vivo was able to correct hyperglycemia in both ALX-induced diabetes and in autoimmune diabetic NOD mice, suggesting that a true beta cell-like reprogramming was occurring, rather than simply activation of the INS promoter and suppression of the GCG promoter in alpha cells. Among all AAV serotypes, we found that 8 and 6 were the best for infecting mouse pancreatic cells (Xiao et al., 2014b). We chose AAV serotype 8 vectors, since we found that serotype 8 had a better infection efficiency in mouse islet cells than serotype 6 (Xiao et al., 2014b). Serotype 6 infects mouse pancreatic ducts cells, while serotype 8 does not, but duct cells were not the focus of the current study (Xiao et al., 2014b). Both serotypes infect mouse acinar cells well (Xiao et al., 2014b).
We used both GCG-Cre; R26RTomato reporter mice and GCGCreERT; R26RTomato reporter mice to lineage-trace alpha cells. In GCG-Cre; R26RTomato reporter mice, although this transgenic GCG promoter that drives Cre is not strong (Herrera, 2000; Shiota et al., 2013), the highly sensitive tomato reporter allowed for more than 70% of the GCG-lineage cells to be successfully labeled. In the control diabetic AAV-GFP-infused mice, very few INS+ cells were detected, and none of them were tagged with tomato red, suggesting that neither the hyperglycemia, nor the viral infection alone, without overexpression of Pdx1 and MafA in target cells, is sufficient to trigger alpha-to-beta cell conversion. Of note, these findings were confirmed in our newly created GCGCreERT; R26RTomato reporter knock-in mice, in which nearly all INS+ cells were tagged with tomato in the ALX and AAV-PM treated mice. Since leakiness may occur in some creERT mice, in which creERT can go into nuclei of the cells and cause recombination without induction by tamoxifen, we examined this issue in this new strain. We found that the pre-labeling of the alpha cells without tamoxifen in these mice is very rare (none of the 2000 examined alpha cells), and thus should not affect the interpretation of the data in the current study.
An hGH minigene has been shown to affect the interpretation of beta cell proliferation and survival studies (Brouwers et al., 2014; Carboneau et al., 2016; Oropeza et al., 2015). However, this problem should not affect the current study, none of the constructs here contained this hGH minigene.
In a previous report, Zhou et al. found conversion of acinar cells into INS-producing cells by overexpression of Pdx1, Ngn3 and MafA (Zhou et al., 2008). Here, we did not detect acinar-to-beta cell conversion with overexpression of Pdx1 and MafA. Thus, Ngn3 may be specifically necessary for acinar-to-endocrine conversion, but may not to be required for reprogramming between different endocrine cell types, as in the conversion from alpha cells to beta-like cells here.
The normal blood glucose restoration and beta cell survival in NOD mice by PM appear to last 4 months prior to re-establishment of autoimmunity. It is impossible to predict how the immune protection in mice might translate to humans. It is conceivable that any immune protection might last a length of time equivalent to the 4 months as a percentage of the mouse lifespan. It is also possible, however, that little or no immune protection may occur in humans. Islet transplants into untreated diabetic normal mice are rapidly rejected within days to weeks, whereas the newly formed beta-cells in our system survive for months. Thus, the window of time we report showing a reversal of hyperglycemia in NOD mice without direct immunomodulation or exogenous INS is a key difference between our approach and similar previously reported strategies. It is unlikely that the return of hyperglycemia in the normalized NOD mice is due to loss of transgene expression, since the few INS+ cells present, as well as the surrounding acinar cells were GFP+, and also because in ALX-treated GCG-CreERT; ROSAtomato mice at 24 weeks, the GFP+ neogenic beta cells persisted and the mice remained euglycemic. Instead, it appears that a return of autoimmunity caused the loss of beta cells, consistent with the presence of many inflammatory cells in the islet region. Thus, the window of protection from autoimmunity may stem from two possible mechanisms. The first possibility is that the ductal viral infusion directly altered the autoimmunity of the NOD mouse, leading to extended survival of the neogenic INS+ cells (viral-induced bystander effect). However, the fact that the control virus infusion had no protective effect argues against this possibility. The delayed, but eventually effective attack on beta-cells in both the adoptive transfer experiments and the NOD/SCID islet transplants, suggests that the autoimmune NOD immune system is intact, but there is a delayed response to the newly formed beta cells. Moreover, we transplanted intact islets into the AAV-GFP-treated NOD mice and the beta cells were completely ablated (no difference from untreated NOD mice), again consistent with no viral bystander effect.
The second possible explanation is that the alpha cell-derived INS+ cells are not recognized well by the autoimmune system. The more variable effectiveness of the AAV-PM treatment at late stages of hyperglycemia may reflect glucotoxicity or alpha cell injury. By RT-qPCR and RNA-seq, we found that expression levels of beta-cell-specific genes in these new INS+ cells were similar, but not identical to true beta-cells. Interestingly, a recent study showed that alpha cells could be converted to beta cells upon loss of both Arx and DNA-methyltransferase 1 (Dnmt1) (Chakravarthy et al., 2017). Here, we also found that DNMT1 was downregulated in the reprogrammed beta cells, suggesting that the two experimental paradigms may share some molecular signaling pathways.
We used GCG-Cre; R26RTomato; MIP-GFP rather than GCGCreERT; R26RTomato; MIP-GFP mice to characterize newly formed INS+ cells by RNA-seq since tamoxifen binding to the estrogen receptor in INS+ cells may result in alterations in gene expression (Liu et al., 2009; Ropero et al., 2002; Tiano et al., 2011). Thus, it may be that these new INS+ cells either lack key antigen(s) to induce an autoimmune attack, or because of their localization to the islet microenvironment, they may somehow avoid a rapid-recurrent autoimmune attack. If the islet niche is critical to the survival of newly introduced beta cells in autoimmune diabetes, it would have important ramifications for any future clinical strategy to deliver exogenous beta-cells to T1D patients.
It is also important to note here that for human therapy we would strive to use a GCG promoter to drive the PM construct to avoid the risks of prolonged transgene expression, since once the alpha cells had converted to beta cells, they would presumably transition to express Pdx1 and MafA from the endogenous locus, not the viral transgene since the GCG promoter should become inactive in beta cells. However, additional work is needed to optimize the generation of an effective, highly specific GCG promoter construct of reduced size to fit into the AAV vector. Moreover, augmentation of the strength of this GCG promoter may be required to allow sufficient expression of the transgene. In addition, the delivery of gene therapeutic virus through the pancreatic duct is potentially easily translatable to humans, since such pancreatic injections are routinely performed in humans through a non-surgical endoscopic procedure known as endoscopic retrograde cholangiopancreatography (ERCP).
Star★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Xiangwei Xiao (Xiangwei.xiao@chp.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
All mouse experiments were approved by the Animal Research and Care Committee at the Children’s Hospital of Pittsburgh and the University of Pittsburgh IACUC, and were carried out in accordance with the approved guidelines. Female C57BL/6, NOD, NOD/SCID, MIP-GFP and R26RTomato (C56BL/6 background) mice were all purchased from the Jackson Lab (Bar Harbor, ME, USA). GCG promoter Cre (GCG-Cre; C56BL/6 background) mice were purchased from MMRRC (USA), which was originally provided by Dr. Pedro Herrera (Herrera, 2000) (University of Geneva, Switzerland). GCG promoter CreERT (GCGCreERT) knock-in mice were generated by gene targeting. The first 66 bases of the preproglucagon coding sequence are replaced by the CreERT2 coding sequence with an SV40 polyadenylation signal in the targeted allele. The BAC transgenic Elastase promoter CreERT reporter (Ela-CreERT) mouse was described before (Ji et al., 2008; Xiao et al., 2013c). Tamoxifen induction of tomato expression in acinar cells in Ela-CreERT; R26RTomato mice, or tomato expression in alpha cells in GCGCreERT; R26RTomato has been described before (Xiao et al., 2013c). A very low tamoxifen dose (2mg), and only one injection was performed, after which the mice were kept for 4 weeks as a washout period before experiment. Female C57BL/6, Ela-CreERT, GCG-Cre, GCGCreERT and NOD/SCID mice were used at 10 weeks of age. Female NOD mice were used when the blood glucose reached a specified level. Exclusion criteria: the only exclusions were NOD mice that failed to develop high blood glucose after 16 weeks of age. Randomization and blind assessment were used in all animal studies. Measurements of mouse blood glucose were performed at 10am after a two-hour fasting period. Intraperitoneal glucose tolerance testing (IPGTT) was performed as previously described (Xiao et al., 2014c; Xiao et al., 2013d).
The beta-cell toxin ALX was injected via the dorsal tail vein at 65 mg/kg body weight, while STZ was injected intraperitoneally at 150 mg/kg body weight, as described before (Xiao et al., 2013a). For destroying beta cells in human islets, STZ (20mmol/l) was added to cultured human islets for 24 hours before further treatment. Fasting blood glucose monitoring and intraperitoneal glucose tolerance test (IPGTT), measurement of human C-peptide, and islet perifusion were performed as described before (Shiota et al., 2013; Xiao et al., 2016; Xiao et al., 2014c).
Pancreatic intraductal viral infusion was performed as described previously (Xiao et al., 2014b), in which 150µl viruses [1012 genome copy particle (GCP)/ml] were infused at a rate of 5µl/min. For labeling proliferating cells, BrdU drinking water was given immediately after viral infusion and renewed weekly until the end of the experiment as described before (Xiao et al., 2014a; Xiao et al., 2013d). Islet transplantation was performed as described before (Xiao et al., 2014c).
Human islets
Human islets were isolated from previously healthy, non-diabetic organ donors by the University of Chicago Transplant Center. Five independent human islet batches from three male donors and two female donors aged ranging from 32 to 55 were used in this study. Each experiment used islets from the same batch to compare different groups. The final data are from a summary of 5 experiments using these 5 batches accordingly.
METHODS DETAILS
Virus production
AAV serotype 8 vectors were generated by transfection of human embryonic kidney 293 cells as described before (Guo et al., 2012a; Guo et al., 2012b). Mouse open reading frames for Pdx1 and MafA were amplified from embryonic pancreatic cDNA. GFP was amplified from pLVX-IRES-ZsGreen (Clontech, Mountain View, CA, USA). AAV-GFP contains a GFP reporter under the cytomegalovirus (CMV) promoter. AAV-PM contains Pdx1, MafA and a GFP reporter under the CMV promoter. The small 2A peptides that connect Pdx1, MafA and GFP in the AAV-PM construct allows efficient, stoichiometric production of discrete protein products within a single vector through a “cleavage” event within the 2A peptide sequence (Guo et al., 2012b). Also, an AAV carrying Pdx1 and MafA only (AAV-PM, no GFP) was prepared for transducing cells in GCG-Cre; R26RTomato; MIP-GFP mice. Transfection was performed with Lipofectamine 2000 reagent (ThermoFisher Scientific, CA, Carlsbad, USA), according to the instructions of the manufacturer. Purification of AAV vectors were described before (Guo et al., 2012a), in which the empty capsid was removed from the sublayer formed after PEG-aqueous partitioning, without requirement for a density gradient. Most of the empty capsid was removed, and the remaining empty capsid was less than 19% (by TEM measurement) in the final purified virus solution. The prepared virus was stored at −80°C. Titration of viral vectors was determined using a dot-blot assay.
RNA isolation, quantitative polymerase chain reaction (RT-qPCR)
RNA extraction and cDNA synthesis have been described before (Xiao et al., 2013a; Xiao et al., 2014a; Xiao et al., 2013c; Xiao et al., 2014c). RT-qPCR primers were all purchased from Qiagen (Valencia, CA, USA). RT-qPCR was performed as described before (Xiao et al., 2013a; Xiao et al., 2014a; Xiao et al., 2013c; Xiao et al., 2014c). Values were normalized against CycloA, which proved to be stable across the samples, and then compared to controls.
Immunocytochemistry, immunohistochemistry and Western blot
Cells cultured in staining-plates were fixed for 2 hours in 4% formalin before immunocytochemical staining. All the mice received heart perfusion to remove red blood cells from the vessels before the pancreas was harvested, as described before (Xiao et al., 2013d). Pancreas samples were then fixed in zinc (BD Biosciences) for 6 hours before an additional 2 hours’ fixation in 4% formalin, then cryo-protected in 30% sucrose overnight, followed by freezing in a longitudinal orientation (from tail to head of the pancreas) and sectioned at 6µm. Tomato and GFP were detected by direct fluorescence. Western blot was performed as described before (Xiao et al., 2016). Nuclear staining was performed with Hoechst solution (HO, Becton-Dickinson Biosciences, San Jose, CA, USA). Confocal images were acquired as previously described (Xiao et al., 2013a; Xiao et al., 2013c).
Adoptive transfer of splenocytes into NOD/SCID mice
Isolation of splenocytes and adoptive transfer of splenocytes into NOD/SCID mice were performed as described before (Delmastro et al., 2012).
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA Sequencing (RNA-seq) and analysis
Total RNA was extracted from alpha-cell-derived INS+ cells (Neo INS+) that were isolated 1 month after AAV-PM infusion in ALX-treated GCG-Cre; R26RTomato; MIP-GFP mice, based on expression of tomato red (alpha-cell lineage) and GFP by flow cytometry. Sorted normal GFP+ beta cells (Beta) and normal TOM+ alpha cells (Alpha) from GCG-Cre; R26RTomato; MIP-GFP mice without any treatment were used as controls. Each sample was assessed using Qubit 2.0 fluorometer and Agilent TapeStation 2200 for RNA quantity and quality. Total RNA libraries were generated using Illumina TruSeq Stranded mRNA sample preparation kit. The mRNA is fragmented, and the fragments are copied into first strand cDNA. These cDNA fragments are then purified and enriched with PCR to create the final cDNA library. Cluster generation and 75 bp single-read dual-indexed sequencing was then performed on Illumina NextSeq 500’s. Sequencing analysis was done using mRNA-seq analysis on Maverix Analytic Platform (Maverix Biomics, Inc, San Mateo, CA, USA). Single reads were mapped to the mouse genome (m10) using STAR in a strand specific manner. Pairwise differential expression was quantified using Cuffdiff. Cufflinks was used to determine FPKM levels for each gene from the STAR alignment and was used as input for Cuffdiff. Read counts were then normalized across all samples and significant differentially expressed genes were determined by adjusted p-value with a threshold of 0.05. RNA-seq data are available at NCBI database (GSE: GSE106709; GSM2849152- GSM2849160).
Quantifications and statistics
For in vitro experiments, each experimental condition entailed 5 repeats (except for RNAseq and in vitro recall assay, which entailed 3 repeats). For in vivo experiments, five mice were used for each group. The sample size was determined according to the published literature. Alpha cell mass, beta cell mass, beta cell number and percentage of proliferating beta cells were quantified as described before (Xiao et al., 2014a). All data were statistically analyzed by one-way ANOVA with a Bonferroni correction, followed by Fisher’s Exact Test. χ-squared test with 1 degree of freedom was applied to compare observed and estimated data. All error bars represent S.D. (standard deviation). Significance was presented as * when p<0.05, and ** when p<0.01. No significance was presented as NS. P value and n value as well as the statistics methods were indicated in the figure legends.
Supplementary Material
Key resources table
| Reagent or Resource | Source | Identifier |
|---|---|---|
| Experimental models: organisms/strains | ||
| Mouse: C57BL/6 | Jackson Lab | Cat#664 |
| Mouse: NOD/SCID | Jackson Lab | Cat#1303 |
| Mouse: NOD | Jackson Lab | Cat#1976 |
| Mouse: MIP-GFP | Jackson Lab | Cat#6864 |
| Mouse: R26RTomato | Jackson Lab | Cat#7909 |
| Mouse: GCG-Cre | MMRRC | (Herrera, 2000) |
| Mouse: GCGCreERT | Gittes Lab | (Shiota et al., 2017) |
| Mouse: Ela-CreERT | Logsdon Lab | (Ji et al., 2008) |
| Antibodies | ||
| Guinea pig anti-INS | Dako | Cat#A0564 |
| Mouse anti-GCG | Sigma-Aldrich | Cat# G2654 |
| Rabbit anti-Pax4 | Santa Cruz Biotechnology | Cat# SC-27835 |
| Rabbit anti-p27 | Santa Cruz Biotechnology | Cat# SC-528 |
| Rat anti-BrdU | Abcam | Cat# Ab6326 |
| Rat anti-Ki-67 | Dako | Cat# M7249 |
| Rat anti-CD45 | BD Biosciences | Cat# 550539 |
| Rabbit anti-MafA | Bethyl Laboratories, Inc. | Cat# IHC-00352 |
| Rabbit anti-Pdx1 | Abcam | Cat# Ab47267 |
| Rabbit anti-CD3 | Abcam | Cat# Ab5690 |
| Rabbit anti-GCG | Cell Signaling | Cat# 2760S |
| Rabbit anti-GAPDH | Cell Signaling | Cat# 2118S |
| Rabbit anti-CCND1 | Santa Cruz Biotechnology | Cat# SC-718 |
| Rabbit anti-CDK4 | Santa Cruz Biotechnology | Cat# SC-260 |
| Cy2, Cy3, or Cy5 conjugated streptavidin-, rabbit-, rat-, goat-, mouse- and guinea pig-specific 2nd antibodies | Jackson ImmunoResearch Labs | n/a |
| Chemicals | ||
| ALX | Sigma-Aldrich | Cat#A7413-10g |
| STZ | Sigma-Aldrich | Cat#85882 |
| Commercial Assays | ||
| Lipofectamine 2000 | ThermoFisher Scientific | Cat#11668019 |
| RNeasy Mini Kit | Qiagen | Cat#74106 |
| Viruses | ||
| AAV-PM | Gittes lab | n/a |
| AAV-GFP | Gittes lab | n/a |
| Oligonucleotides | ||
| CycloA Q-PCR primer | Qiagen | Cat#QT00247709 |
| Pdx1 Q-PCR primer | Qiagen | Cat#QT00102235 |
| Ngn3 Q-PCR primer | Qiagen | Cat#QT00262850 |
| MafA Q-PCR primer | Qiagen | Cat#QT01037638 |
| Pax4 Q-PCR primer | Qiagen | Cat#QT01052772 |
| Nkx6.1 Q-PCR primer | Qiagen | Cat#QT00143318 |
| NeuroD1 Q-PCR primer | Qiagen | Cat#QT00156982 |
| Glut2 Q-PCR primer | Qiagen | Cat#QT00103537 |
| DNMT1 Q-PCR primer | Qiagen | Cat#QT02524613 |
| INS Q-PCR primer | Qiagen | Cat#QT00114289 |
| GCG Q-PCR primer | Qiagen | Cat#QT00124033 |
| RNA-seq data (GSE: GSE106709) | ||
| Primary mouse alpha cells | GSM2849152 | |
| Primary mouse alpha cells | GSM2849153 | |
| Primary mouse alpha cells | GSM2849154 | |
| Primary mouse beta cells | GSM2849155 | |
| Primary mouse beta cells | GSM2849156 | |
| Primary mouse beta cells | GSM2849157 | |
| Primary mouse beta cells reprogrammed from alpha cells | GSM2849158 | |
| Primary mouse beta cells reprogrammed from alpha cells | GSM2849159 | |
| Primary mouse beta cells reprogrammed from alpha cells | GSM2849160 | |
| Software | ||
| Flowjo | Flowjo LLC. | n/a |
| GraphPad Prism 6.0 | GraphPad Software, Inc. | n/a |
| ImageJ | NIH | n/a |
Acknowledgments
The authors would like to acknowledge the generosity and support of Dr. Martin Jendrisak and the entire team of the Gift of Hope Organ & Tissue Donor Network in Chicago for providing the human pancreas tissues used in the present study. We thank Jay Kolls and William Horne from University of Pittsburgh Health Sciences Sequencing Core at Children’s Hospital of Pittsburgh for RNA-seq analysis. XX was supported by a Tenure-track Assistant Professor startup from the Pediatric Division of Children’s Hospital of Pittsburgh. GKG were supported by NIH grant (R01DK098196, R01DK111460 and R01DK112836), Juvenile Diabetes Research Foundation (1-INO-2014-167-A-V) and the Children’s Hospital of Pittsburgh. PW and SR were supported by the University of Chicago DRTC Grant # P30 DK020595, CRC-National Center for Advancing Transitional Sciences of the NIH Grant # UL1TR000430. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Abbreviations
- Ngn3
neurogenin3
- AAV
adeno-associated virus
- PM
Pdx1 and MafA
- ALX
alloxan
- STZ
streptotozocin
- INS
insulin
- GCG
glucagon
- NOD
non-obese diabetes
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Author Contributions:
XX: study design, funding, research data and writing of the manuscript; PG, CS, TZ, GMC, SF, KP, JF and JDP: research data; SR and PW: research data and provision of human islets; GKG: study design, funding, research data, editing of the manuscript.
Conflict of interest:
Xiangwei Xiao, Ping Guo and George Gittes have a US national phase patent pending for intra-pancreatic ductal delivery of reagents through endoscopic retrograde cholangiopancreatography. PCT Application No. PCT/US2015/026532.
References
- Ackermann AM, Gannon M. Molecular regulation of pancreatic beta-cell mass development, maintenance, and expansion. J Mol Endocrinol. 2007;38:193–206. doi: 10.1677/JME-06-0053. [DOI] [PubMed] [Google Scholar]
- Ackermann AM, Wang Z, Schug J, Naji A, Kaestner KH. Integration of ATAC-seq and RNA-seq identifies human alpha cell and beta cell signature genes. Mol Metab. 2016;5:233–244. doi: 10.1016/j.molmet.2016.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akinci E, Banga A, Greder LV, Dutton JR, Slack JM. Reprogramming of pancreatic exocrine cells towards a beta (beta) cell character using Pdx1, Ngn3 and MafA. Biochem J. 2012;442:539–550. doi: 10.1042/BJ20111678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson MA, Leiter EH. The NOD mouse model of type 1 diabetes: as good as it gets? Nat Med. 1999;5:601–604. doi: 10.1038/9442. [DOI] [PubMed] [Google Scholar]
- Baeyens L, Lemper M, Leuckx G, De Groef S, Bonfanti P, Stange G, Shemer R, Nord C, Scheel DW, Pan FC, et al. Transient cytokine treatment induces acinar cell reprogramming and regenerates functional beta cell mass in diabetic mice. Nat Biotechnol. 2014;32:76–83. doi: 10.1038/nbt.2747. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Banga A, Akinci E, Greder LV, Dutton JR, Slack JM. In vivo reprogramming of Sox9+ cells in the liver to insulin-secreting ducts. Proc Natl Acad Sci U S A. 2012;109:15336–15341. doi: 10.1073/pnas.1201701109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramswig NC, Everett LJ, Schug J, Dorrell C, Liu C, Luo Y, Streeter PR, Naji A, Grompe M, Kaestner KH. Epigenomic plasticity enables human pancreatic alpha to beta cell reprogramming. J Clin Invest. 2013;123:1275–1284. doi: 10.1172/JCI66514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bramswig NC, Kaestner KH. Transcriptional regulation of alpha-cell differentiation. Diabetes Obes Metab. 2011;13(Suppl 1):13–20. doi: 10.1111/j.1463-1326.2011.01440.x. [DOI] [PubMed] [Google Scholar]
- Brouwers B, de Faudeur G, Osipovich AB, Goyvaerts L, Lemaire K, Boesmans L, Cauwelier EJ, Granvik M, Pruniau VP, Van Lommel L, et al. Impaired islet function in commonly used transgenic mouse lines due to human growth hormone minigene expression. Cell Metab. 2014;20:979–990. doi: 10.1016/j.cmet.2014.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell PM, Salam A, Ryan EA, Senior P, Paty BW, Bigam D, McCready T, Halpin A, Imes S, Al Saif F, et al. Pretransplant HLA antibodies are associated with reduced graft survival after clinical islet transplantation. Am J Transplant. 2007;7:1242–1248. doi: 10.1111/j.1600-6143.2007.01777.x. [DOI] [PubMed] [Google Scholar]
- Carboneau BA, Le TD, Dunn JC, Gannon M. Unexpected effects of the MIP-CreER transgene and tamoxifen on beta-cell growth in C57Bl6/J male mice. Physiological reports. 2016;4 doi: 10.14814/phy2.12863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelti-Weder C, Shtessel M, Reuss JE, Jermendy A, Yamada T, Caballero F, Bonner-Weir S, Weir GC. Pancreatic duct ligation after almost complete beta-cell loss: exocrine regeneration but no evidence of beta-cell regeneration. Endocrinology. 2013;154:4493–4502. doi: 10.1210/en.2013-1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarthy H, Gu X, Enge M, Dai X, Wang Y, Damond N, Downie C, Liu K, Wang J, Xing Y, et al. Converting Adult Pancreatic Islet alpha Cells into beta Cells by Targeting Both Dnmt1 and Arx. Cell Metab. 2017;25:622–634. doi: 10.1016/j.cmet.2017.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chera S, Baronnier D, Ghila L, Cigliola V, Jensen JN, Gu G, Furuyama K, Thorel F, Gribble FM, Reimann F, et al. Diabetes recovery by age-dependent conversion of pancreatic delta-cells into insulin producers. Nature. 2014;514:503–507. doi: 10.1038/nature13633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chintinne M, Stange G, Denys B, Ling Z, In ‘t Veld P, Pipeleers D. Beta cell count instead of beta cell mass to assess and localize growth in beta cell population following pancreatic duct ligation in mice. PLoS One. 2012;7:e43959. doi: 10.1371/journal.pone.0043959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson SW, Shultz LD, Leiter EH. Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes. 1993;42:44–55. doi: 10.2337/diab.42.1.44. [DOI] [PubMed] [Google Scholar]
- Collombat P, Xu X, Ravassard P, Sosa-Pineda B, Dussaud S, Billestrup N, Madsen OD, Serup P, Heimberg H, Mansouri A. The ectopic expression of Pax4 in the mouse pancreas converts progenitor cells into alpha and subsequently beta cells. Cell. 2009;138:449–462. doi: 10.1016/j.cell.2009.05.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmastro MM, Styche AJ, Trucco MM, Workman CJ, Vignali DA, Piganelli JD. Modulation of redox balance leaves murine diabetogenic TH1 T cells “LAG-3-ing” behind. Diabetes. 2012;61:1760–1768. doi: 10.2337/db11-1591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai BM, Oliver-Krasinski J, De Leon DD, Farzad C, Hong N, Leach SD, Stoffers DA. Preexisting pancreatic acinar cells contribute to acinar cell, but not islet beta cell, regeneration. J Clin Invest. 2007;117:971–977. doi: 10.1172/JCI29988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature. 2004;429:41–46. doi: 10.1038/nature02520. [DOI] [PubMed] [Google Scholar]
- Eizirik DL, Pipeleers DG, Ling Z, Welsh N, Hellerstrom C, Andersson A. Major species differences between humans and rodents in the susceptibility to pancreatic beta-cell injury. Proc Natl Acad Sci U S A. 1994;91:9253–9256. doi: 10.1073/pnas.91.20.9253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon M, Gamer LW, Wright CV. Regulatory regions driving developmental and tissue-specific expression of the essential pancreatic gene pdx1. Dev Biol. 2001;238:185–201. doi: 10.1006/dbio.2001.0359. [DOI] [PubMed] [Google Scholar]
- Georgia S, Bhushan A. Beta cell replication is the primary mechanism for maintaining postnatal beta cell mass. J Clin Invest. 2004;114:963–968. doi: 10.1172/JCI22098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P, El-Gohary Y, Prasadan K, Shiota C, Xiao X, Wiersch J, Paredes J, Tulachan S, Gittes GK. Rapid and simplified purification of recombinant adeno-associated virus. Journal of virological methods. 2012a;183:139–146. doi: 10.1016/j.jviromet.2012.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo P, Xiao X, El-Gohary Y, Paredes J, Prasadan K, Shiota C, Wiersch J, Welsh C, Gittes GK. A simplified purification method for AAV variant by Polyethylene Glycol aqueous two-phase partitioning. Bioengineered. 2012b;4 doi: 10.4161/bioe.22293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang Y, Stein R. MafA and MafB activity in pancreatic beta cells. Trends Endocrinol Metab. 2011;22:364–373. doi: 10.1016/j.tem.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127:2317–2322. doi: 10.1242/dev.127.11.2317. [DOI] [PubMed] [Google Scholar]
- Ji B, Song J, Tsou L, Bi Y, Gaiser S, Mortensen R, Logsdon C. Robust acinar cell transgene expression of CreErT via BAC recombineering. Genesis. 2008;46:390–395. doi: 10.1002/dvg.20411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jimenez V, Ayuso E, Mallol C, Agudo J, Casellas A, Obach M, Munoz S, Salavert A, Bosch F. In vivo genetic engineering of murine pancreatic beta cells mediated by single-stranded adeno-associated viral vectors of serotypes 6, 8 and 9. Diabetologia. 2011;54:1075–1086. doi: 10.1007/s00125-011-2070-3. [DOI] [PubMed] [Google Scholar]
- Kopinke D, Brailsford M, Shea JE, Leavitt R, Scaife CL, Murtaugh LC. Lineage tracing reveals the dynamic contribution of Hes1+ cells to the developing and adult pancreas. Development. 2011;138:431–441. doi: 10.1242/dev.053843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JL, Dubois CL, Hao E, Thorel F, Herrera PL, Sander M. Progenitor cell domains in the developing and adult pancreas. Cell Cycle. 2011a;10:1921–1927. doi: 10.4161/cc.10.12.16010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopp JL, Dubois CL, Schaffer AE, Hao E, Shih HP, Seymour PA, Ma J, Sander M. Sox9+ ductal cells are multipotent progenitors throughout development but do not produce new endocrine cells in the normal or injured adult pancreas. Development. 2011b;138:653–665. doi: 10.1242/dev.056499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kushner JA, Weir GC, Bonner-Weir S. Ductal origin hypothesis of pancreatic regeneration under attack. Cell Metab. 2010;11:2–3. doi: 10.1016/j.cmet.2009.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J, Sugiyama T, Liu Y, Wang J, Gu X, Lei J, Markmann JF, Miyazaki S, Miyazaki J, Szot GL, et al. Expansion and conversion of human pancreatic ductal cells into insulin-secreting endocrine cells. eLife. 2013;2:e00940. doi: 10.7554/eLife.00940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cavelti-Weder C, Zhang Y, Clement K, Donovan S, Gonzalez G, Zhu J, Stemann M, Xu K, Hashimoto T, et al. Long-term persistence and development of induced pancreatic beta cells generated by lineage conversion of acinar cells. Nat Biotechnol. 2014;32:1223–1230. doi: 10.1038/nbt.3082. [DOI] [PubMed] [Google Scholar]
- Liu S, Le May C, Wong WP, Ward RD, Clegg DJ, Marcelli M, Korach KS, Mauvais-Jarvis F. Importance of extranuclear estrogen receptor-alpha and membrane G protein-coupled estrogen receptor in pancreatic islet survival. Diabetes. 2009;58:2292–2302. doi: 10.2337/db09-0257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Herrera PL, Carreira C, Bonnavion R, Seigne C, Calender A, Bertolino P, Zhang CX. Alpha cell-specific Men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology. 2010;138:1954–1965. doi: 10.1053/j.gastro.2010.01.046. [DOI] [PubMed] [Google Scholar]
- Meier JJ, Butler AE, Saisho Y, Monchamp T, Galasso R, Bhushan A, Rizza RA, Butler PC. Beta-cell replication is the primary mechanism subserving the postnatal expansion of beta-cell mass in humans. Diabetes. 2008;57:1584–1594. doi: 10.2337/db07-1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oropeza D, Jouvet N, Budry L, Campbell JE, Bouyakdan K, Lacombe J, Perron G, Bergeron V, Neuman JC, Brar HK, et al. Phenotypic Characterization of MIP-CreERT1Lphi Mice With Transgene-Driven Islet Expression of Human Growth Hormone. Diabetes. 2015;64:3798–3807. doi: 10.2337/db15-0272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan FC, Bankaitis ED, Boyer D, Xu X, Van de Casteele M, Magnuson MA, Heimberg H, Wright CV. Spatiotemporal patterns of multipotentiality in Ptf1a–expressing cells during pancreas organogenesis and injury-induced facultative restoration. Development. 2013;140:751–764. doi: 10.1242/dev.090159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipeleers D, Chintinne M, Denys B, Martens G, Keymeulen B, Gorus F. Restoring a functional beta-cell mass in diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):54–62. doi: 10.1111/j.1463-1326.2008.00941.x. [DOI] [PubMed] [Google Scholar]
- Pipeleers D, Keymeulen B, Chatenoud L, Hendrieckx C, Ling Z, Mathieu C, Roep B, Ysebaert D. A view on beta cell transplantation in diabetes. Ann N Y Acad Sci. 2002;958:69–76. doi: 10.1111/j.1749-6632.2002.tb02948.x. [DOI] [PubMed] [Google Scholar]
- Pipeleers D, Ling Z. Pancreatic beta cells in insulin-dependent diabetes. Diabetes Metab Rev. 1992;8:209–227. doi: 10.1002/dmr.5610080303. [DOI] [PubMed] [Google Scholar]
- Purcell LJ, Mottram PL. Prevention of both rejection and recurrence of autoimmune disease in the NOD/Lt mouse following segmental pancreas transplantation. Transplant Proc. 1995;27:2166–2167. [PubMed] [Google Scholar]
- Ramesh A, Chhabra P, Brayman KL. Pancreatic islet transplantation in type 1 diabetes mellitus: an update on recent developments. Curr Diabetes Rev. 2013;9:294–311. doi: 10.2174/15733998113099990063. [DOI] [PubMed] [Google Scholar]
- Rankin MM, Wilbur CJ, Rak K, Shields EJ, Granger A, Kushner JA. beta-Cells Are Not Generated in Pancreatic Duct Ligation-Induced Injury in Adult Mice. Diabetes. 2013;62:1634–1645. doi: 10.2337/db12-0848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ropero AB, Soria B, Nadal A. A nonclassical estrogen membrane receptor triggers rapid differential actions in the endocrine pancreas. Mol Endocrinol. 2002;16:497–505. doi: 10.1210/mend.16.3.0794. [DOI] [PubMed] [Google Scholar]
- Shiota C, Prasadan K, Guo P, El-Gohary Y, Wiersch J, Xiao X, Esni F, Gittes GK. alpha-Cells are dispensable in postnatal morphogenesis and maturation of mouse pancreatic islets. Am J Physiol Endocrinol Metab. 2013;305:E1030–1040. doi: 10.1152/ajpendo.00022.2013. [DOI] [PubMed] [Google Scholar]
- Shiota C, Prasadan K, Guo P, Fusco J, Xiao X, Gittes GK. Gcg CreERT2 knockin mice as a tool for genetic manipulation in pancreatic alpha cells. Diabetologia. 2017 doi: 10.1007/s00125-017-4425-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solar M, Cardalda C, Houbracken I, Martin M, Maestro MA, De Medts N, Xu X, Grau V, Heimberg H, Bouwens L, et al. Pancreatic exocrine duct cells give rise to insulin-producing beta cells during embryogenesis but not after birth. Dev Cell. 2009;17:849–860. doi: 10.1016/j.devcel.2009.11.003. [DOI] [PubMed] [Google Scholar]
- Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell. 2007;12:817–826. doi: 10.1016/j.devcel.2007.04.011. [DOI] [PubMed] [Google Scholar]
- Thorel F, Damond N, Chera S, Wiederkehr A, Thorens B, Meda P, Wollheim CB, Herrera PL. Normal glucagon signaling and beta-cell function after near-total alpha-cell ablation in adult mice. Diabetes. 2011;60:2872–2882. doi: 10.2337/db11-0876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, Herrera PL. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464:1149–1154. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tiano JP, Delghingaro-Augusto V, Le May C, Liu S, Kaw MK, Khuder SS, Latour MG, Bhatt SA, Korach KS, Najjar SM, et al. Estrogen receptor activation reduces lipid synthesis in pancreatic islets and prevents beta cell failure in rodent models of type 2 diabetes. J Clin Invest. 2011;121:3331–3342. doi: 10.1172/JCI44564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tonne JM, Sakuma T, Munoz-Gomez M, El Khatib M, Barry MA, Kudva YC, Ikeda Y. Beta cell regeneration after single-round immunological destruction in a mouse model. Diabetologia. 2014 doi: 10.1007/s00125-014-3416-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vendrame F, Pileggi A, Laughlin E, Allende G, Martin-Pagola A, Molano RD, Diamantopoulos S, Standifer N, Geubtner K, Falk BA, et al. Recurrence of type 1 diabetes after simultaneous pancreas-kidney transplantation, despite immunosuppression, is associated with autoantibodies and pathogenic autoreactive CD4 T-cells. Diabetes. 2010;59:947–957. doi: 10.2337/db09-0498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir GC, Bonner-Weir S. Islet transplantation as a treatment for diabetes. J Am Optom Assoc. 1998;69:727–732. [PubMed] [Google Scholar]
- Xiao X, Chen Z, Shiota C, Prasadan K, Guo P, El-Gohary Y, Paredes J, Welsh C, Wiersch J, Gittes GK. No evidence for beta cell neogenesis in murine adult pancreas. J Clin Invest. 2013a;123:2207–2217. doi: 10.1172/JCI66323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Fischbach S, Song Z, Gaffar I, Zimmerman R, Wiersch J, Prasadan K, Shiota C, Guo P, Ramachandran S, et al. Transient suppression of transforming growth factor beta receptor signaling facilitates human islet transplantation. Endocrinology. 2016 doi: 10.1210/en.2015-1986. en20151986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Gaffar I, Guo P, Wiersch J, Fischbach S, Peirish L, Song Z, El-Gohary Y, Prasadan K, Shiota C, et al. M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proc Natl Acad Sci U S A. 2014a;111:E1211–1220. doi: 10.1073/pnas.1321347111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Guo P, Chen Z, El-Gohary Y, Wiersch J, Gaffar I, Prasadan K, Shiota C, Gittes GK. Hypoglycemia reduces vascular endothelial growth factor a production by pancreatic Beta cells as a regulator of Beta cell mass. J Biol Chem. 2013b;288:8636–8646. doi: 10.1074/jbc.M112.422949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Guo P, Prasadan K, Shiota C, Peirish L, Fischbach S, Song Z, Gaffar I, Wiersch J, El-Gohary Y, et al. Pancreatic cell tracing, lineage tagging and targeted genetic manipulations in multiple cell types using pancreatic ductal infusion of adeno-associated viral vectors and/or cell-tagging dyes. Nat Protoc. 2014b;9:2719–2724. doi: 10.1038/nprot.2014.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Guo P, Shiota C, Prasadan K, El-Gohary Y, Wiersch J, Gaffar I, Gittes GK. Neurogenin3 Activation Is Not Sufficient to Direct Duct-to-Beta Cell Transdifferentiation in the Adult Pancreas. J Biol Chem. 2013c;288:25297–25308. doi: 10.1074/jbc.M113.484022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Prasadan K, Guo P, El-Gohary Y, Fischbach S, Wiersch J, Gaffar I, Shiota C, Gittes GK. Pancreatic duct cells as a source of VEGF in mice. Diabetologia. 2014c;57:991–1000. doi: 10.1007/s00125-014-3179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao X, Wiersch J, El-Gohary Y, Guo P, Prasadan K, Paredes J, Welsh C, Shiota C, Gittes GK. TGFbeta Receptor Signaling Is Essential for Inflammation-Induced but Not beta-Cell Workload-Induced beta-Cell Proliferation. Diabetes. 2013d;62:1217–1226. doi: 10.2337/db12-1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang YP, Thorel F, Boyer DF, Herrera PL, Wright CV. Context-specific alpha-to-beta-cell reprogramming by forced Pdx1 expression. Genes Dev. 2011;25:1680–1685. doi: 10.1101/gad.16875711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, Grompe M. Generation and regeneration of cells of the liver and pancreas. Science. 2008;322:1490–1494. doi: 10.1126/science.1161431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zaret KS, White MF. Diabetes forum: Extreme makeover of pancreatic alpha-cells. Nature. 2010;464:1132–1133. doi: 10.1038/4641132a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.