

Supplemental Digital Content is available in the text.
Keywords: atherosclerosis, cholesterol, coronary artery disease, foam cells, macrophages
Abstract
Objective—
Rosuvastatin has been widely used in the primary and secondary prevention of coronary heart disease. However, its antiatherosclerotic properties have not been tested in a mouse model that could mimic human coronary heart disease. The present study was designed to test the effects of rosuvastatin on coronary artery atherosclerosis and myocardial fibrosis in SR-B1 (scavenger receptor class B type 1) and apoE (apolipoprotein E) double knockout mice.
Approach and Results—
Three-week-old SR-B1−/−/apoE−/− mice were injected daily with 10 mg/kg of rosuvastatin for 2 weeks. Compared with saline-treated mice, rosuvastatin-treated mice showed increased levels of hepatic PCSK9 (proprotein convertase subtilisin/kexin type-9) and LDLR (low-density lipoprotein receptor) message, increased plasma PCSK9 protein but decreased levels of hepatic LDLR protein and increased plasma total cholesterol associated with apoB (apolipoprotein B) 48-containing lipoproteins. In spite of this, rosuvastatin treatment was associated with decreased atherosclerosis in both the aortic sinus and coronary arteries and reduced platelet accumulation in atherosclerotic coronary arteries. Cardiac fibrosis and cardiomegaly were also attenuated in rosuvastatin-treated SR-B1−/−/apoE−/− mice. Two-week treatment with rosuvastatin resulted in significant decreases in markers of oxidized phospholipids in atherosclerotic plaques. In vitro analysis showed that incubation of bone marrow-derived macrophages with rosuvastatin substantially downregulated cluster of differentiation (CD)36 and inhibited oxidized LDL-induced foam cell formation.
Conclusions—
Rosuvastatin protected SR-B1−/−/apoE−/− mice against atherosclerosis and platelet accumulation in coronary arteries and attenuated myocardial fibrosis and cardiomegaly, despite increased plasma total cholesterol. The ability of rosuvastatin to reduce oxidized phospholipids in atherosclerotic plaques and inhibit macrophage foam cell formation may have contributed to this protection.
Atherosclerosis, a major underlying cause of coronary heart disease (CHD), is a consequence of both dysregulated lipid metabolism as well as inflammatory processes that involve the binding of monocytes to dysfunctional endothelium, their recruitment to susceptible areas of the arterial wall and their differentiation to macrophages and development into foam cells.1 The accumulation of foam cells gives the appearance of fatty streaks in the intima of arterial walls at the early stages of atherosclerosis thereby leading to the formation of atherosclerotic plaques as the disease develops.1 The statins are potent inhibitors of cholesterol synthesis. Several large-scale clinical trials have consistently demonstrated the effects of statins in plasma lipid lowering and in the primary and secondary prevention of CHD.2 Recent studies, however, suggest that the beneficial effects of statins may extend beyond their effects on lowering serum cholesterol levels.3,4 These studies suggest that statins likely protect the cardiovascular system against atherosclerosis via plasma lipid lowering-dependent and lowering-independent mechanisms. Rosuvastatin, a potent statin, has shown several beneficial characteristics, including a long half-life, a favorable safety profile, and low propensity for cytochrome P450 drug interactions.5
The pleiotropic effects of rosuvastatin have been studied using several mouse models exhibiting aspects of atherosclerotic cardiovascular disease in isolation.6–13 The most widely used mouse model of atherosclerosis is the apoE−/− (apolipoprotein E) mouse.14–16 In apoE−/− mice, total plasma cholesterol levels are ≈5× higher than in wild-type mice, because of increased VLDL (very-low–density lipoprotein), IDL (intermediate-density lipoprotein), and LDL (low-density lipoprotein).14–16 ApoE−/− mice fed a normal chow diet develop atherosclerosis by 3 months of age, and atherosclerosis can be accelerated by feeding the mice high-fat or high-cholesterol diets.14–16 Several studies demonstrated that rosuvastatin treatment at varying doses reduced inflammation, thrombosis, and apoptosis in the aortic sinus and proximal aortas of apoE−/− mice, although plasma total cholesterol and triglyceride levels were not altered.6,9,10,12,17–20 On the other hand, other studies reported beneficial effects of rosuvastatin were accompanied by reductions in plasma total cholesterol in apoE−/− mice.7,8 Still, other studies have reported that treatment of apoE−/− mice with other statins, including simvastatin, resulted in increased plasma cholesterol levels in some cases accompanied by increased atherosclerosis.21–23 ApoE−/− mice exhibit severe atherosclerotic plaque formation in the aortic sinus, aortic arch, and brachiocephalic arteries.14–16 Plaques have been observed in coronary arteries of apoE−/− mice, particularly those mice fed high-fat diets for extended periods of time.14–16 However, these are not frequent and these mice do not seem to develop subsequent myocardial infarction and heart dysfunction.24
In contrast, apoE−/− mice that are also deficient in the SR-B1 (scavenger receptor class B type 1) exhibit spontaneous, occlusive coronary artery atherosclerosis, which develops rapidly by 5 weeks of age.25–29 SR-B1 is a cell surface HDL (high-density lipoprotein) receptor, expressed in liver, intestine, steroidogenic tissues (eg, adrenal gland, ovary, and testis), and vascular cells (eg, macrophages and endothelial cells).30 SR-B1 in the liver and steroidogenic tissues mediates selective HDL lipid uptake, and hepatic SR-B1 drives reverse cholesterol transport to remove cholesterol from peripheral tissues for biliary excretion.31,32 SR-B1 has also been shown to enhance cholesterol efflux from cells33,34 and to mediate HDL-dependent activation of cellular signaling pathways in diverse cells including endothelial cells and macrophages.35–39 Mice lacking both SR-B1 and apoE develop increased atherosclerosis in their aortic sinus and occlusive coronary artery atherosclerosis between 3 and 5 weeks of age.25–29 They also exhibit extensive myocardial fibrosis, cardiac functional and conductance abnormalities, and reduced life span (6 to 8 weeks of age) compared with their apoE−/− littermates.25–29 Braun et al27,28 have tested several pharmacological interventions on these phenotypes in SR-B1−/−/apoE−/− mice. Probucol, an antioxidant and lipid-lowering drug, corrected the lipoprotein abnormalities in SR-B1−/−/apoE−/− mice and delayed development of occlusive coronary atherosclerosis, and cardiac pathology, and extended their life span to 36 weeks.28 Ezetimibe, an inhibitor of intestinal cholesterol absorption, lowered LDL cholesterol and reduced aortic sinus, and coronary arterial atherosclerosis and myocardial fibrosis and also increased the survival of SR-B1−/−/apoE−/− mice.27 Similarly, LDL cholesterol was reduced and survival prolonged when SR-B1−/−/apoE−/− mice were treated with SC-435, an apical sodium codependent bile acid transporter inhibitor, which prevents intestinal absorption of bile acid.27 Oral supplementation of drinking water with pomegranate extract reduced oxidative stress and inflammation in coronary arteries of SR-B1−/−/apoE−/− mice and reduced atherosclerosis and myocardial fibrosis and delayed the development of ECG abnormalities, although plasma cholesterol levels were increased.26 These studies suggest that the SR-B1−/−/apoE−/− mouse may be a useful model system in which to analyze mechanisms of pharmacological protection against CHD. To date, however, the effects of statin treatment on development of coronary artery atherosclerosis and associated phenotypes in the SR-B1−/−/apoE−/− mouse have not been tested.
To investigate the effects of rosuvastatin treatment on the development of coronary artery atherosclerosis and myocardial infarction in mice, we treated SR-B1−/−/apoE−/− mice with or without rosuvastatin. Treatment began at 3 weeks of age, preceding the start of atherosclerosis development in the aortic sinus or coronary arteries.26,28 In this study, we show that daily treatment of SR-B1−/−/apoE−/− mice for 2 weeks with rosuvastatin (10 mg/kg of body weight) resulted in increased levels of hepatic LDLR (LDL receptor) and PCSK9 (proprotein convertase subtilisin/kexin type-9) gene expression but reduced levels of hepatic LDLR protein and increased plasma cholesterol associated with VLDL-sized lipoprotein particles. Despite this, rosuvastatin treatment of SR-B1−/−/apoE−/− mice reduced aortic sinus and coronary artery atherosclerosis, platelet accumulation in atherosclerotic coronary arteries, cardiac enlargement, and cardiac fibrosis. Rosuvastatin also reduced the levels of oxidized lipids in atherosclerotic plaques in the aortic sinus and coronary arteries. Rosuvastatin treatment of cultured macrophages from SR-B1−/−/apoE−/− mice reduced levels of cluster of differentiation (CD)36 protein and oxLDL (oxidized low-density lipoprotein)-driven foam cell formation. These reductions in the accumulation of oxidized phospholipids in the walls of arteries and reduced oxLDL uptake by macrophages may have contributed to the ability of rosuvastatin to protect against the development of occlusive coronary artery atherosclerosis and associated CHD in SR-B1−/−/apoE−/− mice. This is the first in vivo study, in a single mouse model, to show statin-mediated reductions in coronary artery atherosclerosis, accumulation of platelets in atherosclerotic coronary arteries and myocardial infarction that are independent of cholesterol lowering. It provides important insights into cholesterol-independent mechanisms of rosuvastatin-mediated protection against CHD.
Materials and Methods
Materials and Methods are available in the online-only Data Supplement.
Results
Rosuvastatin Increased Plasma Cholesterol Levels in SR-B1−/−/ApoE−/− Mice
SR-B1−/−/apoE−/− mice develop severe hypercholesterolemia with a significant increase of VLDL-sized lipoproteins (size fractionated by fast protein liquid chromatography) compared with apoE−/− littermates40 and plasma lipoproteins from SR-B1−/−/apoE−/− mice contain abnormally high levels of unesterified cholesterol,28,29 which may contribute to the reported coronary artery atherosclerosis and cardiac pathology. Similarly, our data showed that the average plasma cholesterol levels in 5-week-old SR-B1−/−/apoE−/− mice was ≈10 mmol/L and that the majority of cholesterol was associated with VLDL-sized fractions (Figure 1A, 1F, and 1G). However, daily treatment of SR-B1−/−/apoE−/− mice with rosuvastatin, beginning at age of 3 weeks and lasting 2 weeks, increased plasma total cholesterol by 33% (Figure 1A). Rosuvastatin treatment did not correct the abnormally high free cholesterol:total cholesterol ratio in SR-B1−/−/apoE−/− mice (Figure 1A). Rosuvastatin treatment increased the plasma levels of apoB48 (apolipoprotein B-48), but not apoB100 (which was present at much lower levels than apoB48) or apoA1 in SR-B1−/−/apoE−/− mice (Figure 1B through 1E). The lipoprotein cholesterol profiles of saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice revealed that rosuvastatin treatment increased cholesterol (Figure 1F and 1G) and apoB48 levels (Figure 1H and 1I) associated with VLDL-sized particles without substantially altering levels of cholesterol or apoB48 associated with IDL/LDL-sized, or cholesterol associated with HDL-sized lipoproteins. Rosuvastatin treatment did not significantly alter levels of apoA1 detected by immunoblotting (Figure 1H and 1J), or apoB100 detected by ELISA in (Figure 1K) in pools of fractions associated with VLDL-sized or IDL/LDL-sized lipoproteins. These results suggest that rosuvastatin treatment resulted in increases in the amount of apoB48 containing lipoproteins that migrated in the VLDL-size range in SR-B1−/−/apoE−/− mice. ApoB48-containing lipoproteins possess only a single molecule of apoB48 per particle.41 Therefore, we interpret this increase in cholesterol and apoB48 protein in the VLDL-sized lipoprotein fraction in rosuvastatin-treated SR-B1−/−/apoE−/− mice as most likely indicating that rosuvastatin treatment increased the concentration of apoB48-containing lipoprotein particles in this size range. However, because we did not separate apoB48-containing from apoA1-containing lipoproteins in the VLDL-sized fractions, we cannot rule out the possibility that rosuvastatin treatment may also have increased cholesterol levels in apoA1-containing (but apoB-free) lipoproteins within the VLDL-sized fraction. The mechanism by which rosuvastatin treatment resulted in increased apoB48 and cholesterol associated with VLDL-sized lipoproteins is currently unclear, but does not seem to be caused by altered levels of known apoB48 receptors (Figure I in the online-only Data Supplement). In humans, statin treatment leads to increased expression of LDLR in liver, promoting the clearance of LDL from blood.42 In contrast, rosuvastatin treatment of SR-B1−/−/apoE−/− mice resulted in an ≈2-fold reduction in LDLR protein levels in liver membrane extracts (Figure 1K and 1L) despite ≈2-fold increased levels of LDLR message in livers detected by RT-PCR (reverse transcription-polymerase chain reaction; Figure 1M). A similar 2-fold increase in PCSK9 mRNA (Figure 1N) in livers and a 60% increase in PCSK9 protein in plasma (Figure 1O) was detected in SR-B1−/−/apoE−/− mice treated with rosuvastatin. These changes in LDLR and PCSK9 message and protein in rosuvastatin-treated SR-B1−/−/apoE−/− mice are consistent with the known coordinate upregulation of LDLR and PCSK9 gene expression by inhibition of cholesterol biosynthesis, and the ability of PCSK9 itself, secreted by the liver, to promote the degradation of LDLR in hepatocytes43,44 (though we noted no differences in the levels of another reported target of PCSK9, LRP-145; Figure I in the online-only Data Supplement). It is not clear if these alterations in PCSK9 or hepatic LDLR protein contribute to the observed increases in apoB48, which is not a ligand of LDLR. Further research is required to understand the mechanisms by which rosuvastatin treatment resulted in increased levels of apoB48 and cholesterol in VLDL-sized lipoproteins in SR-B1−/−/apoE−/− mice.
Figure 1.
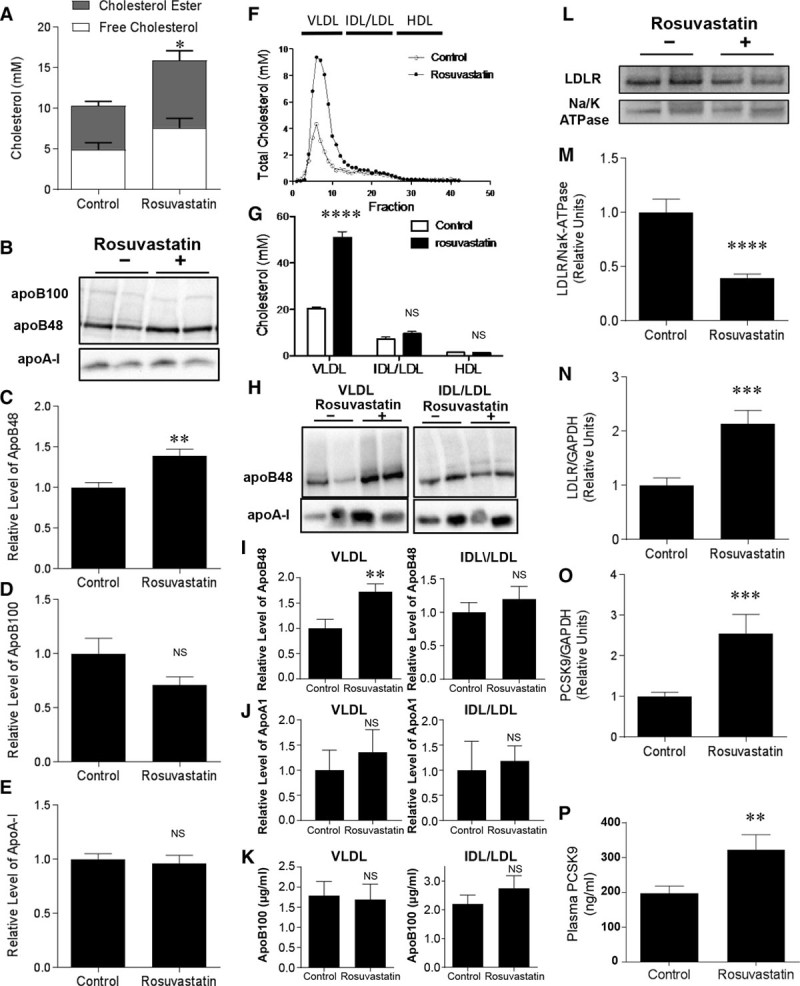
Effects of rosuvastatin on plasma lipoprotein cholesterol in SR-B1−/−/apoE−/− mice. A, Free cholesterol (white bars) and cholesterol ester (gray bars) in plasma samples from control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice (n=10, 10). B, Representative immunoblot of plasma apoB100 (500 kD), apoB48 (250 kD), and apoA1 (25 kD) from control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice. C through E, Quantification of relative amounts of apoB48 (C; n=8, 9), apoB100 (D; n=8, 9), and apoA1 (E; n=5, 4) in plasma from control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice. F, Plasma lipoproteins were fractionated by FPLC (fast protein liquid chromatography), and total cholesterol in each fraction was determined. Representative profiles from control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice are shown. G, Total cholesterol in pooled fractions corresponding to VLDL-, IDL/LDL- and HDL-sized lipoproteins from control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice. (n=6, 6). H, Representative images of immunoblotting of apoB48 and apoA1 in pooled fractions corresponding to VLDL- and IDL/LDL-sized lipoproteins from control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice. I and J, Quantification of relative amounts of apoB48 (I) and apoA1 (J) in pooled fractions corresponding to VLDL- and IDL/LDL-sized lipoproteins from control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice (n=6, 6). K, ELISA measurement of apoB100 in pooled fractions corresponding to VLDL- and IDL/LDL-sized lipoproteins from control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice (n=5, 8). L and M, Representative immunoblot (L) of LDLR and Na/K-ATPase and quantification (M) of relative LDLR protein levels in liver total membranes from control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice (n=8, 9). N and O, RT-PCR (reverse transcription-polymerase chain reaction) analysis of hepatic LDLR (N) and PCSK9 (proprotein convertase subtilisin/kexin type-9; O) transcript levels in control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice (n=9, 10). Transcript levels are expressed as fold change relative to control saline-treated SR-B1−/−/apoE−/− mice and were normalized to GAPDH mRNA. P, PCSK9 protein levels in plasma from control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice (n=12, 10). Mann–Whitney rank sum test was used for all data in except for G, which was analyzed by 2-way ANOVA. *P<0.05, **P<0.02, ***P<0.002, ****P<0.0002, and NS, not statistically significant vs control. apoE indicates apolipoprotein E; HDL, high-density lipoprotein; FPLC, fast protein liquid chromatography; IDL, intermediate-density lipoprotein; LDL, low-density lipoprotein; LDLR, LDL receptor; SR-B1, scavenger receptor class B type 1; and VLDL, very-low–density lipoprotein.
Rosuvastatin Attenuated Atherosclerosis and Cardiac Pathology in SR-B1−/−/ApoE−/− Mice
Saline-treated SR-B1−/−/apoE−/− mice developed substantial atherosclerosis in the aortic sinus (Figure 2A) and coronary arteries (Figure 2D through 2H) by 5 weeks of age, consistent with previous reports.25–29 Despite the increase in lipoprotein cholesterol levels, atherosclerosis was substantially reduced in both the aortic sinus (Figure 2B and 2C) and in coronary arteries (Figure 2I) of rosuvastatin-treated compared with saline-treated SR-B1−/−/apoE−/− mice. Rosuvastatin treatment resulted in an ≈44% reduction in the cross-sectional areas of atherosclerotic plaques in the aortic sinus (Figure 2C; Figure IIA in the online-only Data Supplement). The majority of coronary arteries that developed atherosclerosis in SR-B1−/−/apoE−/− mice were in the upper portion of the heart, more proximal to the aorta (data not shown), which is consistent with the concept that atherosclerosis is a disease involving large- and medium-sized arteries.46 Of coronary arteries observed in this region of hearts from saline-treated SR-B1−/−/apoE−/− mice at 5 weeks of age, 60% exhibited no lipid accumulation (Figure 2D) or only fatty streaks (Figure 2E), whereas ≈40% exhibited raised or completely occluding atherosclerotic plaques (Figure 2F through 2H). However, SR-B1−/−/apoE−/− mice treated with rosuvastatin exhibited substantially fewer atherosclerotic coronary arteries (mean of 14% of total coronary arteries observed; Figure 2I; Figure IIB in the online-only Data Supplement).
Figure 2.
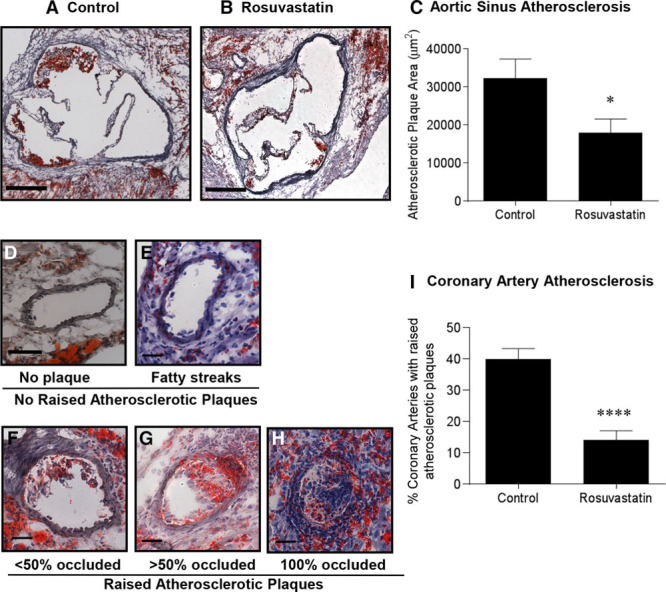
Treatment of SR-B1−/−/apoE−/− mice with rosuvastatin reduced atherosclerosis development in the aortic sinus and coronary arteries. Representative images of oil red O/hematoxylin-stained cross sections of aortic sinus from (A) control saline-treated, and (B) rosuvastatin-treated SR-B1−/−/apoE−/− mice (scale bar=200 µm). C, Quantification of aortic sinus atherosclerotic plaque size (n=10, 10). D through H, Representative sections of oil red O/hematoxylin-stained coronary arteries with various extents of atherosclerosis (D, no plaque; E, fatty streaks; F, <50% occluded; G, >50% occluded; and H, 100% occluded). Scale bar=50 µm. I, Quantification of the percentage of coronary arteries/section with raised atherosclerotic plaques in saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice (n=10, 10). Data were analyzed by the Mann–Whitney rank sum test. *P<0.05 and ****P<0.0001. apoE indicates apolipoprotein E; and SR-B1, scavenger receptor class B type 1.
We reported previously that SR-B1−/−/LDLR−/− mice fed a high-fat, high-cholesterol, cholate-containing diet rapidly developed occlusive coronary artery atherosclerosis and that a substantial percentage of atherosclerotic coronary arteries in these mice were characterized by abundant staining for CD41, a marker of platelet activation, suggesting that coronary artery disease development in these mice may involve thrombus formation in addition to atherosclerosis.47 To determine if similar processes are involved in the development of coronary artery disease in the SR-B1−/−/apoE−/− mice and if they were modulated by rosuvastatin treatment, we performed immunostaining using an antibody to activated CD61/CD41 on platelets in sections of atherosclerotic coronary arteries of saline- and rosuvastatin-treated SR-B1−/−/apoE−/− mice (Figure 3). This revealed that a substantial proportion (50%) of atherosclerotic coronary arteries in 5-week-old SR-B1−/−/apoE−/− mice were positive for CD41 staining (Figure 3J). Both the proportion of atherosclerotic coronary arteries that were positive for CD41 staining (Figure 3J), and the average extent of CD41 staining in atherosclerotic coronary arteries (Figure 3K) were substantially reduced in 5-week-old SR-B1−/−/apoE−/− mice that had been treated for 2 weeks with rosuvastatin. This was observed both for coronary arteries that were virtually completely occluded by atherosclerotic plaques (Figure 3D, 3E, 3H, and 3I) as well as coronary arteries that contained raised atherosclerotic plaques that occluded <50% of the artery lumen (Figure 3A, 3B, 3F, and 3G). In contrast, we detected only 2 instances of atherosclerotic plaques in the aortic sinus that showed CD41-positive staining (not shown), out of ≈27 aortic sinus sections analyzed. Platelets (analyzed by flow cytometry; Figure III in the online-only Data Supplement) were reduced in blood from SR-B1−/−/apoE−/− (double knockout [dKO]) mice compared with those from control apoE−/− single knockout (sKO) mice (Figure 3L). On the other hand platelets from SR-B1−/−/apoE−/− (dKO) mice exhibited higher levels of cell surface P-selectin (Figure 3M) and GPIIbIIIa (PE-JON/A [phycoerythrin-labeled JON/A] binding; Figure 3N) but no differences in cell surface CD36 (Figure IV in the online-only Data Supplement) compared with those from apoE−/− (sKO) mice. This is consistent with reports of thrombocytopenia in SR-B1 sKO mice,48–50 despite increased basal activation of circulating platelets50 and increased susceptibility of SR-B1 sKO mice to arterial50 and deep vein thrombosis.48 Treatment of SR-B1−/−/apoE−/− (dKO) mice with rosuvastatin did not seem to affect either platelet counts in blood or markers of activation under basal conditions (Figure 3L through 3N; Figures III and IV in the online-only Data Supplement). Together, these data suggest that in addition to reducing the degree of coronary artery atherosclerosis, rosuvastatin treatment reduced the accumulation of activated platelets in coronary artery atherosclerotic plaques in SR-B1−/−/apoE−/− mice.
Figure 3.
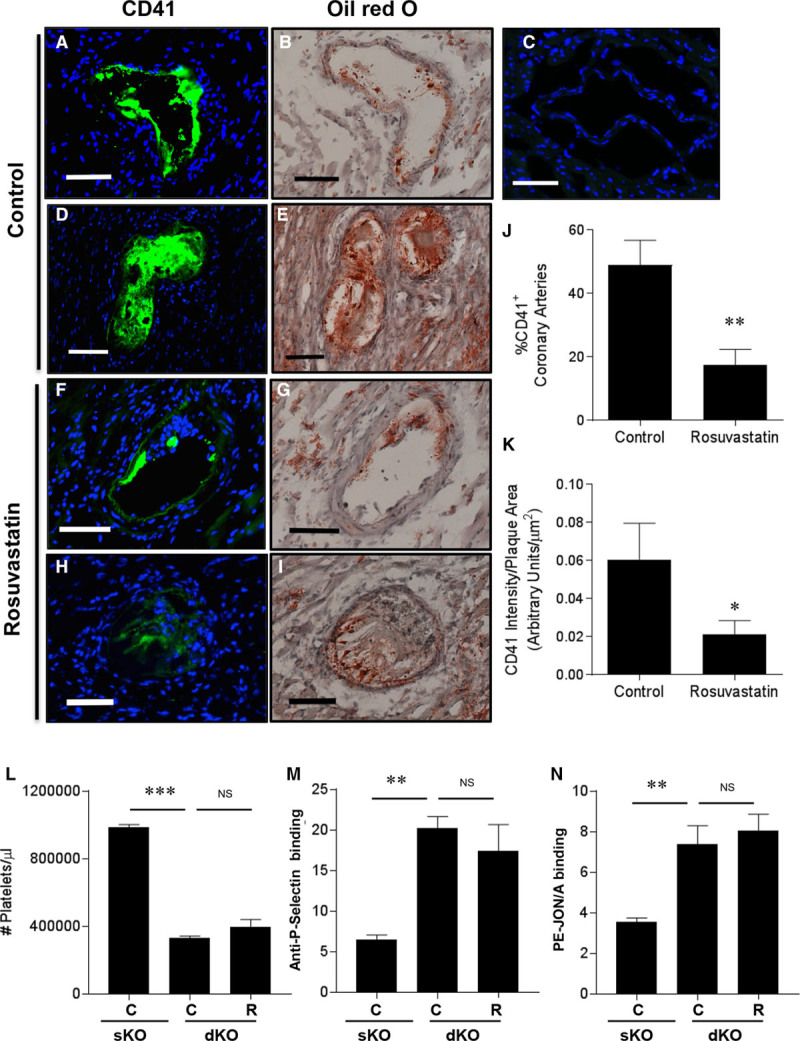
Rosuvastatin treatment reduced CD41 immunostaining in coronary arteries of SR-B1−/−/apoE−/− mice. Indirect immunofluorescence staining (green) for CD41, a marker of activated platelets, is shown in A, D, F, and H; (C) no primary antibody control (nuclear DNA stained blue with DAPI). Oil red O/hematoxylin-stained adjacent sections are shown in B, D, F, and H. A and B, Partially and (D and E) completely occluded coronary arteries from control saline-treated SR-B1−/−/apoE−/− mice. F and G, Partially and (H and I) completely occluded coronary arteries from rosuvastatin-treated SR-B1−/−/apoE−/− mice. Scale bar=50 µm. J, The percentage of CD41-positive atherosclerotic coronary arteries and (K) the intensity of CD41 staining (normalized to plaque area) in positive coronary arteries from control saline- (n=13) and rosuvastatin-treated (n=14) SR-B1−/−/apoE−/− mice. L, Circulating platelet counts, (M) anti-P-selectin antibody binding, or (N) PE-JON/A (phycoerythrin-labeled JON/A; anti-GPIIbIIIa) antibody binding to platelets from control apoE−/− (sKO) or control saline-treated or rosuvastatin-treated SR-B1−/−/apoE−/− (dKO) mice, detected by flow cytometry. Data in M and N are expressed as arbitrary fluorescence units. Data in J and K were analyzed by the Mann–Whitney rank sum test. Data in L through N were analyzed by 1-way ANOVA with the Kruskal–Wallis nonparametric test. *P=0.03, **P<0.01, *** P<0.002, and NS, not statistically significantly different vs control apoE−/− (sKO) or control saline-treated SR-B1−/−/apoE−/− (dKO) mice. apoE indicates apolipoprotein E; C, control saline-treated SR-B1−/−/apoE−/− (dKO) mice; dKO, double knockout; R, rosuvastatin-treated SR-B1−/−/apoE−/− (dKO) mice; sKO, single knockout; and SR-B1, scavenger receptor class B type 1.
Coronary artery atherosclerosis in SR-B1−/−/apoE−/− mice is accompanied by myocardial fibrosis and cardiomegaly.25–29 Consistent with previous reports,25,26,28,29 we found that hearts from 5-week-old SR-B1−/−/apoE−/− mice exhibit extensive collagen deposition, as indicated by blue Masson trichrome staining (healthy myocardium stains red; Figure 4A). Collagen deposition was substantially reduced in hearts from rosuvastatin-treated SR-B1−/−/apoE−/− mice (Figure 4B and 4C). This suggests that by 5 weeks of age, 2-week rosuvastatin treatment reduced cardiac fibrosis over the course of disease development in the SR-B1−/−/apoE−/− mice.
Figure 4.
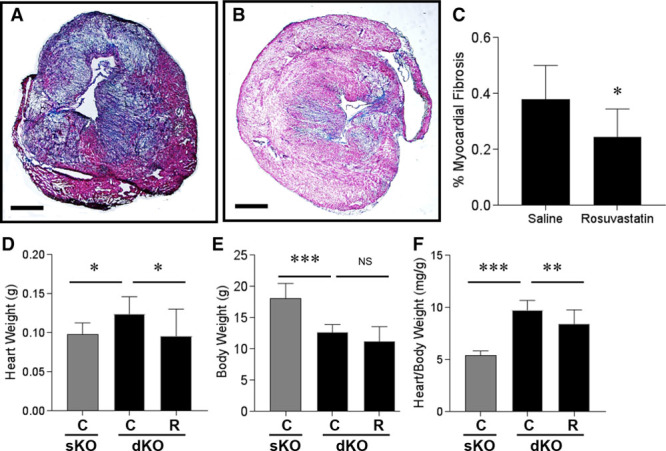
Rosuvastatin treatment attenuated myocardial fibrosis and cardiomegaly in SR-B1−/−/apoE−/− mice. A and B, Representative images of trichrome-stained transverse sections of upper portions of hearts from control saline- and rosuvastatin-treated SR-B1−/−/apoE−/− mice (scale bar=800 µm). Collagen stains blue and healthy myocardium stains red. C, The fibrotic area relative to the total cross-sectional area of the myocardium was quantified in each section for n=10 mice per group. D, Heart weights, (E) body weights, and (F) heart:body weight ratios, for control saline- or rosuvastatin-treated SR-B1−/−/apoE−/− mice (n=10, 10). Heart weights, body weights, and heart:body weight ratios of saline-treated apoE−/− mice (n=10) are shown for reference (gray bars). Data in C was analyzed by the Mann–Whitney rank sum test. Data in D did not pass the statistical test for normality and were analyzed by the Kruskal–Wallis 1-way ANOVA on ranks and Tukey post hoc test. Data in E and F passed statistical tests for normality and equal variance and were analyzed by 1-way ANOVA and Holm–Sidak post hoc test. *P<0.05, **P<0.01, *** P<0.0001, and NS, not statistically significantly different. apoE indicates apolipoprotein E; and SR-B1, scavenger receptor class B type 1.
At 5 weeks of age, SR-B1−/−/apoE−/− mice exhibited significantly greater heart weights and lower body weights than saline-treated apoE−/− littermates, corresponding to significantly higher heart:body weight ratios (Figure 4D through 4F), which is consistent with previous studies.25,26 Rosuvastatin treatment did not affect the body weights but significantly reduced heart weights and heart:body weight ratios in SR-B1−/−/apoE−/− mice, compared with saline treatment (Figure 4D through 4F).
Rosuvastatin Treatment Reduced Levels of Oxidized Phospholipids in Atherosclerotic Plaques
To investigate potential mechanisms involved in the antiatherogenic effects of rosuvastatin in the face of increased lipoprotein cholesterol levels in SR-B1−/−/apoE−/− mice, we analyzed levels of oxidized phospholipids in atherosclerotic plaques in the aortic sinus and coronary arteries of control saline- and rosuvastatin-treated mice. Indirect immunofluorescence staining using the E06 monoclonal antibody (mAb), which detects oxidized phospholipids,51,52 revealed substantial immunostaining in the aortic sinus in both atherosclerotic plaques and in regions of the artery wall that were devoid of atherosclerosis (Figure 5A and 5B) and in coronary arteries (Figure 5F and 5G) of saline-treated control SR-B1−/−/apoE−/− mice. Reduced levels of E06 immunoreactivity were observed in both the aortic sinus (Figure 5C through 5E) and coronary arteries (Figure 5H through 5J) of rosuvastatin-treated SR-B1−/−/apoE−/− mice. Thus, rosuvastatin appeared to reduce levels of E06 mAb-detectable oxidized phospholipids in the walls of arteries in SR-B1−/−/apoE−/− mice. On the other hand, we did not detect differences in the levels of oxLDL detected in plasma of control SR-B1−/−/apoE−/− versus control apoE−/− (sKO) mice or as a result of rosuvastatin treatment of SR-B1−/−/apoE−/− (dKO) mice (Figure 5M). Therefore, rosuvastatin treatment did not seem to alter plasma oxLDL levels, at least in the majority of the SR-B1−/−/apoE−/− mice analyzed, despite the increased plasma total cholesterol and apoB48 levels (Figure 1), but consistent with unaltered levels of cholesterol and apoB48/100 associated with LDL-sized lipoproteins (Figure 1F through 1H). Plasma from control SR-B1−/−/apoE−/− (dKO) mice did, however, have a 45-fold increased mean level of the acute phase inflammatory marker, SAA1 (serum amyloid A1) compared with plasma from control apoE−/− (sKO) mice (Figure 5N), reminiscent of our previous reports of increased plasma interleukin-6 (IL-6) and tumor necrosis factor-α (TNF-α) levels in atherogenic diet-fed SR-B1−/−/LDLR−/− versus LDLR−/− control mice. On the other hand, rosuvastatin treatment did not significantly alter the plasma SAA1 levels in SR-B1−/−/apoE−/− mice (Figure 5N). Similarly, we saw no differences in the proportions of circulating CD3+ (T-), B220+ (B-), or CD11b+ (myeloid) cells from control or rosuvastatin-treated SR-B1−/−/apoE−/− mice (Figure V in the online-only Data Supplement). Therefore, rosuvastatin treatment of SR-B1−/−/apoE−/− mice reduced levels of E06-mAb detectable oxidized phospholipids in atherosclerotic arteries even though oxLDL levels and markers of systemic inflammation in plasma were not altered.
Figure 5.
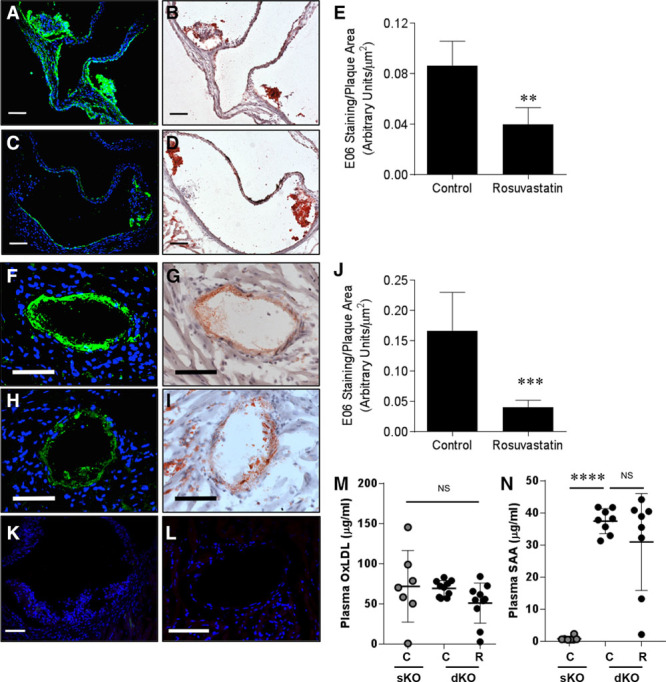
Effects of rosuvastatin on oxidized phospholipids in atherosclerotic plaques of SR-B1−/−/apoE−/− mice. E06 mAb immunostaining for oxidized phospholipids (A, C, F, H) and oil red O/hematoxylin staining (B, D, G, I) in atherosclerotic plaques in the aortic sinus (A through D) and coronary arteries (F through L). Representative images are shown. Green, E06 mAb immunostaining; blue, DAPI counterstaining. Representative negative controls lacking the E06 mAb are shown in K (aortic sinus) and L (coronary artery). Scale bars=50 µm. The intensity of E06 mAb immunostaining was quantified and is expressed normalized to the atherosclerotic plaque area for aortic sinus sections (E) and coronary arteries (J) from control saline-treated (n=13) and rosuvastatin-treated (n=14) SR-B1−/−/apoE−/− mice. M, Plasma oxLDL (n=7, 10, 10) and (N) SAA levels (n=8 per group) as measured by ELISA from control apoE−/− (sKO) and control saline- and rosuvastatin-treated SR-B1−/−/apoE−/− (dKO) mice. Data in E and J were analyzed by the Mann–Whitney rank sum test. Data in M and N were analyzed by the Kruskal–Wallis 1-way ANOVA on ranks with Tukey post hoc test. **P<0.01, ***P<0.001, ****P<0.0001, and NS, not statistically significantly different. apoE indicates apolipoprotein E; dKO, double knockout; oxLDL, oxidized low-density lipoprotein; SAA, serum amyloid A; sKO, single knockout; and SR-B1, scavenger receptor class B type 1.
Rosuvastatin Treatment of Macrophages Decreases CD36 Expression and Foam Cell Formation
To test effects of rosuvastatin on phenotypes of macrophages from SR-B1−/−/apoE−/− mice, bone marrow prepared from SR-B1−/−/apoE−/− mice was differentiated into macrophages in culture and treated with or without rosuvastatin. Rosuvastatin treatment did not affect the induction of IL-6, monocyte chemotactic protein-1, or IL-1β (markers of M1 macrophage polarization) on lipopolysaccharide stimulation, or arginase-1, mannose receptor or Fizz-1 (markers of M2 macrophage polarization) on IL-4 stimulation (Figure VI in the online-only Data Supplement). On the other hand, rosuvastatin treatment protected cultured macrophages from apoptosis induced by the ER stressor, thapsigargin (Figure VII in the online-only Data Supplement). Neither rosuvastatin, oxLDL, nor the combination of rosuvastatin and oxLDL significantly affected the levels of SR-A (scavenger receptor class A) transcripts as detected by RT-PCR (Figure 6A). On the other hand, oxLDL substantially increased CD36 transcripts in bone marrow-derived macrophages from SR-B1−/−/apoE−/− mice (Figure 6B), consistent with previous reports that oxLDL upregulated CD36 expression in macrophages.53–56 Rosuvastatin reduced CD36 gene expression in oxLDL-treated but not untreated cells (Figure 6B). Similarly, rosuvastatin treatment of bone marrow-derived macrophages exposed to oxLDL substantially reduced CD36 protein levels as detected by flow cytometry (Figure 6C through 6E) and indirect immunofluorescence staining (Figure 6F through 6I). To examine the effects of rosuvastatin on foam cell formation, cultured macrophages, treated with oxLDL with or without rosuvastatin, were stained for neutral lipid droplets with oil red O (Figure 6J through 6M). As expected, oxLDL treatment of cultured bone marrow-derived macrophages resulted in substantial formation of intracellular lipid droplets (Figure 6K). In contrast, coincubation with rosuvastatin significantly suppressed lipid accumulation in macrophages (Figure 6L and 6M). Together, these results demonstrate that rosuvastatin suppresses CD36 expression and oxLDL-driven foam cell formation.
Figure 6.
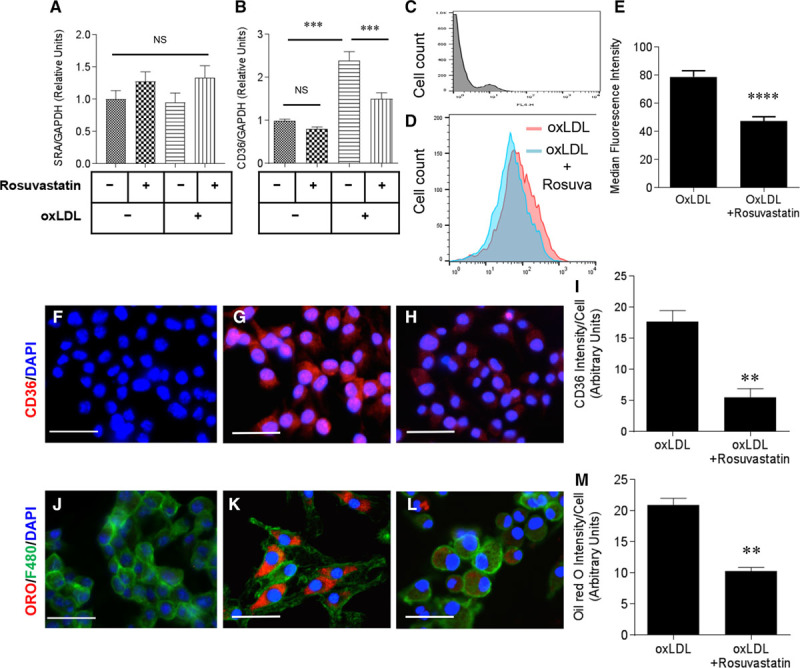
Rosuvastatin reduces cluster of differentiation (CD)36 levels and foam cell formation in macrophages. Bone marrow-derived macrophages were prepared from SR-B1−/−/apoE−/− mice and treated in culture with or without oxLDL (oxidized low-density lipoprotein; 100 µg protein/mL) and rosuvastatin (10 µmol\L) as indicated. A and B, Levels of SR-A (scavenger receptor class A) and CD36 mRNA after 24 h of treatment as indicated. Data are expressed as fold change relative to untreated cells and are normalized to levels of GAPDH mRNA. Cells from 3 mice were analyzed. C through E, CD36 cell surface levels detected by flow cytometry in bone marrow-derived macrophages 24 h after treatment. C, Negative control lacking anti-CD36 primary antibody. D, Representative flow cytometry histograms of cell count vs CD36 fluorescence for bone marrow-derived macrophages treated with oxLDL, with (blue) or without rosuvastatin (pink). E, Median fluorescence intensity for cell surface CD36 analyzed by flow cytometry for bone marrow-derived SR-B1−/−/apoE−/− macrophages treated with oxLDL or both oxLDL and rosuvastatin for 24 h as above (n=10 per group). F through H, Immunofluorescence staining of bone marrow-derived macrophages treated with oxLDL in the absence (G) or presence (H) of rosuvastatin as above (CD36, red; DAPI-stained nuclei, blue). F, Negative control staining lacking the anti-CD36 primary antibody. I, Quantification of the intensity of CD36 immunostaining/cell. J through L, Oil red O staining of bone marrow-derived macrophages treated with oxLDL in the absence (K) or presence (L) of rosuvastatin (oil red O, red; F4/80, green; nuclei, blue). J, Negative control staining lacking oil red O. Scale bars=20 µm (F through H and J through L). M, The level of oil red O staining per cell. Data in I and M represent 3 independent experiments and were confirmed with macrophages derived from bone marrow cells harvested from 2 additional mice. Data in A and B were analyzed by 2-way ANOVA with the Holm–Sidak post test. Data in E, I, and M passed the Shapiro–Wilk test for normality and were analyzed by Student t test. ****P<0.0001, ***P<0.001, **P<0.01, and NS, not statistically significantly different. apoE indicates apolipoprotein E; and SR-B1, scavenger receptor class B type 1.
Discussion
SR-B1−/−/apoE−/− mice do not begin to develop aortic sinus or coronary artery atherosclerosis until after 3 weeks of age.25–28 However, they rapidly develop extensive coronary atherosclerosis, myocardial infarction, and cardiac enlargement by 5 weeks of age when fed a normal chow diet.25–28 Eventually SR-B1−/−/apoE−/− mice die between 6 and 9 weeks of age.25–29 To examine the effects of rosuvastatin treatment on the development of atherosclerosis in the aortic sinus and coronary arteries of these mice, we initiated treatment in mice at 3 weeks of age and euthanized mice at 5 weeks of age, after 2 weeks of daily treatment. By analyzing mice at 5 weeks of age, that is, before onset of spontaneous death, we avoided potential confounding effects of the rapid decline in the health of mice.
Two-week treatment with rosuvastatin resulted in substantial reductions in the development of atherosclerosis in the aortic sinus and in coronary arteries of SR-B1−/−/apoE−/− mice (Figure 2). Despite lower numbers of circulating platelets in SR-B1−/−apoE−/− mice48–50 (Figure 3L) and perhaps consistent with their higher basal activation state compared with those from apoE−/− mice (Figure 3M and 3N), a significant proportion of atherosclerotic plaques in coronary arteries of SR-B1−/−/apoE−/− mice exhibited positive staining for CD41 (Figure 3A through 3K), a marker of activated platelets. On the other hand, CD41 staining was seen only very infrequently in atherosclerotic plaques in the aortic sinus (not shown). This suggests the development of platelet-rich thrombi in atherosclerotic plaques in the coronary arteries of SR-B1−/−/apoE−/− mice and is consistent with similar observations in SR-B1−/−/LDLR−/− mice in which coronary artery atherosclerosis was induced to develop rapidly (within 3 weeks) by feeding the mice a highly atherogenic diet.47 Although statins, including rosuvastatin, have been reported to inhibit platelet activation and thrombosis, independent of lipid-lowering effects,10,42 we did not detect any reduction in basal activation of platelets, or alterations in circulating platelet numbers from SR-B1−/−/apoE−/− mice treated with rosuvastatin compared with those treated with saline (Figure 3L through 3N). Despite this, we observed that fewer atherosclerotic coronary arteries stained positively for CD41 and that in those that were positive, the extent of CD41 staining was reduced in mice treated for 2 weeks with rosuvastatin compared with saline-treated mice (Figure 3A through 3K). This may suggest that rosuvastatin treatment altered properties of the atherosclerotic coronary arteries which reduced the accumulation, retention, and activation of platelets. For example, oxidized phospholipids have been reported to activate platelets and promote thrombosis by binding to CD36 on the platelet surface.57,58 We observed no differences in levels of cell surface CD36 on platelets (Figure IV in the online-only Data Supplement), but did detect reductions in oxidized phospholipids (using the E06 mAb) in atherosclerotic coronary arteries from rosuvastatin- compared with saline-treated SR-B1−/−apoE−/− mice (Figure 5A through 5J). Furthermore, apoptosis of macrophages has been implicated in the destabilization of advanced atherosclerotic plaques by contributing to the development of large necrotic cores.59–62 Rosuvastatin treatment of macrophages in culture protected them against apoptosis (Figure VII in the online-only Data Supplement). Whether rosuvastatin treatment protected vascular cells against cell death and consequently stabilized atherosclerotic plaques in vivo, however, remains to be determined. Further experiments are required to determine the key alterations in the artery wall triggered by rosuvastatin that resulted in the observed reductions in the accumulation and retention of activated platelets in atherosclerotic coronary arteries of SR-B1−/−/apoE−/− mice.
A consequence of coronary artery atherosclerosis in SR-B1−/−/apoE−/− mice is the development of myocardial infarction, characterized by extensive cardiac fibrosis and cardiac enlargement. Treatment with rosuvastatin substantially reduced the extent of cardiac fibrosis in SR-B1−/−/apoE−/− mice (Figure 4A through 4C) and attenuated the pathological increase in heart weights of SR-B1−/−/apoE−/− mice (Figure 4D through 4F). These protective effects of rosuvastatin on cardiac morphology are most likely secondary to its effects on coronary artery atherosclerosis and possibly platelet accumulation in atherosclerotic coronary arteries, although we cannot rule out the possibility that rosuvastatin treatment may have also protected cardiomyocytes directly against ischemia-induced cell death and prevented the fibrotic response, as has been proposed by others.63
To understand the mechanisms by which rosuvastatin treatment reduced atherosclerosis development in SR-B1−/−/apoE−/− mice, we examined the effects of rosuvastatin treatment on plasma and lipoprotein total and unesterified cholesterol levels. Various studies have reported different effects of treatment with statins, including rosuvastatin, on plasma and lipoprotein cholesterol levels in apoE−/− and other mice.6–10,12,17–23,64 For example, some reported reductions in plasma cholesterol levels,7,8,64 whereas others reported unaltered plasma cholesterol and triglyceride levels6,9,10,12,17–20,65,66 in apoE−/− and other mice treated with rosuvastatin. Still other studies reported that treatment of apoE−/− mice with other statins, including simvastatin, resulted in increased plasma cholesterol levels, in some cases accompanied by increased atherosclerosis.21–23 The effects of statin treatment on steady state lipoprotein levels in SR-B1−/−/apoE−/− mice had, thus far, not been reported.
As expected for statin treatment, we detected increased levels of both LDLR and PCSK9 transcripts in livers of rosuvastatin-treated SR-B1−/−/apoE−/− mice (Figure 1N and 1O), consistent with the known effects of statin-induced inhibition of cholesterol biosynthesis and activation of the SREBP-2 (sterol regulatory element-binding protein 2) transcription factor in the liver.43 However, although we observed a corresponding increase in concentration of PCSK9 protein in plasma (Figure 1P), we detected a >50% reduction in the amount of LDLR protein in liver membranes from the same rosuvastatin-treated SR-B1−/−/apoE−/− mice (Figure 1L and 1M). Consistent with our observations, it has been reported previously that treatment of wild-type mice with lovastatin increased PCSK9 protein while reducing LDLR protein in livers67 and that treatment of hyperlipidemic mice with rosuvastatin increased PCSK9 mRNA in liver and protein in plasma.68 Similar effects of different statins on PCSK9 expression have been reported in other model organisms (eg, hamsters) and in humans.69–71 PCSK9 that is either secreted by hepatocytes, or acting intracellularly, can bind to the LDLR and trigger its degradation, reducing LDLR protein levels thereby attenuating the effects of SREBP-2-mediated upregulation of LDLR gene expression, and attenuating statin dependent cholesterol lowering.43 Rosuvastatin substantially increased levels of cholesterol and apoB48 (but not the much less abundant apoB100) in plasma and associated with VLDL-sized lipoprotein particles of SR-B1−/−/apoE−/− mice (Figure 1A through 1J). This is surprising because in the absence of apoE, apoB48 containing lipoproteins are likely not cleared by LDLR, and the levels of apoB48 receptors (LRP-1 and apoB48 receptor72,73) were not altered by rosuvastatin (Figure I in the online-only Data Supplement). The basis for this apparently unique response of SR-B1−/−/apoE−/− mice to rosuvastatin is currently unclear, but may involve the unusual lipid composition/structure of these lipoproteins28,29 which may influence their clearance (or production) in unanticipated ways. Further research will be required to address this.
We observed reduced atherosclerosis in the aortic sinus and coronary arteries, and reduced platelet accumulation in atherosclerotic coronary arteries, accompanied by reduced cardiac fibrosis and attenuated cardiac enlargement in the SR-B1−/−/apoE−/− mice that had been treated with rosuvastatin. These beneficial effects occurred despite the substantially increased levels of apoB48 and total cholesterol in plasma and in fractions associated with VLDL-sized lipoproteins. Rosuvastatin treatment did not reduce markers of systemic inflammation or plasma oxLDL levels, but did reduce levels of oxidized phospholipids detected with the E06 mAb in both the aortic sinus and coronary arteries in SR-B1−/−/apoE−/− mice (Figure 5A through 5L), consistent with a previous report that rosuvastatin treatment reduced oxLDL accumulation in atherosclerotic plaques of obese hyperlipidemic mice.13
CD36 seems to be a major receptor for oxLDL in macrophages, mediating its uptake and subsequent foam cell formation.74 CD36 expression is also upregulated by oxLDL in macrophages.53–56 Rosuvastatin attenuated the oxLDL-mediated upregulation of CD36 message and reduced CD36 protein levels in oxLDL-treated bone marrow-derived macrophages prepared from SR-B1−/−/apoE−/− mice (Figure 6A through 6E). This is consistent with reports that pitavastatin downregulates CD36 expression by reducing PPAR-γ gene expression and increasing PPAR-γ protein phosphorylation.75 It is unknown, however, if rosuvastatin acts through the same mechanism. CD36-mediated uptake of oxLDL by macrophages contributes to oxLDL-driven foam cell formation.55 Consistent with the ability of rosuvastatin to suppress oxLDL-stimulated CD36 gene expression and to reduce CD36 protein levels, rosuvastatin also suppressed oxLDL-mediated foam cell formation in cultured bone marrow-derived macrophages from SR-B1−/−/apoE−/− mice (Figure 6F through 6H). The reduction of oxLDL-driven foam cell formation through downregulation of CD36, suggests a potential mechanism that may contribute to the reduced levels of oxidized phospholipids observed in atherosclerotic plaques and the reduced atherosclerosis seen in the aortic sinus and coronary arteries of SR-B1−/−apoE−/− mice treated with rosuvastatin. In turn the reduced coronary artery atherosclerosis, together with the attenuated platelet accumulation in atherosclerotic coronary arteries may contribute to the reductions in cardiac fibrosis and cardiac enlargement, in the face of increased plasma total cholesterol in rosuvastatin-treated SR-B1−/−/apoE−/− mice.
In conclusion, this is the first study to examine the effects of rosuvastatin treatment in a mouse model that exhibits multiple features of human CHD, including coronary artery atherosclerosis and platelet accumulation, myocardial fibrosis and cardiomegaly. Our findings that rosuvastatin treatment protected against coronary artery atherosclerosis, platelet accumulation in atherosclerotic coronary arteries, cardiac fibrosis, and cardiomegaly, even in the face of elevated cholesterol in plasma and VLDL-sized lipoproteins, suggests that rosuvastatin-mediated protection involved pathways independent of cholesterol lowering.
Acknowledgments
We thank Fatima Igdoura for technical assistance.
Sources of Funding
This research was supported by grants from AstraZeneca Inc (DC-990-0324) and the Canadian Institutes for Health Research (MOP74753) to B.L. Trigatti and from the Heart and Stroke Foundation of Canada (G-15-0009389) and the Canadian Institutes of Health Research (MOP286787) to R.C. Austin. P. Yu was supported by a graduate scholarship from the China Scholarship Council. R.C. Austin is a Career Investigator of the Heart and Stroke Foundation of Ontario and holds the Amgen Canada Research Chair in the Division of Nephrology at St. Joseph’s Healthcare and McMaster University.
Disclosures
This research was funded in part by a grant from AstraZeneca Inc.
Supplementary Material
Nonstandard Abbreviations and Acronyms
- Apo
- apolipoprotein
- CD
- cluster of differentiation
- CHD
- coronary heart disease
- dKO
- double knockout
- HDL
- high-density lipoprotein
- IDL
- intermediate-density lipoprotein
- IL
- interleukin
- LDL
- low-density lipoprotein
- LDLR
- LDL receptor
- mAb
- monoclonal antibody
- oxLDL
- oxidized LDL
- SAA
- serum amyloid A
- sKO
- single knockout
- SRA
- scavenger receptor class A
- SR-B1
- SR class B type 1
- SREBP-2
- sterol regulatory element-binding protein 2
- TNF
- tumor necrosis factor
- VLDL
- very-low–density lipoprotein
The online-only Data Supplement is available with this article at http://atvb.ahajournals.org/lookup/suppl/doi:10.1161/ATVBAHA.117.305140/-/DC1.
Highlights.
This is the first time that rosuvastatin has been tested in a mouse model of spontaneous coronary artery atherosclerosis involving platelet accumulation in atherosclerotic coronary artery plaques and cardiac fibrosis.
Rosuvastatin treatment significantly attenuated the development of atherosclerosis, accumulation of platelets in occluded coronary arteries, cardiac fibrosis, and cardiac enlargement in SR-B1−/−/apoE−/− mice.
These beneficial effects of rosuvastatin occurred despite increased plasma cholesterol levels and seem to involve reduced accumulation of oxidized phospholipids in the walls of affected arteries, and may have involved reduced macrophage foam cell formation.
These findings shed light on pathways by which rosuvastatin protects against coronary artery atherosclerosis and subsequent myocardial infarction independently of cholesterol lowering.
References
- 1.Tuttolomondo A, Di Raimondo D, Pecoraro R, Arnao V, Pinto A, Licata G. Atherosclerosis as an inflammatory disease. Curr Pharm Des. 2012;18:4266–4288. doi: 10.2174/138161212802481237. [DOI] [PubMed] [Google Scholar]
- 2.Maron DJ, Fazio S, Linton MF. Current perspectives on statins. Circulation. 2000;101:207–213. doi: 10.1161/01.cir.101.2.207. [DOI] [PubMed] [Google Scholar]
- 3.Davignon J. Beneficial cardiovascular pleiotropic effects of statins. Circulation. 2004;109(23 suppl 1):III39–III43. doi: 10.1161/01.CIR.0000131517.20177.5a. doi: 10.1161/01.CIR.0000131517.20177.5a. [DOI] [PubMed] [Google Scholar]
- 4.van der Meij E, Koning GG, Vriens PW, Peeters MF, Meijer CA, Kortekaas KE, Dalman RL, van Bockel JH, Hanemaaijer R, Kooistra T, Kleemann R, Lindeman JH. A clinical evaluation of statin pleiotropy: statins selectively and dose-dependently reduce vascular inflammation. PLoS One. 2013;8:e53882. doi: 10.1371/journal.pone.0053882. doi: 10.1371/journal.pone.0053882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cheng-Lai A. Rosuvastatin: a new HMG-CoA reductase inhibitor for the treatment of hypercholesterolemia. Heart Dis. 2003;5:72–78. doi: 10.1097/01.HDX.0000050417.89309.F8. doi: 10.1097/01.HDX.0000050417.89309.F8. [DOI] [PubMed] [Google Scholar]
- 6.Enomoto S, Sata M, Fukuda D, Nakamura K, Nagai R. Rosuvastatin prevents endothelial cell death and reduces atherosclerotic lesion formation in ApoE-deficient mice. Biomed Pharmacother. 2009;63:19–26. doi: 10.1016/j.biopha.2007.11.002. doi: 10.1016/j.biopha.2007.11.002. [DOI] [PubMed] [Google Scholar]
- 7.Grönros J, Wikström J, Brandt-Eliasson U, Forsberg GB, Behrendt M, Hansson GI, Gan LM. Effects of rosuvastatin on cardiovascular morphology and function in an ApoE-knockout mouse model of atherosclerosis. Am J Physiol Heart Circ Physiol. 2008;295:H2046–H2053. doi: 10.1152/ajpheart.00133.2008. doi: 10.1152/ajpheart.00133.2008. [DOI] [PubMed] [Google Scholar]
- 8.Li W, Asagami T, Matsushita H, Lee KH, Tsao PS. Rosuvastatin attenuates monocyte-endothelial cell interactions and vascular free radical production in hypercholesterolemic mice. J Pharmacol Exp Ther. 2005;313:557–562. doi: 10.1124/jpet.104.080002. doi: 10.1124/jpet.104.080002. [DOI] [PubMed] [Google Scholar]
- 9.Monetti M, Canavesi M, Camera M, Parente R, Paoletti R, Tremoli E, Corsini A, Bellosta S. Rosuvastatin displays anti-atherothrombotic and anti-inflammatory properties in apoE-deficient mice. Pharmacol Res. 2007;55:441–449. doi: 10.1016/j.phrs.2007.02.001. doi: 10.1016/j.phrs.2007.02.001. [DOI] [PubMed] [Google Scholar]
- 10.Schäfer A, Fraccarollo D, Eigenthaler M, Tas P, Firnschild A, Frantz S, Ertl G, Bauersachs J. Rosuvastatin reduces platelet activation in heart failure: role of NO bioavailability. Arterioscler Thromb Vasc Biol. 2005;25:1071–1077. doi: 10.1161/01.ATV.0000161926.43967.df. doi: 10.1161/01.ATV.0000161926.43967.df. [DOI] [PubMed] [Google Scholar]
- 11.Schäfer K, Kaiser K, Konstantinides S. Rosuvastatin exerts favourable effects on thrombosis and neointimal growth in a mouse model of endothelial injury. Thromb Haemost. 2005;93:145–152. doi: 10.1160/TH04-07-0415. doi: 10.1160/TH04-07-0415. [DOI] [PubMed] [Google Scholar]
- 12.Schroeter MR, Humboldt T, Schäfer K, Konstantinides S. Rosuvastatin reduces atherosclerotic lesions and promotes progenitor cell mobilisation and recruitment in apolipoprotein E knockout mice. Atherosclerosis. 2009;205:63–73. doi: 10.1016/j.atherosclerosis.2008.11.013. doi: 10.1016/j.atherosclerosis.2008.11.013. [DOI] [PubMed] [Google Scholar]
- 13.Verreth W, De Keyzer D, Davey PC, Geeraert B, Mertens A, Herregods MC, Smith G, Desjardins F, Balligand JL, Holvoet P. Rosuvastatin restores superoxide dismutase expression and inhibits accumulation of oxidized LDL in the aortic arch of obese dyslipidemic mice. Br J Pharmacol. 2007;151:347–355. doi: 10.1038/sj.bjp.0707231. doi: 10.1038/sj.bjp.0707231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Plump AS, Smith JD, Hayek T, Aalto-Setälä K, Walsh A, Verstuyft JG, Rubin EM, Breslow JL. Severe hypercholesterolemia and atherosclerosis in apolipoprotein E-deficient mice created by homologous recombination in ES cells. Cell. 1992;71:343–353. doi: 10.1016/0092-8674(92)90362-g. [DOI] [PubMed] [Google Scholar]
- 15.Zhang SH, Reddick RL, Piedrahita JA, Maeda N. Spontaneous hypercholesterolemia and arterial lesions in mice lacking apolipoprotein E. Science. 1992;258:468–471. doi: 10.1126/science.1411543. [DOI] [PubMed] [Google Scholar]
- 16.Nakashima Y, Plump AS, Raines EW, Breslow JL, Ross R. ApoE-deficient mice develop lesions of all phases of atherosclerosis throughout the arterial tree. Arterioscler Thromb. 1994;14:133–140. doi: 10.1161/01.atv.14.1.133. [DOI] [PubMed] [Google Scholar]
- 17.Giunti S, Calkin AC, Forbes JM, Allen TJ, Thomas MC, Cooper ME, Jandeleit-Dahm KA. The pleiotropic actions of rosuvastatin confer renal benefits in the diabetic Apo-E knockout mouse. Am J Physiol Renal Physiol. 2010;299:F528–F535. doi: 10.1152/ajprenal.00127.2010. doi: 10.1152/ajprenal.00127.2010. [DOI] [PubMed] [Google Scholar]
- 18.Osaka M, Hagita S, Yoshida M. In vivo imaging of leukocyte recruitment to the atheroprone femoral artery reveals anti-inflammatory effects of rosuvastatin. Biomed Res Int. 2013;2013:962369. doi: 10.1155/2013/962369. doi: 10.1155/2013/962369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Patterson KA, Zhang X, Wrobleski SK, Hawley AE, Lawrence DA, Wakefield TW, Myers DD, Diaz JA. Rosuvastatin reduced deep vein thrombosis in ApoE gene deleted mice with hyperlipidemia through non-lipid lowering effects. Thromb Res. 2013;131:268–276. doi: 10.1016/j.thromres.2012.12.006. doi: 10.1016/j.thromres.2012.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shimizu T, Miura S, Tanigawa H, Kuwano T, Zhang B, Uehara Y, Saku K. Rosuvastatin activates ATP-binding cassette transporter A1-dependent efflux ex vivo and promotes reverse cholesterol transport in macrophage cells in mice fed a high-fat diet. Arterioscler Thromb Vasc Biol. 2014;34:2246–2253. doi: 10.1161/ATVBAHA.114.303715. doi: 10.1161/ATVBAHA.114.303715. [DOI] [PubMed] [Google Scholar]
- 21.Cai Q, Du X, Zhou B, Cai C, Kermany MH, Zhang C, Yoo T. Effects of simvastatin on plasma lipoproteins and hearing loss in apolipoprotein E gene-deficient mice. ORL J Otorhinolaryngol Relat Spec. 2009;71:244–250. doi: 10.1159/000236014. doi: 10.1159/000236014. [DOI] [PubMed] [Google Scholar]
- 22.Quarfordt SH, Oswald B, Landis B, Xu HS, Zhang SH, Maeda N. In vivo cholesterol kinetics in apolipoprotein E-deficient and control mice. J Lipid Res. 1995;36:1227–1235. [PubMed] [Google Scholar]
- 23.Wang YX, Martin-McNulty B, Huw LY, da Cunha V, Post J, Hinchman J, Vergona R, Sullivan ME, Dole W, Kauser K. Anti-atherosclerotic effect of simvastatin depends on the presence of apolipoprotein E. Atherosclerosis. 2002;162:23–31. doi: 10.1016/s0021-9150(01)00678-5. [DOI] [PubMed] [Google Scholar]
- 24.Gonzalez L, Yu P, trigatti BL. Mouse models of coronary artery atherosclerosis. J Cardiovasc Disord. 2016;3:1021. [Google Scholar]
- 25.Braun A, Trigatti BL, Post MJ, Sato K, Simons M, Edelberg JM, Rosenberg RD, Schrenzel M, Krieger M. Loss of SR-BI expression leads to the early onset of occlusive atherosclerotic coronary artery disease, spontaneous myocardial infarctions, severe cardiac dysfunction, and premature death in apolipoprotein E-deficient mice. Circ Res. 2002;90:270–276. doi: 10.1161/hh0302.104462. [DOI] [PubMed] [Google Scholar]
- 26.Al-Jarallah A, Igdoura F, Zhang Y, Tenedero CB, White EJ, MacDonald ME, Igdoura SA, Trigatti BL. The effect of pomegranate extract on coronary artery atherosclerosis in SR-BI/APOE double knockout mice. Atherosclerosis. 2013;228:80–89. doi: 10.1016/j.atherosclerosis.2013.02.025. doi: 10.1016/j.atherosclerosis.2013.02.025. [DOI] [PubMed] [Google Scholar]
- 27.Braun A, Yesilaltay A, Acton S, Broschat KO, Krul ES, Napawan N, Stagliano N, Krieger M. Inhibition of intestinal absorption of cholesterol by ezetimibe or bile acids by SC-435 alters lipoprotein metabolism and extends the lifespan of SR-BI/apoE double knockout mice. Atherosclerosis. 2008;198:77–84. doi: 10.1016/j.atherosclerosis.2007.10.012. doi: 10.1016/j.atherosclerosis.2007.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Braun A, Zhang S, Miettinen HE, Ebrahim S, Holm TM, Vasile E, Post MJ, Yoerger DM, Picard MH, Krieger JL, Andrews NC, Simons M, Krieger M. Probucol prevents early coronary heart disease and death in the high-density lipoprotein receptor sr-bi/apolipoprotein e double knockout mouse. Proc Natl Acad Sci U S A. 2003;100:7283–7288. doi: 10.1073/pnas.1237725100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Karackattu SL, Trigatti B, Krieger M. Hepatic lipase deficiency delays atherosclerosis, myocardial infarction, and cardiac dysfunction and extends lifespan in SR-BI/apolipoprotein E double knockout mice. Arterioscler Thromb Vasc Biol. 2006;26:548–554. doi: 10.1161/01.ATV.0000202662.63876.02. doi: 10.1161/01.ATV.0000202662.63876.02. [DOI] [PubMed] [Google Scholar]
- 30.Rigotti A, Miettinen HE, Krieger M. The role of the high-density lipoprotein receptor SR-BI in the lipid metabolism of endocrine and other tissues. Endocr Rev. 2003;24:357–387. doi: 10.1210/er.2001-0037. doi: 10.1210/er.2001-0037. [DOI] [PubMed] [Google Scholar]
- 31.Rigotti A, Trigatti BL, Penman M, Rayburn H, Herz J, Krieger M. A targeted mutation in the murine gene encoding the high density lipoprotein (hdl) receptor scavenger receptor class b type I reveals its key role in hdl metabolism. Proc Natl Acad Sci U S A. 1997;94:12610–12615. doi: 10.1073/pnas.94.23.12610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y, Da Silva JR, Reilly M, Billheimer JT, Rothblat GH, Rader DJ. Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J Clin Invest. 2005;115:2870–2874. doi: 10.1172/JCI25327. doi: 10.1172/JCI25327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ji Y, Jian B, Wang N, Sun Y, Moya ML, Phillips MC, Rothblat GH, Swaney JB, Tall AR. Scavenger receptor BI promotes high density lipoprotein-mediated cellular cholesterol efflux. J Biol Chem. 1997;272:20982–20985. doi: 10.1074/jbc.272.34.20982. [DOI] [PubMed] [Google Scholar]
- 34.Jian B, de la Llera-Moya M, Ji Y, Wang N, Phillips MC, Swaney JB, Tall AR, Rothblat GH. Scavenger receptor class B type I as a mediator of cellular cholesterol efflux to lipoproteins and phospholipid acceptors. J Biol Chem. 1998;273:5599–5606. doi: 10.1074/jbc.273.10.5599. [DOI] [PubMed] [Google Scholar]
- 35.Al-Jarallah A, Chen X, González L, Trigatti BL. High density lipoprotein stimulated migration of macrophages depends on the scavenger receptor class B, type I, PDZK1 and Akt1 and is blocked by sphingosine 1 phosphate receptor antagonists. PLoS One. 2014;9:e106487. doi: 10.1371/journal.pone.0106487. doi: 10.1371/journal.pone.0106487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Jarallah A, Trigatti BL. A role for the scavenger receptor, class B type I in high density lipoprotein dependent activation of cellular signaling pathways. Biochim Biophys Acta. 2010;1801:1239–1248. doi: 10.1016/j.bbalip.2010.08.006. doi: 10.1016/j.bbalip.2010.08.006. [DOI] [PubMed] [Google Scholar]
- 37.Assanasen C, Mineo C, Seetharam D, Yuhanna IS, Marcel YL, Connelly MA, Williams DL, de la Llera-Moya M, Shaul PW, Silver DL. Cholesterol binding, efflux, and a PDZ-interacting domain of scavenger receptor-BI mediate HDL-initiated signaling. J Clin Invest. 2005;115:969–977. doi: 10.1172/JCI200523858. doi: 10.1172/JCI23858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Saddar S, Carriere V, Lee WR, Tanigaki K, Yuhanna IS, Parathath S, Morel E, Warrier M, Sawyer JK, Gerard RD, Temel RE, Brown JM, Connelly M, Mineo C, Shaul PW. Scavenger receptor class B type I is a plasma membrane cholesterol sensor. Circ Res. 2013;112:140–151. doi: 10.1161/CIRCRESAHA.112.280081. doi: 10.1161/CIRCRESAHA.112.280081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Saddar S, Mineo C, Shaul PW. Signaling by the high-affinity HDL receptor scavenger receptor B type I. Arterioscler Thromb Vasc Biol. 2010;30:144–150. doi: 10.1161/ATVBAHA.109.196170. doi: 10.1161/ATVBAHA.109.196170. [DOI] [PubMed] [Google Scholar]
- 40.Trigatti B, Rayburn H, Vinals M, Braun A, Miettinen H, Penman M, Hertz M, Schrenzel M, Amigo L, Rigotti A, Krieger M. Influence of the high density lipoprotein receptor SR-BI on reproductive and cardiovascular pathophysiology. Proc Natl Acad Sci U S A. 1999;96:9322–9327. doi: 10.1073/pnas.96.16.9322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phillips ML, Pullinger C, Kroes I, Kroes J, Hardman DA, Chen G, Curtiss LK, Gutierrez MM, Kane JP, Schumaker VN. A single copy of apolipoprotein B-48 is present on the human chylomicron remnant. J Lipid Res. 1997;38:1170–1177. [PubMed] [Google Scholar]
- 42.Liao JK, Laufs U. Pleiotropic effects of statins. Annu Rev Pharmacol Toxicol. 2005;45:89–118. doi: 10.1146/annurev.pharmtox.45.120403.095748. doi: 10.1146/annurev.pharmtox.45.120403.095748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Horton JD, Cohen JC, Hobbs HH. Molecular biology of PCSK9: its role in LDL metabolism. Trends Biochem Sci. 2007;32:71–77. doi: 10.1016/j.tibs.2006.12.008. doi: 10.1016/j.tibs.2006.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Urban D, Pöss J, Böhm M, Laufs U. Targeting the proprotein convertase subtilisin/kexin type 9 for the treatment of dyslipidemia and atherosclerosis. J Am Coll Cardiol. 2013;62:1401–1408. doi: 10.1016/j.jacc.2013.07.056. doi: 10.1016/j.jacc.2013.07.056. [DOI] [PubMed] [Google Scholar]
- 45.Canuel M, Sun X, Asselin MC, Paramithiotis E, Prat A, Seidah NG. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One. 2013;8:e64145. doi: 10.1371/journal.pone.0064145. doi: 10.1371/journal.pone.0064145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Galkina E, Ley K. Immune and inflammatory mechanisms of atherosclerosis (*). Annu Rev Immunol. 2009;27:165–197. doi: 10.1146/annurev.immunol.021908.132620. doi: 10.1146/annurev.immunol.021908.132620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fuller M, Dadoo O, Serkis V, Abutouk D, MacDonald M, Dhingani N, Macri J, Igdoura SA, Trigatti BL. The effects of diet on occlusive coronary artery atherosclerosis and myocardial infarction in scavenger receptor class B, type 1/low-density lipoprotein receptor double knockout mice. Arterioscler Thromb Vasc Biol. 2014;34:2394–2403. doi: 10.1161/ATVBAHA.114.304200. doi: 10.1161/ATVBAHA.114.304200. [DOI] [PubMed] [Google Scholar]
- 48.Brill A, Yesilaltay A, De Meyer SF, Kisucka J, Fuchs TA, Kocher O, Krieger M, Wagner DD. Extrahepatic high-density lipoprotein receptor SR-BI and apoA-I protect against deep vein thrombosis in mice. Arterioscler Thromb Vasc Biol. 2012;32:1841–1847. doi: 10.1161/ATVBAHA.112.252130. doi: 10.1161/ATVBAHA.112.252130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dole VS, Matuskova J, Vasile E, Yesilaltay A, Bergmeier W, Bernimoulin M, Wagner DD, Krieger M. Thrombocytopenia and platelet abnormalities in high-density lipoprotein receptor-deficient mice. Arterioscler Thromb Vasc Biol. 2008;28:1111–1116. doi: 10.1161/ATVBAHA.108.162347. doi: 10.1161/ATVBAHA.108.162347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Korporaal SJ, Meurs I, Hauer AD, Hildebrand RB, Hoekstra M, Cate HT, Praticò D, Akkerman JW, Van Berkel TJ, Kuiper J, Van Eck M. Deletion of the high-density lipoprotein receptor scavenger receptor BI in mice modulates thrombosis susceptibility and indirectly affects platelet function by elevation of plasma free cholesterol. Arterioscler Thromb Vasc Biol. 2011;31:34–42. doi: 10.1161/ATVBAHA.110.210252. doi: 10.1161/ATVBAHA.110.210252. [DOI] [PubMed] [Google Scholar]
- 51.Hörkkö S, Bird DA, Miller E, Itabe H, Leitinger N, Subbanagounder G, Berliner JA, Friedman P, Dennis EA, Curtiss LK, Palinski W, Witztum JL. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid-protein adducts inhibit macrophage uptake of oxidized low-density lipoproteins. J Clin Invest. 1999;103:117–128. doi: 10.1172/JCI4533. doi: 10.1172/JCI4533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Palinski W, Hörkkö S, Miller E, Steinbrecher UP, Powell HC, Curtiss LK, Witztum JL. Cloning of monoclonal autoantibodies to epitopes of oxidized lipoproteins from apolipoprotein E-deficient mice. Demonstration of epitopes of oxidized low density lipoprotein in human plasma. J Clin Invest. 1996;98:800–814. doi: 10.1172/JCI118853. doi: 10.1172/JCI118853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jedidi I, Couturier M, Thérond P, Gardès-Albert M, Legrand A, Barouki R, Bonnefont-Rousselot D, Aggerbeck M. Cholesteryl ester hydroperoxides increase macrophage CD36 gene expression via PPARalpha. Biochem Biophys Res Commun. 2006;351:733–738. doi: 10.1016/j.bbrc.2006.10.122. doi: 10.1016/j.bbrc.2006.10.122. [DOI] [PubMed] [Google Scholar]
- 54.Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–240. doi: 10.1016/s0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- 55.Shashkin P, Dragulev B, Ley K. Macrophage differentiation to foam cells. Curr Pharm Des. 2005;11:3061–3072. doi: 10.2174/1381612054865064. [DOI] [PubMed] [Google Scholar]
- 56.Tontonoz P, Nagy L, Alvarez JG, Thomazy VA, Evans RM. PPARgamma promotes monocyte/macrophage differentiation and uptake of oxidized LDL. Cell. 1998;93:241–252. doi: 10.1016/s0092-8674(00)81575-5. [DOI] [PubMed] [Google Scholar]
- 57.Berliner JA, Leitinger N, Tsimikas S. The role of oxidized phospholipids in atherosclerosis. J Lipid Res. 2009;50 suppl:S207–S212. doi: 10.1194/jlr.R800074-JLR200. doi: 10.1194/jlr.R800074-JLR200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Podrez EA, Byzova TV, Febbraio M, Salomon RG, Ma Y, Valiyaveettil M, Poliakov E, Sun M, Finton PJ, Curtis BR, Chen J, Zhang R, Silverstein RL, Hazen SL. Platelet CD36 links hyperlipidemia, oxidant stress and a prothrombotic phenotype. Nat Med. 2007;13:1086–1095. doi: 10.1038/nm1626. doi: 10.1038/nm1626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Seimon TA, Wang Y, Han S, Senokuchi T, Schrijvers DM, Kuriakose G, Tall AR, Tabas IA. Macrophage deficiency of p38alpha MAPK promotes apoptosis and plaque necrosis in advanced atherosclerotic lesions in mice. J Clin Invest. 2009;119:886–898. doi: 10.1172/JCI37262. doi: 10.1172/JCI37262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Thorp E, Li G, Seimon TA, Kuriakose G, Ron D, Tabas I. Reduced apoptosis and plaque necrosis in advanced atherosclerotic lesions of Apoe-/- and Ldlr-/- mice lacking CHOP. Cell Metab. 2009;9:474–481. doi: 10.1016/j.cmet.2009.03.003. doi: 10.1016/j.cmet.2009.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tsukano H, Gotoh T, Endo M, Miyata K, Tazume H, Kadomatsu T, Yano M, Iwawaki T, Kohno K, Araki K, Mizuta H, Oike Y. The endoplasmic reticulum stress-C/EBP homologous protein pathway-mediated apoptosis in macrophages contributes to the instability of atherosclerotic plaques. Arterioscler Thromb Vasc Biol. 2010;30:1925–1932. doi: 10.1161/ATVBAHA.110.206094. doi: 10.1161/ATVBAHA.110.206094. [DOI] [PubMed] [Google Scholar]
- 62.Gonzalez L, Trigatti BL. Macrophage apoptosis and necrotic core development in atherosclerosis: a rapidly advancing field with clinical relevance to imaging and therapy. Can J Cardiol. 2017;33:303–312. doi: 10.1016/j.cjca.2016.12.010. doi: 10.1016/j.cjca.2016.12.010. [DOI] [PubMed] [Google Scholar]
- 63.Ludman A, Venugopal V, Yellon DM, Hausenloy DJ. Statins and cardioprotection–more than just lipid lowering? Pharmacol Ther. 2009;122:30–43. doi: 10.1016/j.pharmthera.2009.01.002. doi: 10.1016/j.pharmthera.2009.01.002. [DOI] [PubMed] [Google Scholar]
- 64.Neto-Ferreira R, Rocha VN, Souza-Mello V, Mandarim-de-Lacerda CA, de Carvalho JJ. Pleiotropic effects of rosuvastatin on the glucose metabolism and the subcutaneous and visceral adipose tissue behavior in C57Bl/6 mice. Diabetol Metab Syndr. 2013;5:32. doi: 10.1186/1758-5996-5-32. doi: 10.1186/1758-5996-5-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kim DH, Choi BH, Ku SK, Park JH, Oh E, Kwak MK. Beneficial effects of sarpogrelate and rosuvastatin in high fat diet/streptozotocin-induced nephropathy in mice. PLoS One. 2016;11:e0153965. doi: 10.1371/journal.pone.0153965. doi: 10.1371/journal.pone.0153965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Tian XY, Wong WT, Xu A, Chen ZY, Lu Y, Liu LM, Lee VW, Lau CW, Yao X, Huang Y. Rosuvastatin improves endothelial function in db/db mice: role of angiotensin II type 1 receptors and oxidative stress. Br J Pharmacol. 2011;164(2b):598–606. doi: 10.1111/j.1476-5381.2011.01416.x. doi: 10.1111/j.1476-5381.2011.01416.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Rashid S, Curtis DE, Garuti R, Anderson NN, Bashmakov Y, Ho YK, Hammer RE, Moon YA, Horton JD. Decreased plasma cholesterol and hypersensitivity to statins in mice lacking Pcsk9. Proc Natl Acad Sci U S A. 2005;102:5374–5379. doi: 10.1073/pnas.0501652102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ason B, Tep S, Davis HR, Jr, et al. Improved efficacy for ezetimibe and rosuvastatin by attenuating the induction of PCSK9. J Lipid Res. 2011;52:679–687. doi: 10.1194/jlr.M013664. doi: 10.1194/jlr.M013664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Awan Z, Seidah NG, MacFadyen JG, Benjannet S, Chasman DI, Ridker PM, Genest J. Rosuvastatin, proprotein convertase subtilisin/kexin type 9 concentrations, and LDL cholesterol response: the JUPITER trial. Clin Chem. 2012;58:183–189. doi: 10.1373/clinchem.2011.172932. doi: 10.1373/clinchem.2011.172932. [DOI] [PubMed] [Google Scholar]
- 70.Dong B, Wu M, Li H, Kraemer FB, Adeli K, Seidah NG, Park SW, Liu J. Strong induction of PCSK9 gene expression through HNF1alpha and SREBP2: mechanism for the resistance to LDL-cholesterol lowering effect of statins in dyslipidemic hamsters. J Lipid Res. 2010;51:1486–1495. doi: 10.1194/jlr.M003566. doi: 10.1194/jlr.M003566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Welder G, Zineh I, Pacanowski MA, Troutt JS, Cao G, Konrad RJ. High-dose atorvastatin causes a rapid sustained increase in human serum PCSK9 and disrupts its correlation with LDL cholesterol. J Lipid Res. 2010;51:2714–2721. doi: 10.1194/jlr.M008144. doi: 10.1194/jlr.M008144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Brown ML, Ramprasad MP, Umeda PK, Tanaka A, Kobayashi Y, Watanabe T, Shimoyamada H, Kuo WL, Li R, Song R, Bradley WA, Gianturco SH. A macrophage receptor for apolipoprotein B48: cloning, expression, and atherosclerosis. Proc Natl Acad Sci U S A. 2000;97:7488–7493. doi: 10.1073/pnas.120184097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Véniant MM, Zlot CH, Walzem RL, Pierotti V, Driscoll R, Dichek D, Herz J, Young SG. Lipoprotein clearance mechanisms in LDL receptor-deficient “Apo-B48-only” and “Apo-B100-only” mice. J Clin Invest. 1998;102:1559–1568. doi: 10.1172/JCI4164. doi: 10.1172/JCI4164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Collot-Teixeira S, Martin J, McDermott-Roe C, Poston R, McGregor JL. CD36 and macrophages in atherosclerosis. Cardiovasc Res. 2007;75:468–477. doi: 10.1016/j.cardiores.2007.03.010. doi: 10.1016/j.cardiores.2007.03.010. [DOI] [PubMed] [Google Scholar]
- 75.Han J, Zhou X, Yokoyama T, Hajjar DP, Gotto AM, Jr, Nicholson AC. Pitavastatin downregulates expression of the macrophage type B scavenger receptor, CD36. Circulation. 2004;109:790–796. doi: 10.1161/01.CIR.0000112576.40815.13. doi: 10.1161/01.CIR.0000112576.40815.13. [DOI] [PubMed] [Google Scholar]