

Abstract
The TRIM‐NHL protein Brain tumor (Brat) acts as a tumor suppressor in the Drosophila brain, but how it suppresses tumor formation is not completely understood. Here, we combine temperature‐controlled brat RNAi with transcriptome analysis to identify the immediate Brat targets in Drosophila neuroblasts. Besides the known target Deadpan (Dpn), our experiments identify the transcription factor Zelda (Zld) as a critical target of Brat. Our data show that Zld is expressed in neuroblasts and required to allow re‐expression of Dpn in transit‐amplifying intermediate neural progenitors. Upon neuroblast division, Brat is enriched in one daughter cell where its NHL domain directly binds to specific motifs in the 3′UTR of dpn and zld mRNA to mediate their degradation. In brat mutants, both Dpn and Zld continue to be expressed, but inhibition of either transcription factor prevents tumorigenesis. Our genetic and biochemical data indicate that Dpn inhibition requires higher Brat levels than Zld inhibition and suggest a model where stepwise post‐transcriptional inhibition of distinct factors ensures sequential generation of fates in a stem cell lineage.
Keywords: neurogenesis, stem cells, tumorigenesis, RNA‐binding protein
Subject Categories: Cancer, Development & Differentiation, Neuroscience
Introduction
Stem cells are characterized by the ability to generate both self‐renewing and differentiating progeny. Some of them do this by segregating cell fate determinants into only one of the daughter cells during mitosis where those factors induce differentiation and prevent self‐renewal. Errors in the precise regulation of this balance can cause severe stem cell overgrowth ultimately leading to tumorigenesis 1, 2, 3. Drosophila larval neural stem cells, called neuroblasts (NBs), are a well‐established model for investigating this regulatory mechanism 4, 5, 6. During asymmetric cell division, NBs form apical and basal plasma membrane domains composed of distinct protein sets. Proteins in the apical domain segregate into the self‐renewing cell while proteins located at the basal domain are inherited by the differentiating daughter cell where they specify cell fate identity. The Notch inhibitor Numb, the transcription factor Prospero, and the translational repressor Brain tumor (Brat) have been identified as key components of this basal domain 7, 8, 9. The absence of these factors disturbs neuronal tissue homeostasis and leads to supernumerary NBs in larval brains. Although Numb and Prospero are well understood 10, 11, 12, the precise molecular function of Brat in NBs is currently unclear.
Drosophila NBs are subdivided into type 0, type I, and type II. Type 0 NBs self‐renew and generate a post‐mitotic differentiating neuron 13, 14. Type I NBs self‐renew and generate ganglion mother cells (GMCs), which divide terminally into two neurons or glia cells. Type II NBs also self‐renew, but they generate a transit‐amplifying pool of intermediate neural precursors (INPs) to expand the pool of neurons 15, 16, 17. All NBs express Deadpan (Dpn), a basic helix‐loop‐helix (bHLH) transcriptional repressor related to vertebrate Hes transcription factors 18, 19. Newly born immature INPs (imINPs) first switch off Dpn expression and then turn on another transcription factor called Asense (Ase). Finally, imINPs reinitiate Dpn expression and resume asymmetric division and are then called mature INPs (mINPs) 15, 16, 17, 20, 21. In brat mutants, immature INPs fail to reinitiate Ase and Dpn expression. Instead, they enter a transient cell cycle block and ultimately revert to type II NBs. As a result, they form excessive NBs and ultimately a lethal transplantable brain tumor 17, 21, 22.
Multiple functions for Brat have been demonstrated outside the nervous system. Brat belongs to the TRIM‐NHL family of proteins. It contains two B‐boxes and a coiled‐coil domain at the N‐terminus and a C‐terminal NHL domain 23. Most brat alleles carry mutations in the NHL domain, indicating that this domain is functionally important 9, 17, 23. The NHL domain binds the adaptor protein Miranda (Mira) and the cap‐binding protein d4EHP, an inhibitor of translation 19, 24. Brat is required for establishment of the anterior–posterior body axis by repressing translation of the posterior determinant hunchback (hb) 25. For this, it forms a protein complex with Nanos (Nos) and Pumilio (Pum) that binds to the hb 3′UTR and inhibits translation. Brat also regulates stem cell self‐renewal in the germline by repressing mad and dmyc mRNA together with Pum 26. Recently, it was shown that Brat can also contact mRNA independently of Pum through a specific RNA motif 27, 28, 29. Brat can also interact with the RISC‐complex member Argonaute‐1 30, but unlike for other TRIM‐NHL proteins, a role for Brat in the micro‐RNA pathway has not been found.
How Brat acts in neural stem cell lineages is not completely understood. Brat was suggested to specify INPs by attenuating beta‐catenin/Armadillo activity and thereby inhibiting the self‐renewal factor Klumpfuss (Klu) 21, 31. How this would work molecularly, however, is unclear. To identify the molecular function of Brat in INPs, we developed a screening assay for the earliest transcripts whose abundance changes in brat mutants. We identified Zelda (Zld), previously known as vielfältig 32, a zinc‐finger protein and key activator of early zygotic transcription 33. Zld marks genomic regions in early Drosophila embryos for subsequent transcriptional activation during the maternal‐to‐zygotic transition (MZT) 34, 35. Zld is also involved in wing development 36, but a function in the nervous system has not been described. We show here that Zld is expressed in NBs and allows Dpn re‐expression in mature INPs to promote transit‐amplifying cell division. In brat mutants, Zld expression is sustained and promotes tumor formation and metastatic growth. We also demonstrate that Brat directly binds to the motif (A/U)UGUU(A/G/U) present in the 3′UTRs of both zld and the self‐renewal factor dpn and represses their translation. Interestingly, repression of Zld and Dpn requires different levels of Brat: While low levels of Brat are sufficient to repress Zld, higher Brat levels are required for Dpn repression. We propose a model where different protein concentrations of Brat might regulate progression of the type II NB lineage.
Results
Transcriptome and rescue analysis reveals Zelda as a potential Brat target
To identify new Brat targets involved in brain tumor formation, we analyzed the transcriptome changes that occur in type II NB lineages upon brat RNAi over time. The brat RNAi phenotype corresponds to that of a null allele as Dpn is not repressed (Appendix Fig S1A) and ectopic NBs are formed. To identify the very first mRNA changes, we used the temperature‐sensitive Gal4/Gal80 system to induce brat RNAi in a temporally controlled manner 37 (Fig 1A). 72 h after brat RNAi induction, cell numbers in type II NB lineages are significantly increased when compared to control lineages (Fig 1B, Appendix Figs S1B and S2). 24 h after brat RNAi induction, however, cell numbers in NB lineages were still the same as in wild‐type, although Dpn was already misexpressed (Fig 1B, Appendix Figs S1B–D and S2). Consistently, expression of cell cycle inducers such as Cyclin E, string, E2F transcription factor was unchanged, whereas the cell cycle inhibitor dacapo was moderately increased (Appendix Fig S1G). We isolated mRNA from FACS‐sorted control and 24‐h brat‐depleted type II NB lineages. The FACS protocol was optimized for maintaining a similar cell‐type composition (Appendix Fig S1E). Deep sequencing identified 41 upregulated and 38 downregulated genes upon brat RNAi (FDR1.1; log2 fold change > 0.8; FPKM > 50; Fig 1C, Dataset EV1, Appendix Fig S1F). Among the upregulated genes were five transcription factors, all of which could be confirmed by qRT–PCR (Fig 1D). Besides Dpn and dMyc, which are known to be misregulated in brat mutants 9, 22, these included the transcription factor Zld 32, 33, 34, 35. When inhibited by RNAi together with brat, dpn and zld were the most potent suppressors of adult lethality, suggesting that they might be the most relevant targets of Brat (Fig 1E).
Figure 1. The transcriptional activator Zelda is a potential target of Brat.
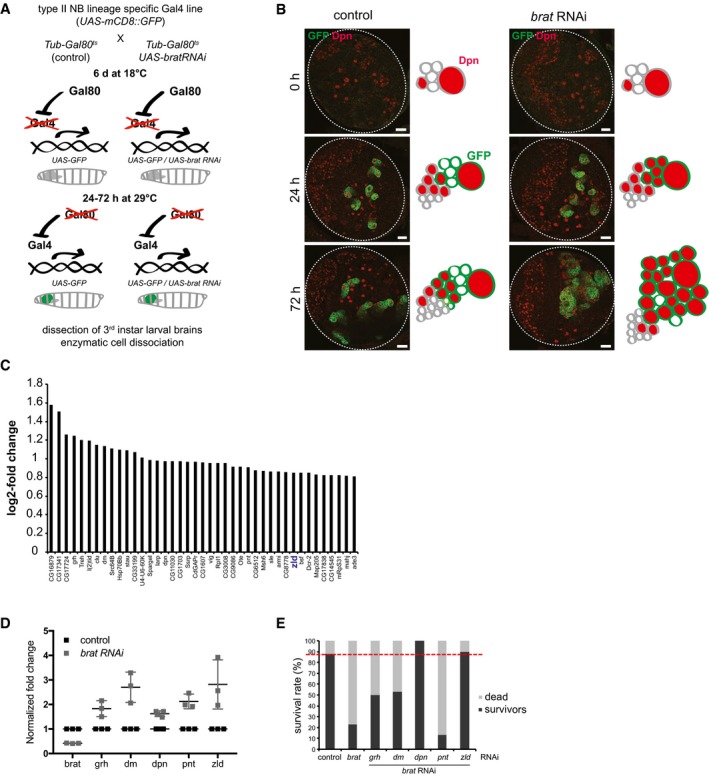
- Cartoon illustrating the time‐ and tissue‐specific induction of brat RNAi.
- Images of brain lobes expressing brat RNAi and/or GFP in type II NB lineages (wor‐Gal4, ase‐Gal80) for 0, 24, or 72 h and stained for Dpn (red). Schematics represent the images.
- Plot showing log2 fold change in the expression of candidate target genes upon brat RNAi (one independent experiment).
- qPCR analysis of candidate target gene expression in FACS‐sorted control and brat RNAi type II lineages (induced with wor‐Gal4, ase‐Gal80) after 24 h of induction.
- brat rescue analysis of candidate target genes (one independent experiment). Survival rate of controls and double RNAi experiments (induced with wor‐Gal4, ase‐Gal80) was determined 2 weeks after adult hatching. Red dashed line indicates the survival rate of the control.
brat tumor growth is impaired upon Zelda knockdown
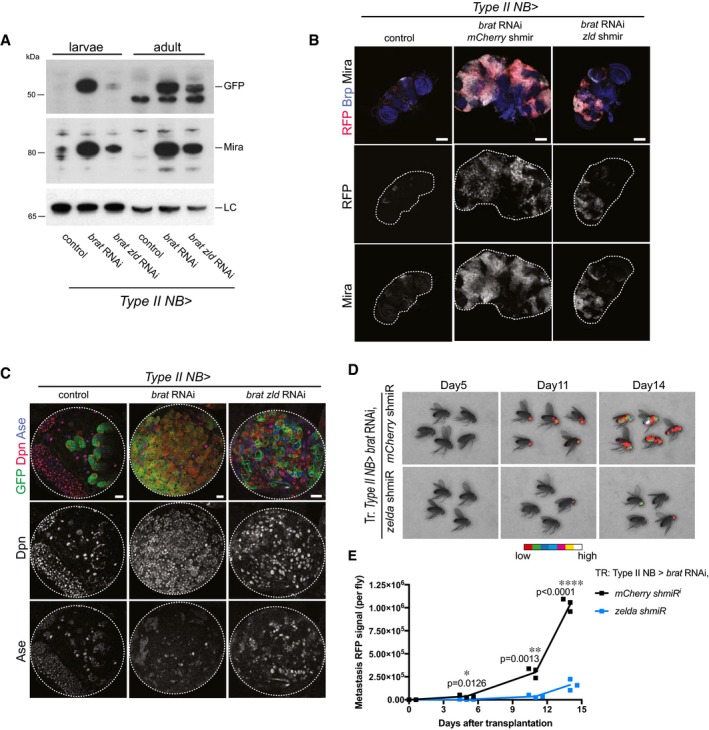
To characterize the role of Zld in tumorigenesis, we analyzed brat zld tumors on three key characteristics: (i) primary brain tumor growth, (ii) lack of differentiation, and (iii) potential for unlimited proliferation. (i) Brain tumors formed upon brat RNAi mainly consist of dividing Mira‐positive ectopic type II NB‐like cells that invade most of the central brain and expand its size. We quantified brat tumor growth by Western blot analysis of Mira and type II NB‐specific GFP expression (worGal4, aseGal80 > UAS‐CD8‐GFP) and observed a significant tumor reduction in brat zld double RNAi compared to brat RNAi (Fig 2A). Consistently, brat zld double RNAi adults developed significantly reduced tumors compared to brat RNAi (Fig 2B, Appendix Fig S3). As a positive control, brat dpn double RNAi adults developed reduced tumors to an even higher extent, further confirming the importance of Dpn expression in brat tumor growth (Appendix Fig S3). (ii) brat RNAi leads to the formation of ectopic type II NB‐like cells that maintain a high‐Dpn, low‐Ase expression pattern (Fig 2C, middle column panels). Upon brat zld double RNAi, however, ectopic NBs expressed both Dpn and Ase or even Ase alone, indicating partial differentiation of these cells (Fig 2C). (iii) brat tumors can undergo unlimited growth and metastasis when transplanted into an adult host 1. To further characterize zld as a tumor‐promoting factor, we developed a method that allows the quantification of transplanted tumor growth. For this, we injected tumors formed upon brat single or brat zld double RNAi into the thorax of wild‐type host flies and assessed their metastatic growth in real time by following tumor‐encoded RFP. Injected brat zld RNAi tumor cells were still able to form metastasis but with much slower kinetics when compared to brat RNAi (Fig 2D and E). We conclude that simultaneous inhibition of Zld reduces the severe overgrowth phenotype observed in brat tumors by inducing differentiation of the ectopic NBs. Our data show that Zld contributes to brat tumor formation and metastatic growth and suggest that Zld might be an additional target for Brat‐mediated repression in imINPs.
Figure 2. Zelda contributes to brat tumor formation.
-
AWestern blot analysis of larval and adult brain tumors. brat tumors display high Mira and GFP levels, whereas upon brat zld double RNAi Mira and GFP levels are reduced in both larval and adult brains compared to control. LC, loading control lamin.
-
BImages of adult brains of control, brat RNAi mCherry shmiR and brat RNAi zld shmiR (all isoforms) (induced with wor‐Gal4, ase‐Gal80). Type II NB lineages are marked with stinger‐RFP (red). Brains are stained for Bruchpilot (Brp, blue) and Mira (white). brat RNAi adult brains are overgrown by Mira‐positive NB‐like cells, whereas upon brat zld double knockdown tumors are reduced in size (images are representative of two (control) to three (other conditions) independent experiments).
-
CImages of larval brain lobes of control, brat RNAi and brat zld double RNAi (induced with wor‐Gal4, ase‐Gal80). Type II NB lineages are marked with membrane‐bound GFP (green). Brains are stained for Dpn (red) and Ase (blue). brat RNAi tumors contain almost only GFP‐positive Dpn‐positive NB‐like cells, whereas upon brat zld double RNAi tumors contain GFP‐marked cells positive for Dpn or Ase, or Dpn and Ase.
-
D, EReal‐time tumor metastasis burden after transplantation of brains expressing nuclear RFP, brat RNAi, mCherry shmiR (upper panels) and zld shmiR (lower panels) from type II NB driver wor‐Gal4, ase‐Gal80. ˜4,000 RFP‐positive cells were injected and RFP signal from whole fly was quantified 5, 11, and 14 days afterward. First metastasis only appeared 6 days later from brat zld RNAi brain injections (day 11 vs. day 5).
Zelda is required for NB lineage formation
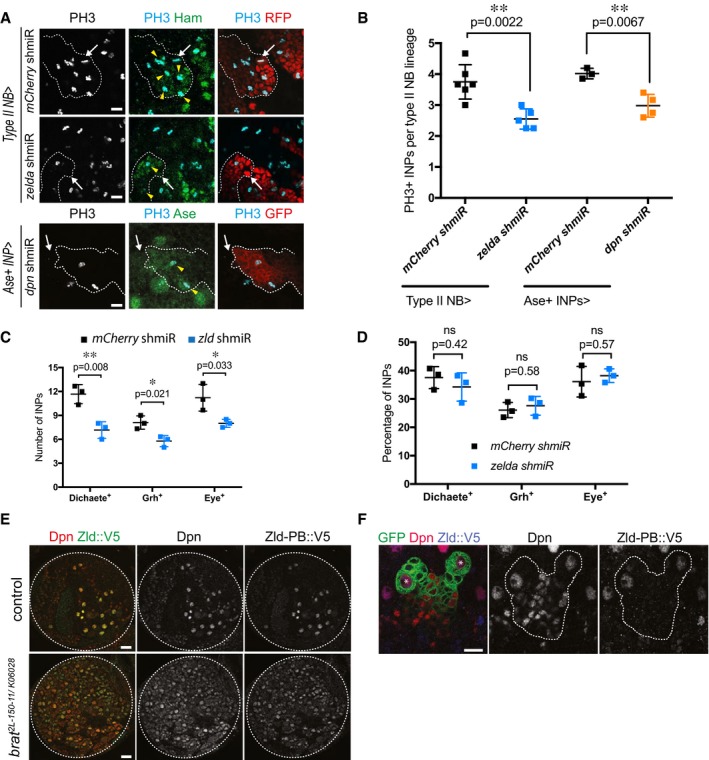
To investigate the role of Zld in type II NB lineages, we used two independent zld RNAi constructs (zld IR and zld shmiR). Like their wild‐type counterparts, zld IR or zld shmiR type II NBs gave rise to Ase‐negative imINPs, which maturated into Ase‐positive imINP (Appendix Fig S6A). In contrast to wild‐type Ase‐positive imINPs however, the mutant cells often failed to re‐express Dpn (Appendix Fig S6A and C). We could rescue this phenotype by overexpressing Zld in zld‐depleted type II NB (Appendix Fig S4A). Additionally, restoring Dpn by overexpression in brat zld‐depleted type II NB could rescue primary tumor growth at larval stages (Appendix Fig S4B) and resulted in restored tumor burden at adult stages as quantified by tumor‐specific RFP signal in the adult heads (Appendix Fig S4C and D). Consistently, qPCR analysis of larval brains expressing zld RNAi from the inscuteable driver confirmed that dpn transcript levels were significantly decreased (Appendix Fig S6B). Interestingly, phospho‐H3 stainings revealed that the ability of imINPs to reinitiate mitosis was significantly reduced upon zld RNAi (Fig 3A and B). The inability of these imINPs to re‐express Dpn is likely responsible for their underproliferation, as depleting Dpn from INPs resulted in the same phenotype (Fig 3A and B). As a result, the number of INPs was significantly decreased in zld RNAi (Fig 3C). Nonetheless, the sequential expression of mINPs temporal identity markers Dichaete, Grainy head, and Eyeless was similarly proportioned in zld RNAi compared to control INPs indicating a normal patterning of these cells (Fig 3D, Appendix Fig S6D–F).
Figure 3. Zelda is required for proliferation potential, but not differentiation patterns of INPs.
-
AClose‐up images of mCherry and zld shmiR type II NB lineages marked with nuclear RFP (red, upper panels) or membrane‐bound GFP (red, lower panels) and stained for INP marker Hamlet or Ase (green) and PH3 (light blue). Note that in zld and dpn shmiR, type II NB lineages contain less proliferative INPs (yellow arrowheads). White arrows designate the position of type II NBs.
-
BQuantification of PH3‐positive INPs in type II NB lineages expressing mCherry or zld shmiR (induced with wor‐Gal4, ase‐Gal80) or mCherry and dpn shmiR (induced with ase‐Gal4) (three or more independent experiments).
-
C, DCell count (C) and relative proportions (D) of Dichaete‐, Grainy head (Grh)‐, and Eyeless (Eye)‐positive INPs of mCherry and zld shmiR (induced with wor‐Gal4, ase‐Gal80) type II NB lineages. While their global number is reduced, the proportions between each temporal state of INP is maintained in zld shmiR type II NB lineages.
-
EBrain lobes of control and brat 2L‐150‐11/K06028 trans‐heterozygous expressing endogenous Zld‐PB::V5 stained for Dpn (red) and V5 (green).
-
FClose‐up images of type II NB lineages marked by membrane‐bound GFP expressing endogenous Zld‐PB::V5 stained for Dpn and V5. Note that Zld‐PB::V5 is only expressed in NBs but absent from their progeny. *type I NB, **type II NB.
Previous analysis has identified the zld transcripts zld‐RB and zld‐RD encoding two distinct proteins, Zld‐PB and Zld‐PD, respectively 38. Zld‐PD is missing three of the four C‐terminal C2H2 zinc fingers that bind to the Zld target site and thus has potentially altered or non‐functional DNA‐binding properties 33, 38, 39. NBs specifically express the active zld‐RB isoform, whereas the inactive zld‐RD isoform is expressed in the differentiating NB progeny 40 (Appendix Fig S7A). FISH analysis using specific probes for the two zld variants confirmed this expression pattern (Appendix Fig S7B and C). Importantly however, whether zld‐RD is actually translated into functional proteins in these cells would remain to be explored. We next assessed the function of the −RB and −RD isoforms independently by performing isoform‐specific RNAi in a brat tumor context. Unlike zld‐RB RNAi, zld‐RD RNAi constructs were unable to rescue brat tumor growth (Appendix Fig S5A–C). Furthermore, overexpressing zld‐RB was sufficient to restore Dpn expression in zld‐deficient type II NB lineage (Appendix Fig S4A). Altogether, these results suggest that zld‐RB but not −RD is required for type II NB lineage proliferation. To distinguish Zld‐PB from Zld‐PD, we used CRISPR/Cas9 technology 41 to insert a V5‐tag at the C‐terminus of zld‐RB (zld‐RB::V5, Appendix Fig S7B and D). The insertion does not affect Zld function as flies homozygous for zld‐RB::V5 are viable and fertile. Zld‐PB::V5 immunostainings confirmed its localization in NBs in both wild‐type and brat mutant larval brains (Fig 3E). The specificity of Zld‐PB::V5 staining was further confirmed by zld RNAi (Appendix Fig S7E). Detailed lineage analysis revealed that Zld‐PB is exclusively present in NB but absent from INPs, with some exceptions where Zld can sometimes still be detected in recently born imINPs (Fig 3F). We observe a similar pattern for other NB‐specific factors such as Dpn (Appendix Fig S9B, yellow arrow). Taken together, our data indicate that active Zld is specifically expressed in NBs and is absent from its progeny. Depleting Zld from the NB leads to a decreased number of INPs, most of which no longer express Dpn.
Brat suppresses Zld‐PB by binding to its 3′UTR
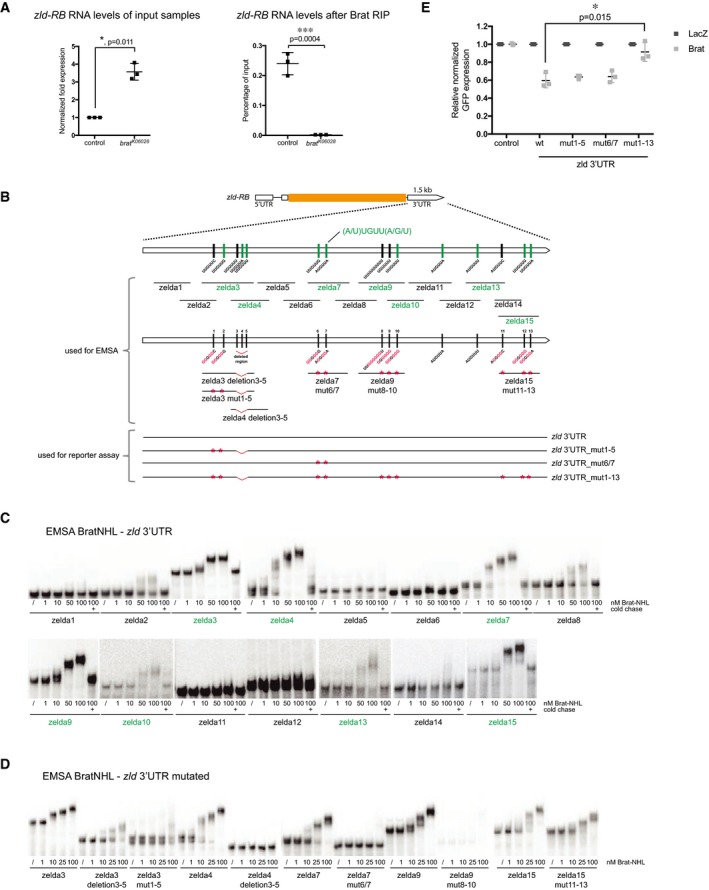
We next investigated whether Zld would be a direct target of Brat. First, we performed RNA immunoprecipitation (RIP) experiments on control and brat K06028 mutant larval brain tissue and observed that Brat can bind to zld‐RB RNA (Fig 4A). Second, in order to investigate the regions of Brat binding within the zld‐RB 3′UTR, we performed an electrophoretic mobility shift assay (EMSA) using the NHL domain of Brat, which was shown to bind mRNA 27, together with various overlapping in vitro transcribed ~150‐nt‐long fragments of the zld‐RB 3′UTR (Fig 4B). Increasing amounts of recombinant Brat‐NHL were incubated with 32P‐labeled RNA fragments and the resulting protein‐RNA complexes analyzed by native gel electrophoresis (Fig 4C). Brat‐NHL did not bind to zld RNA fragments 1, 2, 5, 6, 8, 11, 12, and 14 (Fig 4B and C; “non‐binders” in black), whereas for zld RNA fragments 3, 4, 7, 9, 10, 13, and 15 we observed a clear shift of free RNA to RNA‐protein complex at Brat‐NHL protein concentrations of 50–100 nM (Fig 4B and C; “binders” in green). Interestingly, we observed an almost complete RNA shift at the respective concentrations, indicating high affinity binding. RNA binding was sequence‐specific, as the protein‐RNA complexes could be chased by a 1,000‐fold molar excess of unlabeled binders but not by a 1,000‐fold molar excess of unlabeled t‐RNA, present in all reactions. zld RNA fragments bound by Brat contained a specific motif (A/U)UGUU(A/G/U), the Brat‐binding motif. This motif is very similar to what has recently been identified as a consensus motif for Brat binding 28, 29. To test whether this motif is required for Brat binding to zld RNA, we generated RNA fragments harboring mutations or deletions of the Brat‐binding motif (Fig 4B). Mutations or deletions of the motif sites in zld RNA fragments 3, 4, 7, and 9 reduced or abolished Brat‐NHL binding (Fig 4D). In order to test whether the Brat‐binding motif is required for the repression of zld RNA, we tested the mutated variants of full‐length zld‐RB 3′UTR in a Drosophila S2 reporter assay (Fig 4E). S2 cells were cotransfected with wild‐type or mutated zld‐RB 3′UTR and full‐length Brat. While mutating individual Brat‐binding sites did not interfere with Brat‐mediated repression, mutation of all Brat‐binding sites abolished Brat‐mediated repression of the zld‐RB 3′UTR. Taken together, our data demonstrate that Brat‐NHL directly binds to sequence‐specific motifs in the 3′UTR of zld‐RB, which are required for Brat‐mediated repression.
Figure 4. Brat directly targets the 3′UTR of zld via a specific binding motif.
-
AqPCR analysis of zld‐RB transcript after Brat‐RIP of control and brat K06028 mutant larval brain tissue. Left diagram represents zld‐RB RNA levels of the Input. Right diagram represents zld‐RB RNA levels after Brat‐RIP experiment.
-
BSchematic representation of zld‐RB locus and the zld‐RB 3′UTR fragments used in this study. Sites of the Brat‐binding motif within the zld‐RB 3′UTR are highlighted. Fragments which bind to Brat‐NHL are labeled in green; non‐binding fragments in black. Nucleotide mutations and deletions are marked in red.
-
C, DRecombinant Brat‐NHL was incubated with 32P‐labeled wild‐type (C) or mutated (D) zld RNA fragments as indicated and analyzed by native gel electrophoresis. Note that mutations of the Brat‐binding sites in the zld‐RB 3′UTR greatly impair RNA binding of Brat‐NHL.
-
EDrosophila S2 cells were cotransfected with GFP‐zld‐RB 3′UTR reporters as indicated together with full‐length Brat. RFP was used as a transfection control; LacZ was used as an overexpression control. Note that repression of GFP‐zld‐RB 3′UTR reporter bearing all Brat‐binding site mutations is greatly reduced.
Dpn repression requires high Brat levels
Brat was previously shown to interact with dpn mRNA 29. To compare its binding to dpn and zld RNA, we performed an EMSA experiment using the experimental setup described above (Fig 5A). dpn RNA fragments 1–3 shifted from free RNA to RNA‐protein complexes at Brat‐NHL concentration of 50–100 nM (Fig 5B). In contrast to zld RNA, dpn RNA shifted only partially at a Brat concentration of 50 nM, indicating that Brat binds to dpn RNA with lower affinity. dpn fragments bound by Brat contained the Brat‐binding motif. Consistently, mutations of the motif sites in fragment 1 abolished Brat‐NHL binding (Fig 5C). Interestingly however, mutations of these motifs in fragment 2 and 3 did not prevent Brat‐NHL binding, suggesting the existence of alternative binding specificities. In order to demonstrate that Brat protein–dpn RNA binding can lead to Dpn repression, we co‐expressed a dpn 3′UTR GFP reporter together with full‐length Brat in Drosophila S2 cells. Compared to control, the dpn 3′UTR reporter was repressed upon Brat expression, albeit not to the same extent as the zld 3′UTR reporter (Fig 5D). These data suggest that the translational repression of Dpn requires higher levels of Brat than that of Zld.
Figure 5. Brat represses its targets in a concentration‐dependent manner in INPs.
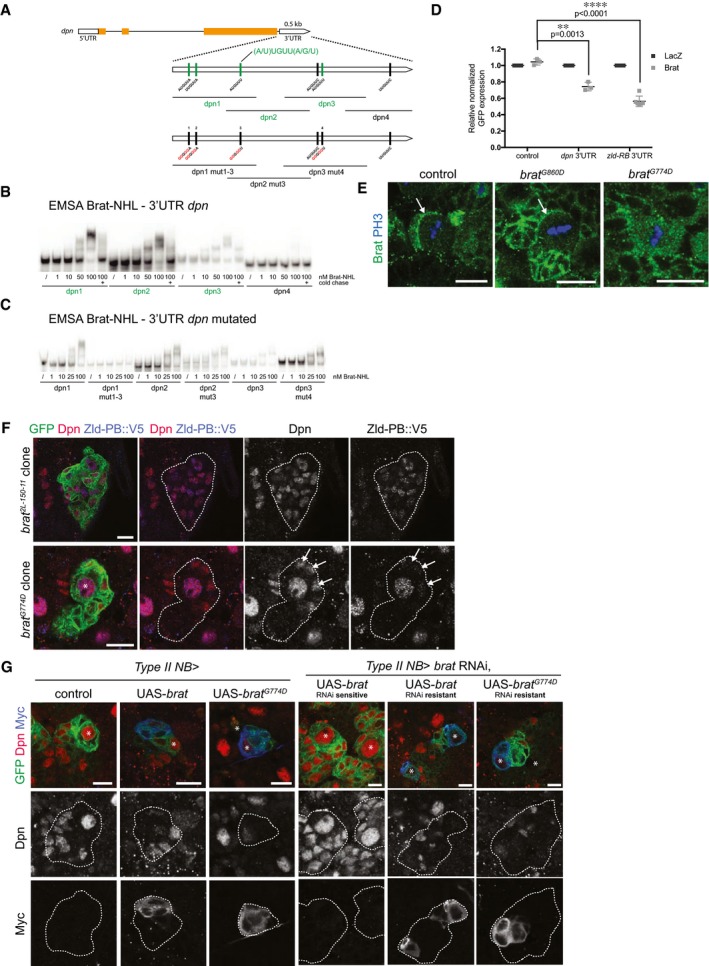
-
ASchematic representation of dpn locus and the dpn 3′UTR fragments used in this study. Sites of the Brat‐binding motif within the dpn 3′UTR are highlighted. Fragments which bind to Brat‐NHL are labeled in green; non‐binding fragments in black. Nucleotide mutations and deletions are marked in red.
-
B, CRecombinant Brat‐NHL was incubated with 32P‐labeled wild‐type (B) or mutated (C) dpn RNA fragments as indicated and analyzed by native gel electrophoresis. Note that mutations of the Brat‐binding sites in the dpn 3′UTR greatly impair RNA binding of Brat‐NHL.
-
DDrosophila S2 cells were cotransfected with GFP‐dpn 3′UTR or GFP‐zld‐RB 3′UTR reporters as indicated together with full‐length Brat. RFP was used as a transfection control; LacZ was used as an overexpression control.
-
EClose‐up images of larval brain NBs stained for PH3 (blue) and Brat (green). In control and brat G860D mutant NBs Brat localizes asymmetrically during mitosis (arrows), whereas in brat G774D mutants Brat remains ubiquitously distributed.
-
FClose‐up images of brat 2L‐150‐11 or brat G774D mutant type II NB lineages marked by membrane‐bound GFP expressing endogenous Zld‐PB::V5 and stained for Dpn (red) and V5 (blue). White arrows indicate immature INPs failing to repress Dpn but not Zld‐PB in a brat G774D clone.
-
GClose‐up images of type II NBs marked with membrane‐bound GFP expressing with wor‐Gal4, ase‐Gal80, (left panels) Myc‐tagged wild‐type brat, or brat G774D or (right panels) RNAi‐sensitive or RNAi‐resistant Myc‐tagged Brat constructs together with brat RNAi and stained for Dpn (red) and Myc (blue). Asterisks designate type II NB.
brat G774D mutants maintain Zld‐PB but not Dpn repression in mINPs
To dissect the relative contribution of Dpn and Zld repression to Brat in vivo function, we used the brat fs1 allele. In brat fs1 (hereafter referred to as brat G774D), glycine 774 in the NHL domain is replaced by Aspartate. Unlike brat 2L‐150‐11 strong loss‐of‐function mutants, brat G774D mutants do not form brain tumors 19, 25. To test the effect of this mutation on Zld and Dpn repression, we analyzed brat G774D MARCM clones. 48 h after heat‐shock induced recombination, control NB clones contained one Dpn‐positive NB, two to three Ase‐negative imINPs, four to five Ase‐positive imINPs and multiple Dpn, Ase double‐positive mINPs (Appendix Fig S8A–C) 42. Clones mutant for the strong loss‐of‐function allele brat 2L‐150‐11 contained multiple Dpn‐positive NB‐like cells and no imINPs or mINPs. brat G774D mutant NB clones, in contrast, contained one Dpn‐positive NB but lacked the first Dpn, Ase double‐negative imINPs stage. Instead, we observed three to four Dpn‐positive, Ase‐negative cells and multiple Dpn, Ase double‐positive mINPs. These Dpn‐positive, Ase‐negative NB daughter cells were located close to the NB and were positive for Mira (Appendix Fig S8E). Importantly, these cells were not more proliferative than WT mINPs and the overall brat G774D clone cell numbers were comparable to WT (Appendix Fig S8F and G). Zld‐PB is absent from NB progeny in controls (Fig 3F). However, although Zld‐PB continued to be expressed in brat null mutant clones, it was still repressed in brat G774D mutant imINPs (Fig 5F). Thus, brat G774D can still repress Zld‐PB but can no longer repress Dpn.
We next aimed to explore the molecular mechanisms underlying the brat G774D mutant phenotype. BratG774D fails to successfully integrate into the Nos/Pum/RNA complex 25. BratG774D also no longer binds to the adaptor protein Mira 19 and therefore cannot localize asymmetrically during NB division (Fig 5E) and is no longer enriched in imINPs. The brat G774D phenotype could be due to the failure of BratG774D to interact with Pum. However, Zld‐PB expression was normal upon pum knockdown or in pum ET1 type II NB clones (Appendix Fig S8D). In addition, neither pum RNAi nor pum ET1 clones affected Dpn repression in imINPs (Appendix Fig S11B), indicating that Brat‐mediated Dpn and Zld repression is Pum‐independent. Alternatively, the brat G774D phenotype could be explained by the inability of BratG774D to segregate asymmetrically, which would result in lower Brat levels in INPs that are sufficient to repress Zld‐PB but no longer repress Dpn. To test this possibility, we overexpressed BratG774D in NBs and analyzed Dpn expression. Consistently, overexpressing wild‐type Brat and BratG774D in NBs both strongly reduced Dpn expression indicating that BratG774D is still capable of repressing Dpn at high protein levels (Fig 5G). We also generated brat RNAi‐resistant constructs to test the ability of BratG774D to rescue tumor formation in a brat RNAi background. Overexpressing wild‐type Brat and BratG774D both rescued brat tumor formation (Fig 5G, Appendix Fig S11C). Taken together, these data suggest that the lack of Dpn repression in brat G774D mutants is due to the defect in asymmetric segregation.
Low Brat levels repress Zld but not Dpn in mINPs
Our results prompted us to test whether the differential activity of Brat toward Zld and Dpn is important for lineage specification. Previous experiments have revealed that Brat levels decrease during INP maturation (a period of 5–6 h 43). Upon NB division, Brat was highly concentrated in imINPs, but only around 40% of the protein level was still present in Dpn‐positive mINPs (Fig 6A, Appendix Fig S9A–C). Consistently, analysis of heat‐shock induced Brat expression showed that protein levels were maximal 1 h after heat shock but strongly reduced 5–10 h after the heat shock (Fig 6B). Thus, Brat protein turnover roughly corresponds to the time of INP maturation. This suggests the intriguing possibility that the decrease in Brat protein levels during INP maturation might allow re‐expression of Dpn but not Zld in mINPs.
Figure 6. High Brat levels are required to repress Dpn.
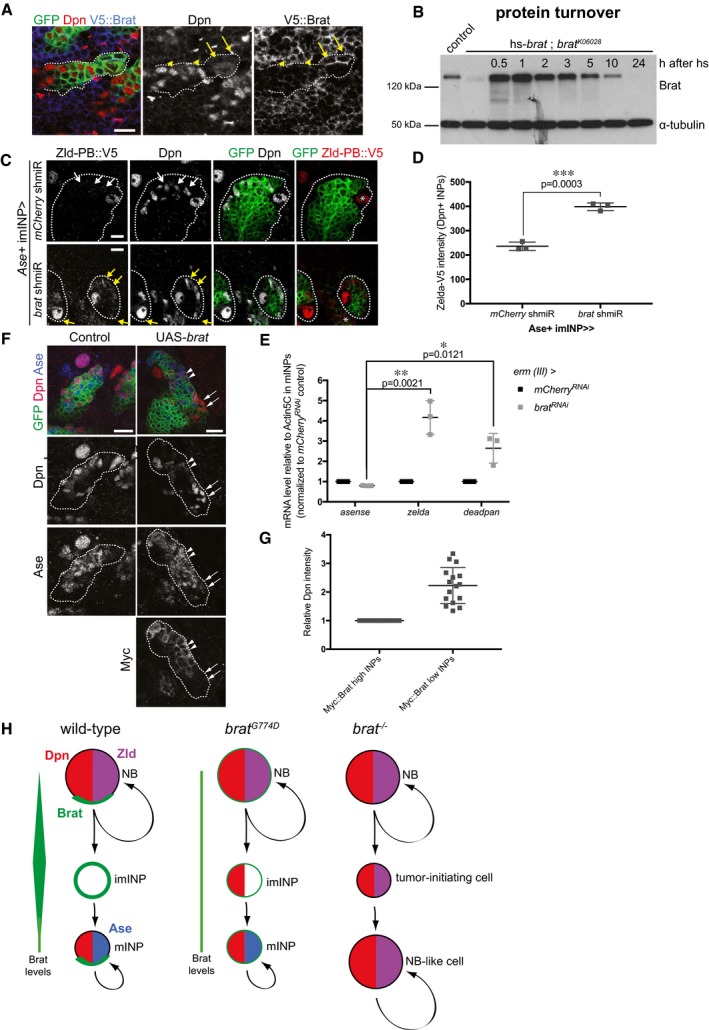
- Close‐up images of larval brains expressing brat shmiR and membrane‐bound GFP from type II NB driver (wor‐Gal4, ase‐Gal80) and stained for Dpn (red) and V5‐Brat (blue). Yellow arrows and arrowheads indicate Dpn‐negatve immature INPs and Dpn‐positive mature INPs, respectively.
- Western blot analysis showing Brat protein turnover. Flies expressing Brat under control of the heat‐shock promoter (hs‐brat) were heat‐shocked for 1 h and Brat expression was determined at indicated time points after heat shock. Note that around 1 h Brat expression reaches its maximum and after 5–10 h, the expression level is strongly reduced.
- Close‐up images of type II NB lineages expressing endogenous V5‐tagged Zld‐PB and expressing membrane‐bound GFP, mCherry, or brat shmiR from Ase+ INPs driver (erm‐Gal4, 3rd chromosome) stained for Dpn and V5 (red). Note brat shmiR INPs re‐express Zld‐PB::V5 (yellow arrows) compared to mCherry shmiR INPs (white arrows).
- Quantification of Zld‐PB::V5 signal (intensity normalized by the INPs’ outlined surface, ImageJ) from mCherry and brat shmiR Dpn‐positive INPs per lineage.
- qPCR analysis of Ase (negative control), zld and dpn transcripts after brat RNAi in FACS‐sorted Ase+ INPs (erm‐Gal4, 3rd chromosome).
- Close‐up images of type II NB lineages expressing membrane‐bound GFP and myc‐tagged Brat from the Ase‐positive imINP driver (erm‐Gal4 on the 3rd chromosome) and stained for Dpn (red) and Myc. mINPs expressing high (arrowheads) and low (arrows) Myc‐Brat are indicated.
- Quantification analysis of Dpn fluorescence intensity measurements in mature INPs expressing high levels of Myc‐Brat (closer to the NB) vs. in INPs expressing low levels of Myc‐Brat (more distant to the NB). White asterisks indicate type II NB.
- Model of Brat‐mediated repression of Zld and Dpn in type II NB lineages.
To support this hypothesis, we used two experimental approaches to titrate Brat levels in INPs. First, we performed brat RNAi in the type II NB progeny using the erm‐Gal4 driver (3rd chromosome). Indeed, brat RNAi in Ase‐positive imINPs was sufficient to partially lift Zld repression in INPs (Fig 6C–E). In a second approach, we used a Myc‐tagged Brat construct (Myc‐Brat) that allowed us to follow Brat protein levels and Dpn and Zld suppression simultaneously in INPs. When expressed from the erm‐Gal4 driver (3rd chromosome), this construct resulted in high levels of Myc‐Brat in both Ase‐positive imINPs and young mINPs (Fig 6F and G). Due to high Brat protein turnover, however, Myc‐Brat levels decreased in older mINPs that are located further away from the NB. As a consequence, young mINPs with high levels of Myc‐Brat showed low Dpn levels whereas older mINPs with lower Myc‐Brat levels had twofold higher Dpn levels (Fig 6F and G). Altogether, these data suggest that the decrease in Brat protein levels during INP maturation allows re‐expression of Dpn but not Zld‐PB in mINPs.
Discussion
Our data suggest a molecular mechanism through which the tumor suppressor Brat could perform its function in type II NB lineages. We first demonstrate that the transcription factor Zld, in addition to Dpn, is a critical target of Brat‐mediated translational repression. Via its NHL domain, Brat directly binds to the dpn and zld 3′UTRs and represses translation via a sequence‐specific binding motif. Several results suggest that distinct levels of Brat are required to suppress these two targets: First, we demonstrate that brat G774D, a hypomorphic mutant quantitatively attenuated in imINPs, is able to repress Zld but not Dpn, but can suppress both targets upon overexpression. Second, we demonstrate that Dpn repression correlates with high Brat protein levels, both in wild‐type imINPs as well as upon overexpression. Finally, we demonstrate that Brat binds to dpn RNA with lower affinity than to zld RNA. Together, our data suggest a model (Fig 6H) where high Brat protein levels repress both dpn and zld RNA in young imINPs immediately after asymmetric cell division. Over time, Brat levels decline allowing for the re‐expression of Dpn but not Zld in INPs. Importantly, our data do not exclude that pre‐ or co‐transcriptional regulation of these factors would occur in parallel of Brat‐mediated post‐transcriptional control.
What could be the role of a Brat level‐dependent inhibition mechanism during type II NB lineage progression? Dpn is well known to be a crucial factor supporting self‐renewal in NB 44, 45, 46 and needs to be repressed in imINPs to prevent reversion into tumor NB‐like cells (illustrated in Appendix Fig S11A). Our data further demonstrate that decreasing Brat levels in mINPs are crucial to allow Dpn re‐expression, which is essential to reinitiate cell division (Fig 3A and B). We have identified Zld as a crucial determinant allowing this re‐expression in INPs. For this, Zld seems to act in type II NBs where its active isoform is expressed but seems dispensable in INPs, where its levels are continuously repressed by Brat (Fig 6C–E).
Importantly however, artificially re‐establishing Zld expression in INPs does not result in ectopic NB formation in a wild‐type or brat G774D background (data not shown). This can be explained by two possibilities. First, although Zld‐PD cannot activate transcription and lacks the relevant domains 39, its RNA is expressed in the NB progeny and could potentially antagonize Zld‐PB function. Whether Zld‐PD is actually translated into functional proteins in these cells would remain to be explored. However, it has been shown that co‐expression of Zld‐PA (identical to PB) and Zld‐PD significantly reduced gene expression, demonstrating that Zld‐PD acts dominantly to suppress Zld‐mediated transcriptional activation. This was explained by competition of the two isoforms for interaction with cofactors required to activate transcription 39. Second, it is possible that low Brat levels maintain the repression of one or several—yet to be characterized—key factors in addition to Zld. Our RNA‐seq data and brat lethality suppression assay highlighted dm and klu as potential additional targets of Brat repression. Thus, the protein level‐dependent Brat‐mediated inhibition mechanism allows a tightly regulated repression machinery during INP cell fate specification and prevents INPs from reverting into ectopic NBs.
Brat does not repress Zld in NBs although it is expressed there. It is possible that an inhibitor or a specific post‐transcriptional modification prevents Brat from acting in NBs. Alternatively, Brat levels normally present in the NBs are too low for translational target gene repression. In INPs, both asymmetric segregation and transcriptional upregulation increase Brat levels and target genes are repressed. Why are zld and dpn RNA differentially sensitive to Brat levels? Brat directly binds to sequence‐specific motifs in the zld and dpn 3′UTR. While the zld 3′UTR contains 10 motif sites, dpn 3′UTR contains only 4. It is possible that the abundance of motif sites within a 3′UTR determines the efficiency of Brat binding and that with a higher number of motif sites within the 3′UTR, the Brat‐binding affinity increases.
What is the molecular activity of Zld in NBs? Zld can bind to the TAGteam elements, which are highly distributed among the genome. Also, Zld was shown to bind to the dpn locus, which contains two TAG motifs in the promoter region and seems to regulate Dpn expression in larval brains. Surprisingly, however, removing Zld only weakly interferes with Dpn expression in the NB, whereas it has very strong consequences for Dpn expression in INPs resulting in their underproliferation. This result is very surprising as the presumably active Zld‐PB protein is present in NBs but not in mINPs. It is possible that Zld‐PD has an active role in the NB progeny. However, it has been demonstrated very nicely that all four zinc fingers are required for TAGteam binding and that Zld‐PD lacking three of the four zinc fingers fails to activate transcription 39. We also cannot exclude that levels of Zld‐PB undetectable with our reagents repress Dpn in INPs. More likely, however, Zld interacts with loci such as Dpn in NBs to license it for transcription. This function would be analogous to what has been described for early embryos where Zld binds to its target regions long before they are transcribed during MZT 34. In this scenario, Zld would modify the its target loci in NBs so that transcription can be re‐initiated in mINPs once Brat levels have declined.
Our transcriptome data showed that genes bearing at least one Brat‐binding motif were significantly enriched among the genes repressed by Brat in type II NB lineages (FDR1.1, FPKM > 1, log2 fold change 1; Appendix Fig S10A). Thus, RNAs harboring a Brat‐binding motif are preferentially repressed in INPs and we suggest that Brat may target a number of genes other than dpn and zld for translational repression. It was proposed that Brat also antagonizes Klu and Arm in imINPs 31. Klu is a self‐renewal factor whose 3′UTR contains three Brat‐binding motifs, and we cannot exclude that it serves as an additional target for Brat.
How does Brat mediate translational repression? Brat can repress hb RNA in a complex with Pum and Nos 25. We show that repression of Dpn or Zld is not altered in pum knockdown or pum mutant clones. This is consistent with what has recently been described for Brat function during embryogenesis 28 and is also supported by our observation that the known Brat/Pum target Nos cannot be detected in NB lineages 40. Thus, Brat most likely acts in NB lineages through a mechanism independent of the Pum/Nos complex. Brat can interact with the cap‐binding protein d4EHP, which inhibits translation by binding the mRNA 5′ cap structure 24, 47. Mutations of amino acid G860, R837 and K882 in the Brat‐NHL domain reduce or abolish interaction with d4EHP 24. While brat G860D no longer represses Dpn, brat R837D and brat K882E still do (Appendix Fig S10B) suggesting that d4EHP binding is not crucial for this activity. Similar results have been previously obtained for Brat‐mediated repression of hb 27. Brat also binds Not1, a subunit of the CCR4‐Not complex catalyzing mRNA deadenylation 48. It is possible that Brat‐mediated translational repression is accompanied by recruitment of CCR4/Not to promote RNA degradation. The translational repressor Smaug 49, 50, 51 has been shown to recruit CCR4/Not to trigger maternal transcript destabilization, and it is plausible that Brat and Smaug act together in translational repression during INP cell fate specification.
The self‐renewing capacity of INPs differs from that of NBs. While NBs divide up to 30 times during their life span, INPs undergo only five to eight rounds of division. So far, all known self‐renewing factors in NBs are first repressed in imINPs but reappear in mINPs (Dpn, N, Klu, HLHmgamma). To our knowledge, active Zld‐PB is the first factor common to all NBs (type I and type II) that is not re‐expressed in INPs but crucial for their proliferation. Zld has been described as a key player in activating the genome at the MZT and to promote timely and robust transcriptional activation 34, 35. We suggest that potential activation of the Dpn locus by Zld in the NB is deactivated in older INPs so that INPs lose their self‐renewing capacity over time. The same may also happen to brat mutant NB‐like cells upon knockdown of zld: The brat tumor contains Ase‐positive differentiated cells and is progressively rather than completely suppressed.
The Trim‐NHL family proteins are conserved among metazoa and are known to play a role in cell fate determination. In humans, only Trim3 acts as a tumor suppressor in glioblastoma that reprograms glioma stem cells toward differentiation by suppressing c‐Myc 52. Also, the mammalian Trim71 promotes reprogramming of differentiated cells into induced pluripotent stem cells by inhibiting the prodifferentiation factor EGR1 53. It would be worth the effort to analyze if the mechanism behind their repressive function is similar to Brat.
Similar to Drosophila, transit‐amplifying lineages can also be found in human brain development and are thought to underlie the evolutionary expansion of the neocortex 54, 55. It would be interesting to evaluate if differences in the presence of a transcriptional activator like Zld also determine the cell behavior of the distinct cell types in human neural stem cell lineages.
Materials and Methods
Fly strains, RNAi, and clonal analysis
Drosophila stocks used in this study were brat RNAi [Transformant ID (TID) 31333 and 105054; Vienna Drosophila RNAi Center (VDRC)]; grh RNAi (TID 106879; VDRC); dm RNAi (TID 106006; VDRC); dpn RNAi (TID 106181; VDRC); pnt RNAi (TID 105390; VDRC); zld IR (TID 38706; VDRC); brat shmiR (BL34646); pum shmiR (BL36676, BL38241); zld‐RB 3′UTR shmiR (BL42016) dpn shmiR (generated in this study, see below); zld shmiRs [“all”, −RB and −RD (1, 2 and 3)] (generated in this study, see below); FRT40A; FRT40A, brat 2L‐150‐11 9; FRT40A, brat fs1 25; FRT40A, brat ts1 23; brat K06028 23; FRT82B; FRT82B pum ET‐1 56; UAS‐3xFlag6xMyc::brat constructs (generated in this study, see below); UAS‐zld‐RB 36, UAS‐dpn (dpn full‐length cDNA cloned into a pUAST and inserted into attP40 site, this study); V5::brat (generated in this study, see below); zld‐RB::V5 (generated in this study, see below). Gal4 driver lines used were UAS‐dcr1; wor‐Gal4, ase‐Gal80; UAS‐mCD8::GFP 57; erm‐Gal4 (II); UAS‐mCD8::GFP 21; UAS‐CD8::GFP; erm‐Gal4 (III) 58, 59. Clones of NBs homozygous for brat 2L‐150‐11, brat fs1, brat ts1 , and pum ET‐1 were generated by Flippase (FLP)/FLP recombination target (FRT)‐mediated mitotic recombination, using elav‐Gal4 (C155) 60. Larvae were heat‐shocked for 1 h at 37°C and dissected as wandering third‐instar larvae. RNAi and shmiR crosses were set up and reared at 29°C, and wandering third‐instar larvae were dissected 5 days after. For the brat rescue double RNAi crosses were set up and reared at 29°C. At least 10 flies of the respective genotype were collected 2 days after adult hatching, kept at 29 degrees, and recounted 2 weeks after.
Generation of shmiR lines
Efficient shRNA prediction was made by implementing an algorithm described previously 61 and modified for 22‐bp shmiRs. An off‐target algorithm was designed to exclude potential off‐targets 62, 63. The synthesized oligos were annealed and cloned into the Walium20 vector according to the protocols of The Transgenic RNAi Project (flyrnai.org). Oligo sequences used for generating dpn and zld shmiRs are listed in Dataset EV2.
Generation of brat overexpression lines
brat coding sequence was recombined into the Gateway pDONR221 vector. Single nucleotide modifications were generated using QuikChange Lightning Site‐directed Mutagenesis Kit (Agilent Technologies). The brat RNAi (TID 105054)‐resistant sequence was generated using a custom‐made Perl script, and the resulting fragment was de novo synthesized (Mr. Gene, Life Technologies). The newly synthesized fragment, covering the attL1 site of the pDONR221 vector and the brat RNAi‐resistant part, was cloned into pDONR221‐brat backbone using the enzymes ApaI and BbvCI. brat modified and unmodified coding sequences were recombined into the Gateway destination vector pUASt containing an N‐terminal 3xFlag‐6xMyc site and an attB site for landing site integration. Plasmids were injected into flies that contain an attP‐landing site with PhiC31 activity at the third chromosome (MM2).
Generation of V5::brat and zld‐RB::V5 using the Cas9/CRISPR system
Endogenous V5::brat and zld::V5 were generated using the Cas9/CRISPR technology as described in 41. Briefly, a donor oligo, which contains the V5 tag and about 60 nucleotides homologues to the brat or zld gene upstream and downstream of the V5‐tag integration site, was synthesized by Integrated DNA Technology (http://eu.idtdna.com/site):
V5::brat:5′GCTGTTACTCCTAAAGGTAAACGGAGCCACCGACGGCACTTACAACAtAAAGATGGGTAAGCCTATACCTAACCCTCTTCTTGGTCTAGATAGCACGGCGAGTTCGCCGACACCATCTCTGGACTCGATGCGGGGCGGGGCGAACTCGATTGAATCATAC
zld::V5:5′ATCAAGAGCGAGTACGTGCAGGAGGAGTTTCAGATGATCGAGAAGAGCATAGAGCTCTACGGTAAGCCTATACCTAACCCTCTTCTTGGTCTAGATAGCACGTGAATGAGTGGGCAGGCCACTGGGTTCTGGGTTTTGAAATGCTCCTAGGCTTTGAGCTTGTCT
Guidance RNAs (brat: CACCGACACCATCTCTGGAC, zld: ATAGAGCTCTACTGAATGAG) were cloned into the CRISPR/Cas9 vector 41 and transfected together with the donor oligo into w1118 flies. Transgenic flies were screened by PCR flanking the V5‐tag region.
Antibodies
Antibodies used in this study were guinea pig anti‐Dpn, 1:1,000, 42; rat anti‐Ase, 1:500, 42; chicken anti‐GFP (Abcam); rabbit anti‐Brat, 1:100, 9; mouse anti‐pH3 (Cell Signaling Technology); mouse anti‐V5 (Sigma‐Aldrich); mouse anti‐Myc (9E10); mouse anti‐Flag M2 (Sigma‐Aldrich); mouse anti‐Lamin ADL67.10 (DSHB); mouse anti‐Bruchpilot nc82, 1:10 (DSHB); guinea pig anti‐Mira, 1:250; rabbit anti‐Hamlet, 1:50 (homemade, 42); rabbit anti‐Dichaete, 1:1,000, 64; mouse anti‐Eyeless, 1:10 (DSHB), rat anti‐Grainy head, 1:1,000, 14.
Immunohistochemistry
For immunofluorescence, larval or adult brains were dissected in PBS, fixed for 20 min in 5% paraformaldehyde (PFA) in PBS (larvae) or in 5% PFA with 0.1% Triton X‐100 (adult), and blocked in 1% normal goat serum (NGS) (larvae) or 5% NGS (adult) in PBS with 0.1% Triton X‐100 (blocking solution), and antibodies were diluted in blocking solution. Brains were mounted in Vectashield (Vector Laboratories). Fluorescence in situ hybridization (FISH) protocol was adapted from 65 and Stellaris protocols. Larval brains were dissected in PBS, fixed for 20 min in 4% PFA in PBS/DEPC, and permeabilized in 70% EtOH for at least 24 h at 4°C. 250 nM final concentration of Stellaris probes in hybridization buffer was used and incubated overnight with larval brains in the dark at 37°C. Brains were mounted in 2× SSC and imaged the same day. Confocal images were acquired on LSM780 microscopes (Carl Zeiss GmbH).
Quantification of Brat, Myc::Brat, Zld‐PB::V5, and Dpn expression
Quantification of protein expression was performed using the Histo feature of the ZEN software. Membrane‐bound GFP was used to outline the respective cell section (at its biggest appearance), and the mean intensity of Brat, Myc::Brat, or Dpn levels were measured.
Brain tumor transplantation and metastasis quantification
L3 brains from UAS‐dcr2/UAS‐brat RNAi; wor‐Gal4, ase‐Gal80/+; UAS‐Stinger::RFP/UAS‐mCherry shmiR or UAS‐dcr2/UAS‐brat RNAi; wor‐Gal4, ase‐Gal80/UAS‐zld shmiR; UAS‐Stinger::RFP/+ were dissected and mechanically disrupted in PBS. RFP+ cells concentration were estimated on a Neubauer cell counter, and ~500 cells were immediately intra‐thoracically injected into 3‐ to 6‐day‐old adult females with a Nanoject II (Drummond). Bright‐field, GFP and RFP pictures of living flies were taken on a Lumar fluorescence stereomicroscope/color SPOT camera repeatedly after transplantation. GFP autofluorescence signal was subtracted from tumor‐specific RFP signal and displayed in false colors red‐green‐blue‐cyan‐magenta‐yellow‐white from least to most intense. Alternatively, the RFP‐specific signal was quantified on ImageJ from the raw intensity displayed in Measure tool on the delineated area of the whole fly. Intensities of all flies in one picture were averaged.
Cell dissociation, FACS, sample preparation, and RNA sequencing
Cell dissociation, FACS, and bioinformatic analysis were done as previously described with minor modifications 40, 66. UAS‐dcr2; wor‐Gal4, ase‐Gal80; UAS‐mCD8::GFP line was used to induce the expression of membrane‐bound GFP and brat RNAi. GFP expression with and without brat RNAi was induced under the control of tub‐Gal80ts for 24 h. GFP‐positive cells were sorted by FACS, and RNA of sorted cells was isolated. Seventy‐six base pair Illumina paired‐end sequencing of Poly‐A‐mRNA libraries was performed on GAIIx. The experiment lacked biological replicates due to difficulties in getting sufficient material to prepare the sequencing library. For the analysis, DESeq was instructed to ignore the condition labels and estimate the variance by treating all the samples as if they were replicates of the same condition (method = “blind”) 67. Sequencing results of selected hits were verified by qPCR. Around 200 larval brains were dissected to obtain sufficient GFP‐positive cells per replicate of the qPCR experiment. First‐strand cDNA was generated using random primers on TRIzol‐extracted total FACS‐sorted cell RNA. qPCR was done using Bio‐Rad IQ SYBR Green Supermix on a Bio‐Rad CFX96 cycler. Expression of each gene was normalized to RpS8, and relative levels were calculated using the method 68. Oligo sequences used for qPCR analysis of FACS‐sorted samples are listed in Dataset EV2.
RNA immunoprecipitation (RIP)
RIP experiment was adopted from 69. Briefly, 100 control and 50 brat K06028 mutant 3rd instar larval brains or 1 ml of control and Brat overexpression S2 cell culture were fixed in 0.5% formaldehyde. Fixed tissue was homogenized in RIP lysis buffer and briefly sonicated with a microtip sonicator. Brain samples were first incubated with 7.5 μg rabbit anti‐Brat antibody and subsequently with Protein G dynabeads. S2 cell samples were incubated with 10 μl anti‐Myc beads. Material elution and cross‐linking reversal was performed by incubating samples in RIP elution. After RNA purification, pellets were resuspended in 20 μl H2O/DEPC and qPCR was performed. See Appendix Supplementary Methods and Dataset EV2.
Drosophila S2 cell reporter assay
dpn and zld 3′UTR including an N‐terminal stop codon were cloned from Drosophila genomic DNA (oligos used for PCR, see Dataset EV2) and recombined first into the Gateway pDONR221 vector and subsequently into the Gateway destination vector pAGW containing an Actin5C promoter and a GFP coding sequence (The Drosophila Gateway vector collection). GFP control reporter: A SV40 terminator sequence was recombined into pAGW. Drosophila S2R+ cells were cotransfected with GFP control/3′UTR reporter, Actin5C‐RFP expression vector (transfection control) and Actin5C‐lacZ/Brat overexpression vector. RFP transfected cells were preselected by FACSCanto II Flow Cytometer, and GFP intensity mean of RFP‐positive cells was measured. Three independent transfections were analyzed. Mycoplasma contamination was not checked.
EMSA experiment
The approximately 150‐nt‐long fragments of the zld and dpn 3′UTR and point mutants thereof were in vitro transcribed from PCR‐amplified DNA templates using oligos listed in Dataset EV2. In vitro transcription, protein purification and native gel electrophoresis were essentially carried out as described in 27. Point mutations were introduced by site‐directed mutagenesis 70 with oligos listed in Dataset EV2.
Statistics
Statistical analyses were performed with GraphPad Prism 7. Unpaired two‐tailed Student's t‐test was used to assess statistical significance between two genotypes/conditions. No statistical methods were used to predetermine the sample size. Sample sizes for experiments were estimated based on previous experience with a similar setup that showed significance. Experiments were not randomized, and investigator was not blinded. No samples or animals were excluded from our analysis.
Data availability
RNA sequencing data have been submitted to NCBI GEO project Accession Number GSE104592.
Author contributions
IR, FB, and JAK designed and interpreted experiments. IR, FB, and VS performed the experiments. IL performed Brat–RNA interaction (EMSA) experiments. TRB performed RNA‐seq data analysis. GM supervised the work of IL. IR and JAK wrote the manuscript with input from all authors.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Appendix
Dataset EV1
Dataset EV2
Source Data for Appendix
{kind=link}
Review Process File
Acknowledgements
We wish to thank Elke Kleiner, Peter Duchek, Sara Farina Lopez, Joseph Francis Gokcezade, and Merve Deniz Abdusellamoglu for technical assistance; all members of the J.A.K. laboratory for discussions; S.G. Tsitilou for sharing reagents; F.B. was supported by an EMBO Long‐Term Fellowship (LTF 1280‐2014). Work in the J.A.K. laboratory is supported by the Austrian Academy of Sciences, the EU Seventh Framework Programme network EuroSyStem, the Austrian Science Fund (grants I_552‐B19 and Z_153_B09), and the advanced grants of the European Research Council 250342 (NeuroSyStem) and 695642 (MiniBrain).
EMBO Reports (2018) 19: 102–117
Reference
- 1. Caussinus E, Gonzalez C (2005) Induction of tumor growth by altered stem‐cell asymmetric division in Drosophila melanogaster . Nat Genet 37: 1125–1129 [DOI] [PubMed] [Google Scholar]
- 2. Knoblich JA (2010) Asymmetric cell division: recent developments and their implications for tumour biology. Nat Rev Mol Cell Biol 11: 849–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Inaba M, Yamashita YM (2012) Asymmetric stem cell division: precision for robustness. Cell Stem Cell 11: 461–469 [DOI] [PubMed] [Google Scholar]
- 4. Homem CCF, Knoblich JA (2012) Drosophila neuroblasts: a model for stem cell biology. Development 139: 4297–4310 [DOI] [PubMed] [Google Scholar]
- 5. Reichert H (2011) Drosophila neural stem cells: cell cycle control of self‐renewal, differentiation, and termination in brain development. Results Probl Cell Differ 53: 529–546 [DOI] [PubMed] [Google Scholar]
- 6. Weng M, Lee C‐Y (2011) Keeping neural progenitor cells on a short leash during Drosophila neurogenesis. Curr Opin Neurobiol 21: 36–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hirata J, Nakagoshi H, Nabeshima Y, Matsuzaki F (1995) Asymmetric segregation of the homeodomain protein Prospero during Drosophila development. Nature 377: 627–630 [DOI] [PubMed] [Google Scholar]
- 8. Knoblich JA, Jan LY, Jan YN (1995) Asymmetric segregation of Numb and Prospero during cell division. Nature 377: 624–627 [DOI] [PubMed] [Google Scholar]
- 9. Betschinger J, Mechtler K, Knoblich JA (2006) Asymmetric segregation of the tumor suppressor brat regulates self‐renewal in Drosophila neural stem cells. Cell 124: 1241–1253 [DOI] [PubMed] [Google Scholar]
- 10. Guo M, Jan LY, Jan YN (1996) Control of daughter cell fates during asymmetric division: interaction of Numb and Notch. Neuron 17: 27–41 [DOI] [PubMed] [Google Scholar]
- 11. Choksi SP, Southall TD, Bossing T, Edoff K, de Wit E, Fischer BE, van Steensel B, Micklem G, Brand AH (2006) Prospero acts as a binary switch between self‐renewal and differentiation in Drosophila neural stem cells. Dev Cell 11: 775–789 [DOI] [PubMed] [Google Scholar]
- 12. Couturier L, Mazouni K, Schweisguth F (2013) Numb localizes at endosomes and controls the endosomal sorting of notch after asymmetric division in Drosophila . Curr Biol 23: 588–593 [DOI] [PubMed] [Google Scholar]
- 13. Karcavich R, Doe CQ (2005) Drosophila neuroblast 7‐3 cell lineage: a model system for studying programmed cell death, Notch/Numb signaling, and sequential specification of ganglion mother cell identity. J Comp Neurol 481: 240–251 [DOI] [PubMed] [Google Scholar]
- 14. Baumgardt M, Karlsson D, Terriente J, Díaz‐Benjumea FJ, Thor S (2009) Neuronal subtype specification within a lineage by opposing temporal feed‐forward loops. Cell 139: 969–982 [DOI] [PubMed] [Google Scholar]
- 15. Bello BC, Izergina N, Caussinus E, Reichert H (2008) Amplification of neural stem cell proliferation by intermediate progenitor cells in Drosophila brain development. Neural Dev 3: 5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Boone JQ, Doe CQ (2008) Identification of Drosophila type II neuroblast lineages containing transit amplifying ganglion mother cells. Dev Neurobiol 68: 1185–1195 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bowman SK, Rolland V, Betschinger J, Kinsey KA, Emery G, Knoblich JA (2008) The tumor suppressors Brat and Numb regulate transit‐amplifying neuroblast lineages in Drosophila . Dev Cell 14: 535–546 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bier E, Vaessin H, Younger‐Shepherd S, Jan LY, Jan YN (1992) deadpan, an essential pan‐neural gene in Drosophila, encodes a helix‐loop‐helix protein similar to the hairy gene product. Genes Dev 6: 2137–2151 [DOI] [PubMed] [Google Scholar]
- 19. Lee C‐Y, Wilkinson BD, Siegrist SE, Wharton RP, Doe CQ (2006) Brat is a Miranda cargo protein that promotes neuronal differentiation and inhibits neuroblast self‐renewal. Dev Cell 10: 441–449 [DOI] [PubMed] [Google Scholar]
- 20. Song Y, Lu B (2011) Regulation of cell growth by Notch signaling and its differential requirement in normal vs. tumor‐forming stem cells in Drosophila . Genes Dev 25: 2644–2658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Xiao Q, Komori H, Lee C‐Y (2012) klumpfuss distinguishes stem cells from progenitor cells during asymmetric neuroblast division. Development 139: 2670–2680 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Janssens DH, Komori H, Grbac D, Chen K, Koe CT, Wang H, Lee CY (2014) Earmuff restricts progenitor cell potential by attenuating the competence to respond to self‐renewal factors. Development 141: 1036–1046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Arama E, Dickman D, Kimchie Z, Shearn A, Lev Z (2000) Mutations in the beta‐propeller domain of the Drosophila brain tumor (brat) protein induce neoplasm in the larval brain. Oncogene 19: 3706–3716 [DOI] [PubMed] [Google Scholar]
- 24. Cho PF, Gamberi C, Cho‐Park YA, Cho‐Park IB, Lasko P, Sonenberg N (2006) Cap‐dependent translational inhibition establishes two opposing morphogen gradients in Drosophila embryos. Curr Biol 16: 2035–2041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sonoda J, Wharton RP (2001) Drosophila brain tumor is a translational repressor. Genes Dev 15: 762–773 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Harris RE, Pargett M, Sutcliffe C, Umulis D, Ashe HL (2011) Brat promotes stem cell differentiation via control of a bistable switch that restricts BMP signaling. Dev Cell 20: 72–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Loedige I, Stotz M, Qamar S, Kramer K, Hennig J, Schubert T, Loffler P, Langst G, Merkl R, Urlaub H et al (2014) The NHL domain of BRAT is an RNA‐binding domain that directly contacts the hunchback mRNA for regulation. Genes Dev 28: 749–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Laver JD, Li X, Ray D, Cook KB, Hahn NA, Nabeel‐Shah S, Kekis M, Luo H, Marsolais AJ, Fung KY et al (2015) Brain tumor is a sequence‐specific RNA‐binding protein that directs maternal mRNA clearance during the Drosophila maternal‐to‐zygotic transition. Genome Biol 16: 94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Loedige I, Jakob L, Treiber T, Ray D, Stotz M, Treiber N, Hennig J, Cook KB, Morris Q, Hughes TR et al (2015) The crystal structure of the NHL domain in complex with RNA reveals the molecular basis of Drosophila brain‐tumor‐mediated gene regulation. Cell Rep 13: 1206–1220 [DOI] [PubMed] [Google Scholar]
- 30. Neumüller RA, Betschinger J, Fischer A, Mechtler K, Cohen SM, Knoblich JA (2008) Mei‐P26 regulates microRNAs and cell growth in the Drosophila ovarian stem cell lineage. Nature 454: 241–245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Komori H, Xiao Q, McCartney BM, Lee C‐Y (2013) Brain tumor specifies intermediate progenitor cell identity by attenuating β‐catenin/Armadillo activity. Development 141: 51–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Staudt N, Fellert S, Chung H‐R, Jäckle H, Vorbrüggen G (2006) Mutations of the Drosophila zinc finger‐encoding gene vielfältig impair mitotic cell divisions and cause improper chromosome segregation. Mol Biol Cell 17: 2356–2365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Liang H‐L, Nien C‐Y, Liu H‐Y, Metzstein MM, Kirov N, Rushlow C (2008) The zinc‐finger protein Zelda is a key activator of the early zygotic genome in Drosophila . Nature 456: 400–403 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Harrison MM, Li X‐Y, Kaplan T, Botchan MR, Eisen MB (2011) Zelda binding in the early Drosophila melanogaster embryo marks regions subsequently activated at the maternal‐to‐zygotic transition. PLoS Genet 7: e1002266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Nien C‐Y, Liang H‐L, Butcher S, Sun Y, Fu S, Gocha T, Kirov N, Manak JR, Rushlow C (2011) Temporal coordination of gene networks by Zelda in the early Drosophila embryo. PLoS Genet 7: e1002339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Giannios P, Tsitilou SG (2013) The embryonic transcription factor Zelda of Drosophila melanogaster is also expressed in larvae and may regulate developmentally important genes. Biochem Biophys Res Comm 438: 329–333 [DOI] [PubMed] [Google Scholar]
- 37. Suster ML, Seugnet L, Bate M, Sokolowski MB (2004) Refining GAL4‐driven transgene expression in Drosophila with a GAL80 enhancer‐trap. Genesis 39: 240–245 [DOI] [PubMed] [Google Scholar]
- 38. Pearson JC, Watson JD, Crews ST (2012) Drosophila melanogaster Zelda and single‐minded collaborate to regulate an evolutionarily dynamic CNS midline cell enhancer. Dev Biol 366: 420–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Hamm DC, Bondra ER, Harrison MM (2015) Transcriptional activation is a conserved feature of the early embryonic factor Zelda that requires a cluster of four zinc fingers for DNA binding and a low‐complexity activation domain. J Biol Chem 290: 3508–3518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Berger C, Harzer H, Burkard TR, Steinmann J, van der Horst S, Laurenson A‐S, Novatchkova M, Reichert H, Knoblich JA (2012) FACS purification and transcriptome analysis of Drosophila neural stem cells reveals a role for klumpfuss in self‐renewal. Cell Rep 2: 407–418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gokcezade J, Sienski G, Duchek P (2014) Efficient CRISPR/Cas9 plasmids for rapid and versatile genome editing in Drosophila . G3 (Bethesda) 4: 2279–2282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Eroglu E, Burkard TR, Jiang Y, Saini N, Homem CCF, Reichert H, Knoblich JA (2014) SWI/SNF complex prevents lineage reversion and induces temporal patterning in neural stem cells. Cell 156: 1259–1273 [DOI] [PubMed] [Google Scholar]
- 43. Homem CCF, Reichardt I, Berger C, Lendl T, Knoblich JA (2013) Long‐term live cell imaging and automated 4D analysis of Drosophila neuroblast lineages. PLoS One 8: e79588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. San‐Juán BP, Baonza A (2011) The bHLH factor Deadpan is a direct target of Notch signaling and regulates neuroblast self‐renewal in Drosophila . Dev Biol 352: 70–82 [DOI] [PubMed] [Google Scholar]
- 45. Zhu S, Wildonger J, Barshow S, Younger S, Huang Y, Lee T (2012) The bHLH repressor Deadpan regulates the self‐renewal and specification of Drosophila larval neural stem cells independently of notch. PLoS One 7: e46724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. San‐Juán BP, Andrade‐Zapata I, Baonza A (2012) The bHLH factors Dpn and members of the E(spl) complex mediate the function of Notch signalling regulating cell proliferation during wing disc development. Biol Open 1: 667–676 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cho PF, Poulin F, Cho‐Park YA, Cho‐Park IB, Chicoine JD, Lasko P, Sonenberg N (2005) A new paradigm for translational control: inhibition via 5′‐3′ mRNA tethering by bicoid and the eIF4E cognate 4EHP. Cell 121: 411–423 [DOI] [PubMed] [Google Scholar]
- 48. Temme C, Zhang L, Kremmer E, Ihling C, Chartier A, Sinz A, Simonelig M, Wahle E (2010) Subunits of the Drosophila CCR4‐NOT complex and their roles in mRNA deadenylation. RNA 16: 1356–1370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Smibert CA, Lie YS, Shillinglaw W, Henzel WJ, Macdonald PM (1999) Smaug, a novel and conserved protein, contributes to repression of nanos mRNA translation in vitro . RNA 5: 1535–1547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Dahanukar A, Walker JA, Wharton RP (1999) Smaug, a novel RNA‐binding protein that operates a translational switch in Drosophila . Mol Cell 4: 209–218 [DOI] [PubMed] [Google Scholar]
- 51. Tadros W, Goldman AL, Babak T, Menzies F, Vardy L, Orr‐Weaver T, Hughes TR, Westwood JT, Smibert CA, Lipshitz HD (2007) SMAUG is a major regulator of maternal mRNA destabilization in Drosophila and its translation is activated by the PAN GU kinase. Dev Cell 12: 143–155 [DOI] [PubMed] [Google Scholar]
- 52. Chen G, Kong J, Tucker‐Burden C, Anand M, Rong Y, Rahman F, Moreno CS, Van Meir EG, Hadjipanayis CG, Brat DJ (2014) Human Brat ortholog TRIM3 is a tumor suppressor that regulates asymmetric cell division in glioblastoma. Can Res 74: 4536–4548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Worringer KA, Rand TA, Hayashi Y, Sami S, Takahashi K, Tanabe K, Narita M, Srivastava D, Yamanaka S (2014) The let‐7/LIN‐41 pathway regulates reprogramming to human induced pluripotent stem cells by controlling expression of prodifferentiation genes. Cell Stem Cell 14: 40–52 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Hansen DV, Lui JH, Parker PRL, Kriegstein AR (2010) Neurogenic radial glia in the outer subventricular zone of human neocortex. Nature 464: 554–561 [DOI] [PubMed] [Google Scholar]
- 55. Lui JH, Hansen DV, Kriegstein AR (2011) Development and evolution of the human neocortex. Cell 146: 18–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Ye B, Petritsch C, Clark IE, Gavis ER, Jan LY, Jan Y‐N (2004) Nanos and Pumilio are essential for dendrite morphogenesis in Drosophila peripheral neurons. Curr Biol 14: 314–321 [DOI] [PubMed] [Google Scholar]
- 57. Neumüller RA, Richter C, Fischer A, Novatchkova M, Neumüller KG, Knoblich JA (2011) Genome‐wide analysis of self‐renewal in Drosophila neural stem cells by transgenic RNAi. Cell Stem Cell 8: 580–593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pfeiffer BD, Jenett A, Hammonds AS, Ngo T‐TB, Misra S, Murphy C, Scully A, Carlson JW, Wan KH, Laverty TR et al (2008) Tools for neuroanatomy and neurogenetics in Drosophila . Proc Natl Acad Sci USA 105: 9715–9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Weng M, Golden KL, Lee C‐Y (2010) dFezf/Earmuff maintains the restricted developmental potential of intermediate neural progenitors in Drosophila . Dev Cell 18: 126–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Lee T, Luo L (1999) Mosaic analysis with a repressible cell marker for studies of gene function in neuronal morphogenesis. Neuron 22: 451–461 [DOI] [PubMed] [Google Scholar]
- 61. Vert J‐P, Foveau N, Lajaunie C, Vandenbrouck Y (2006) An accurate and interpretable model for siRNA efficacy prediction. BMC Bioinformatics 7: 520 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Brennecke J, Stark A, Russell RB, Cohen SM (2005) Principles of microRNA‐target recognition. PLoS Biol 3: e85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Haley B, Foys B, Levine M (2010) Vectors and parameters that enhance the efficacy of RNAi‐mediated gene disruption in transgenic Drosophila . Proc Natl Acad Sci USA 107: 11435–11440 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ma Y, Niemitz EL, Nambu PA, Shan X, Sackerson C, Fujioka M, Goto T, Nambu JR (1998) Gene regulatory functions of Drosophila Fish‐hook, a high mobility group domain Sox protein. Mech Dev 73: 169–182 [DOI] [PubMed] [Google Scholar]
- 65. Raj A, van den Bogaard P, Rifkin SA, van Oudenaarden A, Tyagi S (2008) Imaging individual mRNA molecules using multiple singly labeled probes. Nat Methods 5: 877–879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Harzer H, Berger C, Conder R, Schmauss G, Knoblich JA (2013) FACS purification of Drosophila larval neuroblasts for next‐generation sequencing. Nat Protoc 8: 1088–1099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Anders S, Huber W (2010) Differential expression analysis for sequence count data. Genome Biol 11: R106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Livak KJ, Schmittgen TD (2001) Analysis of relative gene expression data using real‐time quantitative PCR and the 2(‐Delta Delta C(T)) Method. Methods 25: 402–408 [DOI] [PubMed] [Google Scholar]
- 69. Gilbert C, Svejstrup JQ (2006) RNA immunoprecipitation for determining RNA‐protein associations in vivo . Curr Protoc Mol Biol Chapter 27: Unit 27.4 [DOI] [PubMed] [Google Scholar]
- 70. Zheng L, Baumann U, Reymond J‐L (2004) An efficient one‐step site‐directed and site‐saturation mutagenesis protocol. Nucleic Acids Res 32: e115 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix
Dataset EV1
Dataset EV2
Source Data for Appendix
Review Process File
Data Availability Statement
RNA sequencing data have been submitted to NCBI GEO project Accession Number GSE104592.