

Abstract
Purpose
Blue cone monochromacy (BCM) is an X-linked congenital vision disorder characterized by complete loss or severely reduced L- and M-cone function. Patients with BCM display poor visual acuity, severely impaired color discrimination, myopia, nystagmus, and minimally detectable cone-mediated electroretinogram. Recent studies of patients with BCM with adaptive optics scanning laser ophthalmoscopy (AOSLO) showed that they have a disrupted cone mosaic with reduced numbers of cones in the fovea that is normally dominated by L- and M-cones. The remaining cones in the fovea have significantly shortened outer segments but retain sufficient structural integrity to serve as potential gene therapy targets. In this study, we tested whether exogenously expressed human L- and M-opsins can rescue M-cone function in an M-opsin knockout (Opn1mw−/−) mouse model for BCM.
Methods
Adeno-associated virus type 5 (AAV5) vectors expressing OPN1LW, OPN1MW, or C-terminal tagged OPN1LW-Myc, or OPN1MW-HA driven by a cone-specific promoter were injected subretinally into one eye of Opn1mw−/− mice, while the contralateral eye served as the uninjected control. Expression of cone pigments was determined with western blotting and their cellular localization identified with immunohistochemistry. M-cone function was analyzed with electroretinogram (ERG). Antibodies against cone phototransduction proteins were used to study cone outer segment (OS) morphology in untreated and treated Opn1mw−/− eyes.
Results
We showed that cones in the dorsal retina of the Opn1mw−/− mouse do not form outer segments, resembling cones that lack outer segments in the human BCM fovea. We further showed that AAV5-mediated expression of either human M- or L-opsin individually or combined promotes regrowth of cone outer segments and rescues M-cone function in the treated Opn1mw−/− dorsal retina.
Conclusions
Exogenously expressed human opsins can regenerate cone outer segments and rescue M-cone function in Opn1mw−/− mice, thus providing a proof-of-concept gene therapy in an animal model of BCM.
Introduction
Human color vision is mediated by three classes of cone photoreceptor visual pigments with different wavelength sensitivities: short-wavelength (blue), middle-wavelength (green), and long-wavelength (red) [1]. These visual pigments are the primary protein components of cone outer segment disc membranes and acquire their visual sensitivity by covalently binding 11-cis-retinal. In humans, L- and M-cones constitute about 95% of the total cone population. They are densely packed in a hexagonal pattern in the central fovea, the foveola, accounting for our best visual acuity. S-cones are more peripherally located in the retina and are absent from the central human fovea [2,3].
In humans, L-opsin (OPN1LW; Gene ID 5956, OMIM 300822) and M-opsin (OPN1MW; Gene ID 2652, OMIM 300821) genes are tandemly arrayed on the X- chromosome in a head to tail arrangement with a single L-opsin gene in the 5′ position followed by one or more M-opsin genes [4]. Expression of OPN1LW and OPN1MW is regulated by specific proximal promoters and a single upstream locus control region (LCR), ensuring that only one opsin gene is expressed in a single cone photoreceptor [5-7]. It has been shown that only the first two genes in this cluster are normally expressed [6,8]. The L- and M-cone opsins are highly homologous and share 96% amino acid identity. This sequence homology and close genomic proximity predispose the two pigment genes to homologous recombination resulting in gene deletions, duplications, or fusion genes that consist of portions of red and green pigment genes [9-11]. Mutations in the locus control region or harmful mutations in both genes can result in the absence of both functional cone pigments and are the genetic cause of blue cone monochromacy (BCM) [5,12-17]. The two most common causes of BCM are deletions encompassing the LCR or the presence of a deleterious C203R missense mutation either in a single OPN1LW/MW hybrid gene or in multiple OPN1LW/MW genes [5,13,14].
BCM affects 1 in 100,000 individuals, and patients with BCM who must rely on the remaining preserved S-cones and rod photoreceptors display severely impaired color discrimination from birth. Further, patients with BCM typically suffer from reduced visual acuity that may progress to 20/200, myopia, pendular nystagmus, and photoaversion [18,19]. There has been a long history of investigation of the clinical, electrophysiological, and psychophysical aspects in BCM [20-22]. Recently, studies using adaptive optics scanning laser ophthalmoscopy (AOSLO) showed that patients with BCM have a disrupted foveal cone mosaic with reduced numbers of cones. The remaining cones have significantly shortened outer segments but retain sufficient residual structure and viability to serve as targets for gene replacement therapy [23-25].
Previously, we showed that adeno-associated virus (AAV)-mediated expression of mouse M-opsin can rescue M-cone function in an Opn1mw−/− mouse model for BCM [26]. In this study, we provide evidence that the dorsal retina of the Opn1mw−/− mouse does not form cone outer segments, much as observed in the human BCM fovea. In addition, we show that exogenous expression of either human M-opsin or L-opsin individually or together in Opn1mw−/−cones promotes regeneration of cone outer segments and rescues M-cone function, thus providing an experimental basis for moving toward BCM gene therapy clinical trials.
Methods
Animals
All mice used in this study were maintained in the University of Florida Health Science Center Animal Care Service Facilities on a 12 h:12 h light-dark cycle. All animals were maintained under standard laboratory conditions (18–23 °C, 40% to 65% humidity) with food and water available ad libitum. All experiments were approved by the Institutional Animal Care and Use Committee at the University of Florida and conducted in accordance with the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research and National Institutes of Health regulations. The generation and characterization of Opn1mw−/− mice have been described in detail previously [26].
Cloning of AAV vectors containing human L- and M-opsins
Human OPN1MW cDNA was purchased from American Type Culture Collection (Manassas, VA). To facilitate cloning of this cDNA into the AAV vector under the PR2.1 promoter, it was amplified with PCR with primers containing NotI sites at both ends using the forward primer: 5ʹ-GCT AAA GCG GCC GCC ACC ATG GCC CAG CAG TGG AGC CT-3ʹ and the reverse primer: 5ʹ-GCT TAT GCG GCC GCT CAT GCA GGC GAT ACC GAG-3ʹ. To add a hemagglutinin (HA)-tag to the C-terminus of OPN1MW cDNA, PCR was performed with the same forward primer to amplify OPN1MW and the reverse primer: 5ʹ-TTA TGC GGC CGC TCA AGC GTA ATC TGG AAC ATC GTA TGG GTA TGC AGG CGA TAC CGA GGA-3ʹ. To generate the PR2.1-OPN1LW-Myc construct, we used the PR2.1-OPN1LW plasmid as the template, and the forward primer: 5ʹ-TAA AGC GGC CGC CAC CAT GGC CCA GCA GTG GAG C-3ʹ and the reverse primer: 5ʹ-GCT TAT GCG GCC GCT CAC AGA TCC TCT TCT GAG ATG AGT TTT TGT TCG TTA GGC CCT ACC TGG GT-3ʹ to add a Myc tag at the C-terminus. All PCRs were performed in the following conditions: 95 ºC for 3 min, followed by 35 cycles of 95 ºC for 1 min, 57 ºC for 1 min, and 72 ºC for 1 min. The PCR fragments were digested with NotI at both ends and cloned downstream of the PR2.1 promoter to generate PR2.1-hOPN1MW-HA and PR2.1-hOPN1LW-Myc. The PR2.1 promoter containing 2.1 kb of the upstream sequence of human red opsin gene was shown to drive cone-specific expression in several mammalian species [27]. The AAV vector expressing human OPN1LW driven by the PR2.1 promoter (PR2.1-hOPN1LW) was described previously [28]. All AAV vectors were packaged in serotype 5 and purified as described [29]. Serotype 5 is known to express strongly in photoreceptor cells when delivered subretinally. Since Opn1mv is highly expressed, we chose AAV5 and subretinal injection trying to match endogenous Opn1mw expression level.
Subretinal injection
Eyes were dilated with 1% atropine (Akorn, Inc., Lake Forest, IL) and 2.5% phenylephrine hydrochloride solution (Paragon Biotek, Portland, OR) before injection. Trans-corneal subretinal injections were performed with a 33 gauge blunt needle mounted on a 5 ml Hamilton syringe. First, an entering hole was introduced at the edge of the cornea with a 30 gauge disposable needle, and then a Hamilton syringe loaded with 1 µl of viral vector mixed with fluorescein dye (0.1% final concentration) was injected through the corneal opening and delivered into the subretinal space [30,31]. The injection bleb was visualized by a fluorescence positive subretinal signal, and only mice that showed at least 75% bled area were used. Atropine eye drops and neomycin/polymyxin B/dexamethasone ophthalmic ointment (Bausch & Lomb Inc., Tampa, FL) were applied after injection. One eye was injected while the contralateral eye served as a control. One µl of 5 × 109 vector genomes (vg) was injected for all vectors. To inject the PR2.1-hOPN1MW-HA and PR2.1-hOPN1LW-Myc mixture, 0.5 µl of each vector was mixed. Antisedan (Orion Corporation, Espoo, Finland) at 1 mg/kg was given intramuscularly following injection as an anesthetic reversal. All mice were injected at 1 month of age.
Electroretinography
M-cone ERG responses were analyzed using a UTAS Visual Diagnostic System with a Big Shot Ganzfeld dome (LKC Technologies, Gaithersburg, MD). All mice were anesthetized with an intraperitoneal injection of ketamine (72 mg/kg)/xylazine (4 mg/kg). The pupils were dilated with 1% atropine and 2.5% phenylephrine hydrochloride. Mice were first exposed to white light for 5 min at 30 cd/m2 to suppress rod function. Then M-cone ERGs were recorded by stimulation of the green channel middle-wavelength light (510 nm) at intensities of −0.6, 0.4, and 1.4 log cd.s/m2. Twenty-five recordings were averaged for each of the three light intensities.
Statistical analysis
ERG data are presented as average ± standard error of the mean (SEM). Statistical analysis was performed using the paired t test or compared with one-way ANOVA with the Bonferroni post hoc test. Significance was defined as a p value of less than 0.05. Detailed S-cone and rod ERG responses in untreated and mouse M-opsin treated Opn1mw−/− mice were characterized previously [26].
Immunohistochemistry and antibodies
Mice were euthanized in a chamber connected to the CO2 tank until no longer a heartbeat, followed by cervical dislocation, and their eyes were marked at 12 o’clock on the cornea with a burn marker and then enucleated. The eyes were fixed in 4% paraformaldehyde for 30 min, and then the cornea and lens were removed without disturbing the retina. The retinas were further fixed for an additional 2–3 h at room temperature. For frozen retinal sections, the eyecups were rinsed with PBS (1X; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4, pH 7.4) and cryoprotected in 30% sucrose/PBS for 3 h at room temperature, embedded in cryostat compound (Tissue TEK optimum cutting temperature (OCT) compound, Sakura Finetek USA, Inc., Torrance, CA), and frozen at –80 °C. Retinas were cut perpendicularly from dorsal to ventral at 12 μm thickness. For immunohistochemistry, retinal sections were rinsed in PBS and blocked in 3% bovine serum albumin (BSA) and 0.3% Triton X-100 in PBS for 1 h at room temperature. Sections were then incubated with primary antibodies at room temperature overnight. They were washed with PBS three times, incubated with immunoglobulin G (IgG) secondary antibody tagged with Alexa Fluor 594 or Alexa Fluor 488 (Molecular Probes, Eugene, OR) diluted 1:500 in PBS at room temperature for 2 h, and then washed with PBS. Sections were mounted with Vectashield Mounting Medium for Fluorescence (H-1400, Vector Laboratory, Inc., Burlingame, CA) and coverslipped. Human L- and M-opsins are highly homologous, and there are no antibodies that can distinguish between them. Therefore, we used the same antibody to detect human M- and human L-opsin in the treated Opn1mw−/− mouse eyes. All antibodies used in immunohistochemistry are listed in Table 1. Sections were imaged with an Olympus 153 IX81-DSU spinning disk confocal microscope with SlidebookTM 4.2 software (Center Valley, PA) or a Leica TCS 154 SP2 laser scanning confocal fluorescent microscope with Leica LCS software v2.61 (Wetzlar, Germany).
Table 1. Antibodies used in this study.
| Antibodies | Species raised | Dilution | Source |
|---|---|---|---|
| M-opsin |
rabbit |
1:1000 |
Millipore AB5405 |
| M-opsin |
goat |
1:100 |
Santa Cruz SC-22117 |
| S-opsin |
Rabbit |
1:1000 |
Millipore AB5407 |
| S-opsin |
goat |
1:300 |
Santa Cruz SC-14363 |
| HA-Flurescein |
rat |
1:200 |
Roche 11988506001 |
| Myc |
mouse |
1:20 |
9E10, ATCC |
| Cone PDE6α’ |
rabbit |
1:1000 |
Dr. Visvanathan Ramamurthy |
| Cone Tα | rabbit | 1: 2000 | Invitrogen PA5–22340 |
Cone outer segment length measurements
Cone outer and inner segment lengths were measured from masked images taken after PDE6α’ and peanut agglutinin (PNA) staining. The total lengths of the cone inner and outer segments were measured from the distal tip of the cone outer segments to the proximal end of the cone inner segments immediately above the outer nuclear layer. The arithmetic mean length of the cone outer and inner segments in the wild-type dorsal retinas was given a value of 1.0, and the remaining measurements were expressed relative to this value. An arithmetic mean of 90 total cone measurements were obtained from nine topographically different images taken from three different eyes. Error bars represent the SEM (p<0.001).
Western blotting
Untreated, treated, and wild-type retinas (three of each) were carefully dissected. They were homogenized with sonication in a buffer containing 0.23 M sucrose, 5 mM Tris-HCl, pH 7.5, and protease inhibitors (Roche Complete, Basel, Switzerland). After centrifugation, aliquots of the retinal extracts containing equal amounts of protein (50 μg) were analyzed with electrophoresis on 4% to 15% polyacrylamide–sodium dodecyl sulfate (SDS) gel (Bio-Rad, Hercules, CA). The proteins were then transferred to Immobilon-FL membrane (Millipore, St Louis, MO) and probed with the anti-red/green opsin antibody (Millipore, AB5405). An antibody against α-tubulin (Abcam, Cambridge, UK, ab4074) was used as an internal loading control. Visualization of specific bands was performed using an Odyssey Infrared Fluorescence Imaging System (Odyssey; Li-Cor, Lincoln, NE).
Results
Characterization of cone outer segments in Opn1mw−/− mice
Wild-type C57Bl/6 mouse cones coexpress M- and S-opsin in a dorsal-ventral gradient with M-opsin dominant in the dorsal retina and S-opsin dominant in the ventral retina [32,33]. In Opn1mw−/− retinas, we confirmed that M-opsin is not expressed, while S-opsin is expressed predominately in the ventral retina as in wild-type controls [26]. As cone opsins are the primary protein components of cone outer segment disc membranes, we first characterized the consequence of M-opsin loss on cone outer segment morphology in the Opn1mw−/− dorsal retina where M-opsin normally dominates.
Cone outer segment morphology was examined with immunostaining with antibodies (Table 1) against the cone-specific phosphodiesterase 6α’ subunit (PDE6α’; Figure 1A, top panels) and cone transducin α subunit (cTα; Figure 1A, bottom panels) on oriented retinal cryosections. In the Opn1mw−/− ventral retina where S-opsin normally dominates and is unaffected in Opn1mw−/− eyes, the expression patterns of PDE6α’ and cTα were similar to those of the wild-type control and were colocalized with S-opsin. In contrast, in the Opn1mw−/− dorsal retina where M- and S-opsins are missing, PDE6α’ staining was primarily detected in cone inner segments, and cTα expression was completely absent. A direct comparison of the difference in cone outer segment morphology between the Opn1mw−/− dorsal and ventral retina is shown in the dorsal-ventral transition zone (Figure 1B, left panel). In the dorsal region, PDE6α’ staining was observed only in cone inner segments while expression extended into cone outer segments in the ventral hemisphere. We also compared cone outer segment morphology in Opn1mw−/− retinas with PNA labeling that uniquely labels the extracellular glycoprotein matrix of the cone outer and inner segment sheaths [34]. Interestingly, PNA staining showed no difference between the Opn1mw−/− dorsal and ventral retinas as seen at the dorsal-ventral transition zone (Figure 1B, right panel). Therefore, it appears that the outer segments are missing, but the sheaths of the Opn1mw−/− dorsal cones remain normal.
Figure 1.

Cone outer segments are shortened in dorsal retinas of Opn1mw−/− mice. A: PDE6α’ staining (top row) in Opn1mw−/− dorsal cones is observed only in the inner segments, while its staining in ventral cones is concentrated in outer segments shown as colocalization with endogenous S-opsin. PDE6α’ staining is primarily observed in cone outer segments in the wild-type control. cTα staining (bottom row) is absent in the dorsal cones of the Opn1mw−/− retina, while staining is present in cone outer segments in Opn1mw−/− ventral cones as in the wild-type control. As expected, S-opsin staining (red) was primarily observed in ventral retinas in Opn1mw−/− and wild-type mice. B: An image from the Opn1mw−/− dorsal-ventral transition zone showing shortened cone outer segments in the dorsal hemisphere with PDE6α’ staining (left panel). The right panel is the same image superimposed with peanut agglutinin (PNA) staining showing equal lengths of cone outer and inner segment sheaths between the dorsal and ventral hemispheres. COS: cone outer segments; CIS: cone inner segments. Scale bar: 20 µm.
Cone outer segments are restored to normal lengths in treated Opn1mw−/− dorsal retinas
We next explored whether vectored expression of human OPN1LW or OPN1MW restored the cone outer segment defect in the treated Opn1mw−/− dorsal retinas with PDE6α’ and cTα staining. Mice were injected at 1 month of age and euthanized at 6 weeks post-injection. In either vector-treated dorsal retinas, the expression of PDE6α’ in the cone outer segments was lengthened compared to that seen in the untreated dorsal retina (Figure 2A, top row). Additionally, either treatment restored cTα staining to the cone outer segments in the dorsal retinas, in contrast to the lack of cTα staining in the untreated dorsal retina (Figure 2A, bottom row). PDE6α’ and cTα staining in the treated eyes also colocalized with the vector-delivered human L- or M-opsin. The cone outer segment elongation seen with PDE6α’ staining can also be viewed at the dorsal-ventral transition zone where the treated dorsal cone outer segments were equal in length to the ventral cones (Figure 2B). Measurements of the cone outer plus inner segment lengths from images of PDE6α’ staining showed that their length in the treated dorsal cones was approximately twice as long as in the untreated dorsal cones (Figure 2C). Their lengths were now similar to those of the Opn1mw−/− ventral cones, as well as to the cones in wild-type mice. Measurements of the cone outer and inner segment sheath lengths from images of PNA staining showed that the cone sheath lengths remained similar in the untreated dorsal, untreated ventral, treated dorsal, and wild-type retinas confirming that the cone sheaths are not affected (Figure 2C). These results suggest that treatment promoted cone outer segment replenishing with visual pigment and restitution of outer segment length.
Figure 2.

Cone outer segments are restored in the treated Opn1mw−/− dorsal retinas. A: Images from untreated, OPN1MW- or OPN1LW-treated Opn1mw−/− dorsal retinas labeled with PDE6α’ (top row) or cTα (bottom row) showing that cone outer segments were regenerated in the treated retinas. Adeno-associated virus (AAV)–delivered human opsins (red) were observed only in treated eyes, and the human opsins were colocalized with PDE6α’ and cTα in the cone outer segments. Scale bar: 20 µm. B: An image from the dorsal-ventral transition zone in an OPN1LW-treated Opn1mw−/− eye showing equal lengths of cone outer segments with PDE6α’ staining. S-opsin was detected only in the ventral hemisphere. C: Normalized average cone outer and inner segment lengths visualized with PDE6α’ staining (left panel), and normalized average cone outer and inner segment sheath lengths visualized with peanut agglutinin (PNA) staining (right panel) from the untreated Opn1mw−/− dorsal retinas (uTx dorsal), OPN1LW-treated Opn1mw−/− dorsal retinas (Tx dorsal), untreated Opn1mw−/− ventral retinas (uTx ventral), and wild-type controls (WT dorsal, WT ventral). Each bar represents the average of 90 cones from three images each from three individual eyes of different animals. Error bars represent the standard error of the mean (SEM; *p<0.001). COS: cone outer segments; CIS: cone inner segments.
Expression of either human M-opsin or L-opsin restores M-cone function in Opn1mw−/− mice
ERG was performed in the mice at 6 weeks post-injection. ERG responses to M-cone preferred middle-wavelength light stimuli were not different from background in the untreated Opn1mw−/− mice. Treatment with vectors expressing either human M- or L-opsin rescued M-cone ERG responses with a maximum b-wave amplitude of 81.4±36.4 µV (average ± SEM, n = 5) in the M-opsin treated eyes, and 80.0±11.0 µV (n = 5) in the L-opsin treated eyes (Figure 3A). These responses were 70% and 68% of the wild-type level (117±9.00, n = 5), respectively, and statistically significantly higher than that of the untreated contralateral control eyes (p<0.005). There is no difference in the b-wave implicit times between the treated and wild-type controls (as observed in our previous study). The long-term rescue effect was assessed in animals 7 months post-injection, and rescue was still present, similar to our previous observation [26].
Figure 3.

M-cone ERG responses and human M- and L-opsin expression in AAV5-PR2.1-OPN1MW- or AAV5-PR2.1-OPN1LW-treated Opn1mw−/− eyes. A: M-cone electroretinogram (ERG) responses from untreated (UTx), OPN1MW-treated, and OPN1LW-treated Opn1mw-/ eyes and wild-type controls. Each bar represents the mean ± standard error of the mean (SEM) of M-cone b-wave amplitudes recorded at 1.4 log cd.s/m2 (n = 5 for each group, *p<0.005). B: Western blot analysis of OPN1MW and OPN1LW expression in untreated Opn1mw−/− eyes (UTx), OPN1MW-, or OPN1LW- treated Opn1mw−/− eyes and wild-type (WT) untreated control eyes. C: Immunohistochemistry of AAV–mediated OPN1MW and OPN1LW expression in the treated Opn1mw−/− retinas. In the untreated eyes, no M-opsin expression was detected. S-opsin (green) was expressed normally in the ventral retina (bottom row, left panel). The peanut agglutinin (PNA) staining (green) was normal in the dorsal and ventral retinas (top and bottom rows, second column from the left). In the treated eyes, AAV-delivered OPN1MW and OPN1LW were detected in the dorsal and ventral retinas. The vectored opsins were colocalized with PNA in the dorsal retinas (top row, two right panels) and with endogenous S-opsin in the ventral retinas (bottom row, two right panels). Scale bar: 20 µm.
Similar to our previous study [26], we show here that the levels of the AAV-delivered human M- and L-opsin in the treated Opn1mw−/− retinas were about 65% of that in the wild-type control with western blotting (Figure 3B). In the treated Opn1mw−/− retinas, AAV-delivered OPN1MW and OPN1LW were detected in the dorsal and ventral retinas. In the dorsal retina, the delivered human opsins colocalized with PNA out to the tip of the cone outer segments (Figure 3C, top row, third and fourth columns; Table 1). In the ventral retina, the human opsins colocalized with endogenous mouse S-opsin (Figure 3C, bottom row, third, and fourth columns). Immunostaining was performed in multiple treated mice to confirm that the dorsal cones were transduced, and at least part of ERG rescue was driven by them, although focal ERG analysis would be necessary to confirm this conclusion quantitatively.
Coexpression of human M- and L-opsin in Opn1mw−/− mice
Because patients with BCM lack L- and M-opsin, we also characterized the functional consequence and opsin distribution when human L- and M-opsins are expressed together in Opn1mw−/− retinas. As there is no antibody that can distinguish between human L- and M-opsins, we generated AAV5 vectors expressing either C-terminal myc tagged human L-opsin-myc cDNA or C-terminal HA tagged human M-opsin-HA cDNA, again both driven by the cone-specific PR2.1 promoter. Vectors were then delivered either individually or as a mixture containing equal numbers of vector genomes of each. M-cone function analysis of Opn1mw−/− mice treated with either vector individually or as an equal mixture showed significant ERG rescue compared to the untreated controls. The levels of rescue from all treatments are similar to the rescue seen from vectors without tags (Figure 4A). There is no additive effect from the vector mixture presumably because the total number of vector genomes delivered was the same as for each individual vector treatment. In the PR2.1-hOPN1MW-HA-treated eyes, expression of HA tag and M-opsin was detected, and both were colocalized in the cone outer segments (Figure 4B, top panel; Table 1). Similarly, expression of Myc tagged L-opsin was detected and colocalized in the cone outer segments in the PR2.1-hOPN1LW-myc-treated eyes (Figure 4B, bottom panel). In the retinas treated with the vector mixture, most of the cones coexpress human M- and L-opsins as seen with HA and Myc colocalization (Figure 4C). We also noted that individual cones contained different ratios of human M- and L-opsins.
Figure 4.
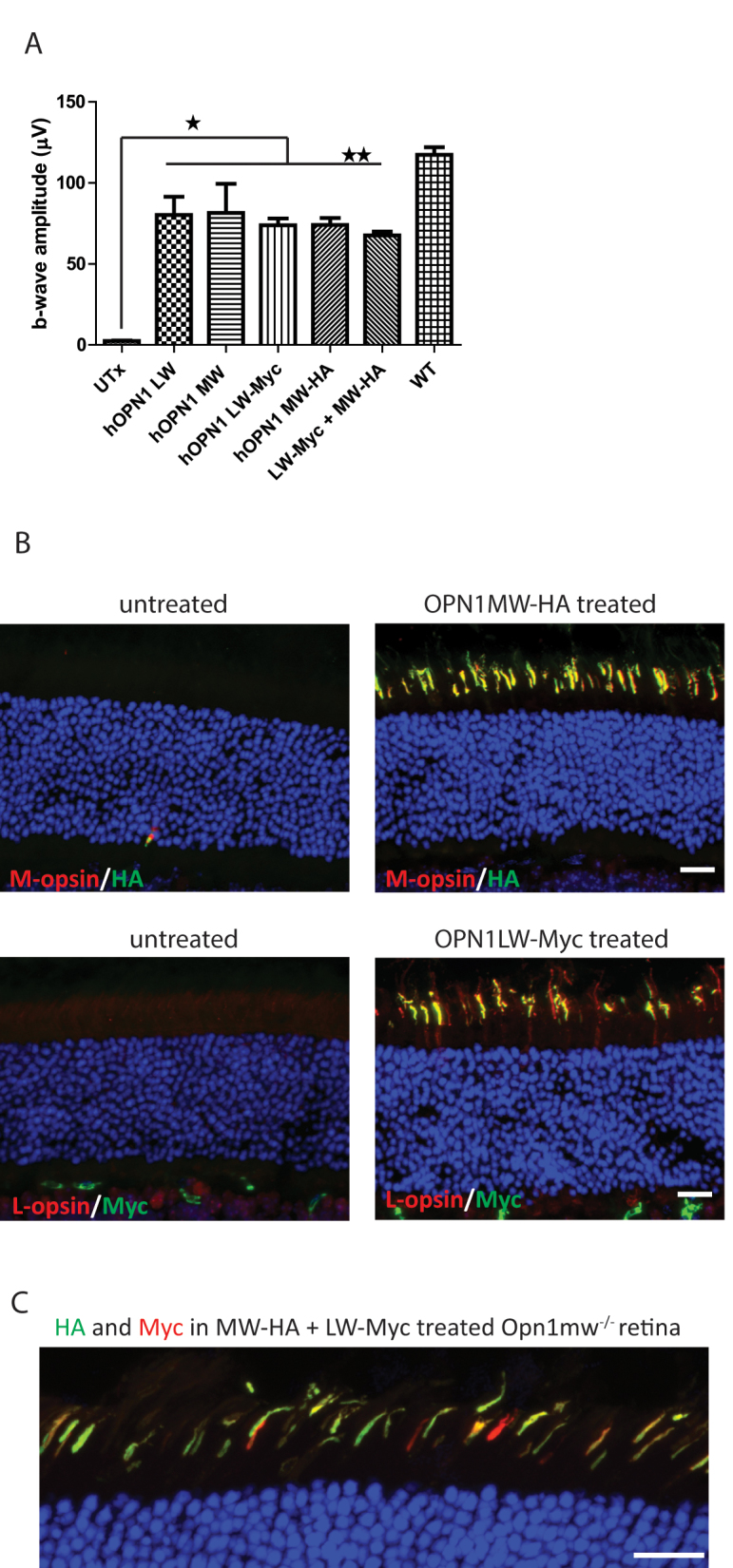
M-cone ERG responses and M- and L-opsin expression in AAV5-PR2.1-OPN1MW-HA or/and AAV5-PR2.1-OPN1LW-Myc-treated Opn1mw−/− eyes. A: M-cone electroretinogram (ERG) b-wave amplitudes showed similar levels of rescue in OPN1LW-, OPN1MW-, OPN1LW-Myc-, OPN1MW-HA-, and OPN1LW-Myc + OPN1MW-HA mixture-treated eyes. Each bar represents the mean ± standard error of the mean (SEM) of M-cone b-wave amplitudes recorded at 1.4 log cd.s/m2. There is no difference among any of the treatments (n = 5, **p>0.05), and they are all statistically significantly higher than those of the untreated controls (*p<0.005). B: M-opsin and HA expression was detected and colocalized in the cone outer segments in the PR2.1-OPN1MW-HA-treated eyes (top row); and L-opsin and Myc expression was detected and colocalized in the cone outer segments in the PR2.1-OPN1LW-Myc-treated eyes (bottom row). C: Representative image showing HA and Myc expression in PR2.1-OPN1LW-Myc + PR2.1-OPN1MW-HA mixture-treated eyes. Scale bar: 20 µm.
No obvious toxicity was observed in eyes treated with vectors containing tagged human opsin sequences, as demonstrated by the normal thickness of the outer nuclear layers. These results suggest that C-terminal tagging of human opsin does not interfere with either the light response function or its cellular localization.
Discussion
In this study, we demonstrate that exogenous expression of human M- or L-opsin individually or together restores M-cone function and promotes regeneration of the cone outer segments in an M-opsin knockout mouse model of BCM. The dorsal retina of Opn1mw−/− mice resembles the human BCM fovea in terms of exhibiting no cone function, abnormally shortened cone outer segments, all hallmarks of the condition. Our observation that the addition of a new human opsin in mouse cones that lack any endogenous opsin can regenerate cone outer segments and rescue cone function provides solid evidence that cones lacking pigments remain viable and can be targeted for gene therapy, thus providing proof-of-concept for treating human BCM.
The amino acid sequence of human M-opsin shows 88% identity and 94% functional conservation to mouse M-opsin. By analogy to rhodopsin, the loop regions facing the cytoplasm involved in interactions with other phototransduction components, such as transducin α’ and arrestin, are highly conserved. The least conserved domains are at the N-termini where hOPN1MW and hOPN1LW have an extra five amino acids (QQWSL) relative to mOPN1MW. In the Opn1mw−/− mouse, this difference appears to have no or minimal effect on structural or functional recovery after gene therapy.
One concern for BCM cone opsin gene therapy is whether the visual cortex of patients with BCM will respond to the new chromatic inputs they have never experienced before. Positive results obtained from adult L-opsin-deficient monkeys after receiving human L-opsin gene augmentation therapy [28] suggest that patients with BCM may also respond. These monkeys were missing a functional L-opsin gene and were therefore born with red-green color deficiency. AAV-mediated expression of human L-opsin in the foveal cones of these adult monkeys led to improved color perception and full color vision–elicited behavior. The restoration of trichromatic visual behavior in these monkeys suggests that new cone inputs to the visual cortex can be generated in adult primates upon the addition of a novel cone wavelength sensitivity.
Another question regarding BCM gene therapy is whether patients with BCM should be treated with M- or L-opsin individually or together as a mixture. We showed that in the retinas treated with an equal vector mixture, although the majority of cones expressed both opsins, there was a great variation in the L/M ratio among even adjacent treated cones. In addition, the total amount of opsin being expressed and its distribution varied in different cones. This is expected due to the stochastic nature of expression from AAV vectors. If we want to achieve a consistent L-/M-opsin ratio in all treated cones in the same retina, we could employ a single vector system using one promoter to express both opsins with a Furin-2A cleavage sequence between the two opsins [35]. However, this approach still would not control the variation in the total amount of protein being expressed in different cones. In our opinion, improving visual acuity rather than restoring color vision is more valuable to patients with BCM; therefore, acuity should be the most important treatment outcome. Thus, in combining the potential for improving visual acuity with minimizing biologic variables and simplifying the interpretation of study results, it would seem best, at least initially, to treat patients with BCM with just one opsin. We suggest that opsin be L-opsin because its response spectrum to light frequencies, when added to the endogenous S-cone frequencies, would give rise to a wider spectrum of perceivable frequencies than an M-opsin plus S-opsin combination. In addition, L-cones are more abundant than M-cones in the human retina [36,37].
A third factor to consider for BCM gene therapy is the fragile structure of the central retinas in these patients. In the LCA2 human clinical trials, subretinal vector injections that detached the foveal area resulted in the loss of the fovea cone outer segments and caused cone cell death. Therefore, it was concluded that subretinal injection under the fovea may cause more damage than benefit and should be approached with prudence [38-40]. This problem might be solved by recent progress in screening and identifying novel AAV capsid mutants with higher penetrating properties after intravitreal injection [41,42]. This class of AAV vectors might have clinical relevance for cone-targeted diseases such as BCM that would require robust, pan-foveal gene expression without retinal detachment.
We also noticed that dorsal cones in Opn1mw−/− mice without any visual pigments remained viable for many months. The cone sheaths appeared to be stable as long as the cone inner segments are intact (data not shown). This is similar to what was observed in the diseased retina in which the cone matrix sheaths remained intact during the degenerative phase of the disease and became compromised only after the cone inner segments disappeared and the rods collapsed [43]. The fate of cones without pigments is in direct contrast to the rods in rhodopsin knockout mice in which rod photoreceptors underwent rapid cell death [44]. The resilience of cones may be explained by the many differences between the structure and function of rods versus cones, such as cones have a lamellated continuously connected membrane, and the turnover rate of cone outer segments is thought to be less rapid than in rods as discussed by Cideciyan et al. [25].
In summary, we showed that the dorsal cones in Opn1mw−/− mouse retinas with little or no opsin remain viable and can be treated with opsin gene augmentation therapy. Treatment with human L- or M-opsin individually or together rescues M-cone function and restores the cone outer segment structure in the mouse. This proof-of-concept in a BCM mouse model provides a solid experimental basis for considering a human BCM gene therapy clinical trial.
Acknowledgments
Supported by NIH grant P30EY021721 (WWH), R01EY08123 (WB), R01EY019298 (WB), P30EY014800-039003 (NEI core grant to the Department of Ophthalmology, University of Utah), NIH 1S10OD020026 (Shared Instrumentation Grant to University of Florida), unrestricted grants to the Departments of Ophthalmology at the University of Florida and the University of Utah from Research to Prevent Blindness (RPB; New York), and WB is a recipient of a RPB Nelson Trust Award, and by grants from BCM Families Foundation, Macula Vision Research Foundation, and AGTC Inc. WWH: P, AGTC. We thank Dr. Visvanathan Ramamurthy for the PDE6α’ antibody, and Doug Smith at the Cell and Tissue Analysis Core at University of Florida for technical assistance with confocal imaging. WWH and the University of Florida have a financial interest in the use of AAV therapies, and WWH owns equity in a company (AGTC Inc.) that might, in the future, commercialize some aspects of this work. Wen-Tao Deng (wdeng@ufl.edu) and Jijing Pang (jpang@ufl.edu) are co-corresponding authors for this paper.
Appendix 1. Representative M-cone ERG waveforms from untreated, OPN1MW-treated, OPN1LW-treated, OPN1MW-HA-treated, OPN1LW-myc-treated Opn1mw-/ eyes and wild-type controls at light intensity of 1.4 log cd.s/m2.
To access the data, click or select the words “Appendix 1”
References
- 1.Nathans J, Thomas D, Hogness DS. Molecular genetics of human color vision: the genes encoding blue, green, and red pigments. Science. 1986;232:193–202. doi: 10.1126/science.2937147. [DOI] [PubMed] [Google Scholar]
- 2.Curcio CA, Allen KA, Sloan KR, Lerea CL, Hurley JB, Klock IB, Milam AH. Distribution and morphology of human cone photoreceptors stained with anti-blue opsin. J Comp Neurol. 1991;312:610–24. doi: 10.1002/cne.903120411. [DOI] [PubMed] [Google Scholar]
- 3.Mustafi D, Engel AH, Palczewski K. Structure of cone photoreceptors. Prog Retin Eye Res. 2009;28:289–302. doi: 10.1016/j.preteyeres.2009.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vollrath D, Nathans J, Davis RW. Tandem array of human visual pigment genes at Xq28. Science. 1988;240:1669–72. doi: 10.1126/science.2837827. [DOI] [PubMed] [Google Scholar]
- 5.Smallwood PM, Wang Y, Nathans J. Role of a locus control region in the mutually exclusive expression of human red and green cone pigment genes. Proc Natl Acad Sci USA. 2002;99:1008–11. doi: 10.1073/pnas.022629799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wang Y, Macke JP, Merbs SL, Zack DJ, Klaunberg B, Bennett J, Gearhart J, Nathans J. A locus control region adjacent to the human red and green visual pigment genes. Neuron. 1992;9:429–40. doi: 10.1016/0896-6273(92)90181-c. [DOI] [PubMed] [Google Scholar]
- 7.Yamaguchi T, Motulsky AG, Deeb SS. Visual pigment gene structure and expression in human retinae. Hum Mol Genet. 1997;6:981–90. doi: 10.1093/hmg/6.7.981. [DOI] [PubMed] [Google Scholar]
- 8.Winderickx J, Battisti L, Motulsky AG, Deeb SS. Selective expression of human X chromosome-linked green opsin genes. Proc Natl Acad Sci USA. 1992;89:9710–4. doi: 10.1073/pnas.89.20.9710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Neitz J, Neitz M. The genetics of normal and defective color vision. Vision Res. 2011;51:633–51. doi: 10.1016/j.visres.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Reyniers E, Van Thienen MN, Meire F, De Boulle K, Devries K, Kestelijn P, Willems PJ. Gene conversion between red and defective green opsin gene in blue cone monochromacy. Genomics. 1995;29:323–8. doi: 10.1006/geno.1995.9998. [DOI] [PubMed] [Google Scholar]
- 11.Gardner JC, Webb TR, Kanuga N, Robson AG, Holder GE, Stockman A, Ripamonti C, Ebenezer ND, Ogun O, Devery S, Wright GA, Maher ER, Cheetham ME, Moore AT, Michaelides M, Hardcastle AJ. X-linked cone dystrophy caused by mutation of the red and green cone opsins. Am J Hum Genet. 2010;87:26–39. doi: 10.1016/j.ajhg.2010.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gardner JC, Michaelides M, Holder GE, Kanuga N, Webb TR, Mollon JD, Moore AT, Hardcastle AJ. Blue cone monochromacy: causative mutations and associated phenotypes. Mol Vis. 2009;15:876–84. [PMC free article] [PubMed] [Google Scholar]
- 13.Kazmi MA, Sakmar TP, Ostrer H. Mutation of a conserved cysteine in the X-linked cone opsins causes color vision deficiencies by disrupting protein folding and stability. Invest Ophthalmol Vis Sci. 1997;38:1074–81. [PubMed] [Google Scholar]
- 14.Nathans J, Davenport CM, Maumenee IH, Lewis RA, Hejtmancik JF, Litt M, Lovrien E, Weleber R, Bachynski B, Zwas F. Molecular genetics of human blue cone monochromacy. Science. 1989;245:831–8. doi: 10.1126/science.2788922. [DOI] [PubMed] [Google Scholar]
- 15.Buena-Atienza E, Ruther K, Baumann B, Bergholz R, Birch D, De Baere E, Dollfus H, Greally MT, Gustavsson P, Hamel CP, Heckenlively JR, Leroy BP, Plomp AS, Pott JW, Rose K, Rosenberg T, Stark Z, Verheij JB, Weleber R, Zobor D, Weisschuh N, Kohl S, Wissinger B. De novo intrachromosomal gene conversion from OPN1MW to OPN1LW in the male germline results in Blue Cone Monochromacy. Sci Rep. 2016;6:28253. doi: 10.1038/srep28253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kohl S, Jagle H, Wissinger B. Achromatopsia. In: Pagon RA, Adam MP, Ardinger HH, Wallace SE, Amemiya A, Bean LJH, Bird TD, Fong CT, Mefford HC, Smith RJH, Stephens K, editors. GeneReviews(R). Seattle (WA)1993. [Google Scholar]
- 17.Nathans J, Maumenee IH, Zrenner E, Sadowski B, Sharpe LT, Lewis RA, Hansen E, Rosenberg T, Schwartz M, Heckenlively JR. Genetic heterogeneity among blue-cone monochromats. Am J Hum Genet. 1993;53:987–1000. [PMC free article] [PubMed] [Google Scholar]
- 18.Michaelides M, Johnson S, Simunovic MP, Bradshaw K, Holder G, Mollon JD, Moore AT, Hunt DM. Blue cone monochromatism: a phenotype and genotype assessment with evidence of progressive loss of cone function in older individuals. Eye (Lond) 2005;19:2–10. doi: 10.1038/sj.eye.6701391. [DOI] [PubMed] [Google Scholar]
- 19.Michaelides M, Hunt DM, Moore AT. The cone dysfunction syndromes. Br J Ophthalmol. 2004;88:291–7. doi: 10.1136/bjo.2003.027102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ayyagari R, Kakuk LE, Coats CL, Bingham EL, Toda Y, Felius J, Sieving PA. Bilateral macular atrophy in blue cone monochromacy (BCM) with loss of the locus control region (LCR) and part of the red pigment gene. Mol Vis. 1999;5:13. [PubMed] [Google Scholar]
- 21.Berson EL, Sandberg MA, Rosner B, Sullivan PL. Color plates to help identify patients with blue cone monochromatism. Am J Ophthalmol. 1983;95:741–7. doi: 10.1016/0002-9394(83)90058-2. [DOI] [PubMed] [Google Scholar]
- 22.Hess RF, Mullen KT, Sharpe LT, Zrenner E. The photoreceptors in atypical achromatopsia. J Physiol. 1989;417:123–49. doi: 10.1113/jphysiol.1989.sp017794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Carroll J, Dubra A, Gardner JC, Mizrahi-Meissonnier L, Cooper RF, Dubis AM, Nordgren R, Genead M, Connor TB, Jr, Stepien KE, Sharon D, Hunt DM, Banin E, Hardcastle AJ, Moore AT, Williams DR, Fishman G, Neitz J, Neitz M, Michaelides M. The effect of cone opsin mutations on retinal structure and the integrity of the photoreceptor mosaic. Invest Ophthalmol Vis Sci. 2012;53:8006–15. doi: 10.1167/iovs.12-11087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carroll J, Rossi EA, Porter J, Neitz J, Roorda A, Williams DR, Neitz M. Deletion of the X-linked opsin gene array locus control region (LCR) results in disruption of the cone mosaic. Vision Res. 2010;50:1989–99. doi: 10.1016/j.visres.2010.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cideciyan AV, Hufnagel RB, Carroll J, Sumaroka A, Luo X, Schwartz SB, Dubra A, Land M, Michaelides M, Gardner JC, Hardcastle AJ, Moore AT, Sisk RA, Ahmed ZM, Kohl S, Wissinger B, Jacobson SG. Human cone visual pigment deletions spare sufficient photoreceptors to warrant gene therapy. Hum Gene Ther. 2013;24:993–1006. doi: 10.1089/hum.2013.153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y, Deng WT, Du W, Zhu P, Li J, Xu F, Sun J, Gerstner CD, Baehr W, Sanford LB, Zhao C, Hauswirth WW, Pang JJ. Gene-based Therapy in a Mouse Model of Blue Cone Monochromacy. Sci Rep. 2017;7:6690. doi: 10.1038/s41598-017-06982-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Q, Timmers AM, Guy J, Pang J, Hauswirth WW. Cone-specific expression using a human red opsin promoter in recombinant AAV. Vision Res. 2008;48:332–8. doi: 10.1016/j.visres.2007.07.026. [DOI] [PubMed] [Google Scholar]
- 28.Mancuso K, Hauswirth WW, Li Q, Connor TB, Kuchenbecker JA, Mauck MC, Neitz J, Neitz M. Gene therapy for red-green colour blindness in adult primates. Nature. 2009;461:784–7. doi: 10.1038/nature08401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zolotukhin S, Potter M, Zolotukhin I, Sakai Y, Loiler S, Fraites TJ, Jr, Chiodo VA, Phillipsberg T, Muzyczka N, Hauswirth WW, Flotte TR, Byrne BJ, Snyder RO. Production and purification of serotype 1, 2, and 5 recombinant adeno-associated viral vectors. Methods. 2002;28:158–67. doi: 10.1016/s1046-2023(02)00220-7. [DOI] [PubMed] [Google Scholar]
- 30.Pang JJ, Boye SL, Kumar A, Dinculescu A, Deng W, Li J, Li Q, Rani A, Foster TC, Chang B, Hawes NL, Boatright JH, Hauswirth WW. AAV-mediated gene therapy for retinal degeneration in the rd10 mouse containing a recessive PDEbeta mutation. Invest Ophthalmol Vis Sci. 2008;49:4278–83. doi: 10.1167/iovs.07-1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pang JJ, Chang B, Kumar A, Nusinowitz S, Noorwez SM, Li J, Rani A, Foster TC, Chiodo VA, Doyle T, Li H, Malhotra R, Teusner JT, McDowell JH, Min SH, Li Q, Kaushal S, Hauswirth WW. Gene therapy restores vision-dependent behavior as well as retinal structure and function in a mouse model of RPE65 Leber congenital amaurosis. Mol Ther. 2006;13:565–72. doi: 10.1016/j.ymthe.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 32.Applebury ML, Antoch MP, Baxter LC, Chun LL, Falk JD, Farhangfar F, Kage K, Krzystolik MG, Lyass LA, Robbins JT. The murine cone photoreceptor: a single cone type expresses both S and M opsins with retinal spatial patterning. Neuron. 2000;27:513–23. doi: 10.1016/s0896-6273(00)00062-3. [DOI] [PubMed] [Google Scholar]
- 33.Haverkamp S, Wassle H, Duebel J, Kuner T, Augustine GJ, Feng G, Euler T. The primordial, blue-cone color system of the mouse retina. J Neurosci. 2005;25:5438–45. doi: 10.1523/JNEUROSCI.1117-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Blanks JC, Johnson LV. Selective Lectin Binding of the Developing Mouse Retina. J Comp Neurol. 1983;221:31–41. doi: 10.1002/cne.902210103. [DOI] [PubMed] [Google Scholar]
- 35.Doronina VA, de Felipe P, Wu C, Sharma P, Sachs MS, Ryan MD, Brown JD. Dissection of a co-translational nascent chain separation event. Biochem Soc Trans. 2008;36:712–6. doi: 10.1042/BST0360712. [DOI] [PubMed] [Google Scholar]
- 36.Hagstrom SA, Neitz J, Neitz M. Variations in cone populations for red-green color vision examined by analysis of mRNA. Neuroreport. 1998;9:1963–7. doi: 10.1097/00001756-199806220-00009. [DOI] [PubMed] [Google Scholar]
- 37.Vimal RL, Pokorny J, Smith VC, Shevell SK. Foveal cone thresholds. Vision Res. 1989;29:61–78. doi: 10.1016/0042-6989(89)90174-0. [DOI] [PubMed] [Google Scholar]
- 38.Hauswirth WW, Aleman TS, Kaushal S, Cideciyan AV, Schwartz SB, Wang L, Conlon TJ, Boye SL, Flotte TR, Byrne BJ, Jacobson SG. Treatment of leber congenital amaurosis due to RPE65 mutations by ocular subretinal injection of adeno-associated virus gene vector: short-term results of a phase I trial. Hum Gene Ther. 2008;19:979–90. doi: 10.1089/hum.2008.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jacobson SG, Cideciyan AV, Ratnakaram R, Heon E, Schwartz SB, Roman AJ, Peden MC, Aleman TS, Boye SL, Sumaroka A, Conlon TJ, Calcedo R, Pang JJ, Erger KE, Olivares MB, Mullins CL, Swider M, Kaushal S, Feuer WJ, Iannaccone A, Fishman GA, Stone EM, Byrne BJ, Hauswirth WW. Gene therapy for leber congenital amaurosis caused by RPE65 mutations: safety and efficacy in 15 children and adults followed up to 3 years. Arch Ophthalmol. 2012;130:9–24. doi: 10.1001/archophthalmol.2011.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Maguire AM, Simonelli F, Pierce EA, Pugh EN, Jr, Mingozzi F, Bennicelli J, Banfi S, Marshall KA, Testa F, Surace EM, Rossi S, Lyubarsky A, Arruda VR, Konkle B, Stone E, Sun J, Jacobs J, Dell’Osso L, Hertle R, Ma JX, Redmond TM, Zhu X, Hauck B, Zelenaia O, Shindler KS, Maguire MG, Wright JF, Volpe NJ, McDonnell JW, Auricchio A, High KA, Bennett J. Safety and efficacy of gene transfer for Leber’s congenital amaurosis. N Engl J Med. 2008;358:2240–8. doi: 10.1056/NEJMoa0802315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dalkara D, Byrne LC, Klimczak RR, Visel M, Yin L, Merigan WH, Flannery JG, Schaffer DV. In vivo-directed evolution of a new adeno-associated virus for therapeutic outer retinal gene delivery from the vitreous. Sci Transl Med. 2013;5:189ra76. doi: 10.1126/scitranslmed.3005708. [DOI] [PubMed] [Google Scholar]
- 42.Kay CN, Ryals RC, Aslanidi GV, Min SH, Ruan Q, Sun J, Dyka FM, Kasuga D, Ayala AE, Van Vliet K, Agbandje-McKenna M, Hauswirth WW, Boye SL, Boye SE. Targeting photoreceptors via intravitreal delivery using novel, capsid-mutated AAV vectors. PLoS One. 2013;8:e62097. doi: 10.1371/journal.pone.0062097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Long KO, Aguirre GD. The cone matrix sheath in the normal and diseased retina: cytochemical and biochemical studies of peanut agglutinin-binding proteins in cone and rod-cone degeneration. Exp Eye Res. 1991;52:699–713. doi: 10.1016/0014-4835(91)90022-7. [DOI] [PubMed] [Google Scholar]
- 44.Humphries MM, Rancourt D, Farrar GJ, Kenna P, Hazel M, Bush RA, Sieving PA, Sheils DM, McNally N, Creighton P, Erven A, Boros A, Gulya K, Capecchi MR, Humphries P. Retinopathy induced in mice by targeted disruption of the rhodopsin gene. Nat Genet. 1997;15:216–9. doi: 10.1038/ng0297-216. [DOI] [PubMed] [Google Scholar]