

Arabidopsis ALKBH10B is a major m6A mRNA demethylase affecting floral transition and vegetative growth.
Abstract
N6-methyladenosine (m6A) is the most abundant, internal, posttranscriptional modification in mRNA among all higher eukaryotes. In mammals, this modification is reversible and plays broad roles in the regulation of mRNA metabolism and processing. Despite its importance, previous studies on the role and mechanism of m6A methylation in Arabidopsis thaliana have been limited. Here, we report that ALKBH10B is a demethylase that oxidatively reverses m6A methylation in mRNA in vitro and in vivo. Depletion of ALKBH10B in the alkbh10b mutant delays flowering and represses vegetative growth. Complementation with wild-type ALKBH10B, but not a catalytically inactive mutant (ALKBH10B H366A/E368A), rescues these effects in alkbh10b-1 mutant plants, suggesting the observed phenotypes are controlled by the catalytic action of ALKBH10B. We show that ALKBH10B-mediated mRNA demethylation affects the stability of target transcripts, thereby influencing floral transition. We identified 1190 m6A hypermethylated transcripts in the alkbh10b-1 mutant involved in plant development. The discovery and characterization of the archetypical RNA demethylase in Arabidopsis sheds light on the occurrence and functional role(s) of reversible mRNA methylation in plants and defines the role of m6A RNA modification in Arabidopsis floral transition.
INTRODUCTION
Plants undergo several developmental transitions during their life cycle, including the transition from an embryonic to postembryonic state, the juvenile-to-adult vegetative switch, and finally the transition from vegetative growth to reproductive development (Bäurle and Dean, 2006). The precise onset of flowering is critical to ensure plant reproductive success. This process is triggered by a combination of responses to both endogenous and exogenous signals that come together and initiate the switch from leaf to flower production in the shoot apical meristem. Histone methylation modification has been found to play epigenetic-based regulatory roles in floral transition (Srikanth and Schmid, 2011; Sung and Amasino, 2004); however, it is unknown whether RNA modifications affect flowering time.
The most prevalent internal mRNA modification, N6-methyladenosine (m6A), is dynamic and has been shown to be reversible in mammals (Jia et al., 2011; Zheng et al., 2013), serving as a type of biological instruction manual that can be “written,” “read,” and “erased” through the action of a complex network of proteins. In mammalian systems, m6A methylation is performed by a multiprotein “writer” complex comprising three key subunits: methyltransferase like 3 (METTL3), METTL14, and Wilms’s tumor 1-associating protein (WTAP) (Liu et al., 2014; Ping et al., 2014). As a mark, m6A is recognized by reader proteins (such as YTH domain family proteins) to regulate RNA metabolism, including mRNA stability, splicing, nuclear export, translation, cap-independent translation, and pri-miRNA processing (Alarcón et al., 2015; Fustin et al., 2013; Meyer et al., 2015; Wang et al., 2014, 2015; Xiao et al., 2016). Furthermore, these mRNA modifications can be “erased” as easily as they were written, by demethylases, the fat mass and obesity-associated protein (FTO) (Fu et al., 2013; Jia et al., 2011), and the alkylated DNA repair protein AlkB homolog 5 (ALKBH5) (Zheng et al., 2013). m6A also functions as a RNA structural switch affecting protein-RNA interactions (Liu et al., 2015). Recent findings have shown m6A is involved in cell circadian rhythms, stem cell pluripotency, and cancer stem cell proliferation (Batista et al., 2014; Fustin et al., 2013; Geula et al., 2015; Zhang et al., 2016), highlighting the functional importance of m6A modification.
While the knowledge on m6A methylation in plants is limited, transcriptome-wide sequencing in two accessions of Arabidopsis thaliana (Can-0 and Hen-16) revealed that m6A is a highly conserved and dynamic modification of mRNA in plants (Luo et al., 2014). Several m6A methyltransferase subunits have been characterized in Arabidopsis: MTA (the plant ortholog of human METTL3), MTB (an ortholog of human METTL14), FIP37 (an ortholog of human WTAP), VIRILIZER (an ortholog of human KIAA1429), and the E3 ubiquitin ligase HAKAI (Zhong et al., 2008; Bodi et al., 2012; Shen et al., 2016; Růžička et al., 2017). Disruption of MTA leads to embryo-lethal and postembryonic development that is arrested between the globular and the heart stage (Bodi et al., 2012; Zhong et al., 2008). FIP37 was first identified as an interacting partner of MTA in Arabidopsis, and fip37 knockout mutants also exhibit an embryo-lethal phenotype due to delays in both endosperm and embryo development (Vespa et al., 2004; Zhong et al., 2008). Recent findings showed that loss of function of FIP37 causes shoot meristem overproliferation and that FIP37 mediates m6A-based methylation of key shoot meristem gene transcripts, suggesting that m6A mRNA modification controls shoot stem cell fate in Arabidopsis (Shen et al., 2016). These results clearly showed that m6A mRNA modification performed by “writers” is essential for plant development, whereas it is unknown whether m6A substituents can be subsequently “erased” in plants, akin to this process in mammals.
Here, we report that ALKBH10B (At4g02940) is an Arabidopsis m6A RNA demethylase able to oxidatively reverse m6A-based mRNA modifications in vitro and in vivo. Depletion of ALKBH10B increases total m6A levels in poly(A)+ RNA and delays floral transition, while overexpression of ALKBH10B decreases m6A levels in poly(A)+ RNA and the resulting plants exhibit an early flowering phenotype. Complementation with wild-type ALKBH10B, but not an inactive mutant ALKBH10B H366A/E368A, rescues these effects in the alkbh10b-1 mutant background, indicating that m6A RNA modification regulates flowering time in Arabidopsis. We found that ALKBH10B-mediated mRNA m6A demethylation affects the mRNA stability of key flowering time regulators, thereby affecting floral transition. Furthermore, we performed a comparative analysis of transcriptome-wide m6A methylomes between Col-0 and alkbh10b-1, and identified 1190 m6A hypermethylated transcripts in alkbh10b-1 involved in various developmental processes. Taken together, our work demonstrates the functional role of reversible mRNA methylation in plants and reveals the critical role of these modifications in the control of floral transition in Arabidopsis.
RESULTS
ALKBH10B Demethylates m6A-Modified RNA in Vitro
To investigate the existence and dynamics of a potential m6A demethylation pathway in plants, we sought to identify demethylase candidates using AlkB orthologs in Arabidopsis as a starting point (Mielecki et al., 2012). BLAST analysis predicted that there are five potential orthologs of human ALKBH5 encoded in the Arabidopsis genome: ALKBH9A, ALKBH9B, ALKBH9C, ALKBH10A, and ALKBH10B (Supplemental Figure 1). We measured gene transcript levels in different Arabidopsis organs and found that ALKBH10B transcripts were abundant in most organs and represented the most highly expressed candidate in flowers (Figure 1). ALKBH9B and ALKBH9C were the next most abundant transcripts. During the revision of this manuscript, Martínez-Pérez et al. (2017) reported that ALKBH9B can remove m6A from single-stranded RNA (ssRNA) in vitro and that its demethylation activity regulates infection of alfalfa mosaic virus through the interaction of ALKBH9B with the coat protein of alfalfa mosaic virus. However, they did not characterize the endogenous substrates of the enzyme in vivo. Here, quantitative analysis of the m6A/A ratio in wild-type (Col-0), alkbh9b, and alkbh9c plants showed that the mutants displayed no significant changes in mRNA m6A methylation in the organs where they were highly expressed (Supplemental Figure 2). Therefore, we focused our subsequent studies on ALKBH10B.
Figure 1.

ALKBH10B Transcripts Are Abundant in Most Arabidopsis Organs.
Relative gene expression levels were determined using RT-qPCR with TUBULIN4 as a reference gene, prior to normalization to ALKBH10B expression levels in seedlings. The amplification efficiencies of the primers we used are approximately equal. Data are represented as means ± se. n = 3×2 (n = biological replicates × technical replicates).
To determine if ALKBH10B acts as an mRNA demethylase (Figure 2A), we recombinantly expressed it in Nicotiana benthamiana and performed demethylation assays with the purified proteins (Figure 2A; Supplemental Figure 3). An in vitro demethylation activity assay using a synthetic 42-mer m6A-modified RNA as a substrate was performed with an equal amount of the recombinant full-length ALKBH10B protein at pH 6.8 at 20°C for 1 h. Liquid chromatography-tandem mass spectrometry (LC-MS/MS) analyses of the nucleosides digested from the reaction products showed that more than 50% of the m6A present in either ssRNA (i.e., nonstructured 42-mer RNA) or the full-length mRNA isolated from Arabidopsis seedlings was effectively removed by recombinant ALKBH10B in vitro (Figures 2B and 2C). Further, the wild-type enzyme showed an ∼20% reduction in demethylation efficiency on the structured 42-mer RNA with a stem loop, in relation to either the nonstructured 42-mer RNA or the full-length mRNA (Figure 2C).
Figure 2.

ALKBH10B Demethylates m6A in Vitro.
(A) Proposed reaction mechanism of oxidative demethylation of m6A to adenosine byALKBH10B.
(B) LC-MS/MS chromatograms of digested substrates revealing that an equal amount of recombinant ALKBH10B protein demethylates 50% of m6A in 42-mer ssRNA in 1 h.
(C) Demethylation activity of an equal amount of recombinant ALKBH10B or the inactive ALKBH10B H366A/E368A using m6A-containing RNA as a substrate. Data are presented as means ± se. n = 3×2 (n = biological replicates × technical replicates).
(D) Comparative analysis of Arabidopsis ALKBH10B- and human ALKBH5-mediated demethylation (%) versus time (min) using structured and nonstructured 42-mer m6A-modified RNA. Data are presented as means ± sd. n = 3×2 (n = biological replicates × technical replicates).
(E) Representative LC-MS/MS chromatograms of m1A and m6A nucleoside standards. The LC-MS/MS chromatograms of m1A and m6A are indicated in red, while the A/U/C/G nucleosides are represented in black.
(F) ALKBH10B-mediated demethylation (%) of m6A- or m1A-modified 42-mer ssRNA versus time. Data are presented as means ± sd. n = 3×2 (n = biological replicates × technical replicates). All reactions were performed at pH 6.8 and 20°C for 1 h.
To confirm the demethylation activity of ALKBH10B, we constructed, expressed, and evaluated the activity of ALKBH10B H366A/E368A, which contains mutations in two conserved FeII binding residues (Supplemental Figure 3). These mutations completely abolished demethylation activity (Figure 2C), which suggests that ALKBH10B mediates oxidative m6A demethylation. We further compared the demethylation activity of ALKBH10B toward m6A modifications in ssRNA (nonstructured 42-mer RNA) and stem-loop RNA (structured 42-mer RNA) with recombinant human ALKBH5 under the same conditions. The activity of Arabidopsis ALKBH10B toward m6A modification present in ssRNA and stem-loop RNA reached saturation at ∼50% and ∼22% of demethylation efficiency in 1 h, while human ALKBH5 demethylated ∼70% and ∼17% of the m6A moieties in 1 h, respectively (Figure 2D). These results showed that recombinant ALKBH10B exhibits slightly lower demethylation activity toward m6A in ssRNA than ALKBH5, whereas they not display significant differences in activity when using m6A modified stem-loop RNA as a substrate. Taken together, these data confirm the in vitro m6A demethylation activity of ALKBH10B.
m6A-Modified mRNA Is the Major Substrate of ALKBH10B
To identify the in vivo physiological substrate of ALKBH10B, we first characterized two independent, homozygous T-DNA insertion mutants in ALKBH10B, designated alkbh10b-1 and alkbh10b-2 (Supplemental Figures 4A and 4B) (Alonso et al., 2003). The mutant line alkbh10b-1 contains a T-DNA insertion in the exon directly before the predicted iron binding sites, while a T-DNA insertion in alkbh10b-2 lies in the intron before the predicted α-ketoglutarate binding sites (Supplemental Figure 4A). RT-PCR results showed that alkbh10b-1 is unable to produce full-length ALKBH10B transcript. However, ∼5% of wild-type levels of full-length ALKBH10B transcript was detected in alkbh10b-2 (Supplemental Figures 4C and 4F), indicating alkbh10b-2 is a weak mutant allele. Thus alkbh10b-1 was predominantly used for subsequent studies.
Different tissues were harvested from the vegetative and reproductive organs of wild-type (Col-0) and alkbh10b-1, respectively. We found that disruption of ALKBH10B led to elevated m6A levels in total mRNA isolated from these tissues (Figure 3A). Next, we set out to confirm the in vivo m6A demethylation activity of ALKBH10B. Wild-type ALKBH10B and the catalytically inactive mutant ALKBH10B H366A/E368A were expressed in alkbh10b-1 mutant plants using the native promoter from ALKBH10B (Supplemental Figure 4). Expression of the wild-type protein in the alkbh10b-1 mutant background led to reduced m6A levels that were similar to those observed in wild-type plants, while complementation of the inactive mutant protein did not significantly reduce mRNA m6A levels (Figure 3A). We also overexpressed ALKBH10B in wild-type (Col-0) plants (Supplemental Figure 4) and observed that total m6A levels were decreased in 35S:ALKBH10B plants when compared with wild-type controls (Figure 3A). Additionally, we also evaluated mRNA m6A levels in the weak mutant allele alkbh10b-2 (Supplemental Figure 5). The mRNA m6A levels were significantly increased in seedlings, cauline leaves, and flower buds isolated from alkbh10b-2; however, more significant m6A/A ratio changes were observed in alkbh10b-1. These results establish the in vivo demethylation activity of ALKBH10B toward m6A in mRNA.
Figure 3.

m6A-Modified mRNA Is the Major Physiological Substrate of ALKBH10B.
(A) Quantification of the m6A/A ratio of mRNA in the indicated plant lines by LC-MS/MS.
(B) to (E) Quantification of the RNA modification ratios (m6A/A, m1A/A, m5C/C. and m1G/G) in tRNA (B), 28S rRNA (C), 18S rRNA (D), and mRNA (E) in wild-type and alkbh10b-1 mutant seedlings.
Data are presented as means ± se. n = 3×3 (n = biological replicates × technical replicates). *P < 0.05 and **P < 0.01.
We further monitored gene expression levels of known components of the methyltransferase complex, including MTA (Zhong et al., 2008), MTB (Růžička et al., 2017), and FIP37 (Shen et al., 2016). We also measured transcript abundance corresponding to other putative m6A demethylases, including ALKBH9B, ALKBH9C, and ALKBH10A in Col-0, and alkbh10b-1, as well as the complementation and overexpression lines. Interestingly, no significant differences in gene expression of any of the aforementioned targets were detected by RT-qPCR analysis. However, minimal decreases in MTA and MTB transcript levels were observed in leaves of alkbh10b-1 plants (Supplemental Figure 6). The data confirmed that the observed changes in the m6A/A ratios in mRNA isolated from these plants were directly mediated by the demethylation activity of ALKBH10B and were not due to the catalytic activity of m6A methyltransferases or other putative demethylases.
Given that m6A modifications are also present in both tRNA and rRNA, we examined m6A methylation levels of tRNA and rRNA in the wild type and alkbh10b-1. Compared with wild-type controls, disruption of ALKBH10B did not significantly change m6A levels in tRNA or 28S and 18S rRNA (Figures 3B to 3D), suggesting ALKBH10B does not globally remove m6A from tRNA and rRNA.
Recently, the mammalian m6A demethylase FTO has been shown to remove N6-methyl groups from another subform of m6A, N6,2′-O-dimethyladenosine (m6Am), which is located at the first nucleotide following the m7G cap (Mauer et al., 2017). Interestingly, m6Am was not present near 5′ caps of Arabidopsis poly(A)+ RNA (Supplemental Figure 7), indicating m6Am near 5′ caps is not a physiological substrate of ALKBH10B in Arabidopsis. N1-methyladenosine (m1A) has been identified as another reversible and dynamic modification in eukaryotic mRNA and tRNA that can be demethylated by human ALKBH3 and ALKBH1, respectively (Dominissini et al., 2016; Li et al., 2016; Liu et al., 2016). Some human AlkB family proteins, such as ALKBH2 and ALKBH3, are able to repair multiple N-methylation modifications (Zheng et al., 2014); therefore, we investigated whether ALKBH10B is active on other RNA methylation modifications in addition to m6A in mRNA. First, we tested the in vitro demethylation activity of recombinant ALKBH10B on m1A modification present on ssRNA. We found that ALKBH10B did not modify m1A in vitro (Figures 2E and 2F). Next, we isolated mRNA, rRNA, and tRNA from seedlings of wild-type and alkbh10b-1 plants and examined the m1A, m5C (5-methylcytosine), and m1G (N1-methylguanosine) methylation levels in mRNA, rRNA, and tRNA. Plants lacking functional ALKBH10B had barely altered m1A, m5C, and m1G levels in mRNA, rRNA, and tRNA (Figures 3B to 3E). These data indicated that ALKBH10B is unable to widely demethylate m1A, m5C, and m1G in mRNA, rRNA, and tRNA. Collectively, these results demonstrated that m6A-modified mRNA is the major physiological substrate of ALKBH10B.
The Demethylation Activity of ALKBH10B Affects Floral Transition and Vegetative Growth
To determine the physiological role of ALKBH10B in planta, we performed phenotypic analysis alkbh10b-1 mutants grown under long-day conditions. A noticeable feature of the mutant plants was late flowering (Figure 4A). While wild-type plants flowered after the emergence of 12 true leaves, alkbh10b mutant plants produced approximately seven additional leaves before the first bud bloomed (Figures 4B and 4C). By contrast, when ALKBH10B was overexpressed in the wild-type background (35S:ALKBH10B), the resulting plants exhibited an early-flowering phenotype producing approximately seven leaves prior to floral induction (Figures 4B and 4C). The phenotypic complementation of the mutant was confirmed by expression of wild-type ALKBH10B in the alkbh10b-1 mutant (Figures 4B and 4C). As expected, similar complementation experiments performed using a construct composed of the catalytically inactive mutant ALKBH10B H366A/E368A showed no changes (Figures 4B and 4C). These results demonstrated that ALKBH10B-catalyzed RNA demethylation is directly related to the observed flowering phenotype.
Figure 4.

Floral Transition Is Disrupted in alkbh10b Mutants.
(A) Late flowering phenotype of alkbh10b-1.
(B) Leaf number (rosette and main stem) of wild type (Col-0), alkbh10b-1, ALKBH10B:ALKBH10B/alkbh10b-1, ALKBH10B:ALKBH10Bm/alkbh10b-1, and 35S:ALKBH10B at flowering time.
(C) Successive leaves per plant at flowering time for the indicated lines.
Data are presented as means ± se. n = 15. *P < 0.05, **P < 0.01, and ***P < 0.001.
ALKBH10B deficiency did not affect leaf morphogenesis (Figure 4C). In the overexpression line (35S:ALKBH10B), cauline leaves had petioles that were similar to those of rosette leaves, which may be correlated with changes in m6A levels in cauline leaves of 35S:ALKBH10B plants (Figure 3A). The mutant plants exhibited other growth defects, including repressed vegetative growth, whereas the overexpression lines did not display noticeable vegetative growth phenotypes (Supplemental Figures 8A and 8B). Ten-day-old alkbh10b-1 seedlings grown on agar plates with 0.5× Murashige and Skoog (MS) medium contained the same number of rosette leaves, but leaves were significantly smaller compared with the wild type (Supplemental Figures 8A, 8C, and 8D). However, leaves of the mutants reached the same final size as those of the wild type before bolting (Figures 4A and 4C), suggesting that ALKBH10B mainly affects growth rate, but not the final size, of Arabidopsis leaves. Complementation of alkbh10b-1 with wild-type ALKBH10B, but not the inactive mutant ALKBH10B H366A/E368A, alleviated the suppressed vegetative growth phenotype and increased leaf growth rate (Supplemental Figures 8A, 8C, and 8D). Furthermore, the alkbh10b-2 mutant exhibited weak late flowering and similar vegetative growth suppression compared with alkbh10b-1 and wild-type plants (Supplemental Figure 9). These results indicated that loss of function of ALKBH10B delays floral transition and represses vegetative growth and also revealed that the observed phenotypes are controlled by the catalytic function of ALKBH10B.
ALKBH10B-Mediated m6A Demethylation Enhances Expression Levels of Key Flowering-Related Genes
To elucidate the mechanisms underlying the late-flowering phenotype of alkbh10b mutant plants, we measured the expression of FLOWERING LOCUS T (FT), which integrates the leaf photoperiod, vernalization, autonomous, gibberellin, and aging pathways (Amasino, 2010; Srikanth and Schmid, 2011), in 14-d-old seedlings. The level of FT mRNA was significantly reduced in alkbh10b-1 plants (Figure 5A), consistent with the late-flowering phenotype observed in the mutants. Compared with wild-type plants, complementation of alkbh10b-1 with wild-type ALKBH10B, but not the inactive mutant ALKBH10B H366A/E368A, recovered FT expression (Figure 5A), indicating that enzymatic activity of ALKBH10B modulates FT mRNA abundance altering floral transition.
Figure 5.

Upregulation of FT, SPL3, and SPL9 Is Correlated with ALKBH10B-Mediated m6A Demethylation.
(A) Diurnal time course of FT expression in seedlings.
(B) m6A levels of FT, SPL3, and SPL9 in wild-type and alkbh10b-1 plants.
(C) Relative mRNA levels of SPL3 and SPL9 in indicated seedlings.
(D) Immunoblot analysis using anti-myc antibody shows the expression of 6×myc-ALKBH10B in ALKBH10B:6×myc-ALKBH10B/alkbh10b-1 plants (input) and in the immunoprecipitated fraction (Anti-myc-IP), but not in wild-type Col-0 and IgG-IP fraction.
(E) RIP-qPCR assays in ALKBH10B:6×myc-ALKBH10B/alkbh10b-1 plants show that 6×myc-ALKBH10B directly binds FT, SPL3, and SPL9 transcripts in vivo.
(F) m6A levels of m6A peaks in FT, SPL3, and SPL9 in the wild type and alkbh10b-1 showing m6A positions demethylated by ALKBH10B.
Data are presented as means ± se. n = 3×2 (n = biological replicates × technical replicates). *P < 0.05 and **P < 0.01 for paired samples; Col-0 versus alkbh10b-1 in dark color and ALKBH10B:ALKBH10B/alkbh10b-1 versus ALKBH10B:ALKBH10Bm/alkbh10b-1 in orange.
We speculated that FT mRNA is modified by m6A methylation, which could be catalyzed by ALKBH10B. To investigate this possibility, we examined m6A levels (defined here as the proportion of m6A methylated transcripts per gene) in FT mRNA using anti-m6A antibody immunoprecipitation (m6A-IP) of intact mRNA followed by qPCR (Dominissini et al., 2012; Molinie et al., 2016). This technique has been validated as a specific, sensitive, and quantitative method for the measurement of m6A levels on individual gene transcripts (Molinie et al., 2016). Approximately 50% of FT mRNA was methylated in alkbh10b-1 mutant plants, compared with <30% in the wild type (Figure 5B), suggesting that disruption of ALKBH10B increases m6A levels in FT mRNA. Given that FT transcription is regulated by multiple genes (Amasino, 2010; Srikanth and Schmid, 2011), we speculated that transcripts of these other genes may also be altered via ALKBH10B-mediated demethylation. Therefore, we screened for changes in mRNA expression and potential m6A methylation for reported FT regulators in wild-type and alkbh10b-1 mutant plants. Among these genes, we found that the transcript levels of SPL3 and SPL9 were significantly decreased and that this change was accompanied by increased m6A levels in the corresponding mRNAs of the alkbh10b-1 mutant at Zeitgeber time (ZT) 13 compared with the wild type (Figures 5B and 5C). Remarkably, the transcript levels of SPL3 and SPL9 were mediated by the demethylation activity of ALKBH10B (Figure 5C). Other genes showed modest differences in transcript levels or m6A methylation levels between these two lines (Supplemental Figures 10 to 12).
To evaluate whether FT, SPL3, and SPL9 are direct targets of ALKBH10B, we performed RNA immunoprecipitation (RIP)-qPCR assays on ALKBH10B:6×myc-ALKBH10B/alkbh10b-1 seedlings. Remarkably, the results revealed a direct interaction of 6×myc-ALKBH10B with FT, SPL3, and SPL9 transcripts in vivo (Figures 5D and 5E). We further determined potential ALKBH10B targeted sites at m6A positions (m6A peak) in FT, SPL3, and SPL9 using m6A-IP-qPCR with fragmented poly(A)+ RNA (Shen et al., 2016; Zhang et al., 2016) in wild-type and alkbh10b-1 plants. We found m6A peaks at both the start and stop codons in FT and in the 3′ UTRs of SPL3 and SPL9, which exhibited significantly higher methylation in alkbh10b-1 compared with the wild type (Figure 5F). Collectively, these data not only demonstrate that ALKBH10B directly binds to and demethylates m6A modifications on FT, SPL3, and SPL9 transcripts, but also suggest that Arabidopsis uses a demethylation-based strategy for posttranscriptional control of transcript levels.
ALKBH10B-Mediated m6A Demethylation Stabilizes FT, SPL3, and SPL9 mRNAs
It has previously been shown that m6A can promote mRNA degradation though binding the m6A reader, YTHDF2, in mammals (Wang et al., 2014). To evaluate this process in Arabidopsis, we applied two independent methods to measure the lifetime of FT mRNA and monitor whether increased methylation stimulated accelerated degradation of FT mRNA in Arabidopsis similar to mammalian systems: (1) blocking transcription with actinomycin D to monitor FT mRNA levels before ZT16 when FT transcript levels are highest during the day and (2) monitoring the degradation rate of FT mRNA during the night because FT transcription is terminated spontaneously after ZT16 confirmed through detection of FT pre-mRNA expression levels (Figures 6A and 6B). Both methods showed that FT mRNA was more rapidly degraded in alkbh10b-1 than in wild-type plants (Figures 6C and 6D), indicating that increased methylation also promotes accelerated mRNA degradation in Arabidopsis, similar to what has been observed in human cells (Wang et al., 2014). Complementation of the alkbh10b-1 mutant with active ALKBH10B enhanced FT mRNA stability (Figure 6C).
Figure 6.

m6A Demethylation by ALKBH10B Increases mRNA Stability of FT, SPL3, and SPL9.
(A) and (B) Measurement of FT mRNA precursors.
(A) RT-PCR analysis confirming the termination time point of FT transcription.
(B) Diurnal time course of FT pre-mRNA.
(C) Degradation of FT transcripts at night. FT mRNA levels in wild-type, alkbh10b-1, ALKBH10B:ALKBH10B/alkbh10b-1, and ALKBH10B:ALKBH10Bm/alkbh10b-1 plants were normalized to ZT16, respectively.
(D) FT, SPL3, and SPL9 mRNA lifetimes in the wild type and alkbh10b-1. The transcription inhibition assays were repeated three times.
(E) RNA gel blot analysis showing the abundance of mature miRNAs (miR156 and miR172) in wild-type and alkbh10b-1 plants. One representative result is shown. 5S rRNA was used as the loading control.
(F) Proposed model of floral transition mediated by ALKBH10B-catalyzed demethylation.
Data are presented as means ± sd. n = 3×3 (n = biological replicates × technical replicates).
Expression of the precursor miRNA-156 (pre-miR156) and the mature miRNA-156 (miR156), the main regulator of SPL genes, caused negligible changes in alkbh10b-1 (Figure 6E; Supplemental Figure 13), indicating that reduced expression levels of SPL3 and SPL9 were not caused by miR156. Transcription inhibition assays showed that SPL3 and SPL9 transcripts were degraded more rapidly in demethylase-defective plants compared with the wild type (Figure 6D). Taken together, these data demonstrate that ALKBH10B-mediated m6A demethylation of FT, SPL3, and SPL9 mRNAs both prolongs the lifetimes of the transcripts and increases transcript levels. Although m6A modifications destabilize mRNA in mammals (Wang et al., 2014; Zhang et al., 2016) and Arabidopsis (Shen et al., 2016), it should be noted that mutagenesis experiments of the m6A site will be needed to elucidate the causal mechanisms of this process and to definitively show that m6A provides mRNA stability in these mRNAs.
ALKBH10B-Mediated m6A Demethylation of FT, SPL3, and SPL9 mRNAs Affects Floral Transition
Disruption of ALKBH10B increased m6A modification levels of FT, SPL3, and SPL9 mRNAs accelerating their degradation (Figure 6D). Taking into consideration that FT is a floral activator, ALKBH10B-mediated FT mRNA decay likely contributes to late flowering. In leaves, SPL9 acts as an activator of miR172 regulating flowering time (Amasino, 2010; Wu et al., 2009), while SPL3 directly binds to the promoter of FT activating its transcription (Kim et al., 2012). Indeed, the levels of both pre-miR172b and the mature miR172 were decreased in alkbh10b-1 plants compared with the wild type (Figure 6E; Supplemental Figure 13). Therefore, in addition to the accelerated mRNA decay due to increased methylation of FT mRNA, the reduced transcript levels of SPL3 and SPL9 caused by the m6A-dependent mRNA decay pathway repress FT and miR172 transcription, resulting in delayed floral transition in alkbh10b-1 mutants (Figure 6F). Furthermore, ectopic expression of FT driven by the FD promoter in the alkbh10b-1 mutant rescued the late-flowering phenotype (Supplemental Figure 14). Our results provide a framework for the molecular basis of m6A RNA modification in Arabidopsis flowering regulation. Other genes directly or indirectly modulated by ALKBH10B also are likely to play a role in the developmental changes observed in alkbh10 mutants.
Disruption of ALKBH10B Leads to Global m6A Hypermethylation
To test the global effects of ALKBH10B-mediated m6A demethylation, we performed m6A-seq and RNA-seq using poly(A)+ RNA from the wild type and alkbh10b-1 (Supplemental Figure 15). We determined the methylation positions (m6A peak) using the published m6A peak-calling algorithm (Dominissini et al., 2012) with an estimated false discovery rate (FDR) <0.01 and an enrichment of ≥2-fold and discarded unconfident peaks with gene expression levels FPKM (fragments per kilobase of transcript per million fragments mapped) <4. We detected 22,406 to 24,314 m6A peaks in each individual biological replicate and classified the peaks in both replicates as “confident peaks” for subsequent analysis (Figure 7A; Supplemental Data Sets 1 to 3). We identified 12,922 putative high-confidence m6A peaks representing the transcripts of 9210 genes in the wild type, and 12,975 m6A peaks representing the transcripts of 8463 genes in alkbh10b-1 (Supplemental Data Sets 2 and 3). Clustering analysis between biological replicates confirmed that our m6A-seq data was robust (Supplemental Figure 15). We used the m6A site differential algorithm (Meng et al., 2013) (FDR < 0.01 and fold change ≥2) to determine hypermethylated m6A peaks in alkbh10b-1. Compared with the wild type, we identified 1276 hypermethylated m6A peaks covering 1190 genes in alkbh10b-1 in both biological replicates (Figures 7B and 7C; Supplemental Data Set 4), which could be directly or indirectly affected by ALKBH10B catalytic activity.
Figure 7.

Global m6A Methylation Is Altered in alkbh10b-1 Plants.
(A) Overlapping m6A peaks identified in the wild type and alkbh10b-1 (R1-R2).
(B) Heat map of IP enrichment values of hypermethylated m6A peaks identified in alkbh10b-1 and wild-type seedlings.
(C) Volcano plot of hypermethylated m6A peaks identified in alkbh10b-1 and the wild type across two biological replicates.
(D) and (E) Metegenomic profiles of read distributions (D) and m6A peak distributions (E) along transcript segments in peak groups.
(F) Fractions and relative enrichment of m6A peaks in each nonoverlapping transcript segment within peak groups.
(G) RRACH consensus motifs in m6A-containing peak regions.
(H) RRACH motif-containing peaks in different groups. *P < 1e-10 and **P < 1e-20. P values were determined by χ2 test.
We next evaluated m6A distribution in wild-type versus alkbh10b-1 plants and investigated hypermethylated m6A peaks in alkbh10b-1. The metagene profiles of m6A peaks or m6A reads in wild-type and alkbh10b-1 seedlings showed that m6A modifications were highly enriched around both the start and stop codons and within the 3′ UTR (Figures 7D and 7E), consistent with the known distribution of Arabidopsis m6A modifications (Luo et al., 2014). We divided these transcripts into five nonoverlapping segments: 5′ UTRs, start codons (200-nucleotide window centered on the start codon), coding sequences (CDSs), stop codons (200-nucleotide window centered on the stop codon), and 3′ UTRs. The observed distribution of m6A peaks indicated that they were most abundant near stop codons (62.61% in the wild type, 56.98% in alkbh10b-1), followed by CDS (17.51% in the wild type, 23.02% in alkbh10b-1), start codons (15.44% in the wild type, 15.95% in alkbh10b-1), and least abundant in 3′ UTRs (3.16% in the wild type, 2.96% in alkbh10b-1) (Figure 7F, upper panel). After segment normalization by the total length of each gene portion, we observed that m6A modification in wild-type and alkbh10b-1 seedlings was significantly enriched around the start and stop codons (Figure 7F, lower panel). The hypermethylated m6A peaks in alkbh10b-1 were highly enriched around the start and stop codons and within the 3′ UTR, with a distinct preference for the CDS compared with the wild type (Figures 7E and 7F). To determine sequence preference of hypermethylated m6A peaks, we analyzed the RRACH m6A consensus sequences (where R represents purine, A is m6A and H is a non-guanine base) in three groups, comprising total m6A peaks in the wild type and alkbh10b-1, and hypermethylated peaks in alkbh10b-1. Over 85% of identified peaks from each group contained at least one RRACH motif and shared the same motif pattern (Figures 7G and 7H).
Subsequently, we performed a SCARLET assay (Liu et al., 2013) and m6A-IP-qPCR of fragmented poly(A)+ RNA (Shen et al., 2016; Zhang et al., 2016) on randomly selected hypermethylated m6A peaks, representing potential ALKBH10B targets, to verify that m6A modification was present at specific m6A sites and to determine if m6A peaks were increased in alkbh10b-1 (Supplemental Figure 16). These results demonstrated that our m6A-seq data were accurate and robust and firmly confirmed that ALKBH10B-mediated m6A modifications were modified from m6A to A.
We identified 165 lower-expressed genes and 181 higher-expressed genes in the alkbh10b-1 mutant compared with the wild type (cutoff criteria with FPKM fold change ≥1.5 and P value <0.05) (Supplemental Data Sets 5 to 7), which was 0.80% and 0.88% of all expressed genes, respectively. In 1190 genes shown to contain hypermethylated peaks, six genes (0.5%) were expressed at lower levels and 26 genes (2.2%) were expressed at higher levels (Supplemental Figure 17A). Genes bearing hypermethylated m6A peaks at stop codons were predominantly upregulated in alkbh10b-1 (Supplemental Figure 17B). These data indicate that m6A affects RNA fate through multiple, complex regulatory pathways in plants that extend beyond m6A-dependent mRNA degradation (Supplemental Figure 18).
To gain functional insights into the observed changes in m6A modifications in alkbh10b-1 mutant plants, we performed Gene Ontology (GO) analysis using the DAVID tool on genes containing hypermethylated m6A peaks. GO analysis revealed that genes covering hypermethylated m6A peaks were positively enriched in several developmental pathways, including regulation of organelle organization, vegetative-to-reproductive phase transition of meristems, organ and trichome morphogenesis, embryo development ending in seed dormancy, and production of ta-siRNAs or miRNAs involved in gene silencing (Supplemental Figure 17C). These gene functions showed clear correspondence with the observed alkbh10b mutant phenotypes and may be modulated directly or indirectly by ALKBH10B action.
DISCUSSION
m6A modification is the most ubiquitous mRNA modification in eukaryotes. Understanding the functional roles of reversible m6A methylation in mammals has been paramount since the discovery of the first human mRNA m6A demethylase FTO (Jia et al., 2011) and the subsequent mapping of transcriptome distributions of m6A modifications in mammalian cells and tissues (Dominissini et al., 2012; Meyer et al., 2012). Several studies regarding Arabidopsis mRNA m6A methyltransferases revealed that m6A modification is critical for normal plant development and regulates shoot stem cell fate in Arabidopsis (Bodi et al., 2012; Shen et al., 2016; Zhong et al., 2008). To deeply explore and understand m6A functions in plants, a key priority is to identify m6A demethylases and uncover the reversibility and dynamics of this process in plants. Here, we have established that m6A mRNA methylation is also reversible in Arabidopsis through analysis of the m6A demethylation activity catalyzed by ALKBH10B in vitro and in vivo. Furthermore, quantification of m6A, m1A, m5C, and m1G modified mRNA, tRNA, and rRNA in wild-type and alkbh10b-1 plants revealed that m6A-modified mRNA is the main physiological substrate of ALKBH10B (Figure 3).
Our results demonstrate that m6A-mediated RNA modification is involved in the complex genetic regulation that controls flowering time. Phenotypic studies showed that loss of function of ALKBH10B led to late flowering and that flowering time is dependent on the catalytic function of ALKBH10B (Figure 4). Mechanistic studies confirmed the occurrence of m6A demethylation-dependent gene regulation of transcripts that control flowering time. Disruption of ALKBH10B increased m6A modification, thereby accelerating mRNA decay of the flowering activator FT and its upregulators SPL3 and SPL9 (Figures 5 and 6). Our results suggest that the reduced levels of SPL3 and SPL9 transcripts caused by the demethylation-dependent mRNA decay pathway repress FT and miR172 transcription, delaying floral transition in alkbh10b mutants. Analogous to histone methylation and miRNAs that modulate floral transition, we discovered a layer of RNA m6A modification-mediated gene regulation involved in floral transition in Arabidopsis. More questions remain to be answered, including whether the relationship between m6A modification and flowering time is autonomous or triggered by endogenous and exogenous signals and how these m6A-related proteins (demethylases and readers) sense the signals.
ALKBH10B deficiency globally enhances m6A hypermethylation, and these important modifications are known to be involved in plant development, especially vegetative-to-reproductive phase transition of the meristem. The functions of these hypermethylated genes are correlated with the observed alkbh10b-1 mutant phenotypes and may be directly or indirectly modulated by ALKBH10B action. The finding that reversible m6A methylation controls plant development, combined with prevalence of mRNA methylation in plants, strongly indicates that m6A methylation represents an epitranscriptomic mark for the regulation of gene expression at the posttranscriptional level in plants (Supplemental Figure 18). Future research should explore the breadth and detailed mechanisms of m6A-dependent regulation in plants, which may provide new, alternative strategies for plant genetic engineering.
METHODS
Plant Materials and Growth Conditions
Arabidopsis thaliana genotypes used included wild-type (Col-0) and the following T-DNA insertion mutant lines: alkbh9b-1 (Salk_055591c), alkbh9b-2 (Salk_111811c), alkbh9c-1 (Salk_021775c), alkbh10b-1 (Salk_004215c), and alkbh10b-2 (Salk_107289c). Mutant lines were obtained from the ABRC, and homozygotes were confirmed (Supplemental Figures 2 and 4 and Supplemental Table 1). Complementation (ALKBH10B:ALKBH10B/alkbh10b-1 and ALKBH10B:ALKBH10Bm/alkbh10b-1) and overexpression lines (35S:ALKBH10B) were obtained by transforming plasmids into the alkbh10b-1 and wild-type (Col-0) backgrounds, respectively. The plant transformation section describes generation of ALKBH10B:ALKBH10B / alkbh10b-1, ALKBH10B:ALKBH10Bm / alkbh10b-1, and 35S:ALKBH10B overexpression lines. All plants were grown at 22°C under long-day conditions (16 h light/8 h dark, white fluorescent tubes, 150 to 200 μmol m−2 s−1). Seedlings and shoot apical meristems were collected from 14-d-old plants grown on half-strength MS nutrient agar plates (PhytoTechnology Laboratories). Juvenile and adult rosette leaves and cauline leaves were harvested at flowering time. Flowers and buds were collected randomly from 6-week-old plants and divided into three biological replicates. Juvenile and maturing siliques were collected from 6-week-old plants. All samples described above were collected at ZT8 and used for LC-MS/MS and RT-qPCR analyses (Figures 1 and 2A; Supplemental Figures1, 4, and 5). Fourteen-day-old seedlings were collected every 3 h and 1 h during the day and night cycles, respectively, and subsequently used for RT-qPCR analysis for the diurnal time course (Figures 5A, 6B, and 6C; Supplemental Figure 7). Fourteen-day-old seedlings at ZT15 were used to quantify m6A mRNA methylation levels (Figure 5B; Supplemental Figure 9). All plant materials were frozen in liquid nitrogen immediately after harvest and stored at −80°C.
Plasmid Construction
ALKBH10B full-length cDNA was obtained from the ABRC. pCAMBIA1300 containing a 2×35S promoter and an N-terminal 6×Myc-tag at the multiple cloning site was used for plasmid construction. Full-length ALKBH10B cDNA was cloned between the PstI and HindIII restriction sites of pCAMBIA1300 to generate 35S-6×Myc-AtAlkBH10B. Site-directed mutagenesis of ALKBH10B to generate the ALKBH10B H366A/E368A catalytically inactive mutant was performed using the QuikChange II XL site-directed mutagenesis kit (Agilent), generating the 35S-6×Myc-ALKBH10Bm construct. A 5ʹ-phosphorylated oligonucleotide containing the DNA sequence of the 6×His-tag was ligated the between XbaI and PstI restriction sites of 35S-6×Myc-ALKBH10B and 35S-6×Myc-ALKBH10Bm to generate 35S-6×Myc-6×His-ALKBH10B and 35S-6×Myc-6×His-ALKBH10Bm plasmids, respectively. PALKBH10B-ALKBH10B and PALKBH10B-ALKBH10Bm were obtained by replacing the sequence from the 2×35S promoter to the end of the 6×Myc-tag in 35S-6×Myc-ALKBH10B and 35S-6×Myc-ALKBH10Bm with the native promoter of ALKBH10B (2000 bp upstream of the ALKBH10B start codon). PALKBH10B-6×Myc-ALKBH10B was obtained by replacing the 2×35S promoter of 35S-6×Myc-ALKBH10B with the native promoter of ALKBH10B, described above. The PFD-FT plasmid was obtained by cloning full-length FT cDNA between the PstI and HindIII restriction sites in pCAMBIA1300 containing the FD promoter and terminator (Abe et al., 2005). All primers were ordered from Sangon Biotech and are listed in Supplemental Table 1. Restriction endonucleases and DNA polymerases were obtained from New England Biolabs.
Plant Transformation
Agrobacterium tumefaciens strain GV3101 was used for plant transformation by the floral dip method (Zhang et al., 2006). T0 seeds were screened for Hygromycin B resistance, and T1 plants were confirmed by PCR analysis of genomic DNA. 35S:ALKBH10B transgenic plants were obtained by transforming the 35S-6×Myc-ALKBH10B construct into 6-week-old Arabidopsis (Col-0). Complementation lines, including ALKBH10B:ALKBH10B / alkbh10b-1, ALKBH10B:6×myc-ALKBH10B/alkbh10b-1, ALKBH10B:ALKBH10Bm/alkbh10b-1, and FD:FT/alkbh10b-1 were obtained by transforming the alkbh10b-1 mutant with PALKBH10B-ALKBH10B, PALKBH10B-6×myc-ALKBH10B, PALKBH10B-ALKBH10Bm, and PFD-FT, respectively.
Phenotypic Analysis
Calculation of seedling leaf area was performed on wild-type and mutant plants (Supplemental Figures 8 and 9) grown on 0.5× MS agar plates, as described above. Briefly, seeds were sown with on plates with enough distance apart to avoid any overlap between developing leaves. Photographs of 10-d-old seedlings were taken and processed with ImageJ software (download from http://imagej.nih.gov/ij/) to calculate leaf area. Cotyledons were counted when calculating the seedling leaf area and to determine seedling leaf number. To determine flowering time, total rosette and cauline leaves on the primary inflorescence stem were counted when the first flower opened, and successive leaves were photographed, excluding cotyledons (Figure 4; Supplemental Figures 9 and 14).
In Planta Protein Expression and Purification
Transient expression of proteins in Nicotiana benthamiana was performed as previously described (Sparkes et al., 2006). Agrobacterium GV3101 transformed with the expression plasmids 35S-6×Myc-6×His-ALKBH10B or 35S-6×Myc-6×His-ALKBH10Bm were infiltrated into N. benthamiana leaves. Plants were grown for an additional 5 d and then the infiltrated zones were excised, ground, frozen in liquid nitrogen, and stored at −80°C. Ground N. benthamiana leaves were suspended in precooled lysis buffer (100 mM Tris-HCl, pH 7.5, 250 mM KCl, 10 mM NaF, 0.1 mM Na3VO4, 0.1% β-ME, and 1× Roche cOmplete protease inhibitor cocktail) and homogenized. The mixture was filtered through Miracloth (Calbiochem) and centrifuged at 18,000g for 30 min. The supernatant was loaded onto Ni-NTA column (GE Healthcare) that was pre-equilibrated with buffer A (100 mM Tris-HCl, pH 7.5, 250 mM KCl, and 0.1% β-ME). The column was washed with wash buffer (buffer A including 50 mM imidazole) and eluted with elution buffer (buffer A with 250 mM imidazole). Eluted fractions were further purified with a Superdex 200 10/300 GL gel-filtration column with GF buffer (50 mM HEPES, pH 6.8, 1M KCl, and 3 mM DTT) and buffer exchanged into assay buffer (50 mM HEPES, pH 6.8, 100 mM KCl, and 3 mM DTT). Purified proteins were concentrated to 5 mg mL−1 and immediately used in subsequent activity assays.
In Vitro Demethylation Assays
Demethylation activity assays were performed as previously reported (Jia et al., 2011) with limited modifications. Briefly, reaction mixtures (50 μL) contained the following components: 0.5 μM oligo RNA with m6A (25 pmol 42-mer RSV RNA AUGGGCCGUUCAUCUGCUAAAAGGm6ACUGCUUUUGGGG-CUUGU) or 500 ng full-length mRNA, 0.5 μM ALKBH10B protein, 50 mM HEPES buffer (pH 6.8), 2 mM l-ascorbic acid, 300 μM (NH4)2Fe(SO4)2·6H2O, 500 μM α-ketoglutarate, and 1 units μL−1 Ribolock RNase inhibitor (Thermo Scientific). Reactions were incubated at 20°C for 1 h and stopped by the addition of 2 mM EDTA, incubated for 15 min at 95°C, and analyzed as described below.
Quantitative Analysis of RNA Modification by LC-MS/MS
For analysis of internal RNA modifications, 100 ng poly(A) RNA or oligo RNA was digested with 1 unit of Nuclease P1 (Wako) in 50 μL buffer containing 10 mM ammonium acetate (pH 5.3) at 42°C for 5 h, followed by the addition of 5.5 μL of 1 M fresh NH4HCO3 and 1 unit of alkaline phosphatase (Sigma-Aldrich). The mixture was incubated at 37°C for an additional 5 h.
For quantification of cap m6Am, 100 ng poly(A) RNA was decapped with 20 units of RNA 5′pyrophosphohydrolase (RppH; M0356; New England Biolabs) in 1×Thermopol buffer (B9004S; New England Biolabs) for 3 h at 37°C, and then treated with Nuclease P1 and alkaline phosphatase as described above.
Digested samples were filtered through 0.22-µm syringe filters prior to UPLC-MS/MS analysis. The nucleosides were separated by UPLC (Shimadzu) equipped with a ZORBAX SB-Aq column (Agilent) and detected by MS/MS using a Triple Quad 5500 (AB SCIEX) mass spectrometer in positive ion mode by multiple reaction monitoring. The MS parameters were optimized for m6A detection, and A/U/C/G signals were weakened due to their abundance relative to m6A modified mRNA. Nucleosides were quantified using the nucleoside-to-base ion mass transitions of m/z 268.0 to 136.0 (A), m/z 282.0 to 150.1 (m6A), m/z 296.0 to 150.0 (m6Am), m/z 282.0 to 150.0 (m1A), m/z 244.0 to 112.0 (C), m/z 258.0 to 126.0 (m5C), m/z 284.0 to 152.0 (G), and m/z 298.0 to 166.0 (m1G). Standard curves were generated by running a concentration series of pure commercial nucleosides (Sigma-Aldrich). Concentrations of nucleosides in samples were calculated by fitting the signal intensities to the standard curves. The m6A/A, m1A/A, m5C/C, and m1G/G ratios were calculated accordingly.
Statistical Analysis
One-way ANOVA followed by LSD post hoc tests on SPSS 20.0 (IBM) was applied for statistical analysis (Supplemental File 1).
RNA Extraction
Total RNA was isolated from ground plant tissues with the Plant RNeasy Mini Kit (Qiagen) or a modified CTAB method. Briefly, well-ground plant tissues were suspended in five volumes of lysis buffer (100 mM Tris-HCl, pH 7.9, 2% CTAB, 2% PVP-30, 2 M NaCl, 25 mM EDTA, 0.05% spermidine, and fresh 2% β-ME) that had been prewarmed to 65°C. The mixtures were heated at 65°C for 10 min and then extracted with chloroform/isoamyl alcohol (24:1) twice. RNAs (>200 nucleotides) were precipitated from the solution using 0.5× volume of 8 M LiCl for 2 h. Total RNA was subsequently treated with RNase-free DNase I (New England Biolabs) to remove DNA contamination for reverse transcription or directly used for mRNA isolation. Poly(A) RNA was isolated from total RNA using oligo(dT)25 Dynabeads (Thermo Fisher Scientific). Removal of contaminating rRNA was achieved using the Ribominus Plant Kit (Thermo Fisher Scientific). The mRNA was qualified using a 2100 Bioanalyzer (Agilent).
Gene Expression Analysis
First-strand cDNA was synthesized from 1 μg of DNase I-treated total RNA using Reverse Transcriptase M-MLV (TaKaRa). RT-qPCR was performed using UltraSYBR Mixture with ROX (CWBIO) on a ViiA 7 Dx (Applied Biosystems). All samples were analyzed in triplicate. TUBULIN2, TUBULIN4, and ACTIN2 were used as reference genes for pre-miRNA (Supplemental Figure 13), tissue differential expression (Figure 1), and other gene expression, respectively. The amount of target was calculated using the following formula: amount of target = 2−ΔΔCT (Livak and Schmittgen, 2001). The half-life of mRNA was referenced by 18S rRNA levels. For RT-PCR, 1 μL of cDNA prepared from 14-d-old seedlings of the wild type (Col-0) or alkbh10b-1 was used as a template for 28 cycles of PCR (95°C for 20s, 55°C for 20s, and 72°C for 30s), followed by analysis by agarose gel (2% w/v) electrophoresis. All primers used are listed in Supplemental Table 1.
Gene-Specific Determination of mRNA m6A Methylation
To determine mRNA methylation level (defined here as the proportion of m6A methylated transcripts per gene) of individual genes in wild-type (Col-0) and alkbh10b-1 mutant plants, we performed an m6A antibody IP enrichment using intact full-length poly(A)+ RNA followed by RT-qPCR analysis (Molinie et al., 2016). The procedure is based on the previously described m6A-seq method (Dominissini et al., 2012; Molinie et al., 2016) with the following modifications. Briefly, 5 μg of full-length mRNA isolated from 14-d-old seedlings of the wild type (Col-0) or alkbh10b-1 collected at ZT15 was incubated with 5 μg of m6A-specific antibody (#202003; Synaptic Systems) overnight at 4°C. The m6A-containing mRNA was pulled down with Protein A Dynabeads that had been preblocked with BSA (Thermo Fisher Scientific). Supernatant were saved and used as m6A-depleted samples or non-m6A mRNA. The RNA-containing beads were eluted twice with elution buffer containing 6.7 mM m6A nucleotide to yield an m6A-enriched sample or m6A-containing mRNA. cDNA was synthesized from 10 ng of the resulting input mRNA, non-m6A mRNA, and m6A-containing mRNA using the Superscript III first-strand synthesis kit (Thermo Fisher Scientific). Twenty microliters of the reaction mixture was diluted 5-fold, and 1 μL was subsequently used for each RT-qPCR reaction.
To determine m6A mRNA methylation, internal gene A was used as the reference. For a certain gene B, the ΔCT of values of (ΔCT,i), non-m6A (ΔCT,n), and m6A-containing mRNA (ΔCT,m) were calculated. The expression levels of gene A and gene B were set as Ea and Eb and the m6A methylation level as Ra and Rb. The amplification efficiencies of the primers for gene B and gene A were tested to be approximately equal. The calculation is based on the 2−ΔΔCT method and Equation 7 (Livak and Schmittgen, 2001).
We calculated the following:
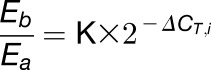 |
(1) |
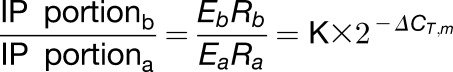 |
(2) |
 |
(3) |
From Equations 3 and 1, we obtain:
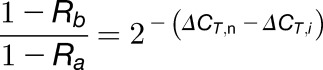 |
(4) |
From Equations 2 and 1, we obtain:
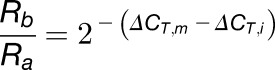 |
(5) |
Rearranging Equation 4 gives the Equation 6:
| (6) |
Rearranging Equation 5 gives Equation 7 or 8:
| (7) |
or
| (8) |
Combining Equations 7 and 6 gives Equation 9:
| (9) |
Rearranging Equation 9 gives Equation 10:
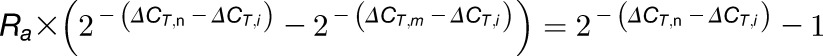 |
(10) |
Rearranging Equation 10 gives Equation 11:
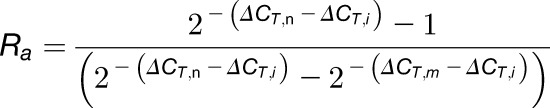 |
(11) |
Finally, dividing Equation 11 by 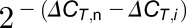 gives Equation 12:
gives Equation 12:
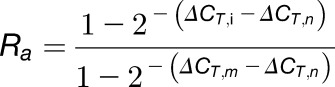 |
(12) |
Similarly, combining Equations 8 and 6 gives Equation 13:
| (13) |
Rearranging Equation 13 gives Equation 14:
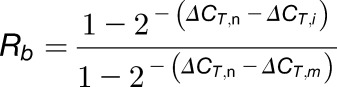 |
(14) |
Thus, we can calculate Ra and Rb using Equations 12 and 14.
A tip for choosing the reference gene A is that either Ra < Rb or Ra > Rb. If Ra≈Rb, 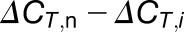 or
or 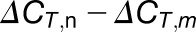 is extremely small (close to 0) (see Equations 4 and 3/2) and even less than the sd of the replicates, which increases the deviation of the calculations. In our experiments, we chose highly methylated gene A (AT3G10390) for calculation of weakly methylated gene B and weakly methylated gene A (AT2G05070) for highly methylated gene B.
is extremely small (close to 0) (see Equations 4 and 3/2) and even less than the sd of the replicates, which increases the deviation of the calculations. In our experiments, we chose highly methylated gene A (AT3G10390) for calculation of weakly methylated gene B and weakly methylated gene A (AT2G05070) for highly methylated gene B.
Transcription Inhibition Assays
The procedure is based on the previously described method (Seo et al., 2011). Briefly, 14-d-old seedlings of the wild type (Col-0) or the alkbh10b-1 mutant were transferred to 0.5× MS medium at ZT13. A final concentration of 200 μM actinomycin D (Sigma-Aldrich) was added to the medium at ZT15. After infiltration for 1 h, 20 seedlings were collected and considered as time 0 controls, and subsequent samples were harvested every 1.5 h in triplicate. To determine remaining mRNA levels using RT-qPCR, 18S rRNA was used as a reference.
Measurement of m6A Methylation Levels of Specific m6A Peaks Using m6A-IP-qPCR of Fragmented Poly(A)+ RNA
The procedure is based on the previously described m6A-seq method (Dominissini et al., 2012). Briefly, 5 µg poly(A)+ RNA was randomly fragmented into ∼250-nucleotide fragments by RNA fragmentation reagents (New England Biolabs). Fragmented mRNA was precipitated and resuspended in 105 μL DEPC-treated water; 5 μL was retained and used as the input sample. The remaining mRNA was incubated with 5 μg m6A-specific antibody (#202003; Synaptic Systems) overnight at 4°C. The m6A-containing fragments were pulled down with Dynabeads Protein A that had been preblocked with BSA (Thermo Fisher Scientific) and eluted with buffer containing 6.7 mM m6A nucleoside, followed by ethanol precipitation. Immunoprecipitated RNA fragments were resuspended in 5 μL DEPC-treated water. Both input and IP samples were reverse-transcribed using the Superscript III first-strand synthesis system (Thermo Fisher Scientific). cDNA (20 μL) was diluted 2-fold, and 1 μL was used for each qPCR reaction. The m6A level of specific mRNA fragments was calculated by the ratio of RNA abundances, IP/input (Zhang et al., 2016).
RIP
RIP was performed according to Köster and Staiger (2014) with minor modifications. Briefly, 2-week-old ALKBH10B:6×myc-ALKBH10B / alkbh10b-1 seedlings were harvested at ZT 15, fixed on ice for 15 min in 1% formaldehyde under vacuum, and terminated with 150 mM glycine for additional 5 min. Two grams of fixed plant material was ground and homogenized in 2 mL of lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM KCl, 5 mM EGTA, 1 mM PMSF, 0.1 unit/μL Ribolock RNase inhibitor, and 1× Roche protease inhibitor cocktail). The extract was then centrifuged at 18,000g for 10 min. Nonidet P-40 was added to the supernatant at a final concentration of 0.1% (v/v). One hundred microliters of lysate was retained and used as the input sample. The remainder was divided into two equal volumes and subsequently immunoprecipitated with 2 μg of anti-myc-tag antibody (#2276; Cell Signaling Technology) or normal rabbit IgG (#2729; Cell Signaling Technology) bound to Dynabeads Protein A, respectively. The input RNA and immunoprecipitated RNAs were extracted with Trizol reagent. The mixtures were then heated at 55°C for 10 min to reverse the RNA-protein cross-link. Equal amounts of RNA from each sample were reverse transcribed with Superscript III first-strand synthesis system (Thermo Fisher Scientific). The relative enrichment of each gene was determined by quantitative real-time PCR.
Protein Immunoblot Analysis
Protein samples were run in a 12% SDS-PAGE for electrophoresis separation and then transferred to a PVDF membrane. The membrane was blocked with TBST (20 mM Tris-HCl, pH 7.4, 150 mM NaCl, and 0.1% Tween-20) containing 5% skimmed milk powder and incubated with anti-myc-tag (71D10) rabbit monoclonal antibody (#2278; Cell Signaling Technology) overnight at 4°C, followed by an incubation with anti-rabbit IgG HRP-linked antibody (#7074; Cell Signaling Technology). The membrane was then washed, and signals were visualized using a 5200 chemiluminescence imaging system (Tanon).
RNA Gel Blot Analysis
RNA gel blot analysis was performed according to previously published protocols (Blevins, 2010; Hu and Zhu, 2017) with minor modifications. Briefly, 20 μg of total RNA isolated from 14-d-old seedlings of wild-type (Col-0) or alkbh10b-1 plants were run in a 18% TBE-urea gel for electrophoresis separation. After electrophoresis, RNA was transferred to Hybond-N+ membrane (GE Healthcare) and immobilized by 150-mJ UV light (wavelength 254 nm). The membrane was prehybridized with ULTRAhyb-Oligo hybridization buffer (Thermo Fisher Scientific) for 4 h prior to overnight incubation with 500 pmol of biotinylated tRNA detection probes. The membrane was washed with washing buffer (2× SSC, 0.5% SDS) five times and then incubated with HRP-labeled streptavidin (Thermo Fisher Scientific) for 1 h. The membrane was then washed three times with washing buffer and signals were visualized using a 5200 chemiluminescence imaging system (Tanon).
High-Throughput m6A Sequencing
This procedure was based on the previously described m6A-seq method (Dominissini et al., 2012). Fourteen-day-old seedlings of the wild type (Col-0) and alkbh10b-1 were used for sequencing. To obtain 10 μg mRNA for each m6A-seq reaction, ∼10 g of 14-d-old seedlings was collected at the ZT15 stage. mRNA was randomly fragmented to ∼100 nucleotides by RNA fragmentation reagents (New England Biolabs) and incubated with 5 μg m6A-specific antibody (#202003; Synaptic Systems) overnight at 4°C. The m6A-containing fragments were pulled down with preblocked Protein A Dynabeads (Thermo Fisher Scientific) and eluted with 6.7 mM m6A nucleoside-containing buffer followed by ethanol precipitation. Immunoprecipitated RNA fragments, and comparable amounts of input, were subjected to sequencing library construction using NEBNext Ultra RNA Library Prep Kit (E7530; New England Biolabs). Sequencing was performed on an Illumina HiSeq 2500. Read numbers for two biological replicates are summarized in Supplemental Data Set 1.
Sequencing Data Analysis
Adaptors and low-quality bases were trimmed from raw sequencing reads using cutadapt (Martin, 2011). Sequencing reads were aligned to the reference genome (TAIR10) (Lamesch et al., 2012) using TopHat (v2.0.14) (Trapnell et al., 2012). If a read was mapped to multiple locations, a random read was then chosen. Gene structure annotations were downloaded from TAIR. The longest isoform was used if multiple isoforms were detected. Aligned reads were extended to 150 bp (average fragment size) and converted from genome-based coordinates to isoform-based coordinates to eliminate the interference from introns in peak calling. The peak calling method was modified from Dominissini et al. (2012). To call m6A peaks, the longest isoform of each gene (TAIR10) was scanned using a 100-bp sliding window with 10-bp steps. To reduce bias from potential inaccurate gene structure annotations and arbitrary usage of the longest isoform, windows with read counts less than 1/20 of the top window in both m6A-IP and input samples were excluded. For each gene, the read counts in each window were normalized by the median count of all windows of that gene. A Fisher’s exact test was used to identify the differential windows between IP and input samples. A window was called as positive if the FDR < 0.01 and log2 (enrichment score) ≥1. Overlapping positive windows were merged. The following four numbers were calculated to obtain the enrichment score of each peak (or window): read count of the IP sample in the current peak/window (a), median read count of the IP sample in all 100-bp windows on the current mRNA (b), read count of the input sample in the current peak/window (c), and median read count of the input sample in all 100-bp windows on the current mRNA (d). The enrichment score of each window was calculated as (a×d)/(b×c). The called peaks with lower abundance mRNA (FPKM < 4) were discarded. We used the peaks in both replicates considered to be “confident peaks” for subsequent analysis. The hypermethylated and hypomethylated m6A peaks in alkbh10b-1 were determined using the m6A site differential algorithm (Meng et al., 2013). The differential peaks in both replicates had to fulfill the criteria of FDR < 0.01 and enrichment fold change ≥2. Gene expression was calculated by Cufflinks (v2.2.1) (Trapnell et al., 2012) using the input sequencing reads. The expression divergence was calculated as the coefficient of variation of log2-transformed FPKMs between orthologous genes. Gene functional annotations (GO enrichment) were performed using DAVID (Huang et al., 2007). Comparison of independent replicates is summarized in Supplemental Figure 15.
SCARLET
Site-specific cleavage and radioactive labeling followed by ligation-assisted extraction and TLC (SCARLET) was performed according Liu et al. (2013). Briefly, 1 μg poly(A)+ RNA was mixed with 3 pmol of corresponding chimeric oligonucleotides in a final volume of 3 μL in 30 mM Tris-HCl (pH 7.5). The mixtures were heated at 95°C for 1 min and then annealed at room temperature for 5 min. One microliter of RNase H mix (2× T4 polynucleotide kinase buffer, 1 unit/μL RNase H; Thermo Fisher Scientific) and 1 μL thermosensitive alkaline phosphatase (1 unit/µL; Thermo Fisher Scientific) were then added. The samples were incubated at 44°C for 1 h and then heated at 75°C for 5 min to inactivate TAP and RNase H. One microliter of 6× T4 PNK mix (1× T4 PNK buffer, 6 units/μL T4 PNK [New England Biolabs], and 28 µCi/μL [γ-32P] ATP [6000 Ci/mmole]) was added to the mixture for radioactive labeling. After incubation for 1 h at 37°C, T4 PNK was inactivated for 5 min at 75°C. Four picomoles of the corresponding Splint and 5 pmol 116-mer DNA oligo (ssDNA-116) was then annealed to radiolabeled fragments. Ligation reactions were performed by incubating samples with 2.5 μL 4× ligation mix (1.4× T4 PNK buffer, 57% DMSO, and 1.9 units/μL T4 DNA ligase) overnight at 37°C. An equal volume of 2× RNA loading buffer (9 M urea, 100 mM EDTA) was added, and RNA was digested by incubation with RNase T1/A mixture (New England Biolabs) at 37°C for 2 h. The ligation products were isolated by 8% denaturing PAGE followed by crushing and soaking of corresponding gel slices, and precipitated in 70% ethanol. The pellet was digested with nuclease P1 (Wako) in a final volume of 5 μL in 10 mM sodium acetate/acetic acid (pH 5.3). The reaction mixtures were then spotted onto cellulose TLC plates. TLC was run with isopropanol:HCl:water (70:15:15, v/v/v) for 14 h. Dried TLC plates were wrapped in plastic film and exposed to a blanked phosphor imager screen. The exposure time was adjusted according to radioactivity of the TLC plate as measured by a Geiger counter.
Accession Numbers
Sequence data in this study can be found in the GenBank/EMBL data libraries under the following accession numbers: ALKBH5, NP_060228.3; AtALKBH9A, AT1G48980; AtALKBH9B, AT2G17970; AtALKBH9C, AT4G36090; AtALKBH10A, AT2G48080; ALKBH10B, AT4G02940; FT, AT1G65480; SPL3, AT2G33810; SPL9, AT2G42200; MTA, AT4G10760; MTB, AT4G09980; and FIP37, AT3G54170. All sequencing data have been deposited into the Gene Expression Omnibus under the accession code GSE79523.
Supplemental Data
Supplemental Figure 1. Sequence alignment of the AlkB domain of human ALKBH5 with five Arabidopsis AlkB family proteins.
Supplemental Figure 2. Characterization of ALKBH9B and ALKBH9C.
Supplemental Figure 3. Quantification of in planta-expressed recombinant ALKBH10B and E. coli-expressed recombinant human ALKBH5.
Supplemental Figure 4. ALKBH10B mutants and transgenic lines.
Supplemental Figure 5. Quantification of the m6A/A ratio of mRNA in the indicated genotypic tissues by LC-MS/MS.
Supplemental Figure 6. Relative mRNA levels of methylation-associated genes in indicated plants.
Supplemental Figure 7. Arabidopsis mRNA does not contain m6Am near its 5′ cap.
Supplemental Figure 8. ALKBH10B affects vegetative growth in Arabidopsis.
Supplemental Figure 9. Phenotypes of alkbh10b mutants.
Supplemental Figure 10. Diurnal time courses of the regulatory genes of FT in wild-type and alkbh10b-1 plants.
Supplemental Figure 11. The relative mRNA levels of the FT regulatory genes in wild-type and alkbh10b-1 plants at ZT4 and ZT13.
Supplemental Figure 12. m6A mRNA methylation levels of the FT regulatory genes in wild-type and alkbh10b-1 mutant plants.
Supplemental Figure 13. The expression level of pre-miR172 was decreased in alkbh10b-1 mutant plants.
Supplemental Figure 14. Phenotype of FD:FT /alkbh10b-1 plants.
Supplemental Figure 15. Correlation of m6A peaks and mRNA FPKM between two biological replicates in wild-type Col-0 and alkbh10b-1.
Supplemental Figure 16. Representative views of hypermethylated m6A peaks in alkbh10b-1 and verification of m6A-IP sequencing results.
Supplemental Figure 17. m6A-hypermethylated transcripts are involved in various biological processes and plant development.
Supplemental Figure 18. A proposed model for reversible m6A methylation of Arabidopsis mRNA.
Supplemental Table 1. Primers and oligonucleotide probes used in this study.
Supplemental Data Set 1. A summary of m6A-seq information.
Supplemental Data Set 2. Confident m6A peaks in wild-type Col-0.
Supplemental Data Set 3. Confident m6A peaks in alkbh10b-1.
Supplemental Data Set 4. Hypermethylated peaks in alkbh10b-1.
Supplemental Data Set 5. Lower-expression genes in alkbh10b-1.
Supplemental Data Set 6. Higher-expression genes in alkbh10b-1.
Supplemental Data Set 7. All expressed genes FPKM in wild-type Col-0 and alkbh10b-1.
Supplemental File 1. ANOVA tables.
Acknowledgments
We thank W. Qian and H. Zhang for providing Arabidopsis vectors and suggestions on plant experiments. We also thank J. Hu and S.F. Reichard for editing the manuscript. This work was supported by the Ministry of Science and Technology of the People's Republic of China (Chinese Ministry of Science and Technology) (MOST2016YFC0900302 and 2017YFA0505201 to G.J. and 2014CB964900 to C.H.) and the National Natural Science Foundation of China (21432002 to P.R.C. and G.J., 21372022 to G.J., and 21210003 to C.H. and G.J.).
AUTHOR CONTRIBUTIONS
H.-C.D. performed the experiments with the help of L.-H.W., C.Z., Y.W., L.C., and Z.L. G.J., C.H., and H.-C.D. designed the experiments and wrote the manuscript with the help of P.R.C.
References
- Abe M., Kobayashi Y., Yamamoto S., Daimon Y., Yamaguchi A., Ikeda Y., Ichinoki H., Notaguchi M., Goto K., Araki T. (2005). FD, a bZIP protein mediating signals from the floral pathway integrator FT at the shoot apex. Science 309: 1052–1056. [DOI] [PubMed] [Google Scholar]
- Alarcón C.R., Goodarzi H., Lee H., Liu X., Tavazoie S., Tavazoie S.F. (2015). HNRNPA2B1 is a mediator of m(6)A-dependent nuclear RNA processing events. Cell 162: 1299–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso J.M., et al. (2003). Genome-wide insertional mutagenesis of Arabidopsis thaliana. Science 301: 653–657. [DOI] [PubMed] [Google Scholar]
- Amasino R. (2010). Seasonal and developmental timing of flowering. Plant J. 61: 1001–1013. [DOI] [PubMed] [Google Scholar]
- Batista P.J., et al. (2014). m(6)A RNA modification controls cell fate transition in mammalian embryonic stem cells. Cell Stem Cell 15: 707–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäurle I., Dean C. (2006). The timing of developmental transitions in plants. Cell 125: 655–664. [DOI] [PubMed] [Google Scholar]
- Blevins T. (2010). Northern blotting techniques for small RNAs. Methods Mol. Biol. 631: 87–107. [DOI] [PubMed] [Google Scholar]
- Bodi Z., Zhong S., Mehra S., Song J., Graham N., Li H., May S., Fray R.G. (2012). Adenosine methylation in Arabidopsis mRNA is associated with the 3′ end and reduced levels cause developmental defects. Front. Plant Sci. 3: 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominissini D., Moshitch-Moshkovitz S., Schwartz S., Salmon-Divon M., Ungar L., Osenberg S., Cesarkas K., Jacob-Hirsch J., Amariglio N., Kupiec M., Sorek R., Rechavi G. (2012). Topology of the human and mouse m6A RNA methylomes revealed by m6A-seq. Nature 485: 201–206. [DOI] [PubMed] [Google Scholar]
- Dominissini D., et al. (2016). The dynamic N(1)-methyladenosine methylome in eukaryotic messenger RNA. Nature 530: 441–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu Y., et al. (2013). FTO-mediated formation of N6-hydroxymethyladenosine and N6-formyladenosine in mammalian RNA. Nat. Commun. 4: 1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fustin J.M., Doi M., Yamaguchi Y., Hida H., Nishimura S., Yoshida M., Isagawa T., Morioka M.S., Kakeya H., Manabe I., Okamura H. (2013). RNA-methylation-dependent RNA processing controls the speed of the circadian clock. Cell 155: 793–806. [DOI] [PubMed] [Google Scholar]
- Geula S., et al. (2015). Stem cells. m6A mRNA methylation facilitates resolution of naïve pluripotency toward differentiation. Science 347: 1002–1006. [DOI] [PubMed] [Google Scholar]
- Hu J., Zhu Y. (2017). Nonradioactive plant small RNA detection using biotin-labeled probes. Methods Mol. Biol. 1640: 211–217. [DOI] [PubMed] [Google Scholar]
- Huang D.W., Sherman B.T., Tan Q., Collins J.R., Alvord W.G., Roayaei J., Stephens R., Baseler M.W., Lane H.C., Lempicki R.A. (2007). The DAVID Gene Functional Classification Tool: a novel biological module-centric algorithm to functionally analyze large gene lists. Genome Biol. 8: R183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia G., Fu Y., Zhao X., Dai Q., Zheng G., Yang Y., Yi C., Lindahl T., Pan T., Yang Y.G., He C. (2011). N6-methyladenosine in nuclear RNA is a major substrate of the obesity-associated FTO. Nat. Chem. Biol. 7: 885–887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J.J., Lee J.H., Kim W., Jung H.S., Huijser P., Ahn J.H. (2012). The microRNA156-SQUAMOSA PROMOTER BINDING PROTEIN-LIKE3 module regulates ambient temperature-responsive flowering via FLOWERING LOCUS T in Arabidopsis. Plant Physiol. 159: 461–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köster T., Staiger D. (2014). RNA-binding protein immunoprecipitation from whole-cell extracts. Methods Mol. Biol. 1062: 679–695. [DOI] [PubMed] [Google Scholar]
- Lamesch P., et al. (2012). The Arabidopsis Information Resource (TAIR): improved gene annotation and new tools. Nucleic Acids Res. 40: D1202–D1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X., Xiong X., Wang K., Wang L., Shu X., Ma S., Yi C. (2016). Transcriptome-wide mapping reveals reversible and dynamic N(1)-methyladenosine methylome. Nat. Chem. Biol. 12: 311–316. [DOI] [PubMed] [Google Scholar]
- Liu F., et al. (2016). ALKBH1-Mediated tRNA Demethylation Regulates Translation. Cell 167: 816–828.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., et al. (2014). A METTL3-METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10: 93–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Dai Q., Zheng G., He C., Parisien M., Pan T. (2015). N6-methyladenosine-dependent RNA structural switches regulate RNA-protein interactions. Nature 518: 560–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu N., Parisien M., Dai Q., Zheng G., He C., Pan T. (2013). Probing N6-methyladenosine RNA modification status at single nucleotide resolution in mRNA and long noncoding RNA. RNA 19: 1848–1856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livak K.J., Schmittgen T.D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Δ Δ C(T)) method. Methods 25: 402–408. [DOI] [PubMed] [Google Scholar]
- Luo G.Z., MacQueen A., Zheng G., Duan H., Dore L.C., Lu Z., Liu J., Chen K., Jia G., Bergelson J., He C. (2014). Unique features of the m6A methylome in Arabidopsis thaliana. Nat. Commun. 5: 5630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17: 10–12. [Google Scholar]
- Martínez-Pérez M., Aparicio F., López-Gresa M.P., Bellés J.M., Sánchez-Navarro J.A., Pallás V. (2017). Arabidopsis m6A demethylase activity modulates viral infection of a plant virus and the m6A abundance in its genomic RNAs. Proc. Natl. Acad. Sci. USA 114: 10755–10760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauer J., et al. (2017). Reversible methylation of m6Am in the 5′ cap controls mRNA stability. Nature 541: 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng J., Cui X., Liu H., Zhang L., Zhang S., Rao M.K., Chen Y., Huang Y. (2013). Unveiling the dynamics in RNA epigenetic regulations. In 2013 IEEE International Conference on Bioinformatics and Biomedicine (BIBM), pp. 139–144, doi/10.1109/BIBM.2013.6732477. [DOI] [Google Scholar]
- Meyer K.D., Patil D.P., Zhou J., Zinoviev A., Skabkin M.A., Elemento O., Pestova T.V., Qian S.B., Jaffrey S.R. (2015). 5′ UTR m(6)A promotes cap-independent translation. Cell 163: 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer K.D., Saletore Y., Zumbo P., Elemento O., Mason C.E., Jaffrey S.R. (2012). Comprehensive analysis of mRNA methylation reveals enrichment in 3′ UTRs and near stop codons. Cell 149: 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mielecki D., Zugaj D.L., Muszewska A., Piwowarski J., Chojnacka A., Mielecki M., Nieminuszczy J., Grynberg M., Grzesiuk E. (2012). Novel AlkB dioxygenases--alternative models for in silico and in vivo studies. PLoS One 7: e30588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinie B., Wang J., Lim K.S., Hillebrand R., Lu Z.X., Van Wittenberghe N., Howard B.D., Daneshvar K., Mullen A.C., Dedon P., Xing Y., Giallourakis C.C. (2016). m(6)A-LAIC-seq reveals the census and complexity of the m(6)A epitranscriptome. Nat. Methods 13: 692–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ping X.L., et al. (2014). Mammalian WTAP is a regulatory subunit of the RNA N6-methyladenosine methyltransferase. Cell Res. 24: 177–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Růžička K., et al. (2017). Identification of factors required for m6 A mRNA methylation in Arabidopsis reveals a role for the conserved E3 ubiquitin ligase HAKAI. New Phytol. 215: 157–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo E., Yu J., Ryu K.H., Lee M.M., Lee I. (2011). WEREWOLF, a regulator of root hair pattern formation, controls flowering time through the regulation of FT mRNA stability. Plant Physiol. 156: 1867–1877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L., Liang Z., Gu X., Chen Y., Teo Z.W., Hou X., Cai W.M., Dedon P.C., Liu L., Yu H. (2016). N6-methyladenosine RNA modification regulates shoot stem cell fate in Arabidopsis. Dev. Cell 38: 186–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sparkes I.A., Runions J., Kearns A., Hawes C. (2006). Rapid, transient expression of fluorescent fusion proteins in tobacco plants and generation of stably transformed plants. Nat. Protoc. 1: 2019–2025. [DOI] [PubMed] [Google Scholar]
- Srikanth A., Schmid M. (2011). Regulation of flowering time: all roads lead to Rome. Cell. Mol. Life Sci. 68: 2013–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung S., Amasino R.M. (2004). Vernalization in Arabidopsis thaliana is mediated by the PHD finger protein VIN3. Nature 427: 159–164. [DOI] [PubMed] [Google Scholar]
- Trapnell C., Roberts A., Goff L., Pertea G., Kim D., Kelley D.R., Pimentel H., Salzberg S.L., Rinn J.L., Pachter L. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7: 562–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vespa L., Vachon G., Berger F., Perazza D., Faure J.D., Herzog M. (2004). The immunophilin-interacting protein AtFIP37 from Arabidopsis is essential for plant development and is involved in trichome endoreduplication. Plant Physiol. 134: 1283–1292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., et al. (2014). N6-methyladenosine-dependent regulation of messenger RNA stability. Nature 505: 117–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Zhao B.S., Roundtree I.A., Lu Z., Han D., Ma H., Weng X., Chen K., Shi H., He C. (2015). N(6)-methyladenosine modulates messenger RNA translation efficiency. Cell 161: 1388–1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G., Park M.Y., Conway S.R., Wang J.W., Weigel D., Poethig R.S. (2009). The sequential action of miR156 and miR172 regulates developmental timing in Arabidopsis. Cell 138: 750–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao W., et al. (2016). Nuclear m(6)A reader YTHDC1 regulates mRNA splicing. Mol. Cell 61: 507–519. [DOI] [PubMed] [Google Scholar]
- Zhang C., Samanta D., Lu H., Bullen J.W., Zhang H., Chen I., He X., Semenza G.L. (2016). Hypoxia induces the breast cancer stem cell phenotype by HIF-dependent and ALKBH5-mediated m6A-demethylation of NANOG mRNA. Proc. Natl. Acad. Sci. USA 113: E2047–E2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X., Henriques R., Lin S.S., Niu Q.W., Chua N.H. (2006). Agrobacterium-mediated transformation of Arabidopsis thaliana using the floral dip method. Nat. Protoc. 1: 641–646. [DOI] [PubMed] [Google Scholar]
- Zheng G., et al. (2013). ALKBH5 is a mammalian RNA demethylase that impacts RNA metabolism and mouse fertility. Mol. Cell 49: 18–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G., Fu Y., He C. (2014). Nucleic acid oxidation in DNA damage repair and epigenetics. Chem. Rev. 114: 4602–4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhong S., Li H., Bodi Z., Button J., Vespa L., Herzog M., Fray R.G. (2008). MTA is an Arabidopsis messenger RNA adenosine methylase and interacts with a homolog of a sex-specific splicing factor. Plant Cell 20: 1278–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]