

Abstract
The recombination-activating gene 1 (RAG1) and RAG2 proteins initiate the V(D)J recombination process, which ultimately enables the generation of T cells and B cells with a diversified repertoire of antigen-specific receptors. Mutations of the RAG genes in humans are associated with a broad spectrum of clinical phenotypes, ranging from severe combined immunodeficiency to autoimmunity. Recently, novel insights into the phenotypic diversity of this disease have been provided by resolving the crystal structure of the RAG complex, by developing novel assays to test recombination activity of the mutant RAG proteins and by characterizing the molecular and cellular basis of immune dysregulation in patients with RAG deficiency.
The plasticity of the adaptive immune system to recognize millions of possible antigens is largely due to the combinatorial joining of variable (V), diversity (D) and joining (J) gene segments that encode the antigen-binding regions of T cell receptors (TCRs) in T cells and B cell receptors (BCRs) in B cells, and to the junctional diversity that can be introduced during the process of V(D)J recombination (BOX 1). Each of the V, D and J gene segments is flanked by recombination signal sequences (RSSs), containing consensus nonamer and heptamer elements that are separated by a spacer of either 12 or 23 nucleotides. Recombination-activating gene 1 (RAG1) and RAG2 (referred to collectively here as RAG genes) encode lymphoid-specific proteins that are expressed during the early stages of T cell and B cell development and initiate the process of V(D)J recombination by introducing DNA double-strand breaks (DSBs) at the junction between the heptamer and a coding element. This results in the formation of sealed hairpin coding ends and blunt signal ends, which are eventually processed and joined by means of the non-homologous end joining pathway (NHEJ pathway) (BOX 1).
Box 1. V(D)J recombination process.
Two recombination-activating gene 1 (RAG1) and two RAG2 molecules form a heterotetramer that binds to recombination signal sequences (RSSs) flanking the variable (V), diversity (D) and joining (J) coding elements of the immunoglobulin and T cell receptor (TCR) genes (see the figure). RSSs are composed of conserved heptamer (5′-CACAGTG-3′) and nonamer (5′-ACAAAAACC-3′) elements, which are separated by a degenerate spacer of either 12 or 23 nucleotides. Efficient DNA cleavage requires synapsis of one 12–RSS and one 23–RSS (the ‘12–23 rule’)97, thus ensuring sequential D-to-J and V-to-DJ joining. Selection of the V, D and J genes that are targeted for recombination is not stochastic but is based on the intrinsic quality of the RSSs97 and on accessibility and epigenetic modifications of the TCR and immunoglobulin loci50,54,98.
Upon binding to a pair of RSSs, RAG1 introduces a nick on one strand of the DNA between the RSS heptamer and the flanking coding element, generating a paired complex. The resulting hydroxyl group on the 3′ end of the coding flank attacks the phosphodiester bond on the opposite DNA strand in a transesterification reaction, forming sealed hairpinned coding ends and blunted signal ends, to which the RAG1–RAG2 heterotetramer remains bound in a cleaved signal complex. Subsequently, DNA- dependent protein kinase catalytic subunit (DNA-PKcs) activates Artemis, which opens the hairpins. Both signal ends and coding ends are then processed by the non-homologous end joining (NHEJ) pathway to enable joining of broken ends. Whereas signal ends containing the RSSs are usually precisely ligated, imprecise joining of coding ends may occur. In particular, asymmetrical opening of the hairpinned coding ends allows incorporation of palindromic sequences (‘P-nucleotides’) during the joining process. Furthermore, terminal deoxynucleotidyl transferase (TdT) may introduce additional nucleotides in the junction, generating ‘N-diversity’. Finally, exonucleolytic cleavage may ‘chew’ nucleotides at the boundary between the two coding ends. This ‘junctional diversity’ contributes to the overall diversity of the TCR and immunoglobulin repertoires. C, constant region; CTD, carboxy-terminal domain; DDBD, dimerization and DNA-binding domain; HMGB, high mobility group protein B; NBD, nonamer-binding domain; PreR, pre-RNase H; RNH, catalytic RNase H; ZnC2, zinc-binding cysteine residue region; ZnH2, zinc-binding histidine residue region.
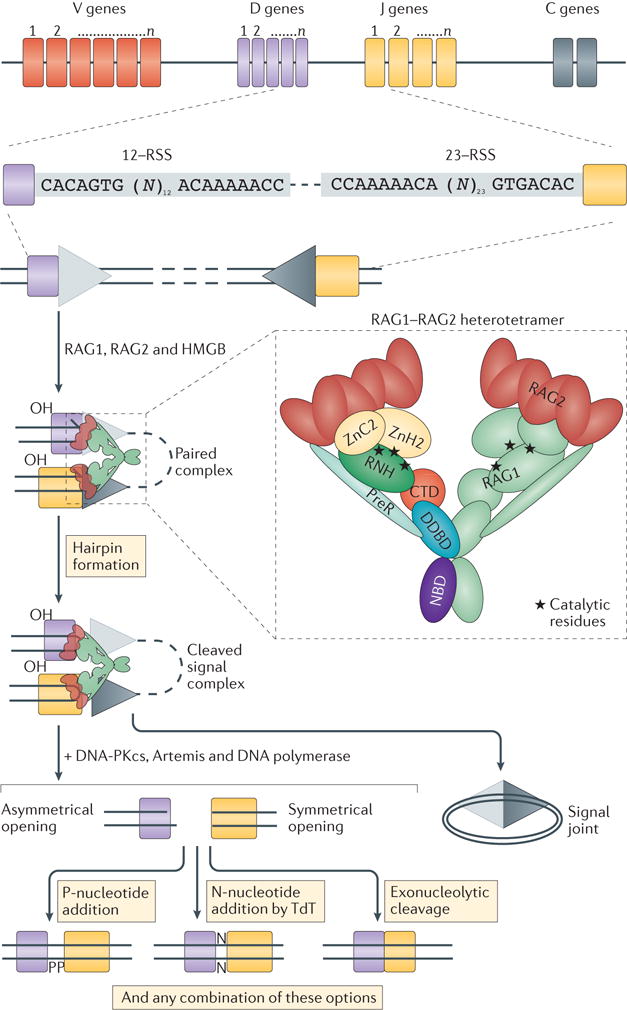
Immunoglobulin or TCR genes
Following cloning of the Rag1 and Rag2 genes1,2 and the demonstration that disruption of these genes in mice prevents V(D)J recombination, which in turn results in a lack of T cells and B cells3,4, it was found that RAG mutations in humans are a prominent cause of severe combined immunodeficiency (SCID) associated with a lack of circulating T cells and B cells (referred to here as T−B− SCID)5. Moreover, hypomorphic RAG mutations that support modest, but residual, recombination activity were identified in infants with Omenn syndrome6, which is a disease that is characterized by immune dysregulation and the presence of oligoclonal, activated T cells infiltrating multiple organs7,8. All of these conditions present early in life with increased susceptibility to severe infections and are inevitably fatal unless treated by haematopoietic stem cell transplantation (HSCT); however, in recent years, RAG mutations have been identified in patients presenting later in childhood or even in young adulthood with CID associated with granulomas and/or autoimmunity (CID–G/AI)9. In this Review, we discuss the molecular and cellular mechanisms that account for the expanding range of clinical and immunological phenotypes of human RAG deficiency, in light of important advances in the characterization of the structure and function of the RAG complex10–12.
Clinical phenotype of RAG deficiency
Mutations of the RAG genes in humans are associated with distinct clinical phenotypes, which are characterized by variable association of infections and auto immunity. In some cases, environmental factors have been shown to contribute to such phenotypic heterogeneity.
T−B− SCID and Omenn syndrome
SCID comprises a heterogeneous group of disorders that are characterized by profound abnormalities in the development and function of T cells (and also B cells in some forms of SCID), and are associated with early-onset severe infections13. This condition is inevitably fatal early in life, unless immune reconstitution is achieved, usually with HSCT14. Among patients with T−B− SCID, two forms of the disease have been identified: some patients have increased cellular radiosensitivity15 — reminiscent of the SCID mouse16,17 — whereas a predominant group of patients have normal cellular radiosensitivity. In 1996, RAG mutations were identified as the main cause of T−B− SCID with normal cellular radiosensitivity5; subsequently, mutations in genes encoding various components of the NHEJ pathway were shown to cause T−B− SCID with increased cellular radiosensitivity18.
A distinct phenotype characterizes Omenn syndrome, which was first described in 1965 (REF. 19). These patients manifest early-onset generalized erythroderma, lymphadenopathy, hepatosplenomegaly, eosinophilia and severe hypogammaglobulinaemia with increased IgE levels, which are associated with the presence of autologous, oligoclonal and activated T cells that infiltrate multiple organs7,8. B cells are typically absent. In these patients, the occurrence of increased IgE levels and eosinophilia indicates that there is skewing of CD4+ T cells to a T helper 2 (TH2) cell phenotype, although the cause of such skewing remains unclear. Consistent with this, in vitro-activated T cells from patients with Omenn syndrome predominantly secrete the TH2 cell cytokines interleukin-4 (IL-4) and IL-5 (REFS 20,21), and serum levels of IL-5 are increased20. Furthermore, selective accumulation of T cells harbouring distinct TCR specificities in different tissues has been demonstrated in patients with Omenn syndrome, suggesting tissue-specific, (self) antigen-driven expansion of T cell populations8,22.
The search for RAG mutations in patients with Omenn syndrome was prompted by the occurrence of Omenn syndrome and T−B− SCID in a pair of siblings7. Indeed, it was demonstrated that hypomorphic RAG mutations that markedly reduce, but do not completely abolish, recombination activity are the most common cause of Omenn syndrome in humans6. In some patients with hypomorphic RAG mutations, a residual presence of autologous T cells was demonstrated without clinical manifestations of Omenn syndrome5,23. This condition is referred to as ‘atypical’ or ‘leaky’ SCID18,24.
A distinct SCID phenotype involving the oligoclonal expansion of autologous γδ T cells (referred to here as γδ T+ SCID) was subsequently reported in infants with RAG deficiency and disseminated cyto megalo virus (CMV) infection25,26. A significant proportion of these patients have detectable B cells, develop auto immune cytopaenias and are at high risk for Epstein–Barr virus-driven lymphoproliferation.
Following the introduction of newborn screening for SCID in the United States, it has become possible to establish that RAG mutations account for 19% of all cases of SCID and SCID-related conditions, and are a prominent cause of atypical SCID and Omenn syndrome in particular27. A much higher frequency of RAG deficiency has been reported in countries with increased rate of parental consanguinity28.
The identification of RAG mutations in patients with SCID and Omenn syndrome has confirmed the indispensable role of the RAG proteins in humans in terms of initiating V(D)J recombination and enabling T cell and B cell development. Moreover, such studies have shown that hypomorphic RAG mutations that allow for residual T cell (and much less so, B cell) development are responsible for the development of Omenn syndrome or atypical SCID phenotypes. Intra- and interfamilial variability of the clinical and immunological phenotypes that are associated with the same RAG mutation23,29,30 has indicated that additional factors, other than the level of RAG-mediated recombination activity alone, contribute to the overall phenotype. These additional factors include the largely stochastic nature of the V(D)J recombination process, as well as the contribution of other genetic modifiers and of environmental factors, as indicated by the expansion of γδ T cells in RAG-mutated infants with CMV infection, as well as by the development of an Omenn syndrome phenotype in an infant with SCID after parainfluenza type 3 virus infection31. Finally, somatic mutations may also modify the disease phenotype. Indeed, oligoclonal expansion of T cells and Omenn syndrome have been reported in an infant with SCID who had developed several second-site RAG1 somatic mutations32.
Novel phenotypes of RAG deficiency
Whereas SCID, atypical SCID and Omenn syndrome are inevitably fatal early in life if untreated, several forms of RAG deficiency with a milder clinical course and delayed presentation have been reported in recent years. In particular, the occurrence of CID–G/AI was reported in three unrelated girls with RAG mutations who manifested granulomas in the skin, mucous membranes and internal organs, and had severe complications after viral infections, including B cell lymphoma9. Despite severe T cell and B cell lymphopaenia and impaired T cell function, these patients had a diverse TCR repertoire and produced specific antibodies. Following this description, several other cases of CID–G/AI with various auto immune manifestations (such as cytopaenias, vitiligo, psoriasis, myasthenia gravis and Guillain–Barré syndrome) have been reported33–40. Additional phenotypes that are associated with RAG deficiency include idiopathic CD4+ T cell lymphopaenia41, common variable immunodeficiency40,42, IgA deficiency43,44, selective deficiency of polysaccharide-specific antibody responses44, hyper-IgM syndrome45 and sterile chronic multifocal osteomyelitis46. Overall, these observations have substantially broadened the clinical and immunological spectrum of human RAG deficiency and have identified immune dysregulation as a prominent manifestation of perturbed RAG function.
Molecular pathology of RAG deficiency
Until recently, the molecular mechanisms accounting for the broad phenotypic spectrum of human RAG deficiency have remained poorly defined. Advances in structural modelling of the RAG complex and the development of novel assays to measure the expression and function of mutant RAG proteins have offered important insights into this phenotypic heterogeneity.
Structure of the RAG complex
The human RAG1 and RAG2 genes are juxtaposed on chromosome 11p13. Both genes contain only one protein-coding exon. Various cis-enhancer elements upstream of RAG2 coordinately activate the transcription of RAG1 and RAG2 during T cell and B cell development47.
The human RAG1 protein (FIG. 1a) is composed of 1,043 amino acids, whereas its mouse orthologue lacks 3 amino acids at the amino terminus. The catalytic core of human RAG1 (amino acids 387–1,011) consists of a nonamer-binding domain (NBD; amino acids 394–460), dimerization and DNA-binding domain (DDBD; amino acids 461–517), pre-RNase H (preR; amino acids 518– 590), catalytic RNase H (RNH; amino acids 591–721), zinc-binding domain (amino acids 722–965) containing two distinct regions with canonical cysteine and histidine zinc-binding residues (ZnC2 and ZnH2, respectively) and the carboxy-terminal domain (CTD; amino acids 966–1,008), which are all crucial for the V(D)J recombination process. The RAG2 protein is composed of 527 amino acids and comprises a core domain (amino acids 1–383) and a non-core region (amino acids 384–527) that includes a plant homeodomain (PHD; amino acids 414–487).
Figure 1. Characterization and distribution of human RAG mutations.
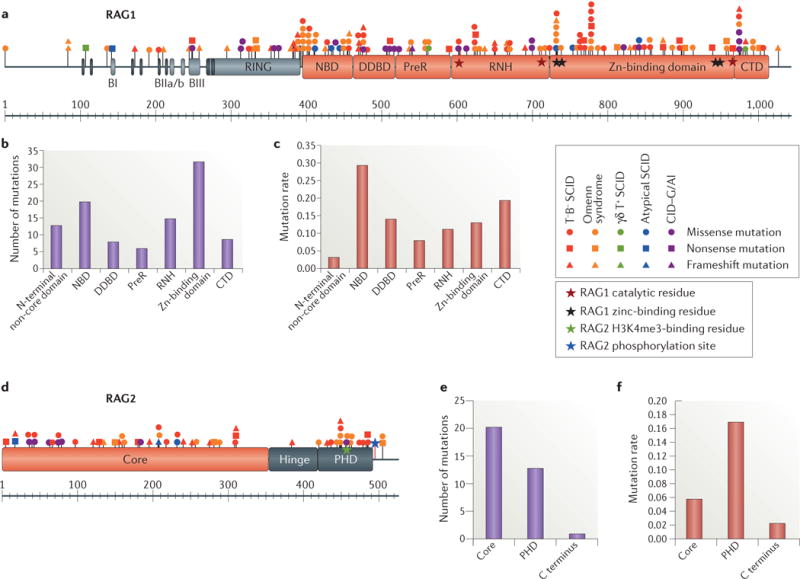
a |Schematic representation of the recombination-activating gene 1 (RAG1) protein with the various mutations colour-coded according to the clinical presentation. See Supplementary information S1 (table) for references. RAG1 catalytic and zinc-binding residues are indicated by red and black stars, respectively. b | Number of RAG1 missense mutations in the various domains. c | Frequency of RAG1 mutations calculated by dividing the number of mutations in a given region by the number of amino acids in that region. d | Schematic representation of the RAG2 protein with the various mutations colour-coded according to the clinical presentation. The trimethylated histone H3 lysine 4 (H3K4me3)-binding residue and T490 phosphorylation site of RAG2 are indicated by green and blue stars, respectively. e | Number of RAG2 missense mutations in the various domains. f | Frequency of RAG2 mutations calculated by dividing the number of mutations in a given region by the number of amino acids in that region. BI, basic I domain; BIIa/b, basic IIa/b domain; BIII, basic III domain; CTD, carboxy-terminal domain; DDBD, dimerization and DNA-binding domain; NBD, nonamer-binding domain; PHD, plant homeodomain; preR, pre-RNase H; RNH, catalytic RNase H.
The crystallographic10 and cryo-electron microscopy12 structures of the heterotetrameric complex of RAG1 and RAG2 core domains have been recently resolved. Two molecules of RAG1 and RAG2 create a Y-shaped structure in which the NBDs of the two RAG1 molecules form the stem, and the DDBDs form the branch point. Then, each of the two RAG1 molecules spreads out, projecting the zinc-binding region to the top but bringing the three conserved catalytic amino acids D603, D711 and E965 (REFS 48,49) together with the CTD into the crevice of the Y-shaped structure (BOX 1). The RAG2 core region folds into a six-bladed β-propeller and associates primarily with the RAG1 domains downstream of the DDBD and inclusive of the CTD10, creating the arms of the Y-shaped structure of the RAG complex (BOX 1). Following DNA binding, the two halves of the complex come closer to each other12. Although RAG2 does not participate in binding to the nonamer element of the RSS, it stabilizes binding of the RAG complex to the heptamer element and interacts with the DNA-coding flank12.
Crystallography and nuclear magnetic resonance studies have also helped to define the structure of the non-core domains of RAG1 and RAG2 (REFS 50,51). The non-core N-terminal region of RAG1 contains a series of basic motifs, among which the basic IIa domain (BIIa; amino acids 219–225) allows interaction with karyopherin subunit α1 (KPNA1; also known as importin subunit α5), which determines RAG1 subnuclear localization52. Following the basic motifs, a C3HC4 RING finger domain and an adjacent zinc finger motif form a single domain that coordinates zinc ions50 and has histone H3 ubiquitin ligase activity, which is required for a normal level of chromosomal V(D)J recombination53. It has been proposed that the interaction of RAG1 with unubiquitylated H3 restrains RAG1 catalytic activity and that H3 ubiquitylation would release RAG1 and allow catalytic function53.
The non-core region of RAG2 has important functions in regulating chromatin accessibility of the RAG complex and in regulating cell cycle-dependent recombination activity. In particular, the non-canonical PHD of RAG2 forms an aromatic channel that allows binding of H3 carrying a trimethylated lysine 4 (H3K4me3)51,54, which is an epigenetic marker of active transcription start sites; thus, RAG2 is proposed to be a ‘reader’ of chromatin accessibility. Binding of the PHD to H3K4me3 promotes recombination activity of the RAG complex55. The cell cycle-dependent expression of the RAG2 protein is mediated by phosphorylation of residue T490 by the cyclin A–CDK2 (cyclin-dependent kinase 2) complex, before G1 to S phase transition in the cell cycle. The phosphorylated RAG2 protein is polyubiquitylated by the SCF (S-phase kinase-associated protein (SKP2)–SKP1–CUL1–F-box) complex and targeted for proteasomal degradation56. Restricting RAG activity to G0/G1 phases of the cell cycle in this manner limits the risk of genotoxicity, and it also has a direct effect on the mechanisms of repair of broken coding and signal ends by favouring NHEJ, which predominates during G0/G1 (REF. 57).
Spectrum and consequences of RAG mutations
In total, 150 RAG1 mutations have been identified in patients, including 103 missense mutations, 18 nonsense mutations and 29 frameshift mutations (FIG. 1a; Supplementary information S1 (table)). Overall, 120 mutations fall in the core domain of RAG1, and 30 mutations fall in the non-core region (FIG. 1a). Disease-associated missense mutations have been predominantly detected in the zinc-binding region of RAG1 core domain (FIG. 1b); however, when normalized to the length of each domain, a higher mutation rate is observed in the NBD, followed by the CTD (FIG. 1c), which indicates that these regions have a low Abelson level of mutation tolerance.
A total of 57 disease-causing mutations have been reported in the RAG2 gene, including 35 missense mutations, 9 nonsense mutations and 13 frameshift mutations (FIG. 1d; Supplementary information S1 (table)). In particular, 38 mutations fall in the core domain compared with 19 in the non-core region of the molecule (FIG. 1e); among these 19 mutations in the non-core region, 16 involve the PHD, which shows a higher mutational rate (FIG. 1f).
In addition to characterizing the various roles of the distinct domains of RAG1 and RAG2, crystallography10 and cryo-electron microscopy12 studies have helped to predict the structural and functional consequences of most mutations. In particular, missense mutations that are associated with more severe forms of RAG deficiency (that is, T−B− SCID and Omenn syndrome) can be divided into four categories: mutations that destabilize the RAG complex, such as the C730F mutation at a zinc-binding site and the adjacent L732F; mutations of polar residues that are exposed to solvent and probably bind DNA, such as arginine, lysine, serine and glutamine residues in the NBD, DDBD and CTD of RAG1; mutations near the conserved catalytic amino acids D603, D711 and E965, which may alter the structure of the catalytic centre and the DNA-binding capability; and mutations that affect the interaction between RAG1 and RAG2, whereby R559S, R561C/H, E669G and R775Q of RAG1 and G35V, R39G and C41W of RAG2 directly affect the binding of RAG2 to the ZnC2 and RNH domains of RAG1 (REF. 10).
By contrast, there are only a small number of RAG1 missense mutations associated with CID–G/AI. These mutations are predominantly located in the DDBD, PreR and CTD (FIG. 2). Although these mutations preserve some recombination activity58, they may affect the quality of the V(D)J recombination process. In particular, it has been demonstrated that the RAG1 zinc-binding helical insertion region contacts the coding DNA during V(D)J recombination59, and the murine R972Q (corresponding to human R975Q) mutation renders the RAG recombinase hypersensitive to selected coding sequences at the hairpin formation step60.
Figure 2. Effects of mutations associated with CID–G/AI on the structure of the RAG complex.
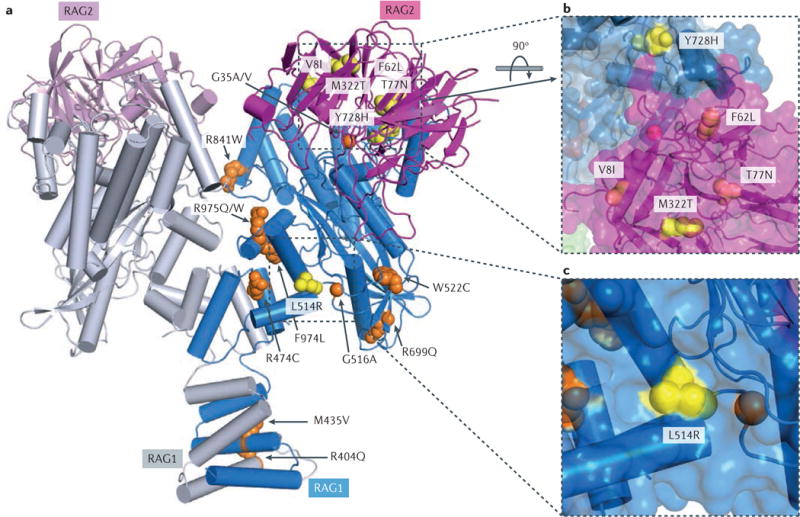
a| Structure of the recombination-activating gene 1 (RAG1)–RAG2 heterotetramer. Two RAG1 core subunits are shown in blue and grey, and two RAG2 core subunits are shown in purple and pink. Side chains of mutations found only in patients with combined immunodeficiency associated with granulomas and/or autoimmunity (CID–G/AI) are shown as yellow spheres, and those found in both patients with CID–G/AI and patients with severe combined immunodeficiency (SCID) or Omenn syndrome are shown in orange. For clarity, mutations are shown in one subunit of RAG1 and RAG2 only. All residues are numbered according to human RAG proteins. b,c | Zoom-in views of mutations that are unique to patients with CID–G/AI. Molecular surfaces are shown together with the ribbon diagram. L514R and Y728H from RAG1 and M322T from RAG2 are partially exposed (shown in yellow), whereas V8I, F62L and T77N from RAG2 are buried inside the protein (shown in purple). These mutations seem to lead to mild structural destabilization of the RAG proteins. The R841W mutation is at the interface of the closed conformation of the RAG complex.
Although they are not strictly required for recombinase activity, the non-core domains of RAG1 and RAG2 have an important regulatory role. Several frameshift mutations in the N terminus of RAG1 have been identified that result in the use of downstream translation initiation sites at positions M183 and M263 and that affect, at least partially, protein intracellular localization61. These mutations allow only modest levels of recombination activity58, and they are associated with severe clinical phenotypes in vivo (Omenn syndrome and γδ T+ SCID)25,61. Two missense mutations in the RAG1 RING domain, R314W and C328Y, have been identified in patients with CID–G/AI and Omenn syndrome, respectively9,23. C328 is one of the residues involved in the coordination of zinc binding; the corresponding murine C325Y mutation alters the structural stability of the RAG1 protein and causes loss of E3 ubiquitin ligase activity and markedly reduced recombination activity62.
Among the 13 missense mutations in the non-core region of RAG2 that have been reported in patients with SCID and Omenn syndrome, 12 affect the RAG2 PHD, and for many of these mutations the structural and functional implications have been studied in vitro63. In particular, C446W and C478Y destabilize the RAG2 protein. Moreover, the W416L, C446W, W453R and C478Y mutant proteins are associated with a marked decrease in DH–JH and Vκ–Jκ rearrangements when introduced into Abelson virus-transformed Rag2−/− pro-B cells63. Interestingly, the W416L, C446W and W453R RAG2 mutants showed abnormal subcellular localization, being partially retained in the cytoplasm63. Overall, these data conclusively demonstrate the crucial role of the RAG2 PHD in V(D)J recombination.
Genotype–phenotype correlation
Evidence of a growing number of variants of the RAG genes and recognition of the broad phenotypic spectrum of RAG deficiency in humans indicate a crucial need to test the pathogenicity of each RAG gene variant individually. The Gene Damaging Index (GDI) is a recently developed, genome-wide, gene-level metric of the mutational damage that has accumulated in the general population, and it provides an estimate of the selective pressure that each gene is subjected to64. Both RAG1 and RAG2 have a medium GDI score, and they are both under moderate purifying selection. Although several algorithms (such as Polyphen and SIFT) exist that may help to predict the pathogenicity of RAG gene variants, none of them is sufficiently robust to validate a disease-causing role. Because the RAG proteins are not expressed in mature lymphoid cells and access to T cell and B cell progenitors from patients is problematic, in vitro functional assays have been developed to enable the functional analysis of newly introduced RAG constructs on suitable recombination substrates (BOX 2). Using this approach, a large series of naturally occurring RAG mutants have been analysed in terms of protein expression and recombination activity, which has enabled a distinction to be made between pathogenic and non-pathogenic variants and has shown, for the first time, that the levels of recombination activity and the clinical phenotype observed in patients are correlated58 (Supplementary information S1 (table)).
Box 2. In vitro assays to measure RAG recombination activity.
Initially, assays to measure the recombination activity of recombination-activating gene (RAG) proteins were based on transfecting mouse lymphoid cells with suitable V(D)J substrate plasmids — containing the prokaryotic transcription terminator sequence flanked by a pair of 12–23 recombination signal sequences (RSSs), upstream of the catalase (CAT) gene encoding chloramphenicol resistance — together with plasmids encoding wild-type or mutant RAG1 and RAG2 proteins99. If the RAG proteins are functional, the terminator sequence is deleted by recombination, allowing expression of the CAT gene. The plasmid DNA is then extracted and used in a bacterial transformation assay, scoring for ampicillin and chloramphenicol resistance5,15. A corresponding assay for human cells has been described100 that allows for the identification of deleterious RAG mutations in patients with severe combined immunodeficiency (SCID) with a lack of circulating T cells and B cells (T−B− SCID)5. However, this assay is time consuming, making it difficult to assess the recombination activity of a large number of RAG mutants.
An alternative assay makes use of another V(D)J substrate plasmid, containing an inverted GFP cassette flanked by RSSs101. The inverted GFP cassette is retrovirally introduced into Rag1−/− or Rag2−/− transformed mouse pro-B cell lines harbouring a B cell leukaemia/lymphoma 2 (Bcl2) transgene. Another retrovirus expressing either wild-type or mutant RAG1 or RAG2 is then used to transduce the pro-B cells containing the inverted GFP cassette flanked by RSSs. To allow for activity of the RAG protein under investigation, the pro-B cells are blocked in the G0/G1 phases of the cell cycle by treatment with imatinib, with the Bcl2 transgene allowing for cell viability, and GFP expression is measured by flow cytometry as a read-out of the recombination activity of the RAG protein102. Although this assay has the advantage of measuring RAG recombination activity on a chromosomal substrate, it has its own limitations. In particular, it tests for recombination activity on a single pair of 12–23 RSSs at a time. By contrast, at the immunoglobulin and T cell receptor loci, the RAG proteins sample a large number of RSSs that differ in their DNA sequence, allowing for stronger or weaker binding and cleavage activity. Furthermore, some regions of RAG1 have been shown to have coding-flank sensitivity59, which indicates that the DNA sequence of the flanking coding element may also have an effect on RAG function. Although these issues can be partly addressed by analysing the recombination activity of human RAG variants at the endogenous Igh locus in transformed pro-B cells58, this remains an artificial setting, as it explores the function of human RAG proteins in a mouse genomic context.
Ultimately, it is important to analyse the impact of RAG mutations on T cell and B cell repertoire diversity and composition in patients. Initially, this goal was achieved by cloning and sequencing individual rearranged V(D)J products and by CDR3 spectratyping (complementarity-determining region 3 spectratyping). Using this approach, oligoclonality and clonal expansions were demonstrated in the TCR repertoire of patients with Omenn syndrome and with other atypical forms of SCID8,22,25. Similarly, a small number of immunoglobulin heavy chain locus (IGH) rearrangements was documented in bone marrow cells from patients with Omenn syndrome65. However, these techniques permit only a broad description of TCR and BCR repertoire composition. More recently, two groups used next-generation sequencing to study the T cell and B cell repertoire in patients with RAG deficiency30,66. In particular, it was shown that patients with various clinical phenotypes, who shared mutations in the N-terminal non-core region of RAG1, manifested a similar block in B cell development, with limited T cell and B cell repertoires and reduced use of the immunoglobulin heavy chain joining 6 (IGHJ6) and immunoglobulin κ-chain joining 5 (IGKJ5) genes30. In another study, diversity of the TCRβ repertoire was markedly reduced in patients with Omenn syndrome, whereas it was largely preserved in a patient with CID–G/AI, although there was skewing of V–J pairing and of amino acid usage in the CDR3 regions of the TCRβ repertoire66. Although the diversity and composition of T cell and B cell repertoires have been analysed in only a small number of patients with mutated RAG genes, these data indicate that RAG mutations that permit lower levels of recombination activity support the generation of a restricted TCR and BCR repertoire, whereas RAG mutations with higher residual levels of recombination may support a broader diversity of the T cell and B cell repertoire, albeit with qualitative differences of individual gene usage.
Immune dysregulation of RAG deficiency
Immune dysregulation has emerged as a key feature of patients with Omenn syndrome and with CID–G/AI; however, until recently, its extent and pathophysiology have remained poorly defined.
In particular, TH2 cell skewing has been observed in patients with Omenn syndrome, but the mechanisms accounting for the hyper-IgE phenotype of this condition and the occurrence of autoimmune cytopaenia in some of these patients remain unclear, given the almost complete lack of peripheral mature B cells. Studies in mouse models of Omenn syndrome and atypical SCID that are due to hypomorphic Rag mutations have demonstrated preferential switching of immature B cells to IgE production67 and the expansion of antibody-and autoantibody-secreting B cell populations in the spleen68,69.
A broad range of serum autoantibodies has been demonstrated particularly in patients with CID–G/AI, which is consistent with the diverse spectrum of clinical manifestations of autoimmunity (alopecia, vitiligo, granulomas, myasthenia gravis, vasculitis and psoriasis) observed in these patients36. In addition, a unique cytokine-specific antibody signature, with neutralizing antibodies specific for interferon-α (IFNα) and IFNω, has been observed in patients with hypomorphic RAG mutations and a history of severe viral infections (in particular, with varicella zoster virus)36. It is not known whether the production of these autoantibodies is a risk factor that precedes the development of severe viral infections or whether autoantibody production is precipitated by an abnormal immune response following infection. A potential role for environmental factors in triggering aberrant immune responses in hosts with partial RAG defects is supported by the recent demonstration that chronic administration of polyinosinic–polycytidylic acid (poly(I:C)) to mice with hypomorphic Rag1 mutations enhanced autoantibody production36. Although these observations indicate that environmental factors may have an important role in unmasking immune dysregulation in patients with partial RAG defects, several data also indicate that these conditions are characterized by primary and secondary defects of central and peripheral T cell and B cell tolerance.
Defects of T cell tolerance
The thymus has a crucial role in controlling autoimmunity. In particular, mature medullary thymic epithelial cells (mTECs) express autoimmune regulator (AIRE), which is a transcription factor that enables the expression of tissue-restricted antigens (TRAs). TRAs are presented by mTECs and thymic dendritic cells to developing T cells, thereby permitting the deletion of autoreactive T cells70 or their conversion to regulatory T (TReg) cells71 (FIG. 3). Maturation of mTECs and the expression of AIRE require crosstalk between thymocytes, innate lymphoid cells and thymic stromal cells72. Abnormalities of thymic architecture, with a loss of cortico-medullary demarcation, and lack of expression of AIRE and of AIRE-dependent TRAs have been demonstrated in patients with SCID, Omenn syndrome and CID–G/AI33,73,74, and in corresponding mouse models75–77. Interestingly, antibodies specific for IFNα and IFNω have also been reported in patients with autoimmune polyendocrinopathy candidiasis and ectodermal dystrophy (APECED)78, a disease that results from AIRE mutations.
Figure 3. RAG deficiency results in impairment of several tolerance checkpoints.
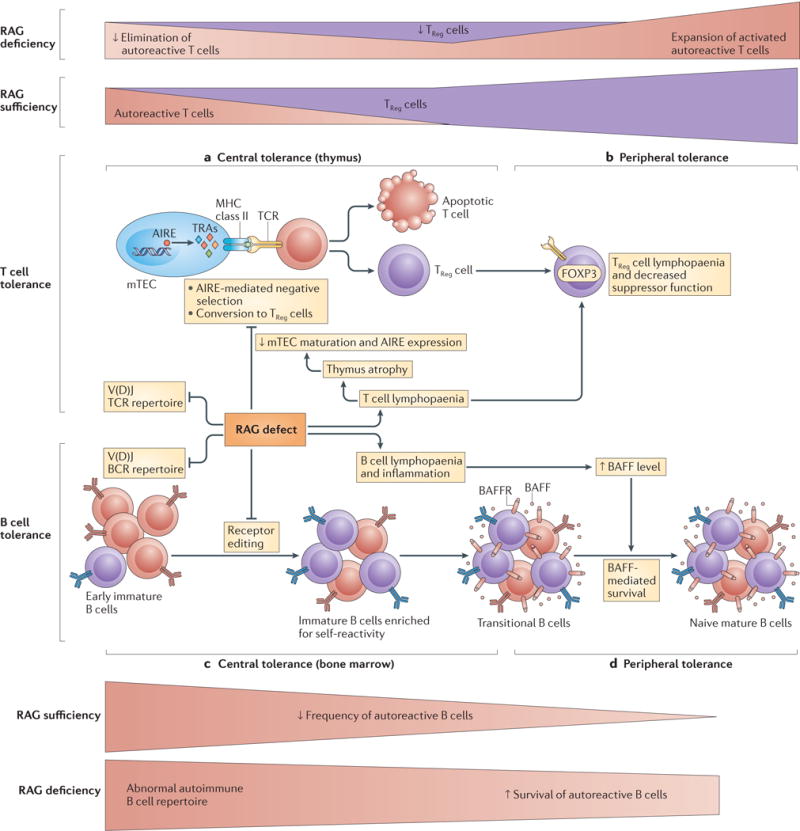
a| Central T cell tolerance. Impaired V(D)J recombination as a result of recombination-activating gene (RAG) deficiency leads to a restricted T cell receptor (TCR) repertoire, T cell lymphopaenia and aberrant thymus architecture. Altered T cell development hinders lymphostromal crosstalk in the thymus and the maturation of medullary thymic epithelial cells (mTECs); the resulting lack of expression of autoimmune regulator (AIRE) and of AIRE-dependent tissue-restricted antigens (TRAs) impairs the negative selection of self-reactive T cells or their conversion to regulatory T (TReg) cells. Autoreactive T cells are exported to the periphery and expand in number. b | Peripheral T cell tolerance. The generation of TReg cells is limited in the thymus, which results in decreased TReg cell count and decreased suppressive function in the periphery. c | Central B cell tolerance. In the bone marrow, decreased V(D)J recombination results in a restricted B cell receptor (BCR) repertoire enriched in autoreactive B cells secondary to impaired receptor editing. d | Peripheral B cell tolerance. In the periphery, B cell lymphopaenia and an inflammatory state induce increased levels of B cell-activating factor (BAFF). In a BAFF-rich environment, the survival of autoreactive cells expressing low levels of BAFF receptor (BAFFR) is favoured. Autoreactive cells are shown in red, non-self-reactive cells in purple. FOXP3, forkhead box P3.
Moreover, reduced generation of TReg cells has been reported in the thymus of patients33,74 and mice75,77 with hypomorphic RAG mutations. Although a variable number of circulating forkhead box P3 (FOXP3)+ T cells may be observed in patients with Omenn syndrome, they have impaired suppressive activity and an abnormal phenotype (that is, CC-chemokine receptor 7 (CCR7)−CD45RA−), and they may therefore be activated memory T cells rather than bona fide TReg cells79. Finally, defects of invariant natural killer T (iNKT) cells80 and the homeostatic proliferation of self-reactive T cells (and B cells) in the context of a limited set of available clones may also contribute to the immune dysregulation of Omenn syndrome.
Defects of B cell tolerance
The demonstration of a broad range of autoantibodies in patients with hypomorphic RAG mutations36 and the proliferation of autoantibody-secreting cells in mouse models of the disease68,69 indicate that B cell tolerance is also disrupted in this condition. Re-expression of RAG proteins in bone marrow immature B cells is required to mediate receptor editing, which is a mechanism that purges self-reactive B cells from the repertoire by promoting the rearrangement of available upstream Vκ and downstream Jκ genes, as well as by inducing rearrangement of the immunoglobulin λ-chain (IGL) locus81. Reduced use of the downstream IGKJ5 gene has been demonstrated in B cells from patients with hypomorphic RAG mutations30. Furthermore, a reduced fraction of Igλ-expressing B cells, together with decreased Vκ to recombining sequence rearrangement (which is another marker of receptor editing) in small pre-BII bone marrow cells, has been reported in peripheral B cells from Rag-mutant mice68,69. Overall, these data indicate that reduced receptor editing of B cells in the bone marrow of patients with Omenn syndrome may contribute to autoantibody production. Finally, by affecting the quality of binding to distinct RSSs, hypomorphic RAG mutations may also affect the selection of antibody coding elements during formation of the pre-immune repertoire, with enrichment for V region genes that are associated with autoimmunity, such as IGHV4–34 (REF. 30). It remains to be studied whether similar findings are observed also in patients with CID–G/AI who carry missense mutations (F974L, R975Q and R975W)9,34,35 that affect a coding flank-sensitive domain of RAG1 (REF. 60).
In addition to defects of central B cell tolerance, abnormalities of peripheral B cell tolerance have been reported in patients with RAG deficiency. In particular, markedly increased B cell-activating factor (BAFF; also known as TNFSF13B) levels have been demonstrated in patients and mice with RAG defects, reflecting B cell lymphopenia and inflammation68,69. Self-reactive B cells have increased dependence on BAFF for survival82, and increased BAFF levels can rescue the survival of autoreactive B cells83. In support of a pathogenic role of increased BAFF levels, in vivo treatment of Rag2-mutant mice with blocking BAFF receptor-specific monoclonal antibody led to the disappearance of autoantibodies and prevented the progression of kidney damage68.
Overall, these data indicate that the occurrence of clinical manifestations of immune dysregulation in patients with hypomorphic RAG mutations reflects both central and peripheral defects of T cell and B cell tolerance, and they suggest that optimal treatment might require interventions aimed at various targets.
RAG deficiency: beyond T cells and B cells
Although the RAG proteins are strictly required only for the development of T cells and B cells, recent data indicate that their expression may also affect the phenotype and function of other lymphoid cells. In particular, studies in mice have shown that RAG expression may occur in common lymphoid progenitors84. Consistent with this, a large proportion (~40%) of mature NK cells present non-productive rearrangements at the TCR or immunoglobulin loci84,85. Such a previous history of RAG expression in cells other than T cells and B cells was not considered to be particularly important, until it was recently reported that NK cells from Rag−/− mice have a more mature phenotype and increased cytotoxic activity, as compared with wild-type NK cells, but fail to proliferate and have decreased capacity to persist as long-lived memory NK cells following in vivo infection with mouse CMV86. Furthermore, NK cells from Rag−/− mice were shown to have a defective ability to repair DNA DSBs, which was associated with lower levels of expression of enzymes involved in the DNA damage response. A decreased ability to repair DNA DSBs was also observed in CD8+ T cells and in innate lymphoid cells of Rag−/− mice86. Overall, these data indicate that RAG expression during the early stages of lymphoid development confers cellular fitness. Whether similar abnormalities also characterize the corresponding cell lineages of patients with RAG deficiency and whether mutations that support different degrees of recombination activity have different effects on lymphoid cell fitness remain to be seen. However, it is interesting to observe that patients with RAG-deficient SCID have a very high rate of graft rejection following unconditioned haploidentical HSCT87, a phenomenon that has been attributed to the presence of NK cells but that has not been reported in other forms of NK+ SCID, such as IL-7 receptor (IL-7R) deficiency or defects of the CD3 chains. Alternatively, the occupation of thymic niches by CD4− CD8− T cell precursors may also contribute to the higher rate of graft failure that is observed after unconditioned haploidentical HSCT in patients with RAG deficiency, but not in patients with SCID with mutations of the IL-2R subunit-γ (IL2RG) and IL7R genes.
Towards gene therapy for RAG deficiency
The mainstay of treatment for patients with severe forms of RAG deficiency (that is, SCID, Omenn syndrome and atypical SCID) is HSCT. Overall survival is ~80% in patients with SCID or Omenn syndrome who receive HSCT from matched related donors but is only 60–70% after haploidentical HSCT with myeloablative conditioning, and the results are even poorer (with high rate of graft failure and insufficient immune reconstitution) after haploidentical transplantation if no conditioning or only serotherapy are used87. Overall, these data indicate the need to develop alternative forms of treatment for patients with RAG deficiency lacking HLA-matched siblings. Gene therapy has been successfully used to treat X-linked SCID88,89 and adenosine deaminase deficiency90,91. In both conditions, a strong selective advantage for gene-corrected cells has been observed in the T cell lineage. Initial attempts to restore immune competence in Rag1−/− mice by transplanting Rag1−/− bone marrow progenitor cells transduced with a Moloney murine leukaemia virus carrying the human RAG1 transgene led to immune reconstitution only when recipient mice were irradiated and providing that a high vector copy number was detected in lymphoid cells92. However, this carries a high risk of insertional mutagenesis; indeed, one treated mouse developed undifferentiated acute lymphoblastic leukaemia. Furthermore, gene therapy-treated mice had lower-than-normal B cell counts93. These data indicate that gene-corrected lymphoid progenitors have only a small selective advantage in the thymus and bone marrow of Rag1−/− mice. Furthermore, relatively inefficient expression of the RAG1 transgene was achieved in these initial attempts. To overcome this problem, self-inactivating lentiviral vectors expressing codon-optimized human RAG1 under the control of various regulatory elements have been developed92,94. Using such vectors, partial reconstitution of T cell immunity has been reported in gene therapy-treated mice; although the number of B cells remained suboptimal, improved antibody responses were observed92. By contrast, using the same vectors, modest reconstitution of both T cells and B cells was reported in another study; moreover, approximately 50% of treated mice developed an Omenn syndrome phenotype, with multi-organ lymphocytic infiltration and increased serum IgE levels94. These conflicting results may reflect different levels of human RAG1 expression by transfected mouse cells or different duration of follow-up of treated mice. Future development of gene therapy for RAG1 deficiency will require novel vectors that can provide optimal levels of transgene expression at a low vector copy number or the development of gene editing strategies that would maintain endogenous regulation of RAG1 expression.
More promising preclinical results have been obtained with gene therapy for RAG2 deficiency95,96. In particular, the use of vectors expressing codon-optimized human RAG2 and containing other modifications to reduce the risk of silencing of the RAG2 transgene resulted in significant improvement of peripheral T cell and B cell immunity in mice even at a relatively low vector copy number, in spite of a subnormal number of single positive T cells in the thymus and the persistence of a higher proportion of pro-B cells in the bone marrow96. These data offer hope for the development of gene therapy for human RAG2 deficiency in the near future, but it remains to be seen whether this strategy will be efficacious in patients with hypomorphic RAG2 mutations, for whom competition between endogenous, uncorrected, lymphoid progenitors and gene-transduced cells might affect the quality of immune reconstitution.
Conclusions
Significant progress has been made in recent years in defining the structure and function of the RAG complex. At the same time, advances in genomic analysis, and the availability of whole-exome sequencing in particular, have shown an unprecedented diversity of clinical and immunological phenotypes associated with RAG mutations. This phenotypic heterogeneity can now be interpreted by interrogating the specific effects of mutations on RAG structure and function. Immune dysregulation has emerged as an important component of partial RAG deficiency. Careful immunological and molecular analysis in patients and in suitable animal models has shown that hypomorphic RAG mutations have an impact on adaptive immune responses by affecting the diversity and composition of the immune repertoire, as well as central and peripheral mechanisms of T cell and B cell tolerance. The development of new mouse models that recapitulate the CID–G/AI phenotype is needed to characterize in greater detail the pathophysiology of this condition. Ultimately, the clinical phenotype of RAG deficiency is determined by how RAG mutations affect immune system development and function, and by immune responses to environmental factors (FIG. 4). Finally, there is a need to define what effects, if any, RAG mutations exert on human NK cells and innate lymphoid cells, and to develop novel, more effective forms of treatment for patients with this condition.
Figure 4. The interaction of genetic, immunological and environmental factors in determining the phenotype of human RAG deficiency.
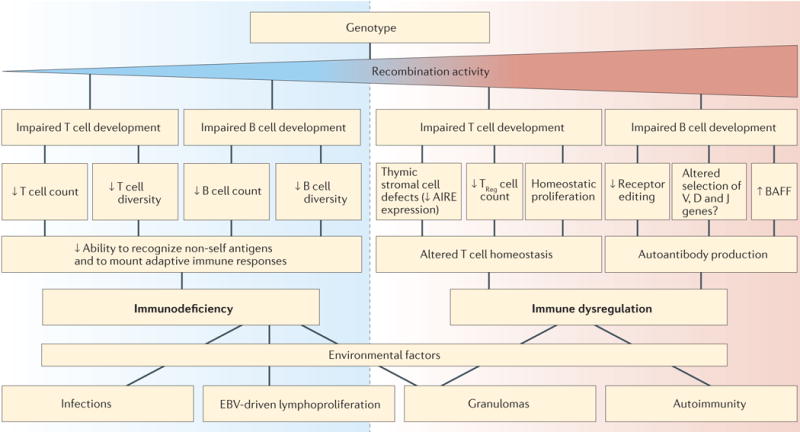
The recombination-activating gene (RAG) genotype determines the levels of recombination activity of the mutant RAG proteins. Mechanisms contributing to immune deficiency are shown on the left (blue) and those associated with autoimmunity are shown on the right (red), for both T cell- and B cell-dependent immune responses. RAG mutations with higher residual recombination activity are more likely to result in immune dysregulation. Ultimately, exposure to environmental triggers affects the immune deficiency and immune dysregulation status of the patient, thereby determining the clinical phenotype. AIRE, autoimmune regulator; BAFF, B cell-activating factor; D, diversity; EBV, Epstein–Barr virus; J, joining; TReg cell, regulatory T cell; V, variable.
Note added in proof
An important role of the microbiota in sustaining inflammation and autoimmunity has been recently shown in a mouse model of Omenn syndrome103. In this model, mucosal B cell deficiency alters the composition of the microbiota and causes bacterial translocation across the intestinal epithelium. Furthermore, loss of T cell tolerance to the commensal microbiota leads to gut inflammation sustained by TH1 and TH17 cells. The administration of antibiotics reverses most of these abnormalities and reduces serum IgE levels.
Supplementary Material
Acknowledgments
L.D.N. was supported by grants from the National Institute of Allergy and Infectious Diseases, US National Institutes of Health (NIH; 5R01AI100887) and the March of Dimes (1-FY13-500). J.E.W. was supported by the NIH grant 5K08AI103035.
Glossary
- Non-homologous end joining pathway (NHEJ pathway)
An error-prone pathway that mediates joining of DNA double-strand breaks without requiring a homologous template. In mammals, the NHEJ pathway involves several proteins — Ku70, Ku80, DNA-dependent protein kinase catalytic subunit (DNA-PKcs), Artemis, Cernunnos (also known as XLF and NHEJ1), X-ray repair cross-complementing protein 4 (XRCC4) and DNA ligase IV. Genetic defects of Artemis are the most common cause of radiosensitive severe combined immunodeficiency (SCID) in humans
- Haematopoietic stem cell transplantation (HSCT)
A therapeutic procedure that involves transfusion of donor HSCs into a recipient. HLA matching between the recipient and the donor determines compatibility. HSCT from a haploidentical donor (such as a parent) is associated with a high risk of graft-versus-host disease, unless T cells are depleted. Chemotherapy is often used before HSCT to eliminate the recipient’s blood cells and favour engraftment of donor-derived HSCs but is not strictly necessary in infants with severe combined immunodeficiency (SCID)
- Cellular radiosensitivity
Susceptibility of cells to the damaging effects of ionizing radiation, resulting in genomic instability, tumour development or cell death. Genetic defects that affect mechanisms of repair of DNA double-strand breaks are associated with increased cellular radiosensitivity
- SCID mouse
An animal model of severe combined immunodeficiency (SCID) that is characterized by lack of T cells and B cells, and increased cellular radiosensitivity. The SCID mouse carries mutations of the Prkdc gene, encoding DNA-dependent protein kinase catalytic subunit (DNA-PKcs)
- Purifying selection
In population genetics, purifying selection refers to the selective removal of deleterious alleles from a given population. Purging of these genetic variants occurs when they cause early death or affect the reproductive fitness of affected individuals
- CDR3 spectratyping (Complementarity-determining region 3 spectratyping)
A PCR-based method that measures the diversity of T cell and B cell repertoires, based on the length of the CDR3 of immunoglobulin and T cell receptor transcripts. A Gaussian distribution of CDR3 length is detected in polyclonal T cells and B cells, whereas a single peak is observed in patients with leukaemia or lymphoma, and an altered distribution may be detected in patients with infections, autoimmune diseases or severe impairment of T cell and/or B cell development
- Autoimmune polyendocrinopathy candidiasis and ectodermal dystrophy (APECED)
A monogenic autoimmune disease caused by mutations of the autoimmune regulator (AIRE) gene, affecting central T cell tolerance. Common manifestations of this disease include autoimmune hypoparathyroidism, Addison disease, type 1 diabetes, candidiasis, alopecia and nail dystrophy
- Codon-optimized
Transgenic products that are generated through a process replacing the original codons with synonymous codons for which a more abundant tRNA is available. This facilitates the rate of translation and ultimately results in the production of higher amounts of the protein
- Whole-exome sequencing
A process by which all exons contained in the genome (collectively comprising the exome) are amplified and subjected to high-throughput sequencing. DNA genetic variants that are present in a given individual are identified by comparing the exome of that subject to the normal reference sequence
Footnotes
Competing interests statement
The authors declare no competing interests.
DATABASES
The Human Gene Mutation database: http://www.hgmd.cf.ac.uk/ac/gene.php?gene=RAG1; http://www.hgmd.cf.ac.uk/ac/gene.php?gene=RAG2
SUPPLEMENTARY INFORMATION
See online article: S1 (table)
References
- 1.Schatz DG, Oettinger MA, Baltimore D. The V(D)J recombination activating gene, RAG-1. Cell. 1989;59:1035–1048. doi: 10.1016/0092-8674(89)90760-5. [DOI] [PubMed] [Google Scholar]
- 2.Oettinger MA, Schatz DG, Gorka C, Baltimore D. RAG-1 and RAG-2, adjacent genes that synergistically activate V(D)J recombination. Science. 1990;248:1517–1523. doi: 10.1126/science.2360047. [DOI] [PubMed] [Google Scholar]
- 3.Mombaerts P, et al. RAG-1-deficient mice have no mature B and T lymphocytes. Cell. 1992;68:869–877. doi: 10.1016/0092-8674(92)90030-g. [DOI] [PubMed] [Google Scholar]
- 4.Shinkai Y, et al. RAG-2-deficient mice lack mature lymphocytes owing to inability to initiate V(D)J rearrangement. Cell. 1992;68:855–867. doi: 10.1016/0092-8674(92)90029-c. [DOI] [PubMed] [Google Scholar]
- 5.Schwarz K, et al. RAG mutations in human B cell-negative SCID. Science. 1996;274:97–99. doi: 10.1126/science.274.5284.97. This is the first study to report that RAG mutations in humans cause SCID. [DOI] [PubMed] [Google Scholar]
- 6.Villa A, et al. Partial V(D)J recombination activity leads to Omenn syndrome. Cell. 1998;93:885–896. doi: 10.1016/s0092-8674(00)81448-8. This is the first demonstration that hypomorphic RAG mutations are a cause of Omenn syndrome. [DOI] [PubMed] [Google Scholar]
- 7.de Saint-Basile G, et al. Restricted heterogeneity of T lymphocytes in combined immunodeficiency with hypereosinophilia (Omenn’s syndrome) J Clin Invest. 1991;87:1352–1359. doi: 10.1172/JCI115139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rieux-Laucat F, et al. Highly restricted human T cell repertoire in peripheral blood and tissue-infiltrating lymphocytes in Omenn’s syndrome. J Clin Invest. 1998;102:312–321. doi: 10.1172/JCI332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Schuetz C, et al. An immunodeficiency disease with RAG mutations and granulomas. N Engl J Med. 2008;358:2030–2038. doi: 10.1056/NEJMoa073966. By identifying patients with delayed-onset disease characterized by granulomas and autoimmunity, this manuscript has broadened the spectrum of human RAG deficiency. [DOI] [PubMed] [Google Scholar]
- 10.Kim MS, Lapkouski M, Yang W, Gellert M. Crystal structure of the V(D)J recombinase RAG1– RAG2. Nature. 2015;518:507–511. doi: 10.1038/nature14174. This study describes the crystal structure of the heterotetrameric complex of RAG1 and RAG2 core domains, offering important insights into RAG complex function and into mechanisms of disease in patients with RAG mutations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shetty K, Schatz DG. Recruitment of RAG1 and RAG2 to chromatinized DNA during V(D)J recombination. Mol Cell Biol. 2015;35:3701–3713. doi: 10.1128/MCB.00219-15. This study investigates the specific requirement of RSS substrates with 12–23 spacers for optimal recognition and binding by RAG1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ru H, et al. Molecular mechanism of V(D)J recombination from synaptic RAG1–RAG2 complex structures. Cell. 2015;163:1138–1152. doi: 10.1016/j.cell.2015.10.055. This report shows by cryo-electron microscopy how the RAG complex interacts with RSSs according to the 12–23 rule. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fischer A, Notarangelo LD, Neven B, Cavazzana M, Puck JM. Severe combined immunodeficiencies and related disorders. Nat Rev Disease Primers. 2015 doi: 10.1038/nrdp.2015.61. http://dx.doi.org/10.1038/nrdp.2015.61. [DOI] [PubMed]
- 14.Pai SY, et al. Transplantation outcomes for severe combined immunodeficiency, 2000–2009. N Engl J Med. 2014;371:434–446. doi: 10.1056/NEJMoa1401177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Nicolas N, et al. A human severe combined immunodeficiency (SCID) condition with increased sensitivity to ionizing radiations and impaired V(D)J rearrangements defines a new DNA recombination/repair deficiency. J Exp Med. 1998;188:627–634. doi: 10.1084/jem.188.4.627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bosma GC, Custer RP, Bosma MJ. A severe combined immunodeficiency mutation in the mouse. Nature. 1983;301:527–530. doi: 10.1038/301527a0. [DOI] [PubMed] [Google Scholar]
- 17.Fulop GM, Phillips RA. The scid mutation in mice causes a general defect in DNA repair. Nature. 1990;347:479–482. doi: 10.1038/347479a0. [DOI] [PubMed] [Google Scholar]
- 18.Woodbine L, Gennery AR, Jeggo PA. The clinical impact of deficiency in DNA non-homologous end-joining. DNA Repair (Amst) 2014;16:84–96. doi: 10.1016/j.dnarep.2014.02.011. [DOI] [PubMed] [Google Scholar]
- 19.Omenn GS. Familial reticuloendotheliosis with eosinophilia. N Engl J Med. 1965;273:427–432. doi: 10.1056/NEJM196508192730806. [DOI] [PubMed] [Google Scholar]
- 20.Schandene L, et al. T helper type 2-like cells and therapeutic effects of interferon-γ in combined immunodeficiency with hypereosinophilia (Omenn’s syndrome) Eur J Immunol. 1993;23:56–60. doi: 10.1002/eji.1830230110. [DOI] [PubMed] [Google Scholar]
- 21.Chilosi M, et al. CD30 cell expression and abnormal soluble CD30 serum accumulation in Omenn’s syndrome: evidence for a T helper 2-mediated condition. Eur J Immunol. 1996;26:329–334. doi: 10.1002/eji.1830260209. [DOI] [PubMed] [Google Scholar]
- 22.Signorini S, et al. Intrathymic restriction and peripheral expansion of the T-cell repertoire in Omenn syndrome. Blood. 1999;94:3468–3478. [PubMed] [Google Scholar]
- 23.Villa A, et al. V(D)J recombination defects in lymphocytes due to RAG mutations: severe immunodeficiency with a spectrum of clinical presentations. Blood. 2001;97:81–88. doi: 10.1182/blood.v97.1.81. [DOI] [PubMed] [Google Scholar]
- 24.Shearer WT, et al. Establishing diagnostic criteria for severe combined immunodeficiency disease (SCID), leaky SCID, and Omenn syndrome: the Primary Immune Deficiency Treatment Consortium experience. J Allergy Clin Immunol. 2014;133:1092–1098. doi: 10.1016/j.jaci.2013.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.de Villartay JP, et al. A novel immunodeficiency associated with hypomorphic RAG1 mutations and CMV infection. J Clin Invest. 2005;115:3291–3299. doi: 10.1172/JCI25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ehl S, et al. A variant of SCID with specific immune responses and predominance of γδ T cells. J Clin Invest. 2005;115:3140–3148. doi: 10.1172/JCI25221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kwan A, et al. Newborn screening for severe combined immunodeficiency in 11 screening programs in the United States. JAMA. 2014;312:729–738. doi: 10.1001/jama.2014.9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Al-Herz W, Al-Mousa H. Combined immunodeficiency: the Middle East experience. J Allergy Clin Immunol. 2013;131:658–660. doi: 10.1016/j.jaci.2012.11.033. [DOI] [PubMed] [Google Scholar]
- 29.Corneo B, et al. Identical mutations in RAG1 or RAG2 genes leading to defective V(D)J recombinase activity can cause either T-B–severe combined immune deficiency or Omenn syndrome. Blood. 2001;97:2772–2776. doi: 10.1182/blood.v97.9.2772. [DOI] [PubMed] [Google Scholar]
- 30.Ijspeert H, et al. Similar recombination-activating gene (RAG) mutations result in similar immunobiological effects but in different clinical phenotypes. J Allergy Clin Immunol. 2014;133:1124–1133. doi: 10.1016/j.jaci.2013.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dalal I, et al. Evolution of a T-B- SCID into an Omenn syndrome phenotype following parainfluenza 3 virus infection. Clin Immunol. 2005;115:70–73. doi: 10.1016/j.clim.2004.08.016. [DOI] [PubMed] [Google Scholar]
- 32.Wada T, et al. Oligoclonal expansion of T lymphocytes with multiple second-site mutations leads to Omenn syndrome in a patient with RAG1-deficient severe combined immunodeficiency. Blood. 2005;106:2099–2101. doi: 10.1182/blood-2005-03-0936. [DOI] [PubMed] [Google Scholar]
- 33.De Ravin SS, et al. Hypomorphic Rag mutations can cause destructive midline granulomatous disease. Blood. 2010;116:1263–1271. doi: 10.1182/blood-2010-02-267583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Avila EM, et al. Highly variable clinical phenotypes of hypomorphic RAG1 mutations. Pediatrics. 2010;126:e1248–e1252. doi: 10.1542/peds.2009-3171. [DOI] [PubMed] [Google Scholar]
- 35.Henderson LA, et al. Expanding the spectrum of recombination-activating gene 1 deficiency: a family with early-onset autoimmunity. J Allergy Clin Immunol. 2013;132:969–971. doi: 10.1016/j.jaci.2013.06.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Walter JE, et al. Broad-spectrum antibodies against self-antigens and cytokines in RAG deficiency. J Clin Invest. 2015;125:4135–4148. doi: 10.1172/JCI80477. This article reports on the autoantibody repertoire in a large series of patients with RAG mutations, including neutralizing IFNα- and IFNω-specific antibodies in patients with CID–G/AI and a history of severe varicella zoster virus infection. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sharapova SO, et al. Late-onset combined immune deficiency associated to skin granuloma due to heterozygous compound mutations in RAG1 gene in a 14 years old male. Hum Immunol. 2013;74:18–22. doi: 10.1016/j.humimm.2012.10.010. [DOI] [PubMed] [Google Scholar]
- 38.Patiroglu T, et al. Atypical severe combined immunodeficiency caused by a novel homozygous mutation in Rag1 gene in a girl who presented with pyoderma gangrenosum: a case report and literature review. J Clin Immunol. 2014;34:792–795. doi: 10.1007/s10875-014-0077-5. [DOI] [PubMed] [Google Scholar]
- 39.Chen K, et al. Autoimmunity due to RAG deficiency and estimated disease incidence in RAG1/2 mutations. J Allergy Clin Immunol. 2014;133:880–882. doi: 10.1016/j.jaci.2013.11.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Buchbinder D, et al. Identification of patients with RAG mutations previously diagnosed with common variable immunodeficiency disorders. J Clin Immunol. 2015;35:119–124. doi: 10.1007/s10875-014-0121-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kuijpers TW, et al. Idiopathic CD4+ T lymphopenia without autoimmunity or granulomatous disease in the slipstream of RAG mutations. Blood. 2011;117:5892–5896. doi: 10.1182/blood-2011-01-329052. [DOI] [PubMed] [Google Scholar]
- 42.Abolhassani H, et al. A hypomorphic recombination-activating gene 1 (RAG1) mutation resulting in a phenotype resembling common variable immunodeficiency. J Allergy Clin Immunol. 2014;134:1375–1380. doi: 10.1016/j.jaci.2014.04.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kato T, et al. RAG1 deficiency may present clinically as selective IgA deficiency. J Clin Immunol. 2015;35:280–288. doi: 10.1007/s10875-015-0146-4. [DOI] [PubMed] [Google Scholar]
- 44.Geier CB, et al. Leaky RAG deficiency in adult patients with impaired antibody production against bacterial polysaccharide antigens. PLoS ONE. 2015;10:e0133220. doi: 10.1371/journal.pone.0133220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chou J, et al. A novel homozygous mutation in recombination activating gene 2 in 2 relatives with different clinical phenotypes: Omenn syndrome and hyper-IgM syndrome. J Allergy Clin Immunol. 2012;130:1414–1416. doi: 10.1016/j.jaci.2012.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Reiff A, et al. Exome sequencing reveals RAG1 mutations in a child with autoimmunity and sterile chronic multifocal osteomyelitis evolving into disseminated granulomatous disease. J Clin Immunol. 2013;33:1289–1292. doi: 10.1007/s10875-013-9953-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kuo TC, Schlissel MS. Mechanisms controlling expression of the RAG locus during lymphocyte development. Curr Opin Immunol. 2009;21:173–178. doi: 10.1016/j.coi.2009.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Landree MA, Wibbenmeyer JA, Roth DB. Mutational analysis of RAG1 and RAG2 identifies three catalytic amino acids in RAG1 critical for both cleavage steps of V(D)J recombination. Genes Dev. 1999;13:3059–3069. doi: 10.1101/gad.13.23.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim DR, Dai Y, Mundy CL, Yang W, Oettinger MA. Mutations of acidic residues in RAG1 define the active site of the V(D)J recombinase. Genes Dev. 1999;13:3070–3080. doi: 10.1101/gad.13.23.3070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Bellon SF, Rodgers KK, Schatz DG, Coleman JE, Steitz TA. Crystal structure of the RAG1 dimerization domain reveals multiple zinc-binding motifs including a novel zinc binuclear cluster. Nat Struct Biol. 1997;4:586–591. doi: 10.1038/nsb0797-586. [DOI] [PubMed] [Google Scholar]
- 51.Matthews AG, et al. RAG2 PHD finger couples histone H3 lysine 4 trimethylation with V(D)J recombination. Nature. 2007;450:1106–1110. doi: 10.1038/nature06431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cortes P, Ye ZS, Baltimore D. RAG-1 interacts with the repeated amino acid motif of the human homologue of the yeast protein SRP1. Proc Natl Acad Sci USA. 1994;91:7633–7637. doi: 10.1073/pnas.91.16.7633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Deng Z, Liu H, Liu X. RAG1-mediated ubiquitylation of histone H3 is required for chromosomal V(D)J recombination. Cell Res. 2015;25:181–192. doi: 10.1038/cr.2015.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Liu Y, Subrahmanyam R, Chakraborty T, Sen R, Desiderio S. A plant homeodomain in RAG-2 that binds hypermethylated lysine 4 of histone H3 is necessary for efficient antigen-receptor-gene rearrangement. Immunity. 2007;27:561–571. doi: 10.1016/j.immuni.2007.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shimazaki N, Tsai AG, Lieber MR. H3K4me3 stimulates the V(D)J RAG complex for both nicking and hairpinning in trans in addition to tethering in cis: implications for translocations. Mol Cell. 2009;34:535–544. doi: 10.1016/j.molcel.2009.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jiang H, et al. Ubiquitylation of RAG-2 by Skp2-SCF links destruction of the V(D)J recombinase to the cell cycle. Mol Cell. 2005;18:699–709. doi: 10.1016/j.molcel.2005.05.011. [DOI] [PubMed] [Google Scholar]
- 57.Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol. 2008;9:297–308. doi: 10.1038/nrm2351. [DOI] [PubMed] [Google Scholar]
- 58.Lee YN, et al. A systematic analysis of recombination activity and genotype-phenotype correlation in human recombination-activating gene 1 deficiency. J Allergy Clin Immunol. 2014;133:1099–1108. doi: 10.1016/j.jaci.2013.10.007. This comprehensive analysis of expression and recombination activity of naturally occurring human RAG1 mutant proteins reveals a genotype–phenotype correlation in this disease. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Mo X, Bailin T, Sadofsky MJA. C-terminal region of RAG1 contacts the coding DNA during V(D)J recombination. Mol Cell Biol. 2001;21:2038–2047. doi: 10.1128/MCB.21.6.2038-2047.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wong SY, Lu CP, Roth DBA. RAG1 mutation found in Omenn syndrome causes coding flank hypersensitivity: a novel mechanism for antigen receptor repertoire restriction. J Immunol. 2008;181:4124–4130. doi: 10.4049/jimmunol.181.6.4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Santagata S, et al. N-terminal RAG1 frameshift mutations in Omenn’s syndrome: internal methionine usage leads to partial V(D)J recombination activity and reveals a fundamental role in vivo for the N-terminal domains. Proc Natl Acad Sci USA. 2000;97:14572–14577. doi: 10.1073/pnas.97.26.14572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Simkus C, Anand P, Bhattacharyya A, Jones JM. Biochemical and folding defects in a RAG1 variant associated with Omenn syndrome. J Immunol. 2007;179:8332–8340. doi: 10.4049/jimmunol.179.12.8332. [DOI] [PubMed] [Google Scholar]
- 63.Couedel C, et al. Analysis of mutations from SCID and Omenn syndrome patients reveals the central role of the Rag2 PHD domain in regulating V(D)J recombination. J Clin Invest. 2010;120:1337–1344. doi: 10.1172/JCI41305. This study reveals the crucial regulatory role of the non-core PHD region of RAG2 by showing that mutations in the PHD compromise recombination activity by affecting protein stability, nuclear localization and/or interaction with histones. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Itan Y, et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc Natl Acad Sci USA. 2015;112:13615–13620. doi: 10.1073/pnas.1518646112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Noordzij JG, et al. The immunophenotypic and immunogenotypic B-cell differentiation arrest in bone marrow of RAG-deficient SCID patients corresponds to residual recombination activities of mutated RAG proteins. Blood. 2002;100:2145–2152. [PubMed] [Google Scholar]
- 66.Yu X, et al. Human syndromes of immunodeficiency and dysregulation are characterized by distinct defects in T-cell receptor repertoire development. J Allergy Clin Immunol. 2014;133:1109–1115. doi: 10.1016/j.jaci.2013.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wesemann DR, et al. Immature B cells preferentially switch to IgE with increased direct Sμ to Sε recombination. J Exp Med. 2011;208:2733–2746. doi: 10.1084/jem.20111155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Cassani B, et al. Homeostatic expansion of autoreactive immunoglobulin-secreting cells in the Rag2 mouse model of Omenn syndrome. J Exp Med. 2010;207:1525–1540. doi: 10.1084/jem.20091928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Walter JE, et al. Expansion of immunoglobulin-secreting cells and defects in B cell tolerance in Rag-dependent immunodeficiency. J Exp Med. 2010;207:1541–1554. doi: 10.1084/jem.20091927. References 68 and 69 demonstrate in animal models and in patients that hypomorphic RAG mutations are associated with abnormalities of central and peripheral B cell tolerance, resulting in the expansion of autoantibody-secreting cell populations. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Anderson MS, et al. Projection of an immunological self shadow within the thymus by the Aire protein. Science. 2002;298:1395–1401. doi: 10.1126/science.1075958. [DOI] [PubMed] [Google Scholar]
- 71.Watanabe N, et al. Hassall’s corpuscles instruct dendritic cells to induce CD4+CD25+ regulatory T cells in human thymus. Nature. 2005;436:1181–1185. doi: 10.1038/nature03886. [DOI] [PubMed] [Google Scholar]
- 72.Akiyama T, Tateishi R, Akiyama N, Yoshinaga R, Kobayashi TJ. Positive and negative regulatory mechanisms for fine-tuning cellularity and functions of medullary thymic epithelial cells. Front Immunol. 2015;6:461. doi: 10.3389/fimmu.2015.00461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cavadini P, et al. AIRE deficiency in thymus of 2 patients with Omenn syndrome. J Clin Invest. 2005;115:728–732. doi: 10.1172/JCI23087. This is the first demonstration that RAG mutations in humans compromise thymic architecture and expression of AIRE and AIRE-dependent TRAs. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Poliani PL, et al. Early defects in human T-cell development severely affect distribution and maturation of thymic stromal cells: possible implications for the pathophysiology of Omenn syndrome. Blood. 2009;114:105–108. doi: 10.1182/blood-2009-03-211029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Rucci F, et al. Abnormalities of thymic stroma may contribute to immune dysregulation in murine models of leaky severe combined immunodeficiency. Front Immunol. 2011;2:15. doi: 10.3389/fimmu.2011.00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marrella V, et al. Anti-CD3ε mAb improves thymic architecture and prevents autoimmune manifestations in a mouse model of Omenn syndrome: therapeutic implications. Blood. 2012;120:1005–1014. doi: 10.1182/blood-2012-01-406827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Marrella V, et al. A hypomorphic R229Q Rag2 mouse mutant recapitulates human Omenn syndrome. J Clin Invest. 2007;117:1260–1269. doi: 10.1172/JCI30928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Meager A, et al. Anti-interferon autoantibodies in autoimmune polyendocrinopathy syndrome type 1. PLoS Med. 2006;3:e289. doi: 10.1371/journal.pmed.0030289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Cassani B, et al. Defect of regulatory T cells in patients with Omenn syndrome. J Allergy Clin Immunol. 2010;125:209–216. doi: 10.1016/j.jaci.2009.10.023. [DOI] [PubMed] [Google Scholar]
- 80.Matangkasombut P, et al. Lack of iNKT cells in patients with combined immune deficiency due to hypomorphic RAG mutations. Blood. 2008;111:271–274. doi: 10.1182/blood-2007-06-096487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Jankovic M, Casellas R, Yannoutsos N, Wardemann H, Nussenzweig MC. RAGs and regulation of autoantibodies. Annu Rev Immunol. 2004;22:485–501. doi: 10.1146/annurev.immunol.22.012703.104707. [DOI] [PubMed] [Google Scholar]
- 82.Lesley R, et al. Reduced competitiveness of autoantigen-engaged B cells due to increased dependence on BAFF. Immunity. 2004;20:441–453. doi: 10.1016/s1074-7613(04)00079-2. [DOI] [PubMed] [Google Scholar]
- 83.Thien M, et al. Excess BAFF rescues self-reactive B cells from peripheral deletion and allows them to enter forbidden follicular and marginal zone niches. Immunity. 2004;20:785–798. doi: 10.1016/j.immuni.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 84.Borghesi L, et al. B lineage-specific regulation of V(D)J recombinase activity is established in common lymphoid progenitors. J Exp Med. 2004;199:491–502. doi: 10.1084/jem.20031800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Pilbeam K, et al. The ontogeny and fate of NK cells marked by permanent DNA rearrangements. J Immunol. 2008;180:1432–1441. doi: 10.4049/jimmunol.180.3.1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Karo JM, Schatz DG, Sun JC. The RAG recombinase dictates functional heterogeneity and cellular fitness in natural killer cells. Cell. 2014;159:94–107. doi: 10.1016/j.cell.2014.08.026. This study demonstrates that RAG expression during the early stages of lymphoid development allows selection of cells with higher fitness and improved ability to respond to infections and cellular stress. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Schuetz C, et al. SCID patients with ARTEMIS versus RAG deficiencies following HCT: increased risk of late toxicity in ARTEMIS-deficient SCID. Blood. 2014;123:281–289. doi: 10.1182/blood-2013-01-476432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hacein-Bey-Abina S, et al. Efficacy of gene therapy for X-linked severe combined immunodeficiency. N Engl J Med. 2010;363:355–364. doi: 10.1056/NEJMoa1000164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hacein-Bey-Abina S, et al. A modified γ-retrovirus vector for X-linked severe combined immunodeficiency. N Engl J Med. 2014;371:1407–1417. doi: 10.1056/NEJMoa1404588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Aiuti A, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- 91.Candotti F, et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 2012;120:3635–3646. doi: 10.1182/blood-2012-02-400937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Pike-Overzet K, et al. Correction of murine Rag1 deficiency by self-inactivating lentiviral vector-mediated gene transfer. Leukemia. 2011;25:1471–1483. doi: 10.1038/leu.2011.106. [DOI] [PubMed] [Google Scholar]
- 93.Lagresle-Peyrou C, et al. Long-term immune reconstitution in RAG-1-deficient mice treated by retroviral gene therapy: a balance between efficiency and toxicity. Blood. 2006;107:63–72. doi: 10.1182/blood-2005-05-2032. [DOI] [PubMed] [Google Scholar]
- 94.van Til NP, et al. Recombination-activating gene 1 (Rag1)-deficient mice with severe combined immunodeficiency treated with lentiviral gene therapy demonstrate autoimmune Omenn-like syndrome. J Allergy Clin Immunol. 2014;133:1116–1123. doi: 10.1016/j.jaci.2013.10.009. [DOI] [PubMed] [Google Scholar]
- 95.Yates F, et al. Gene therapy of RAG-2−/− mice: sustained correction of the immunodeficiency. Blood. 2002;100:3942–3949. doi: 10.1182/blood-2002-03-0782. [DOI] [PubMed] [Google Scholar]
- 96.van Til NP, et al. Correction of murine Rag2 severe combined immunodeficiency by lentiviral gene therapy using a codon-optimized RAG2 therapeutic transgene. Mol Ther. 2012;20:1968–1980. doi: 10.1038/mt.2012.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Feeney AJ, Goebel P, Espinoza CR. Many levels of control of V gene rearrangement frequency. Immunol Rev. 2004;200:44–56. doi: 10.1111/j.0105-2896.2004.00163.x. [DOI] [PubMed] [Google Scholar]
- 98.Teng G, et al. RAG represents a widespread threat to the lymphocyte genome. Cell. 2015;162:751–765. doi: 10.1016/j.cell.2015.07.009. This study demonstrates that RAG1 associates with chromatin at a very large number of promoters and enhancers in developing lymphocytes. During evolution, protection from illegitimate DNA cleavage and genomic instability has been accomplished by reducing the number of cryptic RSS heptamer sequences near RAG1-binding sites that are not associated with TCR and immunoglobulin genes. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hesse JE, Lieber MR, Gellert M, Mizuuchi K. Extrachromosomal DNA substrates in pre-B cells undergo inversion or deletion at immunoglobulin V-(D)-J joining signals. Cell. 1987;19:775–783. doi: 10.1016/0092-8674(87)90615-5. [DOI] [PubMed] [Google Scholar]
- 100.Gauss GH, Lieber MR. Unequal signal and coding joint formation in human V(D)J recombination. Mol Cell Biol. 1987;13:3900–3906. doi: 10.1128/mcb.13.7.3900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Liang HE, et al. The “dispensable” portion of RAG2 is necessary for efficient V-to-DJ rearrangement during B and T cell development. Immunity. 2002;17:639–651. doi: 10.1016/s1074-7613(02)00448-x. [DOI] [PubMed] [Google Scholar]
- 102.Bredemeyer AL, et al. ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 2006;442:466–470. doi: 10.1038/nature04866. [DOI] [PubMed] [Google Scholar]
- 103.Rigoni R, et al. Intestinal microbiota sustains inflammation and autoimmunity induced by hypomorphic RAG defects. J Exp Med. 2016;213:355–375. doi: 10.1084/jem.20151116. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.