

Abstract
Background:
The measurement of disease-relevant biomarkers has become a major component of clinical trial design, but in the absence of rigorous clinical and analytical validation of detection methodology, interpretation of results may be misleading. In Huntington’s disease (HD), measurement of the concentration of mutant huntingtin protein (mHTT) in cerebrospinal fluid (CSF) of patients may serve as both a disease progression biomarker and a pharmacodynamic readout for HTT-lowering therapeutic approaches. We recently published the quantification of mHTT levels in HD patient CSF by a novel ultrasensitive immunoassay-based technology and here analytically validate it for use.
Objective:
This work aims to analytically and clinically validate our ultrasensitive assay for mHTT measurement in human HD CSF, for application as a pharmacodynamic biomarker of CNS mHTT lowering in clinical trials.
Methods:
The single molecule counting (SMC) assay is an ultrasensitive bead-based immunoassay where upon specific recognition, dye-labeled antibodies are excited by a confocal laser and emit fluorescent light as a readout. The detection of mHTT by this technology was clinically validated following established Food and Drug Administration and European Medicine Agency guidelines.
Results:
The SMC assay was demonstrated to be accurate, precise, specific, and reproducible. While no matrix influence was detected, a list of interfering substances was compiled as a guideline for proper collection and storage of patient CSF samples. In addition, a set of recommendations on result interpretation is provided.
Conclusions:
This SMC assay is a robust and ultrasensitive method for the relative quantification of mHTT in human CSF.
Keywords: Assay validation, biomarker, cerebrospinal fluid, huntingtin protein, Huntington’s disease, immunoassay, mutant huntingtin, polyglutamine, ultrasensitive assay
INTRODUCTION
The availability of biomarkers can greatly facilitate the interpretation of clinical trial results, but the use of poorly validated assays to detect them has resulted in a profound lack of useful biomarkers. The biomarker pipeline was recently described as “plagued by problems” and “too prone to failures” by Ioannidis and Bossuyt [1]. In the present work we provide a rigorous analytical clinical validation of an assay we previously demonstrated to be able to detect mutant huntingtin (mHTT) in human cerebrospinal fluid (CSF) of Huntington’s disease (HD) mutation carriers [2].
HD is a neurodegenerative condition caused by the expansion of a CAG nucleotide repeat domain in the HTT gene [3]. This results in the expression of a polyglutamine expanded huntingtin protein (mHTT) that ultimately causes neuronal death [4]. This fact, together with HD being a penetrant monogenic disease, strengthens the concept of decreasing mHTT levels as the most proximal therapeutic strategy for disease modification [5]. To this aim, the quantification of mHTT in CSF, an accessible central nervous system (CNS) related body fluid, may be informative not only as a biomarker for patient stratification, but also as a target engagement pharmacodynamic measure for mHTT-lowering therapeutics, such as RNAi modalities [6, 7]. We recently described a novel single molecule counting (SMC) method capable of detecting and quantifying mHTT in human and animal model CSF [2]; subsequently another method using microbeads-based immunoprecipitation followed by flow cytometry (IP-FCM) [8] was reported. Both techniques are immunoassays based on antibody pairs; in each method, one detects total HTT and the other preferentially detects mHTT mediated by the expanded polyglutamine recognition. The polyglutamine directed antibody is MW1 [9] in both the SMC and IP-FCM assays, whereas the HTT specific antibody 2B7 is used in the SMC assay, and HDB4E10 is used in the IP-FCM assay. To date, the extent of validation that has been carried out for these assays has been focused on the demonstration of selective and specific recognition of the mHTT protein over the wild type protein (wtHTT) by using recombinant protein standards.
The present work aims at providing a detailed analytical clinical validation of the SMC assay based on the 2B7-MW1 antibody pair in order to provide a strong data foundation for its use in future clinical trials. To this end, the 2B7-MW1 assay was preliminarily tested for its specificity for detection of mHTT over wtHTT recombinant protein. Hereafter, the Food and Drug Administration (FDA) and the European Medicines Agency (EMA) bioanalytical method guidelines [10, 11] were applied to technically validate the 2B7-MW1 assay by SMC in human CSF samples. This assay, enabled by the Singulex Erenna platform, is relative quantitative and the mHTT amount present in biological samples is calculated against a purified recombinant mHTT protein standard curve [2]. Our validation of the assay comprised the evaluation of calibration curve performance, accuracy, precision, stability, matrix effects, selectivity, specificity, and reproducibility.
MATERIALS AND METHODS
Human CSF and blood samples
Human CSF and blood samples were collected from healthy and HD patients, at University College London (UCL) by Dr E. Wild and human CSF samples were collected from healthy and HD patients at the Centre for Molecular Medicine and Therapeutics of Vancouver (BC, Canada) by Dr B.R. Leavitt as previously described [2]. All work involving human volunteers was performed in accordance with the Declaration of Helsinki of 1975 and approved by the Central London Research Ethics Committee and the University of British Columbia Clinical Research Ethics Board. All participants provided written informed consent.
In order to perform the assay, the samples and reference proteins were diluted in artificial CSF (aCSF): PBS, 300 mM NaCl, 6 mM KCl, 2.8 mM CaCl2, 1.6 mM MgCl2.
Human blood, serum, and plasma with EDTA K2, EDTA K3, Na-citrate, Na-Heparin and Na-EDTA were purchased from Seralab in order to test the assay selectivity. Hemoglobin A quantification was performed using a commercial ELISA (Bethyl Laboratories) according to the manufacturer’s specification.
HTT silencing in human HD fibroblasts
Three primary fibroblast cells, collected from an HD, JHD, and a compound heterozygous HD patient, having 45/23, 176/23, and 50/40 glutamines respectively, were obtained from The Coriell Institute for Medical Research, Camden, NJ, USA (cat. num. GM09197, GM01085, GM04857). HTT silencing was carried out using HTT-specific siRNA from SIGMA (cat. num. SASI_HS01_00241076) and Lipofectamine 2000 (Invitrogen) as transfecting reagent. Transfected cells were collected 48 h after treatment and lysed. Lysis was done in PBS, 0.4% Triton-X and Protease Inhibitor cocktail tablets (complete from Roche).
Antibodies and recombinant proteins
The MW1 antibody was developed by the late Dr. Paul Patterson [9]. 2B7 antibody generation and characterization were as previously described [12]. The 2B7 antibody was conjugated to magnetic particles (MPs), to a final concentration of 25μg/mg of MPs, and the MW1 antibody was labeled, to a final concentration of 1μg/μl, using the Erenna capture and detection reagent labeling kits from Singulex-Millipore, following the manufacturer’s instructions. Conjugated/labeled antibodies were diluted in Assay Buffer (from Singulex-Millipore), prior to performing the assay, to 1:400 and 1:1000 respectively.
Purified recombinant proteins were obtained from CHDI Foundation and produced as previously published. The large fragment amino terminal human HTT proteins HTT (1-573) Q23, HTT (1-573) Q45, and HTT (1-573) Q73 were produced according to Macdonald et al. [13], and the full length human HTT proteins HTT (1-3144) Q17 and HTT (1-3144) Q46 were produced according to Huang et al. [14].
Immunoassay
The immunoassay was performed as previously described [2]. Briefly, the assay starts with a capture step, where the 2B7 conjugated-MPs are incubated with samples for 1 h, in an Axygen polypropylene V-bottom 96 well plate, previously coated with a solution of 1X Erenna System Buffer (from Singulex-Millipore) 750 mM NaCl, 6% BSA, 0.8% Triton-X and protease inhibitor cocktail (Complete tablets from Roche). Washes were performed on a magnetic rack, using an Elx washer (from Biotek), in 1X Erenna Wash Buffer (from Singulex-Millipore). Afterwards MPs were incubated with MW1 detection antibody for 1 h. Washes were performed again on a magnetic rack, in an Elx washer, in 1X Erenna Wash Buffer. The MPs were then transferred into a new Axygen 96 well plate to eliminate nonspecific binding to the plastic. After buffer aspiration the elution buffer (acidic glycine solution, 0.1 M, pH 2.7) was added to the plate for 5 min of incubation under shaking. The eluted detection antibody was transferred to a Nunc 384-well analysis plate and neutralized with neutralization buffer (Tris, 1 M, pH 9). The plate was spun down, sealed and subsequently analyzed with the Erenna Immunoassay System (Singulex-Millipore).
Data analysis
Data analysis was performed using GraphPad Prism software in order to obtain standard curve fitting and back-calculations on fitting models. The standard curve was obtained without associating any weight to each standard concentration.
RESULTS
Assay technology, antibodies, and detected signals
Singulex Erenna technology provides high sensitivity with a broad dynamic range [15] in a 384-well plate format. The signal detected is a train of events generated when dye-labeled molecules are excited by the confocal laser of the instrument and emit fluorescent light. The instrument measures three outputs based on photon detection. The output best suited to the generation of dilution curves described by the standard protein is event photons (EP), which is the average photon count in all detected events. To perform the immunoassay, two anti-HTT antibodies were used. The 2B7 antibody binds to the N17 region of HTT [12] and was conjugated to magnetic particles for use as the capture antibody. The MW1 antibody recognizes the polyglutamine expansion of mHTT [9] and was labeled with a fluorophore and used as the detection antibody. Both antibodies are well-established in the detection of mHTT by ELISA or time-resolved Förster resonance energy transfer (TR-FRET) in biological samples where the mHTT protein is relatively abundant, such as cellular lysates or tissue homogenates [12, 13, 16].
Calibration curve performance
Calibration standards were prepared by spiking known amounts of purified human recombinant HTT proteins in aCSF, a CSF surrogate matrix that matches the physiological electrolyte concentrations of human CSF. Five forms of human recombinant HTT protein were tested: three large fragment proteins comprising the 1-573 N-terminal amino acids (N573) and bearing polyglutamine expansions of 23, 45 and 73 glutamines in Exon 1; and two full length proteins comprising the 1-3144 amino acids (FL) proteins with 17 and 46 glutamines in Exon 1. Signals obtained by serially diluting the N573 fragments and FL proteins, starting from 4 pM and 25 pM respectively, demonstrated the specificity of the assay in detecting the mutant forms of HTT and its polyglutamine dependency. In fact, the mHTT forms (N573 Q45, Q73 and FL Q73) were each well-detected in the assayed range of concentrations, with detection performance improving with polyglutamine length (i.e., N573 Q73 >N573 Q45). On the contrary, the detection of wild-type HTT forms (N573 Q23 and FL Q17) is ineffective or just above the limit of detection (LoD) at the highest tested concentrations (Fig. 1A).
Fig.1.

Calibration curve performance and definition. Detection of human recombinant (hr) full length (with 17 and 46 glutamines in the polyglutamine stretch) and N573 (with 23, 45, and 73 glutamines in the polyglutamine stretch) HTT protein by the 2B7-MW1 SMC assay. A) Polyglutamine and protein length-dependency in HTT detection were demonstrated analyzing the five proteins. B) A longer series of protein dilutions was analyzed for both the full length Q46 and the N573 Q45 in order to define the LLoQ and ULoQ of the assay. These two values were calculated as the lowest (above the LoD) and the highest points of the standard curve for which % CV and % RE are <25 [19]. They are 6.5 and 10000 fM for the large fragment and 16.5 and 10000 fM for the full length protein. The X and Y axes have logarithmic scales and the curves were fitted with a parameter logistic (5PL) model. Each point is the mean of 3 replicates. Bars represent standard deviation. LoD was calculated as the mean of the blank EP values plus three times the standard deviation.
The calibration curve performance was carried out by using either FL or N573 HTT proteins bearing pathological polyglutamine expansions of similar length (Q46 and Q45, respectively). The dynamic range is defined as lying between the lower and upper limits of quantification (LLoQ and ULoQ): the lowest and highest points of the curve for which the percentage relative error (systematic error or bias from nominal value - % RE) and the random error (imprecision or percentage coefficient of variation - % CV) are≤25% [11, 17]. The determined LLoQ and ULoQ were 6.5 and 8000 fM for the large fragment, and 16.5 and 8000 fM for the full-length protein (Fig. 1B). It has to be noted that, the same concentration of the full length and the large fragment protein, bearing almost the same polyglutamine stretch (Q46 and Q45, respectively), were detected with one log difference in intensity with the large fragment producing a higher signal than the full length (Fig. 1B). This observation suggests that HTT detection is not only influenced by the length of polyglutamine expansion, but also by the size of protein.
In order to identify a proper fitting function, data points relative to the N573 Q45 protein were fitted with linear regression, four-parameter logistic (4PL) and five-parameter logistic (5PL) models [18] (Fig. 2A-C). The values calculated by the three fitting models were compared with the ones measured by the assay determining the % RE and the % CV. The sum of the % RE and the % CV is defined as the percentage total error (% TE). The model that best fits the protein detection is the one where the majority of the dilution points (at least four of six points) do not exceed the 30% of TE; this rule is known as the 4-6-30% rule originally described by DeSilva et al. and adopted by the EMA assay validation guidelines [11, 18, 19]. In our study, the linear regression model was demonstrated to be inadequate since using this model, 50% of the analyzed points show a % TE >30 (Fig. 2A, D). The 4PL model instead was shown to be compliant with the above-mentioned rule for the dilution points within the quantification range of the assay (Fig. 2B, D); the 5PL model was compliant with the rule for all the dilution points (including those under the LLoQ) and was thus used in the following validation steps (Fig. 2C, D).
Fig.2.

Standard curve fitting model evaluation. EP values obtained from the 2B7-MW1 SMC analysis of serial dilutions of N573 Q45 HTT protein were fitted using (A) linear regression, (B) four-parameter logistic (4PL), and (C) 5PL models. Each point is the mean of 3 replicates. Bars represent standard deviation. (D) % TE and % RE were evaluated for each dilution point for the three fitting models. The 4PL and 5PL models achieved less than 30% error for points within the quantification range of the assay, as required by the EMA assay validation guideline. For the 5PL model, all assayed dilutions including the one below the LLoQ, showed error <30%.
Accuracy and precision of the standard curve
The accuracy and precision of the standard curve for detection of mHTT were investigated by within-run (intrabatch) and between-run (interbatch) assessment. Within-run evaluation was carried out by testing three technical replicates of each standard concentration in a single run, while between-run evaluation was performed by at least six independent standard curves assayed on different days. The obtained results show that % RE (Table 1A) and % CV (Table 1B) were less than 20% in all the batch runs for mHTT concentrations between the LLoQ and ULoQ. These results are compliant with the EMA guidelines for bioassay validation [11]. For CV and RE calculation, intrabatch precision was estimated by the pooled intrabatch standard deviation of the mean of the calculated concentrations, while interbatch precision (total random error) was estimated by analysis of variance (ANOVA). Statistical methods and formulas used were as described in detail by DeSilva et al. [18].
Table 1.
Accuracy and precision. Accuracy and precision of the 2B7-MW1 SMC mHTT (N573 Q45) detection assay were investigated within-run (intrabatch), by testing three technical replicates of each standard concentration, and between-run (interbatch), by testing six independent standard curves. % RE (A) and % CV (B) measured for each standard concentration in each run are reported. Both values were demonstrated to be less than 20% for all the concentrations between LLoQ and ULoQ for each run. Intrabatch and Interbatch % CV and % RE were estimated using the statistical methods described by DeSilva et al. [18]. Both interbatch and intrabatch precision and accuracy in %CV and %RE calculation were demonstrated to be <20%
| A. Percent Relative Errors (% RE) of Back-Calculated Standard Concentration | |||||||
| Nominal Concentration (fM) | |||||||
| Batch Run | 4000 | 1600 | 640 | 256 | 102 | 41 | 16 |
| 1 | –1.15 | 2.01 | 5.07 | –10.50 | 7.20 | 4.41 | 9.31 |
| 2 | 1.85 | –3.63 | 0.61 | –1.89 | 9.97 | –6.72 | 1.16 |
| 3 | –0.47 | 4.40 | 2.87 | –7.77 | 4.25 | 4.66 | –6.32 |
| 4 | 2.80 | –4.29 | 10.67 | –11.82 | 10.09 | 1.11 | –3.66 |
| 5 | 7.58 | –10.10 | 4.35 | –0.04 | 7.44 | –6.58 | 1.83 |
| 6 | 0.72 | –4.50 | 8.86 | –7.64 | 4.21 | 1.74 | –1.93 |
| Intrabatch (within-run) Statistics (Pooled): | |||||||
| 1.89 | –2.39 | 5.09 | –6.61 | 7.20 | –0.23 | –0.08 | |
| Interbatch (between-run) Statistics (ANOVA): | |||||||
| 1.89 | –2.68 | 5.40 | –6.61 | 7.20 | –0.23 | 0.06 | |
| ULOQ | LLOQ | ||||||
| B. Percent Coefficient of Variation (% CV) of Back-Calculated Standard Concentration | |||||||
| Nominal Concentration (fM) | |||||||
| Batch Run | 4000 | 1600 | 640 | 256 | 102 | 41 | 16 |
| 1 | 4.80 | 8.22 | 11.26 | 14.32 | 9.89 | 13.18 | 29.71 |
| 2 | 3.59 | 6.53 | 17.70 | 12.72 | 8.54 | 10.97 | 5.27 |
| 3 | 15.98 | 16.42 | 16.01 | 12.97 | 5.24 | 9.10 | 2.58 |
| 4 | 18.42 | 1.83 | 2.22 | 11.65 | 9.65 | 4.39 | 5.08 |
| 5 | 9.61 | 14.89 | 10.41 | 13.05 | 7.53 | 16.06 | 8.81 |
| 6 | 8.81 | 3.38 | 16.54 | 16.67 | 8.40 | 18.17 | 3.61 |
| Intrabatch (within-run) Statistics (Pooled): | |||||||
| 11.55 | 11.43 | 14.02 | 13.66 | 8.35 | 12.81 | 11.92 | |
| Interbatch (between-run) Statistics (ANOVA): | |||||||
| 12.96 | 12.31 | 15.91 | 15.05 | 9.29 | 13.86 | 13.28 | |
| ULOQ | LLOQ | ||||||
Specificity for human endogenous HTT protein
We initially proceeded to investigate whether the assay is suitable for specifically detecting endogenous mHTT protein by conducting an HTT detection specificity study in cells. To this aim we used primary fibroblasts from one juvenile HD (JHD) and two adult-onset HD patients expressing mHTT containing 176/23, 45/23, 50/40 glutamines, respectively. These cells were transfected with an HTT-specific siRNA and a scrambled control siRNA, then lysed and collected after 48 h. HTT silencing was achieved to a level between 50–60% of the scrambled transfected cells, as determined by quantitative RT-PCR measuring HTT mRNA (data not shown). When mHTT protein levels were quantified using the 2B7-MW1 SMC assay on silenced and control cells a decrease of the target protein, similar to the observed mRNA decrease, was detected. This provides a preliminary demonstration of the assay’s validity in complex matrices (Fig. 3).
Fig.3.
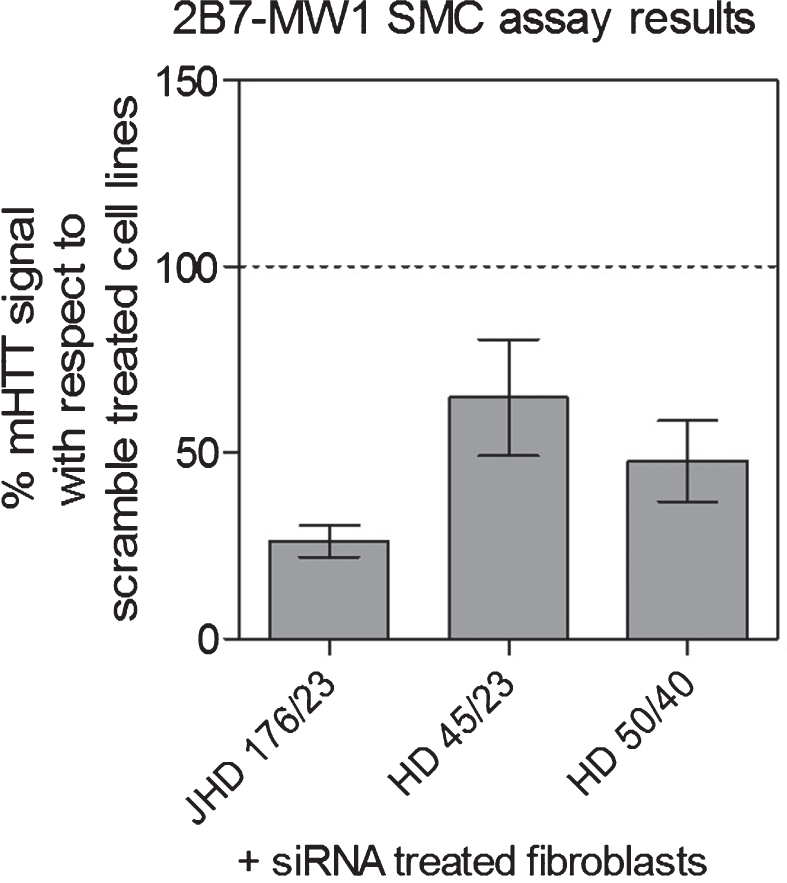
Specificity for human endogenous HTT protein. The 2B7-MW1 SMC assay specificity was demonstrated by analyzing HTT-silenced fibroblasts derived from one JHD and two adult-onset HD subjects. Cells were transfected with an HTT-specific siRNA and a scrambled control. The decrease in mHTT protein levels, as back-calculated on the N573 Q45 standard protein, detected in siRNA treated cell lines, with respect to the scramble treated ones, was found to be coherent with the decrease in HTT mRNA (data not shown).
Analyte stability
To demonstrate that the 2B7-MW1 SMC assay is suitable for the study of biological samples, accuracy and precision have to be assessed on validation samples (VS). VS are real biological samples prepared once and then tested, with the same standard operating procedure as all investigational samples, in every single analyte quantification study to validate the performance of the assay. At least five VS should be used to assess accuracy, precision and the total error of the method: an anticipated ULoQ control (VS1); a high control (VS2); a mid-control (VS3); a control that is more than three times the LLoQ (VS4); and an anticipated LLoQ control (VS5) [11]. These VS were prepared by pooling CSF from healthy and HD donors and, finally, spiking recombinant mHTT protein in order to obtain the five desired mHTT concentrations as presented in Fig. 4A.
Fig.4.

Stability evaluation. Signals obtained from five validation samples (VS) with mHTT levels distributed across the quantification range and their stability. (A) N573 Q45 standard dilutions (black dots) are interpolated with a 5PL model. VS1 anticipates ULoQ, VS2 is a high control, VS3 is a mid-control, VS4 is less than three times the LLOQ and VS5 anticipates LLoQ [11]. Each point is the average of 3 replicates. Bars represent standard deviation. (B, C) Variations in the detection of mHTT in VS detected by the 2B7-MW1 SMC assay on fresh samples after storage at –80°C (first cycle of freeze/thaw), or after 6 h at room temperature (RT), or after the second cycle of freeze/thaw, (B) without preservative or (C) with the addition of 1% Tween. The addition of 1% Tween greatly improved the stability of HTT in all tested conditions (the accuracy % RE remained between 80 and 120 %).
VS were also evaluated for their stability after two freeze-thaw cycles by measuring their variation (% RE) from the initial mHTT concentration. The obtained recovery rate was less than 80% after the first freeze/thaw cycle and decreased even more than 50% after the second cycle (Fig. 4B). One possible determinant of this loss may be the propensity of mHTT to form aggregates by hydrophobic interactions [20]. For this reason, the addition of a detergent was evaluated to prevent mHTT aggregation. Indeed, 1% Tween-20 stabilized the concentration of mHTT in VS after two freeze-thaw cycles to a % RE between 80 and 120% (Fig. 4C). As a consequence, the addition of 1% Tween-20 is recommended to preserve the integrity of mHTT by preventing possible ex vivo precipitation, oligomerization, or aggregation.
Parallelism, dilution linearity, and spike recovery
The parallelism test is used to demonstrate that the endogenous analyte in its biological matrix behaves in a similar immunochemical manner to the standard protein in the same matrix or in a substituted one [19]. In the present work, parallelism was assessed between the calibration standard curve and serially-diluted HD CSF samples in order to exclude possible off-target affinities for other matrix resident analytes and to validate the use of aCSF as substituted matrix for the standard curve [19]. A preliminary evaluation of parallelism was made using the similarity between the curve shape of the CSF dilutions, where at least three points fell within the quantification range, and the standard mHTT curve (Fig. 5A). Parallelism was assessed verifying that the variation (% RE) between back-calculated sample concentrations in a dilution series does not exceed 30% [11]. Results are represented in Fig. 5B, where the calculated concentrations (observed concentration×dilution factor) were divided by the mean of the concentrations and plotted against the inverse of the dilution factor [19]. The calculated ratio was not affected by sample dilution for the four samples which were detected within the quantification range of the assay. Back-calculated mHTT levels were found to not exceed 20% RE in those samples, thus validating the dilution linearity of the assay and consequently its parallelism. Furthermore, the two CSF samples from premanifest HD patients showed a % RE within the 30%, even if they were below the LLoQ, demonstrating that a matrix effect is not observed in real CSF samples using the 2B7-MW1 SMC assay.
Fig.5.

Parallelism evaluation. Six CSF samples were used: four CSF from advanced HD patients and two CSF from pre-manifest HD patients. (A) N573 Q45 standard dilutions (black dots) are interpolated with a 5PL model and shown in each plot together with the signal obtained from 2B7-MW1 SMC analysis of the CSF. X and Y axes have logarithmic scales and the curves were fitted with a 5PL model. Each point is the average of 3 replicates. Bars represent standard deviation. (B) Parallelism was demonstrated by dilution linearity. Calculated mHTT concentrations (observed concentration×dilution factor) were divided by the mean of the concentrations and plotted against the inverse of the dilution factor [19]. The % RE among the dilution points was <20% for all the samples detected over the LLoQ and <30% for the two samples detected under the LLoQ.
mHTT spike recovery was carried out to evaluate the influence of the CSF matrix in mHTT detection by the assay [19]. Recombinant mHTT protein N573 Q45 was spiked and diluted in aCSF and in five real CSFs collected from HD patients. The spike recovery rate percentage was calculated by comparing the assay-determined mHTT amount to a nominal one, after subtracting the sample basal mHTT levels [19, 21]. All mHTT concentrations spiked in the six matrices were recovered within the acceptable range of 80–120 % (Fig. 6).
Fig.6.

Spike recovery evaluation. Seven dilutions of the N573 Q45 recombinant protein were spiked in aCSF and in five CSF samples collected from HD patients. (A) The obtained curves were fitted with a 5PL model. The X and Y axes have logarithmic scales. Each point is the mean of 3 replicates. Bars represent standard deviations. (B) The spike recovery rate of five HTT dilutions was between 80 and 120 % of the nominal concentration in all the tested matrices.
Selectivity
The selectivity of the 2B7-MW1 SMC assay was evaluated in order to demonstrate the suitability of the assay to detect mHTT unequivocally even in the presence of additives and/or contaminants in the target matrix [10, 19]. The former deals with chemicals added to CSF samples to preserve them; the latter is dependent on accidental CSF contamination during sampling by other tissues such as blood. Both the FDA and the EMA guidelines for bioanalytical method validation suggest performing selectivity tests in real biological matrices; nonetheless the volume of human CSF required to complete the study with all the selected contaminants was incompatible with the availability of HD CSF. To overcome this limitation, aCSF was used instead of HD CSF. Our findings for parallelism, dilution linearity and spike recovery, described above, demonstrate the suitability of aCSF as a matrix surrogate in the determination of the assay’s selectivity for mHTT.
Selectivity was assessed by analyzing multiple concentrations of the following potential interfering substances: human plasma pooled from healthy donor with EDTA K2, EDTA K3, sodium citrate, sodium heparin and sodium EDTA (which are commonly used as anti-coagulants and sample preservatives), serum and whole blood (which can be present in CSF due to sampling contamination).
The obtained results demonstrated that up to 10% plasma with sodium heparin or serum did not affect the assay signal. In contrast, plasma with EDTA K2, EDTA K3, sodium citrate, and sodium EDTA concentrations higher than 2, 0.4, 0.3, and 0.08% respectively resulted in signals higher that the LoD (Fig. 7A), suggesting that they should be avoided as CSF preservatives. A summary of all the additive thresholds is reported in Table 2.
Fig.7.

Selectivity evaluation. The selectivity of the 2B7-MW1 SMC assay was assessed against multiple concentrations of potential interfering substances. Different concentrations of eight components were spiked into artificial CSF: (A) plasma with EDTA K2, EDTA K3, sodium citrate, sodium heparin and sodium EDTA, serum and (B) blood collected from six HD subjects and one healthy control. EP signals are reported with respect to the % spike in aCSF in A, or against human hemoglobin concentration in B. Each point is the mean of 3 replicates. Bars represent standard deviations. Numeric results are summarized in Table 2.
Table 2.
Summary of selectivity test results. Contaminant interference in the detection of mHTT and the corresponding threshold concentrations for each tested substance
| Presevative/Contaminant | mHTT detection interference | Acceptance criteria |
| Plasma w EDTA K2 | Yes | ≤2% |
| Plasma w EDTA K3 | Yes | ≤0.40% |
| Plasma w Na-citrate | Yes | ≤0.30% |
| Plasma w Na-Heparin | No | up to 10% |
| Plasma w Na-EDTA | Yes | ≤0.08% |
| Serum | No | Up to 10% |
| Hemoglobin | Yes | ≤2 μg/ml |
While CSF is normally centrifuged after sampling in order to remove blood cells, soluble components of blood and the contents of already-hemolyzed blood cells cannot be removed. The presence of hemolyzed blood was found to impact the assay process by causing the magnetic particles to clump. In addition to this general hemolyzed blood interference, blood contamination may alter mHTT levels in CSF by specifically adding mHTT to the CSF. To assess this possibility, blood from six HD patients was serially diluted in aCSF, starting from a concentration of 0.15%, and analyzed by the 2B7-MW1 Singulex assay. Hemoglobin (HbA) levels for all blood dilutions were measured and employed as an independent means of quantifying blood contamination that could then be used to establish a hemoglobin threshold below which contamination of CSF by mHTT from blood is expected to be negligible.
The correlation between mHTT and hemoglobin levels is reported in Fig. 7B. An appreciable signal was detected at blood spikes greater than 0.002%, corresponding to 2μg/mL of hemoglobin which, as a consequence, was fixed as threshold for mHTT quantification in HD CSF.
All the thresholds of the tested contaminants, which may interfere with mHTT detection by the 2B7-MW1 SMC assay, are summarized in Table 2.
Reproducibility
The reproducibility of an assay is determined by the incurred sample reanalysis (ISR), which is the reanalysis of a portion of subject samples to determine whether the original results are reproducible [10, 11]. The ISR was carried out on 24 HD CSF samples (i.e., 10% of a putative 240-participant study) analyzed in triplicate. Three healthy CSF samples were included as negative controls. The time between the first and the second analysis was set to one week, whereas five weeks elapsed between the first and the third analysis. After collection, samples were aliquoted in single-use vials to avoid multiple freeze-thaw cycles among the different runs. The three control sample results were lower than the LLoQ, confirming the assay specificity. One HD CSF sample result was lower than the LLoQ; thus it was excluded from the ISR study. Hemoglobin levels in the remaining samples were below the 2μg/mL threshold with the exception of one sample, which was excluded from the ISR study (see Table 3). The RE for each run was calculated with respect to the average mHTT concentration. 77.3% of tested samples showed a RE within 30% (Table 3), which is compliant with the assay validation guidelines for large molecules, being greater than the two-thirds (67%) of the incurred samples. [10, 11].
Table 3.
Reproducibility evaluation. Reproducibility of the 2B7-MW1 SMC mHTT detection assay was successfully investigated by testing 22 CSF samples, collected from HD patients, in three independent runs. The mean of mHTT concentrations obtained in each of the three runs is reported, together with CV % and RE % . The RE % was calculated with respect to the average of concentration obtained from the three runs (indicated as AVE). Asterisks indicate samples which failed to meet one of the following criteria: HbA <2μg/ml, CV <30% or RE <30%
| Sample ID | Hemoglobin | Average HTT fM | AVE | SD | CV% | Note | Recovery % w.r.t. AVE | RE % | ||||||
| μg/ml | run #1 | run #2 | run #3 | run #1 | run #2 | run #3 | run #1 | run #2 | run #3 | |||||
| HD 1 | 0.47 | 109.09 | 147.59 | 90.22 | 115.63 | 29.24 | 25.29 | 94.35 | 127.63 | 78.02 | –5.65 | 27.63 | –21.98 | |
| HD 2 | 2.95* | 98.03 | 340.09 | 75.36 | 171.16 | 146.74 | 85.73* | Hb >2 μg/ml | excluded from reproducibility analysis | |||||
| HD 3 | 0.22 | 61.02 | 97.73 | 63.21 | 73.98 | 20.59 | 27.83 | 82.48 | 132.09 | 85.43 | –17.52 | 32.09* | –14.57 | |
| HD 4 | 0.33 | 11.10 | 14.80 | 10.63 | 12.18 | 2.29 | 18.76 | 91.18 | 121.55 | 87.27 | –8.82 | 21.55 | –12.73 | |
| HD 5 | 0.14 | 75.88 | 86.95 | 78.55 | 80.46 | 5.78 | 7.19 | 94.30 | 108.07 | 97.63 | –5.70 | 8.07 | –2.37 | |
| HD 6 | 0.23 | 116.81 | 156.66 | 73.77 | 115.75 | 41.46 | 35.82* | 100.92 | 135.35 | 63.73 | 0.92 | 35.35* | –36.27* | |
| HD 7 | 0.34 | 35.55 | 48.13 | 43.15 | 42.28 | 6.34 | 14.99 | 84.08 | 113.85 | 102.07 | –15.92 | 13.85 | 2.07 | |
| HD 8 | <LoD | 28.55 | 32.15 | 35.42 | 32.04 | 3.44 | 10.73 | 89.10 | 100.35 | 110.55 | –10.90 | 0.35 | 10.55 | |
| HD 9 | 0.09 | 41.35 | 43.25 | 42.50 | 42.37 | 0.96 | 2.26 | 97.60 | 102.09 | 100.30 | –2.40 | 2.09 | 0.30 | |
| HD 10 | 0.43 | 32.63 | 35.75 | 22.70 | 30.36 | 6.81 | 22.44 | 107.48 | 117.74 | 74.78 | 7.48 | 17.74 | –25.22 | |
| HD 11 | 0.15 | 15.23 | 20.00 | 233.15 | 89.46 | 124.46 | 139.12* | 17.03 | 22.36 | 260.62 | –82.97* | –77.64* | 160.62* | |
| HD 12 | 0.40 | 73.27 | 96.72 | 86.82 | 85.60 | 11.77 | 13.75 | 85.59 | 112.98 | 101.42 | –14.41 | 12.98 | 1.42 | |
| HD 13 | 0.09 | 59.93 | 69.33 | 58.37 | 62.54 | 5.93 | 9.48 | 95.82 | 110.85 | 93.33 | –4.18 | 10.85 | –6.67 | |
| HD 14 | 0.33 | 427.62 | 365.50 | 220.00 | 337.71 | 106.57 | 31.56* | 126.63 | 108.23 | 65.15 | 26.63 | 8.23 | –34.85* | |
| HD 15 | 0.34 | 58.15 | 75.93 | 64.50 | 66.19 | 9.01 | 13.61 | 87.85 | 114.71 | 97.44 | –12.15 | 14.71 | –2.56 | |
| HD 16 | 0.06 | 22.20 | 20.53 | 20.00 | 20.91 | 1.15 | 5.49 | 106.17 | 98.19 | 95.64 | 6.17 | –1.81 | –4.36 | |
| HD 17 | 0.18 | 14.55 | 20.37 | 26.57 | 20.50 | 6.01 | 29.32 | 71.00 | 99.37 | 129.63 | –29.00 | –0.63 | 29.63 | |
| HD 18 | 0.04 | 238.00 | 109.52 | 57.64 | 135.05 | 92.85 | 68.75* | 176.22 | 81.10 | 42.68 | 76.22* | –18.90 | –57.32* | |
| HD 19 | 0.10 | 121.61 | 171.77 | 116.60 | 136.66 | 30.51 | 22.33 | 88.99 | 125.69 | 85.32 | –11.01 | 25.69 | –14.68 | |
| HD 20 | 0.07 | 3.50 | 1.06 | 1.23 | 1.93 | 1.36 | 70.43* | <LLoQ | excluded from reproducibility analysis | |||||
| HD 21 | 0.40 | 82.20 | 102.18 | 58.50 | 80.96 | 21.87 | 27.01 | 101.53 | 126.22 | 72.25 | 1.53 | 26.22 | –27.75 | |
| HD 22 | <LoD | 48.13 | 56.75 | 59.30 | 54.73 | 5.85 | 10.70 | 87.95 | 103.70 | 108.35 | –12.05 | 3.70 | 8.35 | |
| HD 23 | <LoD | 149.29 | 186.44 | 149.90 | 161.88 | 21.28 | 13.14 | 92.23 | 115.18 | 92.60 | –7.77 | 15.18 | –7.40 | |
| HD 24 | <LoD | 51.57 | 76.65 | 64.65 | 64.29 | 12.55 | 19.51 | 80.21 | 119.23 | 100.56 | –19.79 | 19.23 | 0.56 | |
| Healthy 1 | <LoD | 3.08 | 4.62 | 3.25 | 3.65 | 0.84 | 23.12 | <LLoQ | excluded from reproducibility analysis | |||||
| Healthy 2 | <LoD | 3.05 | 4.79 | 2.09 | 3.31 | 1.37 | 41.34* | <LLoQ | excluded from reproducibility analysis | |||||
| Healthy 3 | <LoD | 3.30 | 6.02 | 1.13 | 3.48 | 2.45 | 70.20* | <LLoQ | excluded from reproducibility analysis | |||||
DISCUSSION
One of the more promising therapeutic approaches to mitigate or modify the course of HD is based on various techniques to lower the expression of mHTT protein. In order to enable such therapeutic approaches, biomarkers of HTT lowering must be developed to demonstrate that delivery of a HTT lowering agent does indeed lower the amount of HTT protein in the brain of an HD patient. Since the brain cannot be non-invasively used for a pharmacodynamic examination, the analysis of brain-derived proteins enriched in patient CSF can provide a “window into the brain” [6, 22, 23] and represents the most accessible opportunity to biochemically sample the CNS milieu.
The aim of the present work was to clinically validate the approach proposed by Wild and Boggio et al. that is based on the use of the 2B7-MW1 SMC assay as a biomarker for HTT lowering therapeutic approaches. The validation process was carried out following the recommendations of both the FDA and EMA [10, 11]. One partial limitation complicating this validation study was the limited availability of HD patient CSF. Nonetheless, the parallelism, dilution linearity, and spike recovery studies demonstrated that patient CSF matrix can be substituted by aCSF and that the recombinant mHTT protein used to perform the reference standard is detected in a similar biochemical manner to endogenous mHTT protein. The assay was demonstrated to be specific for mHTT with a lower limit of detection of 6.5 fM for the N573 Q45 and 16.5 fM for the FL Q46. It has to be noted that the assay better detects the shorter form of mutant HTT (N573) than the full length one (FL) even though the two were almost identical in terms of polyglutamine expansion. This fact may be relevant when interpreting results, as there are reports showing that endogenous mHTT is present in various truncated forms in cells resulting from proteolytic cleavage as well as proteosomal degradation [24–26].
CSF matrix composition was demonstrated not to interfere with the assay throughout a wide range of dilutions. Still, we recommend the addition of 1% Tween-20 to CSF, at the time of sampling or after the first thawing, to retain the integrity of the biosample by avoiding possible loss of mHTT due to protein precipitation, oligomerization, or aggregation. When various additives were tested for their interference, plasma with EDTA K2, EDTA K3, Na-citrate, or NA-EDTA were found to impact on mHTT quantification; thus their use should be avoided. With regard to contaminant influence, hemolyzed blood was demonstrated to be a potential major issue when hemoglobin exceeds 2μg/ml due to both matrix effect and serum mHTT contamination: thus we recommend that CSF samples containing hemoglobin levels above this threshold be avoided. Finally, the assay was demonstrated to be reliable as mHTT levels in 77.3% of a 22 pre-/early manifest group were consistently quantified over three independent runs.
Despite our demonstration that the 2B7-MW1 SMC assay is accurate and precise, there are some recommendations that should be taken into account when interpreting data produced by the present procedure. First, it has to be considered that the output of mHTT quantification is dependent, to some extent, on two important factors: the poly-glutamine expansion size and the protein size. Therefore, in absence of a set of standard recombinant mHTT proteins with every single possible combination of polyglutamine expansions and protein fragments, the calculated mHTT concentration must be considered as a best estimate rather than an absolute value; as a consequence the assay can be defined as relatively quantitative [17]. A consequence is that the most meaningful results will likely be obtained in paired analyses where a single subject is monitored over time – such as within the setting of a clinical trial. A further consideration is the choice between N573 and FL standard mHTT. Because the performance of these two standards is different, mHTT levels can be compared only when a single standard is used across different runs. Moreover, in consideration of the propensity of mHTT to aggregate, the quality of the standard proteins must be periodically controlled by independent methods such as native PAGE.
The present work demonstrated that the presence of hemolyzed blood is highly detrimental to the assay performance. When the contamination is evident it impairs the assay performance, while at minor contamination (>2μg/ml of hemoglobin) errors in mHTT determination were observed. Therefore, a third recommendation would be to always quantify hemoglobin levels in the CSF prior to running the mHTT quantification in order to exclude contaminated samples from the study.
Finally, to better interpret the mHTT concentration levels, ancillary biomarkers such as the total protein amount in the CSF should be used that provide an additional characterization of the testing matrix [27, 28].
In conclusion, the present work, guided by FDA and EMA principles, demonstrates that the 2B7-MW1 assay is a valid, robust, reproducible, and ultrasensitive method for the relative quantification of mHTT in human CSF.
CONFLICT OF INTEREST
EJW has participated in scientific advisory boards with Hoffmann-La Roche Ltd, Ionis, Shire, GSK and Wave Life Sciences. All honoraria for these advisory boards were paid through UCL Consultants Ltd, a wholly owned subsidiary of UCL. His host clinical institution, University College London Hospitals NHS Foundation Trust, receives funds as compensation for conducting clinical trials for Ionis Pharmaceuticals, Pfizer and Teva Pharmaceuticals.
BRL has participated in scientific advisory boards with Hoffmann-La Roche Ltd, Ionis, Raptor, Teva, uniQure, and LifeMax. Research funding has been provided to his institution by Teva, uniQure, and Lifemax, and he receives funds as compensation for conducting clinical trials for Teva and Ionis Pharmaceuticals.
There are no patents, products in development, or marketed products to declare.
ACKNOWLEDGMENTS
We thank the late Dr. Paul Patterson (CalTech, USA) for the MW1 antibody.
This work is dedicated to Dr. Sergio Altamura who prematurely passed way at the age of 59. At IRBM, Sergio was that leader who feels rewarded by making things possible without asking for more. A rare person who we miss.
EJW is funded by the Medical Research Council (UK), CHDI Foundation Inc and European Huntington’s Disease Network. This work was supported in part by the National Institute for Health Research University College London Hospitals Biomedical Research Centre, the UCL Leonard Wolfson Experimental Neurology Centre.
REFERENCES
- [1]. Ioannidis JPA, Bossuyt PMM. Waste, leaks, and failures in the biomarker pipeline. Clin Chem. 2017;63:963–72. doi: 10.1373/clinchem.2016.254649 [DOI] [PubMed] [Google Scholar]
- [2]. Wild EJ, Boggio R, Langbehn D, Robertson N, Haider S, Miller JRC, et al. Quantification of mutant huntingtin protein incerebrospinal fluid from Huntington’s disease patients. J Clin Invest. 2015;125:1–8. doi: 10.1172/JCI80743DS1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3]. A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. The Huntington’s Disease Collaborative Research Group. Cell. 1993;72:971–83. [DOI] [PubMed] [Google Scholar]
- [4]. Leavitt BR, Wellington CL, Hayden MR. Recent insights into the molecular pathogenesis of Huntington disease. Semin Neurol. 1999;19:385–95. [DOI] [PubMed] [Google Scholar]
- [5]. Aronin N, DiFiglia M. Huntingtin-lowering strategies in Huntington’s disease: Antisense oligonucleotides, small RNAs, and gene editing. Mov Disord. 2014;29:1455–61. [DOI] [PubMed] [Google Scholar]
- [6]. Ross CA, Aylward EH, Wild EJ, Langbehn DR, Long JD, Warner JH, et al. Huntington disease: Natural history, biomarkers and prospects for therapeutics. Nat Rev Neurol. 2014;10:204–16.24614516 [Google Scholar]
- [7]. Wild EJ, Tabrizi SJ. Targets for future clinical trials in Huntington’s disease: What’s in the pipeline? Mov Disord 2014;29:1434–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8]. Southwell AL, Smith SEP, Davis TR, Caron NS, Villanueva EB, Xie Y, et al. Ultrasensitive measurement of huntingtin protein in cerebrospinal fluid demonstrates increase with Huntington disease stage and decrease following brain huntingtin suppression. Sci Rep. 2015;5:12166 doi: 10.1038/srep12166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9]. Ko J, Ou S, Patterson PH. New anti-huntingtin monoclonal antibodies: Implications for huntingtin conformation and its binding proteins. Brain Res Bull. 2001;56:319–29. doi: 10.1016/S0361-9230(01)00599-8 [DOI] [PubMed] [Google Scholar]
- [10]. Food and Drug Administration (2013) Guidance for Industry:Bioanalytical method validation. http://www.labcompliance.de/documents/FDA/FDA-Others/Laboratory/f-507-bioanalytical-4252fnl.pdf
- [11]. European Medicines Agency, Committee for Medicinal Products for Human Use. Guideline on Bioanalytical Method Validation. European Medicines Agency, London, UK. (2011). http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2011/08/WC500109686.pdf
- [12]. Weiss A, Abramowski D, Bibel M, Bodner R, Chopra V, DiFiglia M, et al. Single-step detection of mutant huntingtin in animal and human tissues: A bioassay for Huntington’s disease. Anal Biochem. 2009;395:8–15. doi: 10.1016/j.ab.2009.08.001 [DOI] [PubMed] [Google Scholar]
- [13]. Macdonald D, Tessari MA, Boogaard I, Smith M, Pulli K, Szynol A, et al. Quantification assays for total and polyglutamine-expanded huntingtin proteins. PLoS One. 2014;9:e96854 doi: 10.1371/journal.pone.0096854 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14]. Huang B, Lucas T, Kueppers C, Dong X, Krause M, Bepperling A, et al. Scalable production in human cells and biochemicalcharacterization of full-length normal and mutant huntingtin. PLoS One. 2015;10:1–23. doi: 10.1371/journal.pone.0121055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15]. Todd J, Freese B, Lu A, Held D, Morey J, Livingston R, et al. Ultrasensitive flow-based immunoassays using single-molecule counting. Clin Chem. 2007;53:1990–5. doi: 10.1373/clinchem.2007.091181 [DOI] [PubMed] [Google Scholar]
- [16]. Fodale V, Kegulian NC, Verani M, Cariulo C, Azzollini L, Petricca L, et al. Polyglutamine- and temperature-dependent conformationalrigidity in mutant huntingtin revealed by immunoassays andcircular dichroism spectroscopy. PLoS One. 2014;9:e112262 doi: 10.1371/journal.pone.0112262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17]. Lee JW, Devanarayan V, Barrett YC, Weiner R, Allinson J, Fountain S, et al. Fit-for-purpose method development and validation for successful biomarker measurement. Pharm Res. 2006;23:312–28. doi: 10.1007/s11095-005-9045-3 [DOI] [PubMed] [Google Scholar]
- [18]. Desilva B, Smith W, Weiner R, Kelley M, Smolec J, Lee B, et al. Recommendations for the bioanalytical method validation of ligand-binding assays to support pharmacokinetic assessments of macromolecules. Pharm Res. 2003;20:1885–900. doi: 10.1023/B:PHAM.0000003390.51761.3d [DOI] [PubMed] [Google Scholar]
- [19]. Lee JW. Method validation and application of protein biomarkers:Basic similarities and differences from biotherapeutics. Bioanalysis. 2009;1:1461–74. doi: 10.4155/bio.09.130 [DOI] [PubMed] [Google Scholar]
- [20]. Legleiter J, Mitchell E, Lotz GP, Sapp E, Ng C, DiFiglia M, et al. Mutant huntingtin fragments form oligomers in a polyglutamine length-dependent manner in vitro and in vivo. J Biol Chem. 2010;285:14777–90. doi: 10.1074/jbc.M109.093708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21]. Andreasson U, Perret-Liaudet A, van Waalwijk van Doorn LJC, Blennow K, Chiasserini D, Engelborghs S, et al. A practical guide to immunoassay method validation. Front Neurol. 2015;6:1–8. doi: 10.3389/fneur.2015.00179 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22]. Reiber H. Proteins in cerebrospinal fluid and blood: Barriers, CSF flow rate and source-related dynamics. Restor Neurol Neurosci. 2003;21:79–96. [PubMed] [Google Scholar]
- [23]. Blennow K, Hampel H, Weiner M, Zetterberg H. Cerebrospinal fluid and plasma biomarkers in Alzheimer disease. Nat Rev Neurol. 2010;6:131–44. [DOI] [PubMed] [Google Scholar]
- [24]. Landles C, Sathasivam K, Weiss A, Woodman B, Moffitt H, Finkbeiner S, et al. Proteolysis of mutant huntingtin produces an exon 1 fragment that accumulates as an aggregated protein in neuronal nuclei in Huntington disease. J Biol Chem. 2010;285:8808–23. doi: 10.1074/jbc.M109.075028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25]. DiFiglia M, Sapp E, Chase KO, Davies SW, Bates GP, Vonsattel JP, et al. Aggregation of huntingtin in neuronal intranuclearinclusions and dystrophic neurites in brain. Science. 1997;277:1990–3. doi: 10.1126/science.277.5334.1990 [DOI] [PubMed] [Google Scholar]
- [26]. Vonsattel JPG, Keller C, Cortes Ramirez EP. Huntington’s disease - neuropathology. H Handb Clin Neurol. 2011;100:83–100. doi: 10.1016/B978-0-444-52014-2.00004-5 [DOI] [PubMed] [Google Scholar]
- [27]. Hühmer AF, Biringer RG, Amato H, Fonteh AN, Harrington MG. Protein Analysis in Human Cerebrospinal Fluid: Physiological Aspects, Current Progress and Future Challenges. Dis Markers. 2006;22:3–26. doi: 10.1155/2006/158797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28]. Schutzer SE, Liu T, Natelson BH, Angel TE, Schepmoes AA, Purvine SO, et al. Establishing the proteome of normal human cerebrospinal fluid. PLoS One. 2010;5:e10980 doi: 10.1371/journal.pone.0010980 [DOI] [PMC free article] [PubMed] [Google Scholar]