

Abstract
Benzocyclobutenones 1a–1g undergo cycloreversion at 150 °C in m-xylene solvent to form transient α-oxo-ortho-quinodimethanes or “ortho-quinoid ketene methides,” which engage in intermolecular [4+2] cycloadditions with isatins 2a–2f to form 2-oxindole spirolactones 3a–3l. This process tolerates an array of different functional groups and substitution patterns, and is applicable to unprotected isatins 2b–2f bearing free NH-functionalities. The superior performance of isatins compared to other carbonyl based dienophiles was demonstrated and rationalized with the aid of quantum chemical calculations.
Graphical Abstract
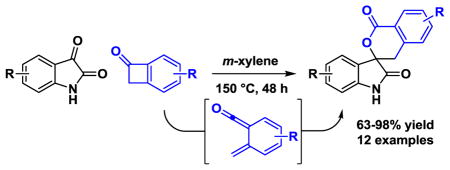
Due to ring-strain and their ability to undergo cycloreversion to form highly reactive ortho-quinodimethanes,1 benzocyclobutenes have emerged as versatile building blocks in chemical synthesis.2 In the course of developing catalytic processes that convert lower alcohols to higher alcohols via hydrogen auto-transfer,3 we recently discovered a suite of redox-independent ruthenium(0) catalyzed [4+2] cycloadditions between diols, α-ketols or diones and diverse unsaturated partners,4 including benzocyclobutenones.5 During investigation of the mechanism of the latter process, it was found that benzocyclobutenones engage in thermally promoted hetero-Diels-Alder cycloadditions with isatins. As the resulting oxindole spirolactones represent a medicinally relevant class of bioactive compounds5 and thermally promoted hetero-Diels-Alder reactions of isatins with ortho-quinodimethanes were unknown,1,2,7,8,9 the optimization and evaluation of scope for this process was undertaken. Here, we disclose that upon heating at 150 °C in m-xylene, diverse benzocyclobutenones engage in highly efficient intermolecular [4+2] cycloaddition with unprotected isatins by way of transient α-oxo-ortho-quinodimethanes to form 2-oxindole spirolactones (Figure 1).
Figure 1.

hetero-Diels-Alder cycloadditions of isatins.
Initial experiments were aimed at identifying optimal conditions for the thermal [4+2] cycloaddition of benzocyclobutenone 1a (100 mol %) and N-benzyl isatin 2a (100 mol %) in m-xylene solvent (0.8 M). A lower temperature threshold of 150 °C was required to promote cycloreversion of the benzocyclobutenone such that reasonable reaction rates were observed. Under these conditions, the spirolactone 3a was formed in 49% yield. Analysis of the crude reaction mixture revealed that while benzocyclobutenone 1a was completely consumed, significant quantities of isatin 2a were still present. This observation suggests that apart from the desired hetero-Diels-Alder reaction pathway, the α-oxo-ortho-quinodimethane reacts through alternate manifolds to form undefined side products. To compensate, an excess of benzocyclobutenone 1a (150 mol%) was employed, which enabled formation of spirolactone 3a in 81% yield.
Under these optimized conditions, the thermal [4+2] cycloaddition of benzocyclobutenone 1a (150 mol %) with diverse isatins 2a–2f (100 mol %) was explored (Table 1). The corresponding spirolactones 3a–3f, which derive from isatins bearing substituents in the 5-, 6-, or 7-position, were formed in good to excellent yields. Notably, it was found that N-benzyl substitution is not required. The unprotected N-H derivatives 2b–2f engage in highly efficient cycloadditions. Variation of the benzocyclobutenone also was explored (Table 2). The unsubstituted benzocyclobutenone 1b and a diverse range of polysubstituted benzocyclobutenones 1c–1g each underwent thermal [4+2] cycloaddition to form the respective spirolactones 3g–3l in good to excellent yields. The structural assignment of spirolactones 3a–3l is made in analogy to that for cycloadduct 3h, which was determined by single crystal x-ray diffraction analysis (Figure 2).
Table 1.
Cycloaddition of benzocyclobutenone 1a with isatins 2a–2f to form spirolactones 3a–3f.a

|
Yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
Table 2.
Cycloaddition of benzocyclobutenones 1b–1f with isatin 2b to form spirolactones 3g–3l.a

|
Yields are of material isolated by silica gel chromatography. See Supporting Information for further experimental details.
Figure 2.
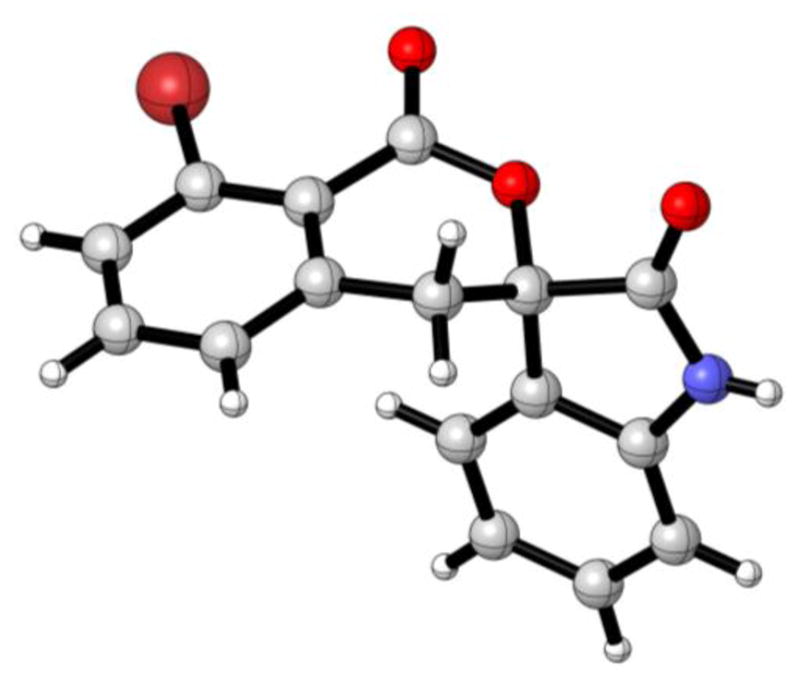
Molecular solid state structure of compound 3h as determined by single crystal x-ray diffraction.a
aSee Supporting Information for tabulated crystallographic data.
Pursuant to this evaluation of reaction scope, the feasibly of utilizing less activated carbonyl partners was assessed. The electron deficient, electroneutral and electron rich aldehydes p-trifluoromethyl benzaldehyde, benzaldehyde and p-anisaldehyde were exposed to benzocyclobutenone 1a under standard conditions, however, products of [4+2] cycloaddition were not observed. The aliphatic aldehyde octanal and even ethyl pyruvate, which incorporates a vicinal dicarbonyl motif, failed to undergo cycloaddition. These results were surprising, as the thermal [4+2] cycloaddition of the parent benzocyclobutenone 1b with benzaldehyde and ethyl pyruvate is reported to occur at 160 °C, albeit in the absence of solvent.7a
Our observations demonstrate that isatins are significantly more efficient partners for [4+2] cycloaddition compared to other carbonyl dienophiles in the context of benzocyclobutenone derived α-oxo-ortho-quinodimethanes. To better understand the origins of their superior reactivity, quantum chemical calculations were conducted (Table 3). It is well known that the rate of Diels-Alder reactions is strongly influenced by the energy gap between the frontier molecular orbitals of the diene and dienophile, as well as the shapes of these orbitals.10 Looking at the energies of the reaction partners, the smallest frontier orbital energy gaps are, in all considered systems, observed between the LUMO of the carbonyl compound and the HOMO of the diene. The investigated reactions are therefore “normal electron demand” hetero-Diels-Alder reactions. Among the indicated carbonyl compounds, a consideration of LUMO energies reveals that isatin is by far the most electrophilic. Therefore, the smallest gap in energy between frontier molecular orbitals is found for α-oxo-ortho-quinodimethane and isatin. Apart from these energetic criteria, the shape of the frontier molecular orbitals of the reactants (Figure 3), that is, the diene moiety of the α-oxo-ortho-quinodimethane and carbonyl moiety of the isatin, are such that good orbital overlap for the hetero-Diels-Alder reaction is anticipated. Thus, the experimentally observed and theoretically predicted reactivities are in alignment.
Table 3.
Calculated frontier molecular orbital energies of representative diene and carbonyl compounds.a
| Diene or Dienophile | EHOMO [eV] | ELUMO [eV] |
|---|---|---|
| (s-cis)-1,3-butadiene | −6.53 | −1.27 |
| α-oxo-ortho-quinodimethane | −5.19 | −1.81 |
| acetaldehde | −7.22 | −0.90 |
| benzaldehyde | −7.23 | −2.03 |
| (s-trans)-ethyl pyruvate | −7.13 | −2.18 |
| isatin | −6.84 | −2.95 |
Calculated at the B3LYP-d3(BJ)/cc-pVTZ level of theory at fully optimized geometries.
Figure 3.

Shape of the HOMO of the parent α-oxo-ortho-quinodimethane (left) and the LUMO of isatin (right).a
aCalculated at the B3LYP-d3(BJ)/cc-pVTZ level of theory at fully optimized geometries.
In summary, we report the first hetero-Diels-Alder reactions of benzocyclobutenones with isatins to form oxindole spirolactones. These [4+2] cycloadditions occur through cycloreversion of the benzocyclobutenones to form transient α-oxo-ortho-quinodimethanes. Isatins were shown to be superior dienophiles compared to other carbonyl compounds. These experimental observations are readily understood upon consideration of the calculated frontier molecular orbital energies for certain diene and carbonyl dienophile partners.
Experimental Section
General Information
Unless otherwise noted, all reactions were performed using oven-dried glassware under an atmosphere of dry Ar. Thermal hetero-Diels-Alder reactions were conducted in pressure tubes (13 x 100 mm) sealed with PTFE-lined caps. Analytical thin-layer chromatography (TLC) was carried out using 0.25 mm commercial silica gel plates. Visualization was conducted with UV light and then treatment with a cerium ammonium molybdate or p-anisaldehyde solution followed by heating. Isolation of reaction products was conducted via flash column chromatography using 40–63 μm silica gel. Unless otherwise noted, reagents were purchased from commercial sources and used as received. Benzocyclobutenones 1a and 1b,11 1c,12 1d–f13 and 1g14 were prepared according to literature procedures. 1H Nuclear magnetic resonance spectra were recorded using a 400 or 500 MHz spectrometer. Coupling constants are reported in Hertz (Hz) and protons chemical shifts (δ) are reported in parts per million (ppm) downfield from tetramethylsilane and are referenced to the proton resonance of the residual protic solvent (CHCl3, δ = 7.26 ppm; acetone, δ = 2.05 ppm; DMSO, δ = 2.50 ppm). 13C Nuclear magnetic resonance spectra were recorded using a 125 MHz spectrometer and chemical shifts are reported as parts per million (ppm) relative to the carbon resonances of residual protic solvent (CHCl3, δ = 77.16 ppm; acetone: δ = 29.84 ppm; DMSO: δ = 39.52 ppm). High-resolution mass spectra (HRMS) are reported as m/z (relative intensity) using time-of-flight (TOF) analyzers. Accurate masses are reported for the molecular ion (M+H, M+Na) or a suitable fragment ion.
General Procedure for the synthesis of 2-Oxindole Spirolactones
A resealable pressure tube (13 x 100 mm) was charged with reactant isatin (0.20 mmol, 100 mol%), and reactant benzocyclobutenone (0.30 mmol, 150 mol%). The tube was sealed with a rubber septum and purged with argon for 10 min. m-Xylene (0.25 mL, 0.80 M with respect to the isatin reactant) was injected and the rubber septum was replaced with a PTFE-lined screw cap. The tube was placed in a 150 °C oil bath for 48 h. After cooling to room temperature, the crude reaction mixture was subjected to flash column chromatography (SiO2) to furnish the product of cycloaddition.
1-Benzyl-8′-methoxyspiro[indoline-3,3′-isochroman]-1′,2-dione (3a)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, hexanes:acetone = 1:1 to 0:1) provided the title compound (62 mg, 0.16 mmol) in 81 % yield as a white solid. TLC (SiO2): Rf = 0.32 (hexanes : EtOAc = 1:1, UV). 1H NMR (500 MHz, DMSO-d6): δ 7.62 (dd, J = 7.5, 8.5 Hz, 1H), 7.38–7.26 (m, 6H), 7.18 (d, J = 8.6 Hz, 1H), 7.02 (d, J = 7.8 Hz, 1H), 6.99 (d, J = 4.2 Hz, 2H), 6.95 (d, J = 7.5 Hz, 1H), 4.92 (d, J = 15.8 Hz, 1H), 4.88 (d, J = 15.8 Hz, 1H), 3.90 (s, 3H), 3.55 (d, J = 16.5 Hz, 1H), 3.45 (d, J = 16.5 Hz, 1H).13C NMR (100 MHz, DMSO-d6): δ 172.3, 160.4, 159.4, 142.1, 138.8, 135.7, 135.4, 130.8, 128.8 (2C), 127.6, 127.2 (2C), 127.1, 124.0, 123.1, 120.1, 112.5, 111.9, 110.1, 79.4, 56.0, 42.8, 33.9. HRMS (ESI-TOF) m/z: [M + K]+ Calcd for C24H19KNO4 424.0946; Found 424.0953. FTIR (neat): 1720, 1598 cm−1. Melting Point: 232 °C.
8′-Methoxyspiro[indoline-3,3′-isochroman]-1′,2-dione (3b)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, hexanes : acetone = 1:1 to 0:1) provided the title compound (42 mg, 0.16 mmol) in 81% yield as an off-white solid. TLC (SiO2): Rf = 0.17 (hexanes: EtOAc = 1:1, UV).1H NMR (500 MHz, DMSO-d6): δ 10.75 (bs, 1H), 7.6 (dd, J = 7.5, 8.6 Hz, 1H ), 7.31 (dt, J = 1.56, 7.7 Hz, 1H), 7.16 (d, J = 8.6 Hz, 1H), 6.96–6.86 (m, 4H), 3.89 (s, 3H), 3.43 (d, J = 16.6 Hz, 1H), 3.36 (d, J = 16.6 Hz, 1H).13C NMR (125 MHz, DMSO-d6): δ 173.7, 160.3, 159.5, 141.7, 138.9, 135.3, 130.8, 127.6, 124.0, 122.3, 120.0, 112.6, 111.8, 110.5, 79.8, 56.0, 33.9. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H13NNaO4 318.0737; Found 318.0739. FTIR (neat): 3271, 1740, 1706, 1615 cm−1. Melting Point: 313 °C (decomposition).
8′-Methoxy-5-methylspiro[indoline-3,3′-isochroman]-1′,2-dione (3c)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, hexanes : acetone = 1:1 to 0:1) provided the title compound (48 mg, 0.18 mmol) in 89 % yield as a beige solid. TLC (SiO2): Rf = 0.18 (hexanes: ethyl acetate 1:1, UV). 1H NMR (500 MHz, DMSO-d6): δ 10.64 (bs, 1H), 7.59 (dd, J = 7.4, 8.58 Hz, 1H), 7.15 (d, J = 8.58 Hz, 1H), 7.12 (d, J = 7.4 Hz, 1H), 6.91 (d, J = 7.4 Hz, 1H), 6.81–6.77 (m, 2H), 3.89 (s, 3H), 3.45 (d, J = 16.6 Hz, 1H), 3.31 (d, J = 16.6 Hz, 1H), 2.16 (S, 3H). 13C NMR (100 MHz, DMSO-d6): δ 173.9, 160.3, 159.6, 139.1, 138.9, 135.2, 131.2, 131.0, 127.7, 124.7, 120.0, 112.6, 111.8, 110.2, 79.8, 56.0, 33.9, 20.6. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H15NNaO4 332.0893; Found 332.0895. FTIR (neat): 3295, 1740, 1706 cm−1. Melting Point: 297 °C (decomposition).
6-bromo-8′-methoxyspiro[indoline-3,3′-isochroman]-1′,2-dione (3d)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, hexanes : acetone = 1:1 to 0:1) provided the title compound (59 mg, 0.18 mmol) in 88% yield as a beige solid. TLC (SiO2): Rf = 0.24 (hexanes : EtOAc = 1:1, UV). 1H NMR (500 MHz, DMSO-d6): δ 10.09 (bs, 1H), 7.6 (dd, J = 7.46, 8.56 Hz, 1H), 7.18–7.12 (m, 2H), 7.05 (d, J = 1.88 Hz, 1H), 6.9 (d, J = 7.65 Hz, 1H), 6.87 (d, J = 8.03 Hz, 1H), 3.88 (s, 3H), 3.48 (d, J = 16.6 Hz, 1H), 3.35 (d, J = 16.6 Hz, 1H).13C NMR (125 MHz, DMSO-d6): δ 174.1, 160.8, 159.8, 143.8, 139.2, 135.9, 127.4, 126.5, 125.5, 124.0, 120.5, 113.9, 112.4, 79.9, 56.5, 34.1. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H12BrNNaO4 395.9842; Found 395.9848. FTIR (neat): 3257, 1744, 1706 cm−1. Melting Point: 317 °C (decomposition).
7-Fluoro-8′-methoxyspiro[indoline-3,3′-isochroman]-1′,2-dione (3e)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, hexanes : acetone = 1:1 to 0:1) provided the title compound (49 mg, 0.18 mmol) in 88% yield as an off white solid. TLC (SiO2): Rf = 0.25 (hexanes : EtOAc = 1:1, UV). 1H NMR (500 MHz, DMSO-d6): δ 11.29 (bs, 1H), 7.61 (dd, J = 7.6, 8.6 Hz, 1H), 7.26 (dd, J = 8.5, 10.4 Hz, 1H), 7.17 (d, J = 8.6 Hz, 1H), 6.99–6.94 (m, 1H), 6.91 (d, J = 7.6 Hz, 1H), 6.77 (d, J = 7.6 Hz, 1H), 3.89 (s, 3H), 3.49 (d, J = 16.6 Hz, 1H), 3.39 (d, J = 16.6 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): δ 173.5, 160.3, 159.2, 146.5 (d, JFC= 247 Hz), 138.7, 135.4, 130.4, 128.9 (d, JFC= 13 Hz), 123.4 (d, JFC= 5.3 Hz), 120.1 (d, JFC= 19.3 Hz), 117.9 (d, JFC= 16.9 Hz), 112.4, 111.9, 79.7, 56.0, 33.7. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C17H12NNaO4 336.0643; Found 336.0639. FTIR (neat): 3230, 1746, 1717 cm−1. Melting Point: 315 °C (decomposition).
8′-Methoxy-5-(trifluoromethoxy)spiro[indoline-3,3′-isochroman]-1′,2-dione (3f)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, hexanes : acetone = 1:1 to 0:1) provided the title compound (53 mg, 0.14 mmol) in 72% yield as a beige solid. TLC (SiO2): Rf = 0.21 (hexanes: ethyl acetate = 1:1, UV). 1H NMR (500 MHz, DMSO-d6): δ 10.91 (bs, 1H), 7.6 (dd, J = 7.5, 8.6 Hz, 1H), 7.35 (dd, J = 2.5, 8.3 Hz, 1H), 7.16 (d, J = 8.3 Hz, 1H), 7.10 (d, J = 2.5 Hz, 1H), 6.99 (d, J = 8.6 Hz, 1H), 6.91 (d, J = 7.7 Hz, 1H), 3.88 (s, 3H), 3.63 (d, J = 16.6 Hz, 1H), 3.34 (d, J = 16.6 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): δ 174.0, 160.3, 159.3, 143.3, 140.9, 138.6, 135.3, 129.1, 124.1, 120.1 (q, JFC= 256 Hz), 119.9, 118.1, 112.5, 111.8, 111.6, 79.6, 56.0, 33.5. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H12F3NNaO5 402.0560; Found 402.0566. FTIR (neat): 3290, 1743, 1713 cm−1. Melting Point: 266 °C.
Spiro[indoline-3,3′-isochroman]-1′,2-dione (3g)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, hexanes : acetone = 9:1 to 0:1) provided the title compound (52 mg, 0.19 mmol) in 98% yield as a white solid. TLC (SiO2): Rf = 0.50 (DCM : acetone = 9:1). 1H NMR (400 MHz, DMSO-d6): δ 10.79 (br. s, 1H), 8.02–7.99 (m, 1H), 7.70–7.65 (m, 1H), 7.53–7.48 (m, 1H), 7.40–7.38 (m, 1H), 7.36–7.32 (m, 1H), 7.13–7.11 (m, 1H), 7.00–6.96 (m, 1H), 6.94–6.91 (m, 1H), 3.64 (d, J = 17.0 Hz, 1H), 3.40 (d, J = 17.1 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): δ 173.9, 163.6, 141.6, 136.7, 134.2, 131.0, 128.9, 128.2, 127.8, 127.6, 124.4, 124.3, 122.4, 110.5, 80.6, 32.8. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C16H11NO3Na 288.0631; Found 288.0641. FTIR (neat): 3271, 1727, 1705 cm−1. Melting Point: 246–248 °C.
8′-Bromospiro[indoline-3,3′-isochroman]-1′,2-dione (3h)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, DCM : EtOAc = 1:0 to 9:1) provided the title compound (68 mg, 0.20 mmol) in 98% yield as a white solid. TLC (SiO2): Rf = 0.57 (DCM : EtOAc = 4:1, UV). 1H NMR (1H NMR (400 MHz, acetone-d6): δ 9.68 (s, 1H), 7.80 (d, J = 8.0 Hz, 1H), 7.55–7.49 (m, 1H), 7.44 (d, J = 7.5 Hz, 1H), 7.39–7.33 (m, 1H), 7.12–7.08 (m, 1H), 7.04–6.99 (m, 2H), 3.69 (d, J = 16.7 Hz, 1H), 3.53 (d, J = 16.7 Hz, 1H). 13C NMR (125 MHz, acetone-d6): δ 174.3, 161.0, 142.7, 140.7, 135.5, 135.0, 131.9, 128.9, 128.5, 125.4, 125.2, 124.3, 123.5, 111.5, 80.7, 35.7. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C16H10BrNO3Na 365.9736; Found 356.9737. FTIR (neat): 3333, 1742, 1714 cm−1. Melting Point: 254–256 °C.
Spiro[[1,3]dioxolo[4,5-h]isochromene-7,3′-indoline]-2′,9(6H)-dione (3i)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, hexanes : acetone = 9:1 to 0:1) provided the title compound (46 mg, 0.15 mmol) in 74% yield as a white solid. TLC (SiO2): Rf = 0.40 (DCM : acetone = 9:1). 1H NMR (400 MHz, DMSO-d6): δ 10.77 (br. s, 1H), 7.36–7.32 (m, 1H), 7.16 (d, J = 7.8 Hz, 1H), 7.12 (d, J = 7.4 Hz, 1H), 6.99–6.96 (m, 1H), 6.91 (d, J = 7.8 Hz, 1H), 6.79 (d, J = 7.8 Hz, 1H), 6.24 (app. d, J = 4.1 Hz, 2H), 3.45 (d, J = 16.5 Hz, 1H), 3.29 (d, J = 16.7 Hz, 1H). 13C NMR (125 MHz, DMSO-d6): δ 173.7, 160.2, 148.8, 147.9, 141.6, 130.9, 128.7, 127.5, 124.3, 122.4, 120.3, 112.9, 110.5, 107.4, 102.8, 80.9, 32.7. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C17H12NO5 310.0710; Found 310.0716. FTIR (neat): 3322, 1726, 1705 cm−1. Melting Point: 296–298 °C.
6′,8′-Dimethoxyspiro[indoline-3,3′-isochroman]-1′,2-dione (3j)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, acetone : hexanes = 1:9 to 1:0) provided the title compound (60 mg, 0.18 mmol) in 92% yield as a beige solid. TLC (SiO2): Rf = 0.30 (DCM : acetone = 9:1, UV). 1H NMR (400 MHz, DMSO-d6): δ 10.76 (br. s, 1H), 7.32–7.28 (m, 1H), 6.94–6.87 (m, 3H), 6.64 (d, J = 2.3 Hz, 1H), 6.54 (d, J = 2.3 Hz, 1H), 3.88 (s, 3H), 3.85 (s, 3H), 3.32 (s, 2H). 13C NMR (125 MHz, DMSO-d6): δ 173.7, 164.5, 162.5, 159.2, 141.6, 140.9, 130.8, 127.8, 124.1, 122.3, 110.5, 105.6, 105.5, 98.0, 79.4, 56.0, 55.7, 34.2. HRMS (ESI-TOF) m/z: [M + H]+ Calcd for C18H16NO5 326.1023; Found 326.1028. FTIR (neat): 3253, 1734, 1704 cm−1. Melting Point: 265–267 °C.
5′,8′-Dimethoxyspiro[indoline-3,3′-isochroman]-1′,2-dione (3k)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, DCM : EtOAc = 1:0 to 4:1) provided the title compound (62 mg, 0.19 mmol) in 95% yield as a white solid. TLC (SiO2): Rf = 0.25 (DCM : EtOAc = 4:1, UV). 1H NMR (500 MHz, acetone-d6) δ 9.60 (s, 1H), 7.35–7.28 (m, 2H), 7.12 (d, J = 9.2 Hz, 1H), 7.02–6.97 (m, 2H), 6.95 (t, J = 7.6 Hz, 1H), 3.88 (s, 3H), 3.80 (s, 3H), 3.36 (d, J = 17.1 Hz, 1H), 3.32 (d, J = 17.1 Hz, 1H). 13C NMR (125 MHz, acetone-d6): δ 173.7, 159.2, 154.8, 149.8, 141.7, 130.7, 128.4, 126.5, 124.2, 122.4, 117.1, 114.6, 112.3, 110.5, 79.6, 55.9, 55.7, 29.3. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H15NO5Na 348.0842; Found 348.0847. FTIR (neat): 3300, 1748, 1708 cm−1. Melting Point: 283–285 °C.
7′-Bromo-5′,8′-dimethoxyspiro[indoline-3,3′-isochroman]-1′,2-dione (3l)
The reaction was conducted in accordance with the general procedure. Flash column chromatography (SiO2, DCM : EtOAc = 1:0 to 9:1) provided the title compound (51 mg, 0.13 mmol) in 63% yield as a white solid. TLC (SiO2): Rf = 0.58 (DCM : EtOAc = 4:1, UV).1H NMR (500 MHz, acetone-d6): δ 9.63 (s, 1H), 7.55 (s, 1H), 7.36 (t, J = 7.7 Hz, 1H), 7.23 (d, J = 7.4 Hz, 1H), 7.07–6.97 (m, 2H), 3.89 (s, 3H), 3.87 (s, 3H), 3.42 (d, J = 17.4 Hz, 1H), 3.31 (d, J = 17.4 Hz, 1H).13C NMR (125 MHz, acetone-d6): δ 174.7, 160.0, 153.5, 152.3, 142.6, 131.8, 128.7, 127.0, 125.2, 123.6, 121.4, 121.1, 118.6, 111.5, 80.7, 61.9, 57.0, 30.2. HRMS (ESI-TOF) m/z: [M + Na]+ Calcd for C18H14BrNO5Na 425.9948; Found 425.9958. FTIR (neat): 3291, 1740, 1711 cm−1. Melting Point: 248–250 °C.
Supplementary Material
Acknowledgments
The Robert A. Welch Foundation (F-0038) and the NIH-NIGMS (RO1-GM069445) and the Center for Green Chemistry and Catalysis for partial support of this research. The Alexander von Humboldt foundation is acknowledged for support through the Feodor-Lynen postdoctoral fellowship program (T. W.).
Footnotes
Notes.
The authors declare no competing financial interest.
Supporting Information. 1H and 13C NMR spectra, single crystal X-ray diffraction data for compound 3h and computational details. This material is available free of charge via the internet at http://pubs.acs.org
References
- 1.For selected reviews on the chemistry of ortho-quinone methides and ortho-quinodimethanes, see: Oppolzer W. Synthesis. 1978:793.Charlton JL, Alauddin MM. Tetrahedron. 1987;43:2873.Martin N, Seoane C, Hanack M. Org Prep Proc. 1991;23:237.Segura JL, Martin N. Chem Rev. 1999;99:3199. doi: 10.1021/cr990011e.Van De Water RW, Pettus TRR. Tetrahedron. 2002;58:5367.Yoshida H, Ohshita J, Kunai A. Bull Chem Soc Jpn. 2010;83:199.Pathak TP, Sigman MS. J Org Chem. 2011;76:9210. doi: 10.1021/jo201789k.
- 2.For selected reviews on the synthesis and reactivity of benzocyclobutenes and benzocyclobutenones, see: Klundt IL. Chem Rev. 1970;70:471.Thummel RP. Acc Chem Res. 1980;13:70.Mehta G, Kotha S. Tetrahedron. 2001;57:625.Sadana AK, Saini RK, Billups WE. Chem Rev. 2003;103:1539. doi: 10.1021/cr010022j.Flores-Gaspar A, Martin R. Synthesis. 2013:563.Souillart L, Cramer N. Chem Rev. 2015;115:9410. doi: 10.1021/acs.chemrev.5b00138.Chen P-h, Dong G. Chem Eur J. 2016;22:18290. doi: 10.1002/chem.201603382.Fumagalli G, Stanton S, Bower JF. Chem Rev. 2017;117:9404. doi: 10.1021/acs.chemrev.6b00599.
- 3.For recent reviews on alcohol mediated C-C coupling, see: Ketcham JM, Shin I, Montgomery TP, Krische MJ. Angew Chem, Int Ed. 2014;53:9142. doi: 10.1002/anie.201403873.Dechert-Schmitt AMR, Schmitt DC, Gao X, Itoh T, Krische MJ. Nat Prod Rep. 2014;31:504. doi: 10.1039/c3np70076c.Perez F, Oda S, Geary LM, Krische MJ. Top Curr Chem. 2016;374:365. doi: 10.1007/s41061-016-0028-0.Nguyen KD, Park BY, Luong T, Sato H, Garza VJ, Krische MJ. Science. 2016;354:aah5133. doi: 10.1126/science.aah5133.Kim SW, Zhang W, Krische MJ. Acc Chem Res. 2017;50:2371. doi: 10.1021/acs.accounts.7b00308.
- 4.For a review on ruthenium(0) catalyzed cycloaddition of 1,2-diols, ketols or diones via alcohol-mediated hydrogen transfer, see: Sato H, Turnbull BWH, Fukaya K, Krische MJ. Angew Chem Int Ed. 2017;56 doi: 10.1002/anie.201709916.
- 5.Bender M, Turnbull BWH, Ambler BR, Krische MJ. Science. 2017;357:779. doi: 10.1126/science.aao0453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.For selected reviews on the synthesis and biological properties of spirooxindoles, see: Trost BM, Brennan MK. Synthesis. 2009:3003.Badillo JJ, Hanhan NV, Franz AK. Curr Opin Drug Disc Devel. 2010;13:758.Singh GS, Desta ZY. Chem Rev. 2012;112:6104. doi: 10.1021/cr300135y.Dalpozzo R, Bartoli G, Bencivenni G. Chem Soc Rev. 2012;41:7247. doi: 10.1039/c2cs35100e.Ball-Jones NR, Badillo JJ, Franz AK. Org Biomol Chem. 2012;10:5165. doi: 10.1039/c2ob25184a.Santos MMM. Tetrahedron. 2014;70:9735.Yu Bin, Yu D-Q, Liu H-M. Eur J Med Chem. 2015;97:673. doi: 10.1016/j.ejmech.2014.06.056.Yu B, Zheng YC, Shi XJ, Qi PP, Liu HM. Anti-Cancer Agents Med Chem. 2016;16:1315. doi: 10.2174/1871520615666151102093825.Ye N, Chen H, Wold EA, Shi PY, Zhou J. ACS Infect Dis. 2016;2:382. doi: 10.1021/acsinfecdis.6b00041.
- 7.For intermolecular hetero-Diels-Alder reactions of carbonyl compounds with ortho-quinodimethanes and their synthetic equivalents, see: Schiess P, Eberle M, Huys-Francotte M, Wirz J. Tetrahedron Lett. 1984;25:2201.Griesbeck AG, Stadtmueller S. Chem Ber. 1993;126:2149.Chino K, Takata T, Endo T. Synth Commun. 1996;26:2145.Hentemann MF, Allen JG, Danishefsky SJ. Angew Chem Int Ed. 2000;39:1937.Ueno S, Ohtsubo M, Kuwano R. Org Lett. 2010;12:4332. doi: 10.1021/ol101792a.Takaki K, Fujii T, Yonemitsu H, Fujiwara M, Komeyama K, Yoshida H. Tetrahedron Lett. 2012;53:3974.Chen X, Yang S, Song BA, Chi YR. Angew Chem Int Ed. 2013;52:11134. doi: 10.1002/anie.201305861.Janssen-Müller D, Singha S, Olyschläger T, Daniliuc CG, Glorius F. Org Lett. 2016;18:4444. doi: 10.1021/acs.orglett.6b02335.
- 8.For hetero-Diels-Alder reactions of isatins, see: Cassani C, Melchiorre P. Org Lett. 2012;14:5590. doi: 10.1021/ol302711w.Cui HL, Tanaka F. Chem Eur J. 2013;19:6213. doi: 10.1002/chem.201300595.Cui HL, Chouthaiwale PV, Yin F, Tanaka F. Org Biomol Chem. 2016;14:1777. doi: 10.1039/c5ob02393a.Cui HL, Chouthaiwale PV, Yin F, Tanaka F. Asian J Org Chem. 2016;5:153. doi: 10.1039/c5ob02393a.
- 9.For selected reviews on hetero-Diels-Alder reactions, see: Waldmann H. Synthesis. 1994:535.Tietze LF, Kettschau G. Top Curr Chem. 1997;189:1.Jørgensen KA. Angew Chem Int Ed. 2000;39:3558.Jørgensen KA. Eur J Org Chem. 2004:2093.Lin L, Liu X, Feng X. Synlett. 2007:2147.Pellissier H. Tetrahedron. 2009;65:2839.Blond G, Gulea M, Mamane V. Curr Org Chem. 2016;20:2161.Sogani N, Bansal RK. Curr Catal. 2017;6:3.
- 10.For selected contributions on frontier molecular orbital studies of the Diels Alder reaction, see: Herndon WC. Chem Rev. 1972;72:157.Houk KN. J Am Chem Soc. 1973;95:4092.Houk KN. Acc Chem Res. 1975;8:361.McCarrick MA, Wu YD, Houk KN. J Org Chem. 1993;58:3330.
- 11.Stevens RV, Bisacchi GS. J Org Chem. 1982;47:2393. [Google Scholar]
- 12.Gokhale A, Schiess P. Helvetica Chimica Acta. 1998;81:251. [Google Scholar]
- 13.Liebeskind LS, Lescosky LJ, McSwain CM., Jr J Org Chem. 1989;54:1435. [Google Scholar]
- 14.Azadi-Ardakani M, Wallace TW. Tetrahedron. 1988;44:5939. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.