

Abstract
Virally encoded proteins have evolved to perform multiple functions, and the core protein (HBc) of the hepatitis B virus (HBV) is a perfect example. While HBc is the structural component of the viral nucleocapsid, additional novel functions for the nucleus-localized HBc have recently been described. These results extend for HBc, beyond its structural role, a regulatory function in the viral life cycle and potentially a role in pathogenesis. In this article, we review the diverse roles of HBc in HBV replication and pathogenesis, emphasizing how the unique structure of this protein is key to its various functions. We focus in particular on recent advances in understanding the significance of HBc phosphorylations, its interaction with host proteins and the role of HBc in regulating the transcription of host genes. We also briefly allude to the emerging niche for new direct-acting antivirals targeting HBc, known as Core (protein) Allosteric Modulators (CAMs).
Keywords: Hepatitis B virus, HBV, HBV core antigen, HBc, HBc structure, HBc phosphorylations, PLK1, Core allosteric modulators, CAMs
1. Introduction
Hepatitis B virus (HBV) is a major hepatotropic human pathogen, with at least 2 billion people having been exposed to date to the virus according to World Health Organization (Revill and Yuan, 2013). HBV exposure leads to chronic infection in 85–95% of infected neonates/children and 5–15% of infected adults (Hadziyannis, 2011). Chronic infections are associated with a higher risk of developing liver cancer (Revill and Yuan, 2013), with 50% of hepatocellular carcinoma (HCC) attributed to HBV. Viral persistence is due to the ability of the virus to escape the host immune system, and due to establishment of an episome in the nucleus of infected cells, known as covalently closed circular double-stranded DNA (cccDNA). This cccDNA resembles the host genome, as it assumes a chromosome-like structure, and serves as main template for viral transcription. However, cccDNA is devoid of telomeres and replication origins, implying that it can be lost when cells divide; consequently, its reservoir is maintained by recycling of neosynthesized, relaxed-circular DNA (rcDNA) containing nucleocapsids. For an in-depth discussion of cccDNA biology see recent reviews (Allweiss and Dandri, 2017; Lucifora and Protzer, 2016). CccDNA is a 3.2 kb molecule that expresses at least 6 overlapping RNAs, from 4 open reading frames (ORF), leading to production of 7–8 proteins, including viral polymerase (P), regulatory X protein (HBx), envelop proteins (S, M, and L), HBeAg, and core/capsid (HBc). In this review, we focus our discussion on the function of HBc.
HBc, a small versatile protein of 21 kDa, has been classically viewed as a structural protein that self-assembles to form the viral nucleocapsid. In turn, the nucleocapsid initially contains the pregenomic RNA (pgRNA), subsequently converted into rcDNA by reverse transcription. The versatile nature of HBc is primarily a consequence of post-translational modifications occurring at its C-terminal domain (CTD), and thus, this protein directs several viral processes. Accumulated evidence supports that HBc functions extend well beyond this structural role. HBc plays a role in nearly every stage of the HBV life cycle, including: subcellular trafficking and release of HBV genome, RNA metabolism, capsid assembly and transport, and reverse transcription (Fig. 1) (Basagoudanavar et al., 2007; Nassal, 1992; Perlman et al., 2005; Rabe et al., 2003; Zlotnick et al., 2015). Moreover, recent findings implicate an active role for HBc in epigenetic regulation of the viral and host genomes. Collectively, this multiplicity of functions elevates HBc as an excellent target for mechanism-based antiviral therapeutics (Durantel and Zoulim, 2016; Zlotnick et al., 2015). This review will highlight how the unique structure of HBc, its capacity to multimerize, as well as its post-translational modifications, can explain its multiple roles in the HBV life cycle.
Fig. 1.
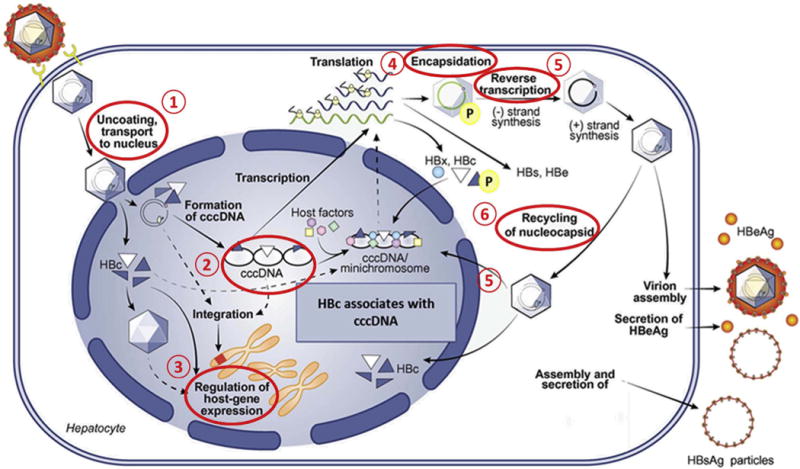
Stages of the HBV life cycle regulated by HBc. 1) HBc regulates transport and nuclear release of the viral genome (Blondot et al., 2016). 2) HBc associates with cccDNA (Guo et al., 2011). 3) HBc modulates host gene expression (Xie et al., 2017). 4) HBc is required for pgRNA encapsidation (Nassal, 1992). 5) HBc is required for reverse transcription (Lewellyn and Loeb, 2011), and 6) persistence of cccDNA involves recycling of nucleocapsids into the nucleus (Nassal, 2015).
2. HBc: from translation to capsid structure and self-assembly
The core protein is encoded by the HBV pgRNA which also encodes the viral polymerase, and serves as template for rcDNA synthesis via reverse-transcription. The viral polymerase acts as a molecular switch to regulate these two otherwise competitive processes allowing the virus to replicate efficiently (Ryu et al., 2010, 2008). The 21 kDa HBc protein is the building block or capsomer of the HBV genome-containing icosahedral capsid. The first crystal structure, resolved by Wynne and colleagues, revolutionized our understanding of HBc biology (Wynne et al., 1999). HBc is a helical protein with five alpha-helices and lacking β-sheets. Helices 2 and 3, making a long alpha-helical hairpin, are responsible for the HBc folding pattern. Interestingly, most nucleocapsid proteins that form icosahedral shells usually fold into barrel structures instead of helical ones, e.g., those seen in the HIV-1 capsid (Wynne et al., 1999). A hydrophobic region makes up the base of the protein. HBc assembles first into dimers via alpha-helical hairpins, which form a four-helix bundle, assuming an inverted T shape. A disulfide bridge between the two Cys-61 residues, stabilizes the association, although this cystine formation is not necessary for dimer assembly (Nassal, 1992; Zheng et al., 1992; Zlotnick et al., 1996). Residues Tyr-132, Arg-127, Pro-129, and Ile-139 appear to be involved in dimer formation (Wynne et al., 1999). Following dimer formation, trimers of dimers assemble into hexamers, eventually forming icosahedral capsid particles (Birnbaum and Nassal, 1990). Multimerization of 120 HBc dimers, i.e., 240 capsomers, leads to production of a 22 nm, icosahedral capsid particle, without need of other viral proteins (Birnbaum and Nassal, 1990). The dimers form the icosahedral capsid with a triangulation number T = 4. Of note, a small fraction of capsids consist of only 90 dimers with a triangulation number T = 3 (Crowther et al., 1994). However, it is unknown whether virions comprised of smaller capsids are infectious or merely represent dead-end products caused by aberrant assembly.
Focusing on the HBc primary sequence, the full-length protein consists of 183 amino acids with distinct N- and C-terminal domains connected by a hinge region (Nassal, 1992). The N- terminal domain of HBc, spanning amino acid residues 1–140, contains the capsid assembly domain, as truncated HBc 1-140 is sufficient for self-assembly (Birnbaum and Nassal, 1990). On the other hand, the CTD of HBc, spanning amino acids 141–183, is dispensable for in vitro viral assembly. Notably, capsids lacking the last 34 amino acids maintain assembly capacity in vitro, but do not support genome replication in cells (Nassal, 1992), suggesting a role for the CTD in this process. Of the 34 amino acid residues comprising the CTD, 14 are arginines, and at least seven other residues are either serines or threonines, putative phospho-acceptor sites.
Studies to understand the role of HBc CTD in HBV replication have led to the proposal that electrostatic interactions between the highly basic CTD in assembled capsids and the viral RNA/DNA regulate reverse transcription of HBV DNA (Newman et al., 2009). Charge-driven destabilization of capsids, by either rcDNA maturation and/or changes in post-translational modifications of CTD, for example by phosphorylation, could explain capsid disassembly at the nuclear pores (Basagoudanavar et al., 2007; Perlman et al., 2005; Rabe et al., 2009). In addition, results of a study by Lewellyn and Loeb favor an hypothesis that CTD could serve as a nucleic acid chaperone domain, i.e., CTD could mediate changes in nucleic acid structure hence enabling pregenomic RNA (pgRNA) reverse transcription into rcDNA (Lewellyn and Loeb, 2011). These two models are not necessarily mutually exclusive, since the function of HBc as chaperone may be dependent on the overall charge of its CTD.
A recent study employing cryo-EM scanning has determined the high-resolution 3D structure of assembled HBV capsids (Yu et al., 2013). It was proposed that HBc CTD shuttles between the interior and exterior of capsids, as evidenced by the presence of CTDs either on the exterior of some capsids or RNA-bound in the interior of other capsids (Yu et al., 2013). This model is attractive as it provides a mechanism by which HBc can be accessible for interaction with host proteins involved in intracellular trafficking, capsid maturation, or capsid disassembly. In this model, the positively charged CTD interacts transiently with the negatively charged Asp2 and Glu43 that line the local three-fold channel, presumably allowing movement of CTD in and out of capsids (Yu et al., 2013). In congruence with the cryo-EM study, biochemical binding assays demonstrate a nucleic acid sensitive and transient exposure of the CTD tail (Chen et al., 2011), further confirming the presence of CTD on the exterior of capsids. Specifically, RNA-filled capsids failed to bind serine-arginine protein kinase (SRPK), a host chaperone that binds full length HBc capsids, but not HBc 1-149 capsids (Chen et al., 2011), confirming the dynamic exposure of CTDs is mediated by CTD-nucleic acid interactions. These results also agree with biochemical studies showing the CTD of assembled capsids is not only sensitive to proteases, but also accessible to host proteins (Chen et al., 2011; Gallina et al., 1989). The exact mechanism of this dynamic CTD shuttling remains unknown, although a role of phosphorylation has been suggested by site directed mutagenesis of HBc (Selzer et al., 2015). However, further detailed analyses of the effect of CTD phosphorylations and dephosphorylations in CTD shuttling are required.
3. HBc, a nucleic acid chaperone
Nucleic acid chaperones are proteins that catalyze structural rearrangement of genetic material into a functional conformation (Semrad, 2011). The binding of nucleic acid chaperones to their targets is characteristically weak and transient, lacks sequence specificity, and does not require ATP hydrolysis (Semrad, 2011). An array of viral proteins displays nucleic acid chaperone activity. For example, nucleocapsid proteins of HIV (Levin et al., 2005), dengue virus (Pong et al., 2011), and hepatitis delta virus (Huang and Wu, 1998) are described as nucleic acid chaperones. The CTD of HBc contains an arginine-rich domain resembling histone tails with more than 50% of the residues being arginine (Lewellyn and Loeb, 2011). The CTD binds to nucleic acids in vivo due to its positive charge (Nassal, 1992). In addition to its characteristic positive charge, CTD is also structurally disordered (Yu et al., 2013), another key feature of nucleic acid chaperones (Chu et al., 2014). The localization of CTD to the interior of capsids (Zlotnick et al., 1997) and its ability to bind nucleic acids in vivo support the model that CTD works as a nucleic acid chaperone. This chaperone activity was recently confirmed in vitro using recombinant full length HBc, as well as CTD synthetic peptides (Chu et al., 2014). HBc CTD facilitated annealing and unwinding of DNA and enhanced hammerhead ribozyme cleavage in vitro, rendering HBc CTD a bona fide nucleic acid chaperone (Chu et al., 2014). However, the high degree of functional complexity associated with the CTD, makes it challenging to demonstrate the nucleic acid chaperone activity of HBc in vivo, without disturbing other HBc functions.
4. HBc localization: a dynamic process involving active-transport
Reports of subcellular localization of HBc in vivo demonstrated both cytoplasmic as well as nuclear distribution. Histologic analysis of tissues from HBV-infected patients indicated a distribution of HBc/capsids in both cytosolic and nuclear compartments. Sharma et al., as well as Akiba et al., described a predominant nuclear distribution (Akiba et al., 1987; Sharma et al., 2002) while others observed mainly a cytosolic core localization (Michalak and Nowoslawski, 1982; Petit and Pillot, 1985). In vitro analysis has shown HBc subcellular localization is in fact cell-cycle dependent (Yeh et al., 1993). The amount of core protein in the nucleus increases in G1 phase, diminishes in S phase, and increases again to detectable levels in non-proliferating cells (Yeh et al., 1993). HBc shuttles between cytoplasm and nucleus rapidly and continuously (Li et al., 2010). These latter points could explain differences described above. Although HBc is 21 kDa, suggesting passive transport to and from the nucleus, this trapping, as exists for human T-lymphotropicvirus-1 (HTLV-1) (Goff, 2007) and human papillomaviruses (Aydin et al., 2014), would be fairly inefficient due to the quasi-quiescent state of hepatocytes. Given that HBc is a phosphorylation target of well-characterized cell cycle regulated kinases, such as cyclin-dependent kinase 2 (CDK2) (Ludgate et al., 2012) and polo-like kinase 1 (PLK1) (Diab et al., 2017), it is highly plausible that such phosphorylation may contribute to HBc/capsid localization. Investigating this question in a physiologically relevant infection model will most certainly expand our understanding of the dynamics of HBc subcellular localization.
Furthermore, it remains unresolved whether HBc enters the nuclear pore complex (NPC) as a dimer or as a mature capsid. Thus, signals modulating HBc import and export from the nucleus are probably distinct from those that guide transport of mature capsids. An early in vitro model for capsid transport by Kann and colleagues suggested mature capsid disassembles into HBc dimers at the cytosolic side of the NPC, releasing the rcDNA-pol complex (Kann et al., 1997). Subsequently, HBc dimers bind importin-β and are transported through a NPC. At this point, HBc dimers dissociate from transport factors and could remain free in the nucleus, eventually playing a role as transcription factors, as described below, or reassemble as empty-capsid forms. On the other hand, as viral polymerase does not contain an importin-β binding (IBB) domain, formation of importin-α/β is required to import the genome complex (Kann et al., 1997). Once at the nuclear side, the rcDNA-pol complex dissociates from importin α/β. Since the viral genome comprises pathogen-associated molecular patterns (PAMPs), it could trigger sensing by cytosolic pattern recognition receptors (PRRs) (Paludan, 2013). This model of genomic nuclear import, showing a release of viral rcDNA in the cytosol could explain the weak innate immunity activation upon HBV infection (Faure-Dupuy et al., 2017; Lucifora et al., 2014; Vanwolleghem et al., 2015). However, this model is based on Woodchuck Hepatitis Virus where capsids were denatured using urea treatment in order to release the rcDNA-pol complex (Kann et al., 1997). Under such conditions, changes in conformation of the viral polymerase could enable its interaction with importin-α.
A more recent alternative model (see Blondot et al., 2016 for in-depth review), suggests a direct interaction between mature capsids and an essential component of the NPC’s basket, the nucleoporin 153 (Nup153). It is known that importin-α interacts directly with Nup153, promoting importin-α/β-mediated nuclear import (Ogawa et al., 2012). Of note, it was shown earlier that mature capsids are transported to nuclear pores by an importin α/β complex (Kann et al., 2007). The new model provides two options for mature capsids going through nuclear pores: first, direct binding to Nup153 at the basket of the NPC (cargo size 36 nM) or via coating by importin-α/β and binding to Nup153 (cargo size 38 nM). According to this model, the HBc CTD is not necessary to anchor the capsid at the nuclear basket since importin α/β is required for capsid transport. While both models intend to explain the mechanism of capsid transport through nuclear pores, neither reveals the trigger of such transport.
Studies of the CTD tail revealed a role in directing HBc subcellular localization (Blondot et al., 2016; Li et al., 2010). Several attempts have been made to map the nuclear localization sequence (NLS) to the arginine repeats of CTD. However, the results are conflicting, probably due to the redundancy of arginine repeats within the CTD (Liao and Ou, 1995; Rabe et al., 2003). Recently, an exhaustive mutagenesis analysis of the CTD localized the NLS sequence to the 1st and 3rd arginine repeats, while the 2nd and 4th repeats contained the cytoplasmic retention signals (CRS) (Fig. 2) (Li et al., 2010). The presence of more than one NLS in a protein is known to maximize import efficiency (Mears et al., 1995). Other viral proteins, such as ICP27, of herpes simplex virus type 1 (HSV-1) (Mears et al., 1995) and NS1 of Influenza (Melen et al., 2012), also contain multiple NLS sites. However, one must also consider the accessibility of these sequences in view of the dynamic structural changes of assembled HBc in viral capsids.
Fig. 2.

Arginine-rich motifs of different viral proteins exhibit an abundance of arginine residues within short 10–20 residues. The arginine-rich domain of HBc has distinct nuclear localization (NLS) and cytoplasmic retention signals (CRS) (Li et al., 2010).
The dynamic positioning of HBc CTD in assembled capsids makes it quite challenging to define clearly the different signals regulating HBc transport. Nuclear export of HBc resembles ICP27 of HSV-1 and EBNA1 of Epstein Barr virus (EBV), in that they all bind to the RNA export factor TAP, and all contain the characteristic arginine rich domains (Fig. 2) (Yang et al., 2014). Since subcellular localization of ICP27 and EBNA1 are regulated by arginine methylation (Souki et al., 2009), it is reasonable to envisage a similar mechanism driving HBc localization. However, it is still unknown if the arginine repeats of CTD are methylated, an hypothesis worthy of future investigation. Of note, arginine methyltransferase 5 (PRMT5) was reported as a negative effector of HBV replication and transcription via epigenetic repression of cccDNA transcription and interference with pregenomic RNA encapsidation (Zhang et al., 2017). Interestingly, PRMT5 was shown to bind HBc, and HBc-deficient virus (HBV-ΔHBc) had less PRMT5 bound cccDNA when compared to the wildtype HBV (Zhang et al., 2017).
The contribution of HBc CTD to capsid localization is only beginning to be resolved. The ambiguity is based on the long-standing notion that the CTD localizes to the interior of capsids based on cryo-EM studies (Zlotnick et al., 1997), and has a well-documented role in pgRNA packaging (Nassal, 1992). However, as previously discussed, more recent studies suggest a dynamic model in which HBc CTD shuttles between the interior and exterior of the capsid (Chen et al., 2011; Li et al., 2010; Selzer et al., 2015).
Transport of cargo through the nuclear envelope is often facilitated by regulatory phosphorylation/dephosphorylation of specific residues (Nardozzi et al., 2010). Earlier studies reported phosphorylated HBc was only observed in the cytoplasm, based on subcellular fractionation of in vivo 32P-radiolabed cell lysates (Yeh et al., 1993). However, the molecular mechanism regulating subcellular localization of monomeric/dimeric vs. assembled HBc in capsids remains ambiguous. The potential contribution of HBc phosphorylation to the nuclear import of capsids comes from known import mechanisms for other viral proteins, including EBNA-1 (Kitamura et al., 2006) and SV40 T-antigen (Hubner et al., 1997). Specifically, phosphorylation of EBNA-1 increases its affinity for the import adaptor importin-α5 which mediates the nuclear transport via interaction with importin-β (Kitamura et al., 2006). SV-40 large T antigen has the best characterized NLS, and phosphorylation upstream of the classic NLS enhances nuclear import (Hubner et al., 1997). Thus, the phosphorylation status of HBc CTD could facilitate interaction with nuclear pore complex components. Indeed, in pulldown experiments using HBc CTD-GST fusion proteins, serine to alanine substitution at the three serine/proline (S/P) sites (Fig. 3) showed increased binding to importin-α (Liu et al., 2015), suggesting phosphorylation impedes trafficking of HBc to the nucleus. While the in vivo relevance of these observations remains to be determined, other groups have reported that HBc phosphorylated in vitro by PKC in assembled capsids, preferentially bound importin-β (Kann et al., 1999). However, the phosphorylation sites of HBc required for interaction with importin-β have not been mapped (Kann et al., 1999). Furthermore, it is unresolved whether protein kinase C (PKC) is the in vivo kinase that phosphorylates HBc CTD (Daub et al., 2002). Thus, further studies are necessary to address this issue, employing well-characterized and functionally documented mutants of HBc.
Fig. 3.

HBc C-terminal domain (CTD) is a phosphorylation substrate for CDK2 (SP sites at positions 155, 162 and 170) and PLK1 (phosphorylation sites at positions 168, 176 and 178), as shown by Ludgate et al. (2012) and Diab et al. (2017), respectively.
Alternatively, as shown by Schmitz et al., capsid maturation could be a driving force of HBV nuclear import (Schmitz et al., 2010). At the nuclear pore, HBc capsids dissociate into dimers followed by entry into the nucleus (Rabe et al., 2009). Mature capsids directly interact with Nup153, an essential component of the nuclear basket that facilitates nuclear import via importin-β (Schmitz et al., 2010). Interaction of mature capsids with Nup153 resulted in capsid disintegration followed by release of viral genomes into the nucleus (Schmitz et al., 2010). Interestingly, capsids with immature genomes, i.e., RNA-filled capsids, accumulate at the nuclear pore. Recently, it was reported that serine to glutamate substitutions at the 3 S/P sites of HBc CTD (Fig. 3) reduced CTD exposure to the exterior of capsids suggesting a role for these phosphorylations in directing CTD exposure– inside or outside the capsid (Selzer et al., 2015). However, the mechanism responsible for distinguishing mature capsids vs. RNA-filled capsids remains unclear (Hu and Liu, 2017). Phosphorylation of HBc, shuttling of the HBc CTD to the interior vs. exterior of capsids, and maturation of the genome, may regulate exposure of different localization signals to host transport machinery. Such a multilayer and dynamic regulation network can best explain the difficulty in deciphering the mechanism of HBc subcellular localization during the HBV life cycle. To understand these important dynamic regulatory networks, it is essential to gain better understanding of HBc structure, HBc interacting partners, and HBc post-translational modifications.
5. HBc phosphorylation
Since the HBV genome does not encode a protein kinase, endogenous/cellular kinases must mediate post-translational HBc modifications (Lanford and Notvall, 1990). PKC (Kann and Gerlich, 1994; Wittkop et al., 2010), serine arginine protein kinase 1 (SRPK1) (Daub et al., 2002) and CDK2 (Ludgate et al., 2012) have been suggested as endogenous kinases that associate with or are packaged within viral capsids. The robust phosphorylation of HBc has been demonstrated in vitro by phospho-proteomic approaches (Chen et al., 2011), and in vivo by labeling with 32P-orthophosphate (Jung et al., 2014). The HBc CTD contains multiple serine/threonine phospho-acceptor sites which are remarkably well-conserved among related viruses (Jung et al., 2014). Phospho-proteomic analyses of HBc have identified at least seven conserved serine and threonine sites that are phosphorylated in vivo (Chen et al., 2011). Notably, proline-directed serine (S/P) sites at positions 155, 162 and 170, are highly conserved and several S/P kinases have been reported to phosphorylate these sites (Fig. 3) (Daub et al., 2002; Ludgate et al., 2012). Other putative kinases, including: SRPKs, Glyceraldehyde-3-phosphate dehydrogenase protein kinase (GAPD-protein kinase), PKC, and an unknown kinase of 46 kDa, have also been reported (Daub et al., 2002; Duclos-Vallee et al., 1998; Kann et al., 1999; Ludgate et al., 2012) (Table 1). Yet none of these studies have mapped phosphorylation to specific sites, with the exception of SRPK1 and SRPK2 at the 3 S/P sites (Daub et al., 2002). However, the exact contribution of the SRPK-mediated phosphorylation to viral replication is enigmatic, because, although overexpression of SRPK1 or SRPK2 impaired HBV replication, this effect was independent of kinase activity (Zheng et al., 2005). These results support that SRPK-mediated phosphorylation of HBc observed in vitro is due to promiscuous substrate specificity of these kinases (Ludgate et al., 2012), and that SRPKs exert other functions affecting viral replication. For example, SRPKs could act as non-canonical chaperones in capsid assembly (Chen et al., 2011). Regarding PKC-mediated HBc phosphorylation, inactivation of PKC led to defective capsid envelopment, but had no effect on genome maturation (Wittkop et al., 2010). There is, however, lack of consensus regarding PKC being the candidate host kinase since specific inhibition of PKC had no effect on CTD phosphorylation (Daub et al., 2002; Ludgate et al., 2012). CDK2 was shown to phosphorylate the core protein of HBV both in vitro and in vivo (Ludgate et al., 2012). Intriguingly, CDK2 inhibitors had no observable effect on viral replication (Ludgate et al., 2012). The absence of such effect could be due to lack of specificity of the chemical inhibitors (Sakurikar and Eastman, 2016) or, more likely, due to redundancy among host kinases in phosphorylating HBc. Knockdown cell lines for CDK2 or other kinases via expression of shRNAs could elucidate their contribution to HBc phosphorylation and viral replication.
Table 1.
Kinases that phosphorylate HBc.
| Kinase | Effect on viral replication | Immuno-detected in capsids | Phosphorylation sites | References |
|---|---|---|---|---|
| PKC | Required for envelopment | Yes | ND | Kann and Gerlich, 1994; Wittkop et al., 2010 |
| GAPD-PK | ND | No | ND | Duclos-Vallee et al., 1998 |
| SRPK1/2 | Decrease replication | ND | S155,S162,S170 | Daub et al., 2002 |
| CDK2 | No effect | Yes | S155,S162,S170 | Ludgate et al., 2012 |
| PLK1 | Enhance replication | ND | S168,S176,S178 | Diab et al., 2017 |
Although the kinases that phosphorylate HBc are still to be determined, phosphorylations of HBc do regulate its function at many levels (Gazina et al., 2000; Jung et al., 2014; Kann et al., 1999; Ludgate et al., 2011). Specifically, phosphorylation and de-phosphorylation of the HBc protein is required for capsid maturation as demonstrated by mass spectrometric analyses and site-directed mutagenesis studies of HBc (Basagoudanavar et al., 2007; Perlman et al., 2005). Phosphorylation of different serine residues of HBc CTD regulates reverse transcription, pgRNA encapsidation, DNA synthesis, subcellular localization, and virion secretion (Basagoudanavar et al., 2007; Gazina et al., 2000; Lan et al., 1999; Perlman et al., 2005). HBc phosphorylation is also required for the chaperone activity of CTD (Chu et al., 2014). It was also reported that the HBc phosphorylation status influences capsid stability (Ludgate et al., 2016), since capsids with glutamate phosphomimetic substitutions at the 3 S/P sites (S/P to E/P) were more stable than wild type capsids. It is likely that during various stages in the life cycle of HBV or as a function of the metabolic and proliferative status of the infected hepatocyte, distinct kinases target HBc phosphorylation, perhaps in an overlapping manner. The identification of those kinases, and the mechanisms by which they regulate HBV replication, is of great significance and will provide valuable targets for mechanism-based therapeutics.
To this end, our recent study has identified the mitotic PLK1 as another cellular kinase that phosphorylates HBc in vitro (Diab et al., 2017). PLK1 substrates are usually primed by phosphorylation at S/P directed sites mediated by CDKs, including CDK1 or 2 (Elia et al., 2003). Recombinant HBc, as well as immuno-purified HBc from transfected mammalian cells serves as robust PLK1 substrate in vitro (Diab et al., 2017). We have mapped the phospho-acceptor sites to S168, S176, and S178 (Fig. 3) (Diab et al., 2017), all of which have been identified as in vivo phospho-acceptor sites by an unknown kinase (Jung et al., 2014). Remarkably, inhibition of PLK1 by specific chemical inhibitors or siRNA-mediated knockdown of PLK1 inhibited viral replication, suggesting HBc phosphorylation by PLK1 is functionally important in virus biosynthesis (Diab et al., 2017). Significantly, we made the interesting observation, in accordance with the known substrate preference of PLK1, that alanine substitutions at the 3 S/P sites (S/P to A/P) resulted in loss of PLK1 phosphorylation in vitro. By contrast, PLK1 phosphorylation was robustly recovered in phospho-mimetic mutants, containing serine to aspartic acid substitutions at all three S/P sites (Diab et al., 2017). This suggests that CDK2 functions as the priming kinase for HBc, mediating the phosphorylation of the S/P sites which in turn enables the subsequent phosphorylation by PLK1 (Elia et al., 2003). Based on our results that PLK1 is a positive effector of HBV replication, a mechanistic understanding of the role of PLK1 phosphorylation of HBc in HBV replication has promising therapeutic potential. So far, PLK1 is the sole host-kinase whose chemical inhibition results in impaired HBV replication in vivo (i.e., in liver humanized mouse model) at concentrations comparable to other known anti-HBV agents (Diab et al., 2017). Finally, based on this dependence of PLK1 phosphorylation of HBc on its prior phosphorylation by CDK2, we reason combination treatment with inhibitors for CDK2 and PLK1 kinases will be effective in suppressing these modifications and thus effectively inhibit virus biosynthesis. This hypothesis is particularly attractive since safety of PLK1 (Gjertsen and Schoffski, 2015) and CDK2 (Asghar et al., 2015) inhibitors has been established in clinical trials for different types of human cancer.
6. HBc and interaction with host factors
HBc modulates nearly every stage of the HBV life cycle. With such extensive and intricate functions, HBc has evolved to modulate several host-protein functions. For example, HBc dimers interact with HSP90, and HSP90 facilitates formation of HBV capsids both in vitro and in vivo (Shim et al., 2011). On the other hand, the host HSP40 protein also binds, but de-stabilizes HBc by accelerating its degradation (Sohn et al., 2006). The binding of HBc to other cellular factors, such as NIRF, an E3 ubiquitin ligase, is implicated in HBc degradation (Qian et al., 2012). Thus, dynamic interactions of HBc with host chaperones regulate both HBc stability and capsid assembly (Qian et al., 2012; Shim et al., 2011; Sohn et al., 2006).
Viruses hijack host RNA processing machinery (Salvetti and Greco, 2014) and HBV is no exception. HBc utilizes the nuclear export factor 1 (NXF1/p15) and transcription export (TREX) machinery for nuclear export as a ribonucleoprotein complex along with the viral pgRNA (Yang et al., 2014); however, TREX-mediated export of HBc and pgRNA are independent of each other (Yang et al., 2014). Other viruses that hijack the NXF1/p15 pathway for mRNA processing and export include EBV (Juillard et al., 2009) and HSV-1 (Johnson et al., 2009). Interestingly, depletion of different TREX components has no apparent effect on accumulation of viral DNA, suggesting redundancy among host factors utilized by the virus.
Another class of host proteins commonly targeted by viruses is serine, arginine rich-proteins (Howard and Sanford, 2015) which, along with the kinases (SRPKs) that phosphorylate them, play central roles in alternative RNA splicing (Howard and Sanford, 2015). Intriguingly, HBc CTD resembles serine, arginine-rich proteins and it is not surprising that SRPKs associate with and potentially phosphorylate HBc (Chen et al., 2011; Daub et al., 2002; Zheng et al., 2005). Interestingly, mass spectrometry analyses of HBc interacting proteins has identified SRPK1 as well as several known SRPK1 interacting partners, including: SRSF1, SRSF9, DHX9, CDKN2A, DDX21, and DDX50 (Diab et al. unpublished data). The functional significance of these HBc interactions in the viral life cycle is unknown and worthy of investigation. We speculate that HBV usurps the host RNA-processing machinery for production and processing of its own viral RNA. It is also likely that such interactions may allow HBc to interfere with host gene expression. This has been evident in the case of E1E4 protein of the human papilloma type 1 virus (HPV1), which not only binds to SRPK1, but also inhibits phosphorylation of its host serine arginine substrates (Prescott et al., 2014). A yeast two-hybrid screen of a human liver cDNA library identified the human protein GIPC1 as another HBc interaction partner (Razanskas and Sasnauskas, 2010), but the significance of such interaction remains to be determined. GIPC1 contains a PDZ domain, a protein-protein interaction domain (reviewed in Hung and Sheng, 2002). PDZ-containing host proteins are commonly targeted by viral proteins; examples include the Tax protein of the T-cell leukemia virus type 1 (Rousset et al., 1998) and the E6 protein of the human papilloma virus type 18 (Favre-Bonvin et al., 2005).
It is reasonable to speculate that HBc exerts its various roles by interacting with specific host factors at different stages of the viral life cycle in a phosphorylation dependent manner. A better dissection of host factors that interact with HBc could facilitate development of new antiviral strategies or help elucidate core assembly modulators (CAMs).
7. HBc as a regulator of transcription
In chronically infected patients, HBc is detected in both cytoplasm and nucleus of hepatocytes (Chu and Liaw, 1987). In vitro models of HBV infection also show nuclear as well as cytoplasmic localization of HBc. Nuclear localization of HBc, in biopsies from HBV infected patients, has been linked to high viremia and deficient immune response (Chu and Liaw, 1987; Nguyen et al., 2009). These results prompted investigations of the nuclear function of HBc, in relation to virus biosynthesis and influence upon host gene expression.
In the nucleus of infected cells, the viral cccDNA assumes chromatin-like structure in association with histone and non-histone proteins, along with viral HBx and HBc proteins (Bock et al., 2001; Lucifora and Protzer, 2016). Association of HBc with the cccDNA mini-chromosome was reported to alter nucleosome spacing (Bock et al., 2001). Furthermore, it was reported that HBc preferentially associated with CpG island 2 of the HBV genome that overlaps enhancers (Enh) I and II involved in viral transcription; HBc binding to CpG island 2 positively correlated with the rcDNA/cccDNA ratio and serum levels of HBV DNA in infected patients (Guo et al., 2011). HBc may also act as transcriptional activator of the HBV pre-Core promoter by enhancing binding of NF-kB upstream of viral Enh II (Kwon and Rho, 2002). However, how HBc modulates the function of the cccDNA mini-chromosome remains to be determined. Since HBc is also essential for viral DNA replication, a definitive genetic approach involving mutagenesis or depletion of HBc is not feasible as a means to delineate its role in cccDNA mini-chromosome functions. Therefore, novel approaches are required to target nuclear HBc, influencing its capacity to associate with the cccDNA mini-chromosome, without affecting HBc cytoplasmic functions.
Regarding effects of HBc on host gene expression, HBc enhanced cAMP-Response-Element (CRE)-mediated transcription via the CRE/CREB/CBP pathway (Xiang et al., 2015) in a manner similar, but weaker to previously reported HBx-mediated CRE activation (Andrisani, 1999; Williams and Andrisani, 1995). HBc may also function as a transcriptional repressor of host genes. For example, binding of HBc to E2F1 reduced the DNA-binding ability of E2F1 at the p53 promoter, thereby inhibiting p53 transcription (Kwon and Rho, 2003). In hepatoma cell lines expressing the core protein, HBc blocked the human death receptor 5 (DR5) by repressing its promoter, and inhibition of DR5 desensitized hepatocytes to TRAIL-induced apoptosis (Du et al., 2009). These results suggest a role for HBc in development of chronic infection by preventing hepatocyte death via blocking DR5 expression. The gene regulatory function of HBc may have evolved as a means whereby the virus may evade host immunity. HBc has been shown to downregulate IFN-induced host antiviral responses in hepatoma cell lines, by interacting with the promoter of MxA, an INF-inducible gene widely implicated in antiviral response (Fernandez et al., 2003). However, these conclusions must be further validated by determining in vivo association of HBc with the MxA promoter, in a physiological cellular context of HBV infection. Interestingly, MxA has been shown to inhibit HBV replication by direct interaction with HBc, preventing capsid formation (Li et al., 2012), and providing yet another example of antagonism between viral and host proteins. Ectopic expression of HBc in HepG2 cells and primary hepatocyte cultures sensitized cells to TNF-α induced apoptosis, by disrupting the interaction between mitogen-activated protein kinase kinase 7 (MKK7) and receptor of activated protein kinase C 1 (RACK1) (Jia et al., 2015). This suggests a direct role of HBc in driving liver pathogenesis in chronically infected patients. These results require validation in a rigorous experimental model.
In continuing this effort to understand the effect of HBc on host gene expression, recent studies have generated the genome-wide profile of HBc in HBV-infected hepatocytes, employing chromatin immunoprecipitation microarray studies (ChIP-on-chip). These studies revealed that promoters of nearly 3100 host genes exhibited increased binding to HBc (Guo et al., 2012). However, further studies are necessary to understand the functional significance of HBc binding to the host genome in affecting HBV-mediated disease pathogenesis. Toward this goal, Lucifora et al. recently demonstrated interaction of nuclear deaminases APOBEC3A and APOBEC3B with HBc-bound cccDNA in host nucleus, leading to cccDNA deamination and degradation (Lucifora et al., 2014). Whether HBc recruits these nuclear deaminases to host genes is unknown and of great importance, given the role of APOBEC proteins as cancer driver genes in humans (Harris, 2015; Morganella et al., 2016; Nik-Zainal et al., 2012). Muli-omics analyses of HBc transfected cells linked HBc over expression up-regulated glycolysis and amino acid metabolism, providing further insights into the role of HBc in the molecular pathogenesis of HBV-mediated tumorigenesis (Xie et al., 2017).
8. HBc as a target for clinical intervention against chronic HBV infection
Due to the multiple functions of HBc in the HBV life cycle, this protein has become a preferential target for developing novel, direct-acting agents against HBV replication. Molecules targeting capsid assembly have been known for some time with the discovery BAY41-4109, a heteroaryldihydropyrimidine derivative (Deres et al., 2003), and AT130, a phenylpropenamide derivative (Delaney et al., 2002). This class of inhibitors were subsequently abbreviated core assembly/allosteric modulators (CAMs), due to their mode of action, which is mainly based on allosteric modulation (Zlotnick et al., 2015). Interestingly, these molecules were shown to be active against nucleoside analogue resistant strains, which render them very attractive when viral resistance was a genuine clinical challenge (Billioud et al., 2011; Delaney et al., 2002; Wang et al., 2012). Moreover, CAMs are active against most HBV genotypes (Berke et al., 2017). Surprisingly, CAM therapy has not reached the clinic –probably due to safety or pharmacological issues. Since this early time, active research on structural and biological features of the HBc protein has led to the discovery of many other CAMs. Currently there are 3 types of CAMs according to their different molecular architectures, including heteroaryldihydropyrimidine, phenylpropenamide, and sulfamoylbenzamides derivatives (Fig. 4) (Zlotnick et al., 2015). These CAMs are thought to inhibit nucleocapsid-associated reverse transcription of pgRNA into rcDNA by preventing encapsidation of pgRNA, by accelerating formation of HBV nucleic-acid-free, yet normal-in-shape capsids, or by promoting formation of abnormal capsids. This defines two classes of CAMs based on these in vitro/in tubo structural criteria. In addition to their role in nucleocapsid formation and resulting conversion of pgRNA into rcDNA, CAMs have other modes of action. CAMs have been shown to prevent, in a post-entry manner, formation of cccDNA (Berke et al., 2017). This could be due to retention of rcDNA at the nuclear basket; in this respect, CAMs could bind to nucleocapsid and stabilize the structure, thus preventing its disassembly. CAMs also prevent the release from hepatocytes of RNA-containing and genome-free viral particles (Lam et al., 2015; AASLD abstract). Very recently it has been reported that specific CAMs prevent release of the HBe antigen (Lahlali et al., HBV meeting 2017). HBeAg, a secreted antigen sharing primary structure with HBc, has potential immune-modulatory functions. An active and fast inhibition of HBeAg secretion by CAMs could promote HBeAg seroconversion, an immunological process that occurs during natural history of HBV infection, and considered an important clinical end-point during therapy.
Fig. 4.
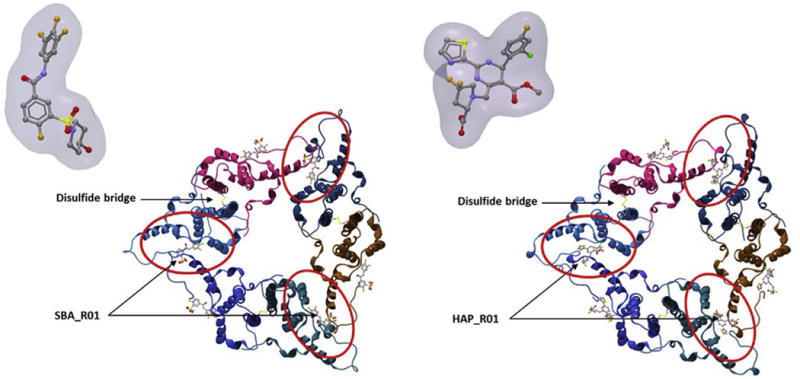
Top view of an HBc hexamer in complex with CAM sulfamoylbenzamide SBA_R01, PDB: 5T2P (Left) and CAM heteroaryldihydropyrimidine HAP_R01 PDB: 5WRE (Zhou et al., 2017) (Right).
However, bearing in mind some of the other functions described in this review (i.e. regulation of host gene expression, modulation of host and viral RNA metabolism, etc.), one might anticipate CAMs could play a role in HBV replication inhibition and HBV-induced pathogenesis. To date several CAMs are undergoing clinical trials: NVR 3–778 (NCT02112799; Novira Therapeutics), ABI-H0731 (NCT02908191; Assembly Biosceinces), JNJ-56136379 (NCT02662712; Janssen), and GLS-4 (trial with China-CFDA; Morphothiadine Mesilate, HEC Pharm), and many others are the focus of pre-clinical evaluations.
9. Unanswered questions and future directions
As discussed in this review, HBc is involved in various aspects of HBV life cycle: formation of the viral capsid, pgRNA encapsidation followed by its reverse transcription, and as a component of the viral mini-chromosome, contributing to its chromatin structure. The unique structure of HBc CTD and its modifications, mainly phosphorylation, are essential for viral biosynthesis. Accordingly, HBc appears to be an ideal target for mechanism-based therapeutics. Yet many questions remain unanswered when it comes to HBc biology. A definitive answer to the identity of the kinases responsible of HBc phosphorylation in vivo is yet to be determined. Additionally, systematic investigation of HBc-host interactions, in the context of physiologic infection as well as well as details of its molecular properties, will expand our knowledge of how HBc is regulated by post-translational modifications, as well as how HBc regulates host functions. Moreover, given the unique structure of the CTD, the seemingly promiscuous affinity of HBc for RNA and nucleic acids in general, (Birnbaum and Nassal, 1990; Nassal, 1992), suggests potential interactions between HBc and host RNA metabolism. This corroborates our recent findings of long-noncoding (lnc) RNAs NEAT1 and NEAT2 as HBc interacting partners in cellular models of HBV replication and infection (Diab et al., HBV meeting 2017). Ribonucleoprotein immunoprecipitation (RIP) of HBc confirmed HBc associates with NEAT1 and NEAT2, known scaffolds of ribonucleoprotein complexes that constitute nuclear speckles (Hutchinson et al., 2007). Employing affinity purification of HBc, combined with mass spectrometry-based proteomics, we identified nuclear speckle proteins heterogeneous nuclear ribonucleoprotein M (HNRNPM), splicing factor proline/glutamine-rich (SFPQ), and non-POU domain containing protein (NONO) as HBc interacting partners. Thus, we propose that HBc, as an RNA-interacting protein, associates and regulates host RNAs and specifically lncRNAs. Accumulating evidence supports a regulatory role of lncRNAs in cellular gene expression (Hutchinson et al., 2007). Whether HBc/lncRNA interactions also exerts a role in hepatocyte physiology and virus biosynthesis is presently unknown (Zhang et al., 2016) and these topics merit further investigation.
Acknowledgments
Supported by NIH grant DK044533-19 to OA, the Chateaubriand Fellowship from the French Embassy in Washington DC to AD, and grants from ANRS (French national agency for research on AIDS and viral hepatitis; several grants from CSS4) and INSERM core grants to FZ and DD. We also acknowledge funding by the DEVweCAN LABEX (ANR-10-LABX-0061) of the “Université de Lyon”, within the program “Investissements d’Avenir” (ANR-11-IDEX-0007) operated by the French National Research Agency (ANR) to FZ and DD. The authors thank Dr. RL Hullinger for his critical review and editing of this manuscript.
References
- Akiba T, Nakayama H, Miyazaki Y, Kanno A, Ishii M, Ohori H. Relationship between the replication of hepatitis B virus and the localization of virus nucleocapsid antigen (HBcAg) in hepatocytes. J Gen Virol. 1987;68(Pt 3):871–877. doi: 10.1099/0022-1317-68-3-871. http://dx.doi.org/10.1099/0022-1317-68-3-871. [DOI] [PubMed] [Google Scholar]
- Allweiss L, Dandri M. The role of cccDNA in HBV maintenance. Viruses. 2017;9 doi: 10.3390/v9060156. http://dx.doi.org/10.3390/v9060156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrisani OM. CREB-mediated transcriptional control. Crit Rev Eukaryot Gene Expr. 1999;9:19–32. [PubMed] [Google Scholar]
- Asghar U, Witkiewicz AK, Turner NC, Knudsen ES. The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat Rev Drug Discov. 2015;14:130–146. doi: 10.1038/nrd4504. http://dx.doi.org/10.1038/nrd4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aydin I, Weber S, Snijder B, Samperio Ventayol P, Kuhbacher A, Becker M, Day PM, Schiller JT, Kann M, Pelkmans L, Helenius A, Schelhaas M. Large scale RNAi reveals the requirement of nuclear envelope breakdown for nuclear import of human papillomaviruses. PLoS Pathog. 2014;10:e1004162. doi: 10.1371/journal.ppat.1004162. http://dx.doi.org/10.1371/journal.ppat.1004162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basagoudanavar SH, Perlman DH, Hu J. Regulation of hepadnavirus reverse transcription by dynamic nucleocapsid phosphorylation. J Virol. 2007;81:1641–1649. doi: 10.1128/JVI.01671-06. http://dx.doi.org/10.1128/JVI.01671-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berke JM, Tan Y, Verbinnen T, Dehertogh P, Vergauwen K, Vos A, Lenz O, Pauwels F. Antiviral profiling of the capsid assembly modulator BAY41-4109 on full-length HBV genotype A-H clinical isolates and core site-directed mutants in vitro. Antivir Res. 2017;144:205–215. doi: 10.1016/j.antiviral.2017.06.016. http://dx.doi.org/10.1016/j.antiviral.2017.06.016. [DOI] [PubMed] [Google Scholar]
- Billioud G, Pichoud C, Puerstinger G, Neyts J, Zoulim F. The main hepatitis B virus (HBV) mutants resistant to nucleoside analogs are susceptible in vitro to non-nucleoside inhibitors of HBV replication. Antivir Res. 2011;92:271–276. doi: 10.1016/j.antiviral.2011.08.012. http://dx.doi.org/10.1016/j.antiviral.2011.08.012. [DOI] [PubMed] [Google Scholar]
- Birnbaum F, Nassal M. Hepatitis B virus nucleocapsid assembly: primary structure requirements in the core protein. J Virol. 1990;64:3319–3330. doi: 10.1128/jvi.64.7.3319-3330.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondot ML, Bruss V, Kann M. Intracellular transport and egress of hepatitis B virus. J Hepatol. 2016;64:S49–S59. doi: 10.1016/j.jhep.2016.02.008. http://dx.doi.org/10.1016/j.jhep.2016.02.008. [DOI] [PubMed] [Google Scholar]
- Bock CT, Schwinn S, Locarnini S, Fyfe J, Manns MP, Trautwein C, Zentgraf H. Structural organization of the hepatitis B virus minichromosome. J Mol Biol. 2001;307:183–196. doi: 10.1006/jmbi.2000.4481. http://dx.doi.org/10.1006/jmbi.2000.4481. [DOI] [PubMed] [Google Scholar]
- Chen C, Wang JCY, Zlotnick A. A kinase Chaperones Hepatitis b virus capsid assembly and captures capsid dynamics in vitro. PLoS Pathog. 2011;7 doi: 10.1371/journal.ppat.1002388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu CM, Liaw YF. Intrahepatic distribution of hepatitis B surface and core antigens in chronic hepatitis B virus infection. Hepatocyte with cytoplasmic/membranous hepatitis B core antigen as a possible target for immune hepatocytolysis. Gastroenterology. 1987;92:220–225. doi: 10.1016/0016-5085(87)90863-8. [DOI] [PubMed] [Google Scholar]
- Chu TH, Liou AT, Su PY, Wu HN, Shih C. Nucleic acid chaperone activity associated with the arginine-rich domain of human hepatitis B virus core protein. J Virol. 2014;88:2530–2543. doi: 10.1128/JVI.03235-13. http://dx.doi.org/10.1128/JVI.03235-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowther RA, Kiselev NA, Bottcher B, Berriman JA, Borisova GP, Ose V, Pumpens P. Three-dimensional structure of hepatitis B virus core particles determined by electron cryomicroscopy. Cell. 1994;77:943–950. doi: 10.1016/0092-8674(94)90142-2. [DOI] [PubMed] [Google Scholar]
- Daub H, Blencke S, Habenberger P, Kurtenbach A, Dennenmoser J, Wissing J, Ullrich A, Cotten M. Identification of SRPK1 and SRPK2 as the major cellular protein kinases phosphorylating hepatitis B virus core protein. J Virol. 2002;76:8124–8137. doi: 10.1128/JVI.76.16.8124-8137.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney WE, Edwards R, Colledge D, Shaw T, Furman P, Painter G, Locarnini S. Phenylpropenamide derivatives AT-61 and AT-130 inhibit replication of wildtype and lamivudine-resistant strains of hepatitis B virus in vitro. Antimicrob Agents Chemother. 2002;46:3057–3060. doi: 10.1128/AAC.46.9.3057-3060.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deres K, Schroder CH, Paessens A, Goldmann S, Hacker HJ, Weber O, Kramer T, Niewohner U, Pleiss U, Stoltefuss J, Graef E, Koletzki D, Masantschek RNA, Reimann A, Jaeger R, Gross R, Beckermann B, Schlemmer KH, Haebich D, Rubsamen-Waigmann H. Inhibition of hepatitis B virus replication by drug-induced depletion of nucleocapsids. Science. 2003;299:893–896. doi: 10.1126/science.1077215. http://dx.doi.org/10.1126/science.1077215. [DOI] [PubMed] [Google Scholar]
- Diab AM, Foca A, Fusil F, Lahlali T, Jalaguier P, Amirache F, N’Guyen L, Isorce N, Cosset FL, Zoulim F, Andrisani OM, Durantel D. Polo-like-kinase 1 is a proviral host-factor for hepatitis B virus replication. Hepatology. 2017 doi: 10.1002/hep.29236. http://dx.doi.org/10.1002/hep.29236. [DOI] [PMC free article] [PubMed]
- Du J, Liang X, Liu Y, Qu Z, Gao L, Han L, Liu S, Cui M, Shi Y, Zhang Z. Hepatitis B virus core protein inhibits TRAIL-induced apoptosis of hepatocytes by blocking DR5 expression. Cell Death Differ. 2009;16 doi: 10.1038/cdd.2008.144. http://dx.doi.org/10.1038/cdd.2008.144. [DOI] [PubMed] [Google Scholar]
- Duclos-Vallee JC, Capel F, Mabit H, Petit MA. Phosphorylation of the hepatitis B virus core protein by glyceraldehyde-3-phosphate dehydrogenase protein kinase activity. J Gen Virol. 1998;79(Pt 7):1665–1670. doi: 10.1099/0022-1317-79-7-1665. [DOI] [PubMed] [Google Scholar]
- Durantel D, Zoulim F. New antiviral targets for innovative treatment concepts for hepatitis B virus and hepatitis delta virus. J Hepatol. 2016;64:S117–S131. doi: 10.1016/j.jhep.2016.02.016. http://dx.doi.org/10.1016/j.jhep.2016.02.016. [DOI] [PubMed] [Google Scholar]
- Elia AEH, Rellos P, Haire LF, Chao JW, Ivins FJ, Hoepker K, Mohammad D, Cantley LC, Smerdon SJ, Yaffe MB. The molecular basis for phosphode-pendent substrate targeting and regulation of Plks by the Polo-box domain. Cell. 2003;115:83–95. doi: 10.1016/s0092-8674(03)00725-6. [DOI] [PubMed] [Google Scholar]
- Faure-Dupuy S, Lucifora J, Durantel D. Interplay between the hepatitis B virus and innate immunity: from an understanding to the development of therapeutic concepts. Viruses. 2017;9 doi: 10.3390/v9050095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Favre-Bonvin A, Reynaud C, Kretz-Remy C, Jalinot P. Human papillomavirus type 18 E6 protein binds the cellular PDZ protein TIP-2/GIPC, which is involved in transforming growth factor beta signaling and triggers its degradation by the proteasome. J Virol. 2005;79:4229–4237. doi: 10.1128/JVI.79.7.4229-4237.2005. http://dx.doi.org/10.1128/JVI.79.7.4229-4237.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez M, Quiroga JA, Carreno V. Hepatitis B virus downregulates the human interferon-inducible MxA promoter through direct interaction of precore/core proteins. J Gen Virol. 2003;84:2073–2082. doi: 10.1099/vir.0.18966-0. http://dx.doi.org/10.1099/vir.0.18966-0. [DOI] [PubMed] [Google Scholar]
- Gallina A, Bonelli F, Zentilin L, Rindi G, Muttini M, Milanesi G. A recombinant hepatitis B core antigen polypeptide with the protamine-like domain deleted self-assembles into capsid particles but fails to bind nucleic acids. J Virol. 1989;63:4645–4652. doi: 10.1128/jvi.63.11.4645-4652.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazina EV, Fielding JE, Lin B, Anderson Da. Core protein phosphorylation modulates pregenomic RNA encapsidation to different extents in human and duck hepatitis B viruses. J Virol. 2000;74:4721–4728. doi: 10.1128/jvi.74.10.4721-4728.2000. http://dx.doi.org/10.1128/JVI.74.10.4721-4728.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjertsen BT, Schoffski P. Discovery and development of the Polo-like kinase inhibitor volasertib in cancer therapy. Leukemia. 2015;29:11–19. doi: 10.1038/leu.2014.222. http://dx.doi.org/10.1038/leu.2014.222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goff SP. Host factors exploited by retroviruses. Nat Rev Microbiol. 2007;5:253–263. doi: 10.1038/nrmicro1541. http://dx.doi.org/10.1038/nrmicro1541. [DOI] [PubMed] [Google Scholar]
- Guo YH, Li YN, Zhao JR, Zhang J, Yan Z. HBc binds to the CpG islands of HBV cccDNA and promotes an epigenetic permissive state. Epigenetics. 2011;6:720–726. doi: 10.4161/epi.6.6.15815. [DOI] [PubMed] [Google Scholar]
- Guo Y, Kang W, Lei X, Li Y, Xiang A, Liu Y, Zhao J, Zhang J, Yan Z. Hepatitis B viral core protein disrupts human host gene expression by binding to promoter regions. BMC Genomics. 2012;13:1–12. doi: 10.1186/1471-2164-13-563. http://dx.doi.org/10.1186/1471-2164-13-563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadziyannis SJ. Natural history of chronic hepatitis B in Euro-Mediterranean and African countries. J Hepatol. 2011;55:183–191. doi: 10.1016/j.jhep.2010.12.030. http://dx.doi.org/10.1016/j.jhep.2010.12.030. [DOI] [PubMed] [Google Scholar]
- Harris RS. Molecular mechanism and clinical impact of APOBEC3B-catalyzed mutagenesis in breast cancer. Breast Cancer Res. 2015;17:8. doi: 10.1186/s13058-014-0498-3. http://dx.doi.org/10.1186/s13058-014-0498-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard JM, Sanford JR. The RNAissance family: SR proteins as multifaceted regulators of gene expression. Wiley Interdiscip Rev RNA. 2015;6:93–110. doi: 10.1002/wrna.1260. http://dx.doi.org/10.1002/wrna.1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Liu K. Complete and incomplete hepatitis B virus particles: formation, function, and application. Viruses. 2017;9 doi: 10.3390/v9030056. http://dx.doi.org/10.3390/v9030056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang ZS, Wu HN. Identification and characterization of the RNA chaperone activity of hepatitis delta antigen peptides. J Biol Chem. 1998;273:26455–26461. doi: 10.1074/jbc.273.41.26455. [DOI] [PubMed] [Google Scholar]
- Hubner S, Xiao CY, Jans DA. The protein kinase CK2 site (Ser111/112) enhances recognition of the simian virus 40 large T-antigen nuclear localization sequence by importin. J Biol Chem. 1997;272:17191–17195. doi: 10.1074/jbc.272.27.17191. [DOI] [PubMed] [Google Scholar]
- Hung AY, Sheng M. PDZ domains: structural modules for protein complex assembly. J Biol Chem. 2002;277:5699–5702. doi: 10.1074/jbc.R100065200. http://dx.doi.org/10.1074/jbc.R100065200. [DOI] [PubMed] [Google Scholar]
- Hutchinson JN, Ensminger AW, Clemson CM, Lynch CR, Lawrence JB, Chess A, Mattick J, Makunin I, Morey C, Avner P, Kelley R, Kuroda M, Avner P, Heard E, Clemson C, McNeil J, Willard H, Lawrence J, Willingham A, Orth A, Batalov S, Peters E, Wen B, Aza-Blanc P, Hogenesch J, Schultz P, Clemson C, Hall L, Byron M, McNeil J, Lawrence J, Lamond A, Spector D, Fu X, Maniatis T, Hall L, Smith K, Byron M, Lawrence J, Moen J, Smith K, Lawrence J, Wei X, Somanathan S, Samarabandu J, Berezney R, Huang S, Deerinck T, Ellisman M, Spector D, Herman R, Weymouth L, Penman S, Carter K, Taneja K, Lawrence J, Lawrence J, Carter K, Xing X, Politz J, Tuft R, Prasanth K, Baudendistel N, Fogarty K, Lifshitz L, Langowski J, Spector D, Pederson T, Kent W, Ashburner M, Misra S, Roote J, Lewis S, Blazej R, Davis T, Doyle C, Galle R, George R, Harris N, Hartzell G, Harvey D, Hong L, Houston K, Hoskins R, Johnson G, Martin C, Moshrefi A, Palazzolo M, Reese M, Spradling A, Tsang G, Wan K, Whitelaw K, Celniker S, Loebel D, Tsoi B, Wong N, Tam P, Prasanth K, Prasanth S, Xuan Z, Hearn S, Freier S, Bennett C, Zhang M, Spector D, Beissbarth T, Speed T, Dahlberg J, Lund E, Geirsson A, Lynch R, Paliwal I, Bothwell A, Hammond G, Guru S, Agarwal S, Manickam P, Olufemi S, Crabtree J, Weisemann J, Kester M, Kim Y, Wang Y, Emmert-Buck M, Liotta L, Spiegel A, Boguski M, Roe B, Collins F, Marx S, Burns L, Chandrasekharappa S, Ji P, Diederichs S, Wang W, Boing S, Metzger R, Schneider P, Tidow N, Brandt B, Buerger H, Bulk E, Thomas M, Berdel W, Serve H, Muller-Tidow C, Peyman J, Jareborg N, Birney E, Durbin R, Hong Y, Ontiveros S, Strauss W, Bejerano G, Pheasant M, Makunin I, Stephen S, Kent W, Mattick J, Haussler D, Edgar R, Domrachev M, Lash A, Jongeneel C, Iseli C, Stevenson B, Riggins G, Lal A, Mackay A, Harris R, O’Hare M, Neville A, Simpson A, Strausberg R, Carter K, Bowman D, Carrington W, Fogarty K, McNeil J, Fay F, Lawrence J, Ensminger A, Chess A, Singh N, Ebrahimi F, Gimelbrant A, Ensminger A, Tackett M, Qi P, Gribnau J, Chess A, Huang S, Spector D, Nickerson J, Krochmalnic G, Wan K, Penman S, Schmidt U, Richter K, Berger A, Lichter P, Moen P, Johnson C, Byron M, Shopland L, de la Serna I, Imbalzano A, Lawrence J, Shopland L, Johnson C, Byron M, McNeil J, Lawrence J, Fox A, Bond C, Lamond A, Saha S, Murthy S, Rangarajan P, Reich M, Ohm K, Angelo M, Tamayo P, Mesirov J, Johnson C, McNeil J, Carter K, Lawrence J, Tam R, Shopland L, Johnson C, McNeil J, Lawrence J, Blencowe B, Issner R, Nickerson J, Sharp P. A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 2007;8:39. doi: 10.1186/1471-2164-8-39. http://dx.doi.org/10.1186/1471-2164-8-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia B, Guo M, Li G, Yu D, Zhang X, Lan K, Deng Q. Hepatitis B virus core protein sensitizes hepatocytes to tumor necrosis factor-induced apoptosis by suppression of the phosphorylation of mitogen-activated protein kinase kinase 7. J Virol. 2015;89:2041–2051. doi: 10.1128/JVI.03106-14. http://dx.doi.org/10.1128/JVI.03106-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson LA, Li L, Sandri-Goldin RM. The cellular RNA export receptor TAP/NXF1 is required for ICP27-mediated export of herpes simplex virus 1 RNA, but the TREX complex adaptor protein Aly/REF appears to be dispensable. J Virol. 2009;83:6335–6346. doi: 10.1128/JVI.00375-09. http://dx.doi.org/10.1128/JVI.00375-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Juillard F, Hiriart E, Sergeant N, Vingtdeux-Didier V, Drobecq H, Sergeant A, Manet E, Gruffat H. Epstein-Barr virus protein EB2 contains an N-terminal transferable nuclear export signal that promotes nucleocytoplasmic export by directly binding TAP/NXF1. J Virol. 2009;83:12759–12768. doi: 10.1128/JVI.01276-09. http://dx.doi.org/10.1128/JVI.01276-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung J, Hwang SG, Chwae YJ, Park S, Shin HJ, Kim K. Phosphoacceptors threonine 162 and serines 170 and 178 within the carboxyl-terminal RRRS/T motif of the hepatitis B virus core protein make multiple contributions to hepatitis B virus replication. J Virol. 2014;88:8754–8767. doi: 10.1128/JVI.01343-14. http://dx.doi.org/10.1128/JVI.01343-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann M, Bischof A, Gerlich WH. In vitro model for the nuclear transport of the hepadnavirus genome. J Virol. 1997;71:1310–1316. doi: 10.1128/jvi.71.2.1310-1316.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann M, Gerlich WH. Effect of core protein phosphorylation by protein kinase C on encapsidation of RNA within core particles of hepatitis B virus. J Virol. 1994;68:7993–8000. doi: 10.1128/jvi.68.12.7993-8000.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann M, Schmitz A, Rabe B. Intracellular transport of hepatitis B virus. World J Gastroenterol. 2007;13:39–47. doi: 10.3748/wjg.v13.i1.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kann M, Sodeik B, Vlachou A, Gerlich WH, Helenius A. Phosphorylation-dependent binding of hepatitis B virus core particles to the nuclear pore complex. J Cell Biol. 1999;145:45–55. doi: 10.1083/jcb.145.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura R, Sekimoto T, Ito S, Harada S, Yamagata H, Masai H, Yoneda Y, Yanagi K. Nuclear import of Epstein-Barr virus nuclear antigen 1 mediated by NPI-1 (Importin alpha5) is up- and down-regulated by phosphorylation of the nuclear localization signal for which Lys379 and Arg380 are essential. J Virol. 2006;80:1979–1991. doi: 10.1128/JVI.80.4.1979-1991.2006. http://dx.doi.org/10.1128/JVI.80.4.1979-1991.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon JA, Rho HM. Transcriptional repression of the human p53 gene by hepatitis B viral core protein (HBc) in human liver cells. Biol Chem. 2003;384:203–212. doi: 10.1515/BC.2003.022. http://dx.doi.org/10.1515/BC.2003.022. [DOI] [PubMed] [Google Scholar]
- Kwon JA, Rho HM. Hepatitis B viral core protein activates the hepatitis B viral enhancer II/pregenomic promoter through the nuclear factor kappaB binding site. Biochem Cell Biol. 2002;80:445–455. doi: 10.1139/o02-133. [DOI] [PubMed] [Google Scholar]
- Lam A, Ren S, Vogel R, Espiritu C, Kelly M, Lau V, et al. Inhibition of hepatitis B virus replication by the HBV core inhibitor NVR 3-778. Hepatology (Baltimore, Md) 2015:62. S1: 223A.
- Lan YT, Li J, Liao W, Ou J. Roles of the three major phosphorylation sites of hepatitis B virus core protein in viral replication. Virology. 1999;259:342–348. doi: 10.1006/viro.1999.9798. http://dx.doi.org/10.1006/viro.1999.9798. [DOI] [PubMed] [Google Scholar]
- Lanford RE, Notvall L. Expression of hepatitis B virus core and precore antigens in insect cells and characterization of a core-associated kinase activity. Virology. 1990;176:222–233. doi: 10.1016/0042-6822(90)90247-o. [DOI] [PubMed] [Google Scholar]
- Levin JG, Guo J, Rouzina I, Musier-Forsyth K. Nucleic acid chaperone activity of HIV-1 nucleocapsid protein: critical role in reverse transcription and molecular mechanism. Prog Nucleic Acid Res Mol Biol. 2005;80:217–286. doi: 10.1016/S0079-6603(05)80006-6. http://dx.doi.org/10.1016/S0079-6603(05)80006-6. [DOI] [PubMed] [Google Scholar]
- Lewellyn EB, Loeb DD. The arginine clusters of the carboxy-terminal domain of the core protein of hepatitis B virus make pleiotropic contributions to genome replication. J Virol. 2011;85:1298–1309. doi: 10.1128/JVI.01957-10. http://dx.doi.org/10.1128/JVI.01957-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li HC, Huang EY, Su PY, Wu SY, Yang CC, Lin YS, Chang WC, Shih C. Nuclear export and import of human hepatitis B virus capsid protein and particles. PLoS Pathog. 2010;6:e1001162. doi: 10.1371/journal.ppat.1001162. http://dx.doi.org/10.1371/journal.ppat.1001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Zhang L, Chen L, Feng W, Xu Y, Chen F, Liu X, Chen Z, Liu W. MxA inhibits hepatitis B virus replication by interaction with hepatitis B core antigen. Hepatology. 2012;56:803–811. doi: 10.1002/hep.25608. http://dx.doi.org/10.1002/hep.25608. [DOI] [PubMed] [Google Scholar]
- Liao W, Ou JH. Phosphorylation and nuclear localization of the hepatitis B virus core protein: significance of serine in the three repeated SPRRR motifs. J Virol. 1995;69 doi: 10.1128/jvi.69.2.1025-1029.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Ludgate L, Yuan Z, Hu J. Regulation of multiple stages of hepadnavirus replication by the carboxyl-terminal domain of viral core protein in trans. J Virol. 2015;89:2918–2930. doi: 10.1128/JVI.03116-14. http://dx.doi.org/10.1128/JVI.03116-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucifora J, Protzer U. Attacking hepatitis B virus cccDNA - the holy grail to hepatitis B cure. J Hepatol. 2016;64:S41–S48. doi: 10.1016/j.jhep.2016.02.009. http://dx.doi.org/10.1016/j.jhep.2016.02.009. [DOI] [PubMed] [Google Scholar]
- Lucifora J, Xia Y, Reisinger F, Zhang K, Stadler D, Cheng X, Sprinzl MF, Koppensteiner H, Makowska Z, Volz T, Remouchamps C, Chou WM, Thasler WE, Huser N, Durantel D, Liang TJ, Munk C, Heim MH, Browning JL, Dejardin E, Dandri M, Schindler M, Heikenwalder M, Protzer U. Specific and nonhepatotoxic degradation of nuclear hepatitis B virus cccDNA. Science. 2014;343:1221–1228. doi: 10.1126/science.1243462. http://dx.doi.org/10.1126/science.1243462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludgate L, Adams C, Hu J. Phosphorylation state-dependent interactions of hepadnavirus core protein with host factors. PLoS One. 2011;6:e29566. doi: 10.1371/journal.pone.0029566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludgate L, Liu K, Luckenbaugh L, Streck N, Eng S, Voitenleitner C, Delaney WE, Hu J. Cell-free hepatitis B virus capsid assembly dependent on the core protein C-Terminal domain and regulated by phosphorylation. J Virol. 2016 doi: 10.1128/JVI.00394-16. http://dx.doi.org/10.1128/JVI.00394-16. [DOI] [PMC free article] [PubMed]
- Ludgate L, Ning X, Nguyen DH, Adams C, Mentzer L, Hu J. Cyclin-dependent kinase 2 phosphorylates s/t-p sites in the hepadnavirus core protein C-terminal domain and is incorporated into viral capsids. J Virol. 2012;86:12237–12250. doi: 10.1128/JVI.01218-12. http://dx.doi.org/10.1128/JVI.01218-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mears WE, Lam V, Rice SA. Identification of nuclear and nucleolar localization signals in the herpes simplex virus regulatory protein ICP27. J Virol. 1995;69:935–947. doi: 10.1128/jvi.69.2.935-947.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melen K, Tynell J, Fagerlund R, Roussel P, Hernandez-Verdun D, Julkunen I. Influenza A H3N2 subtype virus NS1 protein targets into the nucleus and binds primarily via its C-terminal NLS2/NoLS to nucleolin and fibrillarin. Virol J. 2012;9:167. doi: 10.1186/1743-422X-9-167. http://dx.doi.org/10.1186/1743-422X-9-167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michalak T, Nowoslawski A. Crystalline aggregates of hepatitis B core particles in cytoplasm of hepatocytes. Intervirology. 1982;17 doi: 10.1159/000149295. http://dx.doi.org/10.1159/000149295. [DOI] [PubMed] [Google Scholar]
- Morganella S, Alexandrov LB, Glodzik D, Zou X, Davies H, Staaf J, Sieuwerts AM, Brinkman AB, Martin S, Ramakrishna M, Butler A, Kim HY, Borg A, Sotiriou C, Futreal PA, Campbell PJ, Span PN, Van Laere S, Lakhani SR, Eyfjord JE, Thompson AM, Stunnenberg HG, van de Vijver MJ, Martens JWM, Borresen-Dale AL, Richardson AL, Kong G, Thomas G, Sale J, Rada C, Stratton MR, Birney E, Nik-Zainal S. The topography of mutational processes in breast cancer genomes. Nat Commun. 2016;7:11383. doi: 10.1038/ncomms11383. http://dx.doi.org/10.1038/ncomms11383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nardozzi JD, Lott K, Cingolani G. Phosphorylation meets nuclear import: a review. Cell Commun Signal. 2010;8:32. doi: 10.1186/1478-811X-8-32. http://dx.doi.org/10.1186/1478-811X-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nassal M. HBV cccDNA: viral persistence reservoir and key obstacle for a cure of chronic hepatitis B. Gut. 2015;64:1972–1984. doi: 10.1136/gutjnl-2015-309809. http://dx.doi.org/10.1136/gutjnl-2015-309809. [DOI] [PubMed] [Google Scholar]
- Nassal M. The arginine-rich domain of the hepatitis B virus core protein is required for pregenome encapsidation and productive viral positive-strand DNA synthesis but not for virus assembly. J Virol. 1992;66:4107–4116. doi: 10.1128/jvi.66.7.4107-4116.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman M, Chua PK, Tang FM, Su PY, Shih C. Testing an electrostatic interaction hypothesis of hepatitis B virus capsid stability by using an in vitro capsid disassembly/reassembly system. J Virol. 2009;83:10616–10626. doi: 10.1128/JVI.00749-09. http://dx.doi.org/10.1128/JVI.00749-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen T, Desmond P, Locarnini S. The role of quantitative hepatitis B serology in the natural history and management of chronic hepatitis B. Hepatol Int. 2009;3:5–15. doi: 10.1007/s12072-009-9149-7. http://dx.doi.org/10.1007/s12072-009-9149-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nik-Zainal S, Alexandrov LB, Wedge DC, Van Loo P, Greenman CD, Raine K, Jones D, Hinton J, Marshall J, Stebbings LA, Menzies A, Martin S, Leung K, Chen L, Leroy C, Ramakrishna M, Rance R, Lau KW, Mudie LJ, Varela I, McBride DJ, Bignell GR, Cooke SL, Shlien A, Gamble J, Whitmore I, Maddison M, Tarpey PS, Davies HR, Papaemmanuil E, Stephens PJ, McLaren S, Butler AP, Teague JW, Jonsson G, Garber JE, Silver D, Miron P, Fatima A, Boyault S, Langerod A, Tutt A, Martens JWM, Aparicio SAJR, Borg A, Salomon AV, Thomas G, Borresen-Dale AL, Richardson AL, Neuberger MS, Futreal PA, Campbell PJ, Stratton MR. Mutational processes molding the genomes of 21 breast cancers. Cell. 2012;149:979–993. doi: 10.1016/j.cell.2012.04.024. http://dx.doi.org/10.1016/j.cell.2012.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogawa Y, Miyamoto Y, Oka M, Yoneda Y. The interaction between importin-alpha and Nup153 promotes importin-alpha/beta-mediated nuclear import. Traffic. 2012;13:934–946. doi: 10.1111/j.1600-0854.2012.01367.x. http://dx.doi.org/10.1111/j.1600-0854.2012.01367.x. [DOI] [PubMed] [Google Scholar]
- Paludan SR. Immune Sensing of DNA. 2013 doi: 10.1016/j.immuni.2013.05.004. http://dx.doi.org/10.1016/j.immuni.2013.05.004. [DOI] [PMC free article] [PubMed]
- Perlman DH, Berg Ea, O’connor PB, Costello CE, Hu J. Reverse transcription-associated dephosphorylation of hepadnavirus nucleocapsids. Proc Natl Acad Sci U S A. 2005;102:9020–9025. doi: 10.1073/pnas.0502138102. http://dx.doi.org/10.1073/pnas.0502138102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petit MA, Pillot J. HBc and HBe antigenicity and DNA-binding activity of major core protein P22 in hepatitis B virus core particles isolated from the cytoplasm of human liver cells. J Virol. 1985;53 doi: 10.1128/jvi.53.2.543-551.1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pong WL, Huang ZS, Teoh PG, Wang CC, Wu HN. RNA binding property and RNA chaperone activity of dengue virus core protein and other viral RNA-interacting proteins. FEBS Lett. 2011;585:2575–2581. doi: 10.1016/j.febslet.2011.06.038. http://dx.doi.org/10.1016/j.febslet.2011.06.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prescott EL, Brimacombe CL, Hartley M, Bell I, Graham S, Roberts S. Human papillomavirus type 1 E1^E4 protein is a potent inhibitor of the serine-arginine (SR) protein kinase SRPK1 and inhibits phosphorylation of host SR proteins and of the viral transcription and replication regulator E2. J Virol. 2014;88:12599–12611. doi: 10.1128/JVI.02029-14. http://dx.doi.org/10.1128/JVI.02029-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian G, Jin F, Chang L, Yang Y, Peng H, Duan C. NIRF, a novel ubiquitin ligase, interacts with hepatitis B virus core protein and promotes its degradation. Biotechnol Lett. 2012;34:29–36. doi: 10.1007/s10529-011-0751-0. http://dx.doi.org/10.1007/s10529-011-0751-0. [DOI] [PubMed] [Google Scholar]
- Rabe B, Delaleau M, Bischof A, Foss M, Sominskaya I, Pumpens P, Cazenave C, Castroviejo M, Kann M. Nuclear entry of hepatitis B virus capsids involves disintegration to protein dimers followed by nuclear reassociation to capsids. PLoS Pathog. 2009;5:e1000563. doi: 10.1371/journal.ppat.1000563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabe B, Vlachou A, Pante N, Helenius A, Kann M. Nuclear import of hepatitis B virus capsids and release of the viral genome. Proc Natl Acad Sci U S A. 2003;100:9849–9854. doi: 10.1073/pnas.1730940100. http://dx.doi.org/10.1073/pnas.1730940100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Razanskas R, Sasnauskas K. Interaction of hepatitis B virus core protein with human GIPC1. Arch Virol. 2010;155:247–250. doi: 10.1007/s00705-009-0561-z. http://dx.doi.org/10.1007/s00705-009-0561-z. [DOI] [PubMed] [Google Scholar]
- Revill P, Yuan Z. New insights into how HBV manipulates the innate immune response to establish acute and persistent infection. Antivir Ther. 2013;18:1–15. doi: 10.3851/IMP2542. http://dx.doi.org/10.3851/IMP2542. [DOI] [PubMed] [Google Scholar]
- Rousset R, Fabre S, Desbois C, Bantignies F, Jalinot P. The C-terminus of the HTLV-1 Tax oncoprotein mediates interaction with the PDZ domain of cellular proteins. Oncogene. 1998;16:643–654. doi: 10.1038/sj.onc.1201567. http://dx.doi.org/10.1038/sj.onc.1201567. [DOI] [PubMed] [Google Scholar]
- Ryu DK, Ahn BY, Ryu WS. Proximity between the cap and 5′ epsilon stem-loop structure is critical for the suppression of pgRNA translation by the hepatitis B viral polymerase. Virology. 2010;406:56–64. doi: 10.1016/j.virol.2010.07.005. http://dx.doi.org/10.1016/j.virol.2010.07.005. [DOI] [PubMed] [Google Scholar]
- Ryu DK, Kim S, Ryu WS. Hepatitis B virus polymerase suppresses translation of pregenomic RNA via a mechanism involving its interaction with 5′ stem-loop structure. Virology. 2008;373:112–123. doi: 10.1016/j.virol.2007.11.010. http://dx.doi.org/10.1016/j.virol.2007.11.010. [DOI] [PubMed] [Google Scholar]
- Sakurikar N, Eastman A. Critical reanalysis of the methods that discriminate the activity of CDK2 from CDK1. Cell Cycle. 2016;15:1184–1188. doi: 10.1080/15384101.2016.1160983. http://dx.doi.org/10.1080/15384101.2016.1160983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvetti A, Greco A. Viruses and the nucleolus: the fatal attraction. Biochim Biophys Acta. 2014;1842:840–847. doi: 10.1016/j.bbadis.2013.12.010. http://dx.doi.org/10.1016/j.bbadis.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz A, Schwarz A, Foss M, Zhou L, Rabe B, Hoellenriegel J, Stoeber M, Pante N, Kann M. Nucleoporin 153 arrests the nuclear import of hepatitis B virus capsids in the nuclear basket. PLoS Pathog. 2010;6:e1000741. doi: 10.1371/journal.ppat.1000741. http://dx.doi.org/10.1371/journal.ppat.1000741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selzer L, Kant R, Wang JCY, Bothner B, Zlotnick A. Hepatitis B virus core protein phosphorylation sites affect capsid stability and transient exposure of the C-terminal domain. J Biol Chem. 2015;290:28584–28593. doi: 10.1074/jbc.M115.678441. http://dx.doi.org/10.1074/jbc.M115.678441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semrad K. Proteins with RNA chaperone activity: a world of diverse proteins with a common task-impediment of RNA misfolding. Biochem Res Int. 2011;2011:532908. doi: 10.1155/2011/532908. http://dx.doi.org/10.1155/2011/532908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma RR, Dhiman RK, Chawla Y, Vasistha RK. Immunohistochemistry for core and surface antigens in chronic hepatitis. Trop Gastroenterol. 2002;23:16–19. [PubMed] [Google Scholar]
- Shim HY, Quan X, Yi YS, Jung G. Heat shock protein 90 facilitates formation of the HBV capsid via interacting with the HBV core protein dimers. Virology. 2011;410:161–169. doi: 10.1016/j.virol.2010.11.005. http://dx.doi.org/10.1016/j.virol.2010.11.005. [DOI] [PubMed] [Google Scholar]
- Sohn SY, Kim SB, Kim J, Ahn BY. Negative regulation of hepatitis B virus replication by cellular Hsp40/DnaJ proteins through destabilization of viral core and X proteins. J Gen Virol. 2006;87:1883–1891. doi: 10.1099/vir.0.81684-0. http://dx.doi.org/10.1099/vir.0.81684-0. [DOI] [PubMed] [Google Scholar]
- Souki SK, Gershon PD, Sandri-Goldin RM. Arginine methylation of the ICP27 RGG box regulates ICP27 export and is required for efficient herpes simplex virus 1 replication. J Virol. 2009;83:5309–5320. doi: 10.1128/JVI.00238-09. http://dx.doi.org/10.1128/JVI.00238-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanwolleghem T, Hou J, van Oord G, Andeweg AC, Osterhaus ADME, Pas SD, Janssen HLA, Boonstra A. Re-evaluation of hepatitis B virus clinical phases by systems biology identifies unappreciated roles for the innate immune response and B cells. Hepatology. 2015;62:87–100. doi: 10.1002/hep.27805. http://dx.doi.org/10.1002/hep.27805. [DOI] [PubMed] [Google Scholar]
- Wang XY, Wei ZM, Wu GY, Wang JH, Zhang YJ, Li J, Zhang HH, Xie XW, Wang X, Wang ZH, Wei L, Wang Y, Chen HS. In vitro inhibition of HBV replication by a novel compound, GLS4, and its efficacy against adefovir-dipivoxil-resistant HBV mutations. Antivir Ther. 2012;17:793–803. doi: 10.3851/IMP2152. http://dx.doi.org/10.3851/IMP2152. [DOI] [PubMed] [Google Scholar]
- Williams JS, Andrisani OM. The hepatitis B virus X protein targets the basic region-leucine zipper domain of CREB. Proc Natl Acad Sci U S A. 1995;92:3819–3823. doi: 10.1073/pnas.92.9.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittkop L, Schwarz A, Cassany A, Grün-Bernhard S, Delaleau M, Rabe B, Cazenave C, Gerlich W, Glebe D, Kann M. Inhibition of protein kinase C phosphorylation of hepatitis B virus capsids inhibits virion formation and causes intracellular capsid accumulation. Cell Microbiol. 2010;12:962–975. doi: 10.1111/j.1462-5822.2010.01444.x. http://dx.doi.org/10.1111/j.1462-5822.2010.01444.x. [DOI] [PubMed] [Google Scholar]
- Wynne SA, Crowther RA, Leslie AG. The crystal structure of the human hepatitis B virus capsid. Mol Cell. 1999;3:771–780. doi: 10.1016/s1097-2765(01)80009-5. [DOI] [PubMed] [Google Scholar]
- Xiang A, Ren F, Lei X, Zhang J, Guo R, Lu Z, Guo Y. The hepatitis B virus (HBV) core protein enhances the transcription activation of CRE via the CRE/CREB/CBP pathway. Antivir Res. 2015;120:7–15. doi: 10.1016/j.antiviral.2015.04.013. http://dx.doi.org/10.1016/j.antiviral.2015.04.013. [DOI] [PubMed] [Google Scholar]
- Xie Q, Fan F, Wei W, Liu Y, Xu Z, Zhai L, Qi Y, Ye B, Zhang Y, Basu S, Zhao Z, Wu J, Xu P. Multi-omics analyses reveal metabolic alterations regulated by hepatitis B virus core protein in hepatocellular carcinoma cells. Sci Rep. 2017 doi: 10.1038/srep41089. http://dx.doi.org/10.1038/srep41089. [DOI] [PMC free article] [PubMed]
- Yang CC, Huang EY, Li HC, Su PY, Shih C. Nuclear export of human hepatitis B virus core protein and pregenomic RNA depends on the cellular NXF1-p15 machinery. PLoS One. 2014;9:e106683. doi: 10.1371/journal.pone.0106683. http://dx.doi.org/10.1371/journal.pone.0106683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh CT, Wong SW, Fung YK, Ou JH. Cell cycle regulation of nuclear localization of hepatitis B virus core protein. Proc Natl Acad Sci U S A. 1993;90:6459–6463. doi: 10.1073/pnas.90.14.6459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu X, Jin L, Jih J, Shih C, Zhou ZH. 3.5Å cryoEM structure of hepatitis B virus core assembled from full-length core protein. PLoS One. 2013;8:e69729. doi: 10.1371/journal.pone.0069729. http://dx.doi.org/10.1371/journal.pone.0069729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Xing Z, Mani SKK, Bancel B, Durantel D, Zoulim F, Tran EJ, Merle P, Andrisani O. RNA helicase DEAD box protein 5 regulates Polycomb repressive complex 2/Hox transcript antisense intergenic RNA function in hepatitis B virus infection and hepatocarcinogenesis. Hepatology. 2016;64:1033–1048. doi: 10.1002/hep.28698. http://dx.doi.org/10.1002/hep.28698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Chen J, Wu M, Zhang X, Zhang M, Yue L, Li Y, Liu J, Li B, Shen F, Wang Y, Bai L, Protzer U, Levrero M, Yuan Z. PRMT5 restricts hepatitis B virus replication through epigenetic repression of covalently closed circular DNA transcription and interference with pregenomic RNA encapsidation. Hepatology. 2017;66:398–415. doi: 10.1002/hep.29133. http://dx.doi.org/10.1002/hep.29133. [DOI] [PubMed] [Google Scholar]
- Zheng J, Schodel F, Peterson DL. The structure of hepadnaviral core antigens. Identification of free thiols and determination of the disulfide bonding pattern. J Biol Chem. 1992;267 [PubMed] [Google Scholar]
- Zheng Y, Fu XD, Ou JHJ. Suppression of hepatitis B virus replication by SRPK1 and SRPK2 via a pathway independent of the phosphorylation of the viral core protein. Virology. 2005;342:150–158. doi: 10.1016/j.virol.2005.07.030. [DOI] [PubMed] [Google Scholar]
- Zhou Z, Hu T, Zhou X, Wildum S, Garcia-Alcalde F, Xu Z, Wu D, Mao Y, Tian X, Zhou Y, Shen F, Zhang Z, Tang G, Najera I, Yang G, Shen HC, Young JAT, Qin N. Heteroaryldihydropyrimidine (HAP) and sulfamoylbenzamide (SBA) inhibit hepatitis B virus replication by different molecular mechanisms. Sci Rep. 2017;7:42374. doi: 10.1038/srep42374. http://dx.doi.org/10.1038/srep42374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnick A, Cheng N, Conway JF, Booy FP, Steven AC, Stahl SJ, Wingfield PT. Dimorphism of hepatitis B virus capsids is strongly influenced by the C-terminus of the capsid protein. Biochemistry. 1996;35:7412–7421. doi: 10.1021/bi9604800. http://dx.doi.org/10.1021/bi9604800. [DOI] [PubMed] [Google Scholar]
- Zlotnick A, Cheng N, Stahl SJ, Conway JF, Steven AC, Wingfield PT. Localization of the C terminus of the assembly domain of hepatitis B virus capsid protein: implications for morphogenesis and organization of encapsidated RNA. Proc Natl Acad Sci U S A. 1997;94:9556–9561. doi: 10.1073/pnas.94.18.9556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zlotnick A, Venkatakrishnan B, Tan Z, Lewellyn E, Turner W, Francis S. Core protein: a pleiotropic keystone in the HBV lifecycle. Antivir Res. 2015;121:82–93. doi: 10.1016/j.antiviral.2015.06.020. http://dx.doi.org/10.1016/j.antiviral.2015.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]