

Abstract
Colorectal cancer (CRC) is a heterogeneous disease with distinct molecular and clinical features, which reflects the wide range of prognostic outcomes and treatment responses observed among CRC patients worldwide. Our understanding of the CRC epigenome has been largely developed over the last decade and it is now believed that among thousands of epigenetic alterations present in each tumor, a small subgroup of these may be considered as a CRC driver event. DNA methylation profiles have been the most widely studied in CRC, which includes a subset of patients with distinct molecular and clinical features now categorized as CpG island methylator phenotype (CIMP). Major advances have been made in our capacity to detect epigenetic alterations, providing us with new potential biomarkers for diagnostic, prognostic and therapeutic purposes. This review aims to summarize our current knowledge about epigenetic alterations occurring in CRC, underlying their potential future clinical implications in terms of diagnosis, prognosis and therapeutic strategies for CRC patients.
Keywords: colorectal cancer, CIMP, epigenetic alterations, biomarkers, prognosis, treatment
1. Introduction
Colorectal cancer (CRC) is a considerable health issue worldwide. Globally, it is the third most common cancer, with an incidence of 1.4 million cases and about 700.000 deaths in 2012 (1). Unfortunately, it is predicted that the number of cases will rise by more than 60% by 2030 with an incidence of 2,2 million new cases and 1,1 million deaths (2).
Over the last decades, improved screening strategies and more effective therapies have led to a decrease in mortality rates in different countries. It has also led to an increase in the median overall survival (OS) for metastatic colorectal cancer (mCRC) patients, which has now reached more than 30 months. Nevertheless, more powerful diagnostic tools and more effective and personalized treatment are urgently needed in daily clinical practice.
In 1990, Fearon and Vogelstein proposed a model for the genetic basis of CRC (3), and since then the development and progression of CRC have been widely studied, leading to a profound knowledge about genetic and epigenetic mechanisms that play specific roles in this process. Indeed, the original multistep model considered tubular and tubulovillous adenomas as the premalignant lesions of CRC, arising mainly via APC mutations or deletions and leading to chromosomal instability. It is now recognized that approximately 30% of CRCs develop from the serrated pathway, that includes hyperplastic polyps (HPs), sessile serrated adenoma (SSA), traditional serrated adenoma (TSA). This pathway is associated with microsatellite instability, aberrant DNA hypermethylation, and BRAF mutation.
Approximately 90% of CRCs develop sporadically, and only a few cases (less than 10%) are hereditary. Familial adenomatous polyposis (FAP), hereditary non-polyposis colorectal cancer (HNPCC), MUTYH-associated polyposis (MAP), Peutz-Jeghers syndrome (PJS) and Serrated polyposis syndrome (SPS) are the main hereditary causes of CRC.
Nowadays, three major pathways for CRC development have been characterized, the most common of which is the chromosomal instability (CIN), that represents 70-80% of tumors. The second most common is the microsatellite instability (MSI) pathway, accounting for 5-20% of tumors, according to the stage of disease. The last group is the CpG island methylation phenotype (CIMP), identified by Toyota and colleagues in 1999 (4), which represents about 15% of CRCs. Recent approaches which enable comprehensive genomewide analysis of the methylome have provided extensive knowledge about aberrant methylation in different types of tumors. By focusing on the state-of-the-art of epigenetic alterations in CRC, our review will contribute to this considerably growing research field, leading to potential changes in crucial clinical aspects, such as early diagnosis, prognosis and treatment.
2. Molecular pathways leading to CRC
2.1 Chromosomal instability (CIN)
CIN is a hallmark characteristic of most CRC cases (80–85%), and it is characterized by extensive abnormality in chromosome number (aneuploidy) and loss of heterozygosity (LOH). CIN can be observed in several forms, including chromosomal numerical abnormalities, small sequence modifications such as base deletions or insertions; chromosomal rearrangements and gene amplification (5). CRC-related tumor suppressor genes are thought to be altered in the early phase of cancer development, and adenomatous polyposis coli (APC) mutation is the first step in the translation of normal mucosa to neoplastic tissue, leading to the activation of WNT pathway. Subsequent mutations that occur in genes, such as KRAS, TP53, SMAD4 and type II TGF-β receptor (TGFBR2), lead to the progression from polyp to cancer.
Since Vogelstein proposed the adenoma-cancer model, our knowledge about molecular pathogenesis of CRC has markedly increased. Although conventional tubular and tubulovillous adenomas are well-recognized as precursor lesions of the chromosomal instability pathway, recently a new premalignant form has been recognized: serrated polyps. The serrated neoplasia pathway develops by the accumulation of insertion or deletion mutations throughout the genome, leading to microsatellite instability-high (MSI-H) adenocarcinomas, BRAF or KRAS mutation, and CIMP that can be either low level (CIMP-L) or high level (CIMP-H) (reviewed in 6).
2.2 Microsatellite Instability (MSI)
MSI is a hypermutable phenotype caused by the loss of DNA mismatch repair (MMR) activity due to either mutations or epigenetic silencing of MLH1, MSH2, MSH6, and PMS2 genes. Most MSI CRCs have lost expression of MLH1, mainly due to acquired hypermethylation of the promoter of the MLH1 gene, which occurs in tumors with the CpG island methylator phenotype (CIMP) (7). The familial form of MSI CRC is hereditary non-polyposis CRC (HNPCC, or Lynch syndrome), which is caused by germline mutations in the mismatch repair genes MLH1, PMS2, MSH6, or MSH2, and accounts for about 3-5 % of all CRC cases.
About 15% of sporadic CRCs have MSI as a mechanism of development and progression. These patients show distinct characteristics which are important for clinical practice. Tumors with MMR deficiency exhibit a high frequency of microsatellite instability, because these regions are more susceptible to DNA mutations when MMR genes are compromised.
2.3 CpG island methylator phenotype (CIMP)
CIMP colon cancer is a unique molecular subgroup, characterized by a global genome hypermethylation in specific DNA regions, called CpG island. These are sequences greater than 200–500 bases in length with greater than 50% CpG content (8). Usually, CpG islands overlap the promoter region of 60–70% of genes and tend to be protected from methylation; however, they can become aberrantly methylated in cancer. Methylation of CpG islands within the promoter region causes transcriptional silencing, although it seems that only few methylated genes show a decreased gene expression in CRC. Many studies have expanded the idea of CpG islands to “CpG island shores,” which are also abnormally methylated in cancer. CpG island shores are regions of DNA with a low density of CpG dinucleotides that are up to 2 Kb upstream of a CpG island. The methylation of CpG island shores is correlated with transcriptional inactivation and expression of splice variants (9). More details will be addressed below.
3. DNA methylation
Epigenetics is commonly defined as changes in gene functions that are heritable during cell division and that cannot be explained by alteration in DNA sequence. Different epigenetic mechanisms are considered to have a role in cancer development, such as DNA methylation, histone modifications, nucleosome positioning and non-coding RNAs, specifically microRNA expression (10). In fact, the most widely studied epigenetic alteration in cancer is aberrant DNA methylation.
DNA methylation in mammals occurs primarily at CpG residues (fig. 1): genome wide, 60–80% of the CpG residues are methylated. However, in CpG islands and active regulatory regions, only 10% of the CpGs are methylated (11).
Figure 1. DNA methylation and DNA demethylation pathways.
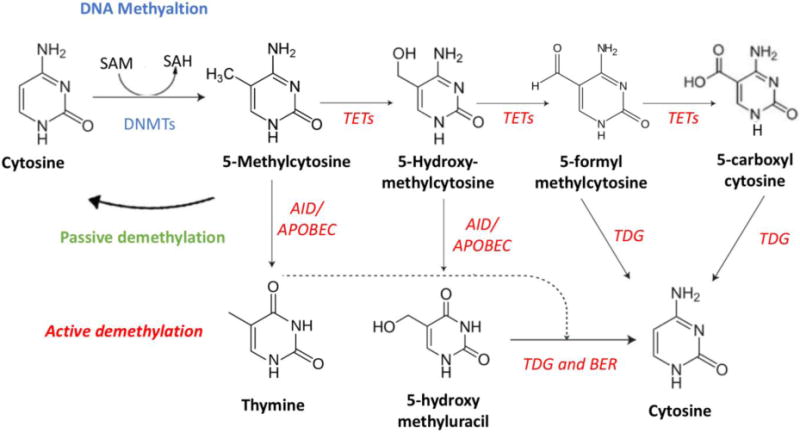
DNA methylation (high left corner – blue characters) and DNA demethylation (active - red italic characters; passive - green characters) pathways. See text for details.
SAM: S-Adenosyl methionine; SAH: S-adenosylhomocysteine; DNMT: DNA methyltransferase; AID: activation-induced deaminase
APOBEC: apolipoprotein B mRNA-editing enzyme complex; TDG: thymine-DNA glycosylase; BER: Base excision repair
The methylation of the 5-carbon on cytosine residues (5mC) in CpG dinucleotides was the first described covalent modification of DNA and is the most extensively characterized epigenetic modification. DNA methylation is critical for genome stability, proper embryonic development, X chromosome inactivation, genomic imprinting, and the silencing of retrotransposons (12).
The role of DNA methylation in controlling the activities of gene promoters, whether CpG islands or non-CpG islands, has been extensively investigated over the past 3 decades. Thanks to the improvement of genome-wide mapping technologies and the possibility to analyze DNA methylation in different genomic regions, it is now believed that the function of DNA methylation varies depending on the context, underlying a level of complexity that should be addressed in the future (13).
The methylation process involved a family of enzymes called DNA methyltransferase (DNMTs). They are responsible for the transfer of a methyl group from S-adenosyl-L- methionine (SAM), to the 5-position of cytosine residues in DNA. DNMTs gene family consists of five members: DNMT1, DNMT2, DNMT3A, DNMT3B and DNMT3L. In fact, DNMT3L does not possess any inherent enzymatic activity. Generally, the favored substrate for DNMTs is a CpG dinucleotide sequence. In normal mammalian cells, most CpGs are methylated, with unmethylated CpGs being typically present only in regions of DNA called CpG islands (8). More controversial is the methylation that does not occur in CpG island, called gene-body methylation. In these regions, DNA methylation seems to be correlated with transcriptional activation, instead of genome silencing (14). Indeed, gene-body DNA methylation increases gene expression and the rapid establishment of this is dependent on the presence of the DNMT3B. Since DNA methylation inhibitors (such as azacitidine and decitabine) induce DNA demethylation across all genomic features, these drugs may affect both the hypermethylated DNA gene promoter and gene-body methylation: this process can lead not only to the direct reactivation of tumor suppressor genes but also to the downregulation of oncogenes and metabolic genes. Therefore, gene body DNA methylation might be an intriguing additional target for epigenetics therapy in the future.
DNMT1 is responsible for maintenance methylation, while DNMT3A and DNMT3B regulate de novo methylation. Maintenance methylation occurs during DNA replication, and refers to the replication of the methylation pattern of the unreplicated strand of DNA onto the newly replicated strand of DNA; de novo methylation refers to the methylation of DNA without the use of a DNA template that carries an existing methylation pattern (15). The regulation of these enzymes occurs at different levels: transcriptional, translational and post-translational. For example, p53 transcriptionally suppresses DNMTs through binding with Sp1 protein to the DNMT promoters. RB transcriptionally suppresses DNMT1/3A through binding with E2F1 protein to the DNMT1 and 3A promoters. FOXO3a binds to the FOXO3a DNA element of the DNMT3B promoter to repress DNMT3B transcription. In addition, overexpressed MDM2 may induce DNMT1, DNMT3A, and DNMT3B expression by negative control over p53, RB and FOXO3a (16).
DNA methylation-mediated transcriptional silencing can be obtained via multiple mechanisms. One is the direct inhibition of cis-binding elements, such as the transcriptional factors activating protein 2 (AP-2), E2 promoter binding factor (E2F), Core Binding Factor (CBF), nuclear factor kappa light-chain-enhancer of B-cells (NF-kB), cAMP response element-binding protein (CREB) and CCAAT enhancer-binding protein C/EBF (8). One other important mechanism is the alteration of chromatin structure through the interaction with proteins such as: the zinc finger proteins Kaiso, ZBTB4 and ZBTB38; the SET- and RING-finger associated proteins, UHRF1 and UHRF2; and methyl-CpG domain-binding protein (MBD), including MeCP2, MBD1, MBD2, MBD3, and MBD4 (17). They recruit proteins which eventually lead to a compacted chromatin environment that represses gene expression.
It has been previously demonstrated that epigenetic alterations, and specifically DNA methylation, occur in the early phase of tumor development and in the transition from normal mucosa to adenomatous polyp. These findings suggested that epigenotype development occurs at an earlier stage than carcinoma formation, and is already completed at the adenoma stage. Recent comprehensive genome-wide methylations analysis revealed that DNA methylation may be a useful tool for the diagnosis, prognosis and prediction of response to therapy in CRC (18).
4. DNA demethylation
On the one hand, DNA hypermethylation in CpG islands has been shown to promote CRC by silencing the expression of tumor suppressor genes. On the other hand, global DNA hypomethylation is now considered a common characteristic of CRC. In fact, since it has been discovered in 1983 (19) there is ample evidence to highlight its role in promoting genomic instability and proto-oncogenes activation.
While there is considerable knowledge about DNA methylation mechanisms and their respective genes, the pathway surrounding DNA demethylation has to be fully described yet (Fig. 1). Since 2009, a crucial family of enzymes involved in oxidizing 5mC has been characterized, known as the TET family (20). The three mammalian proteins TET, namely TET1, TET2 and TET3, are Fe2+- and 2-oxoglutarate-dependent dioxygenases that successively oxidize 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), and then can further oxidize 5hmC to 5-formylcytosine (5fC) and 5-carboxylcytosine (5caC) in DNA (reviewed in 21). In addition, TET activity is increased by vitamin C, which induces a TET- dependent DNA demethylation.
Different demethylation pathways have been proposed, both passive and active. In the passive pathway, 5hmC is not recognized by DNMT1 during replication, therefore DNMT1, that is responsible for maintenance methylation, is not able to replicate the methylation pattern in the new daughter DNA strand, leading to a loss of DNA methylation after DNA replication. In the active pathway, 5hmC can be deaminated by activation-induced deaminase (AID)/apolipoprotein B mRNA-editing enzyme complex (APOBEC) and transformed in 5-hydroxymethyluracil (5hmU), which can be replaced with cytosine by base excision repair (BER) mechanism (22). This involves at least 11 different DNA glycosylases, such as thymine-DNA glycosylase (TDG), Single-Strand-Selective Monofunctional Uracil-DNA Glycosylase 1 (SMUG1), Methyl-CpG Binding Domain 4 (MBD4). Finally, 5caC and 5fC are specifically recognized and excised by TDG (23).
As mentioned above, α-ketoglutarate (α-KG) is needed for the right function of TETs protein. α-KG is provided by isocitrate dehydrogenase (IDH) enzymes through oxidation of isocitrate. It has been discovered that IDH mutation leads to generate 2-hydroxyglutarate (2-HG) (24), which eventually inhibits the TETs dioxygenases (25). IDH-1 and IDH-2 mutations have been identified in rare tumors, such as glioma, but also in chondrosarcoma, cholangiocarcinoma, a small proportion of prostate cancers, and angioimmunoblastic T-cell lymphomas (AITL) (26), while TET mutations are found in leukemias, where they were originally discovered (27).
Although IDH and TET mutations do not seem to be common in CRC, recently TET1 expression downregulation has been noted to be an early event in cell transformation and to be related to colon cancer growth by leading to a constitutive activation of the WNT pathway (28). Many other studies are warranted to identify the impact of TET enzymes in CRC carcinogenesis and to figure it out if they may be a potential target for future treatment.
It has been shown that long interspersed nucleotide element-1 (LINE-1) methylation measured by pyrosequencing is reproducible and well correlated with global DNA methylation level (29). LINE-1 is a retrotransposon that has been widely correlated with important CRC features involved in its carcinogenesis: an inverse relation has been demonstrated between LINE-1 hypomethylation and MSI-H and CIMP-H; whereas 18q loss of heterozygosity (LOH) is associated with LINE1 hypomethylation. More importantly, LINE-1 methylation levels have been related to prognosis: hypomethylation confers poor prognosis in terms of colon cancer-specific mortality (CCSM) and overall mortality (OM) (30). In addition, LINE-1 hypomethylation in microsatellite stable (MSS) / CIMP+ stage II and III CRC had predictive value for benefit from adjuvant chemotherapy with oral fluoropyrimidines (31). Although these results seem to be promising, these findings need further validations in order to be considered for clinical purposes. However, it suggests the DNA demethylation, as well as DNA methylation, may play a critical role in CRC carcinogenesis, prognosis and response to treatment.
5. The CpG Island Methylator Phenotype (CIMP)
Since 1999, DNA methylation in CpG islands has been recognized as one of the three major CRC oncogenesis pathways and the CpG Island Methylator Phenotype (CIMP) represents a distinct group of CRCs, characterized by specific epidemiology, histology, molecular features and prognosis (32). In the last two decades, several ways to classify CRCs using DNA methylation pattern have been proposed. Toyota and colleagues, when first discovered this subset of CRCs (4), they identified MINT clones, p16, THBS1, and hMLH1 genes as the main targets of DNA methylation. In addition, the MLH1-promoter hypermethylation is the fundamental molecular basis for MSI in sporadic MSI CRC, therefore most MSI CRCs are CIMP+. Subsequent studies revealed the main features of CIMP+ CRCs: preferentially located in the right colon, related to female sex and older age, harboring BRAF V600E mutation, associated to MSI-H by MLH1 epigenetic silencing due to promoter DNA hypermethylation, diploid copy number and the absence of TP53 (33).
Ogino et al. (34) identified CIMP-low subgroup of CRCs. Using real-time polymerase chain reaction (MethyLight), they quantified DNA methylation in five CIMP-specific gene promoters (CACNA1G, CDKN2A, CRABP1, MLH1, and NEUROG1), and CIMP-low CRC were defined as 1/5 to 3/5 methylated promoters: this subgroup is characterized by an association with male sex and KRAS-mutation, suggesting that CIMP-low may be considered a different subtype of CRC from CIMP-high (with >4/5 methylated promoters) and CIMP-0 (with 0/5 methylated promoters).
Shen and coworkers (35) highlighted three distinct groups of CRCs namely CIMP1, CIMP2, and CIMP-negative, based on an unsupervised hierarchical clustering of the DNA methylation data of 27 genes (including MINTs, p16, SOCS1, RUNX3 among others): CIMP1 are characterized by MSI (80%) and BRAF mutations (53%); CIMP2 is associated with 92% KRAS mutations; CIMP-negative cases have a high rate of p53 mutations (71%) and lower rates of MSI (12%) or BRAF-mut (2%) or KRAS-mut (33%).
Yagi and colleagues (36) demonstrated that CRC can be classified into three different epigenotypes: high-, intermediate-, and low-methylation genotypes (HME, IME and LME). HME is strongly correlated with BRAF mutation (71%) and MSI-H (76%), while IME showed correlation with KRAS mutation (63%).
Finally, Hinoue et al. (37) identified four DNA methylation-based subgroups of CRC using model-based cluster analyses: CIMP-high and CIMP-low overlapped with the previous classification showing association with MLH1 DNA hypermethylation, BRAFV600E mutation and KRAS mutations, respectively. Non-CIMP tumors were separated into two distinct clusters. One non-CIMP subgroup is distinguished by a significantly higher frequency of TP53 mutations and frequent occurrence in the distal colon, while the fourth group exhibits a low frequency of both cancer-specific DNA hypermethylation and gene mutations and were significantly enriched for rectal tumors.
Until now, a global consensus does not exist to define CIMP status. In fact, two distinct genes panels have been proposed: the Ogino scoring panel includes 8 genes (38) (RUNX3, CACNA1G, IGF2, MLH1, NEUROG1, CRABP1, SOCS1, and CDKN2A), whereas the Weisenberger panel includes 5 of the above-mentioned genes (CACNA1G, IGF2, NEUROG1, RUNX3 and SOCS1) (39). A recent study has demonstrated that there is a statistically significant variation in the frequency of CIMP depending on the panel of genes used. This highlights that a universal agreement on a qualitative, quantitative and technical level is warranted in order to improve the clinical translation of the role of CIMP in CRC patients (40).
CIMP status is a promising prognostic marker for CRCs, even if contradictory results have been reported by several studies, mainly due to an overlap between the CIMP+ phenotype and the microsatellite instability phenotype, associated in 50% of cases with BRAF mutation (41). In the adjuvant setting, different studies showed CIMP+ to be associated with shorter OS as compared to CIMP− (42) in patients treated with surgery alone (43) and significantly worse disease free survival (DFS) for CIMP+ subgroup with BRAF mutation and proximal tumor location (44). Five other studies have investigated the prognostic value of CIMP+ mixing stage II and III CRCs: three showed a decrease in DFS in the CIMP+ group (45, 46, 47), although no significant difference in DFS between CIMP+ and CIMP− was noticed in the other two (48, 49). Donada et al. (50) specifically studied the prognostic impact of CIMP in stage II CRC and found an OS benefit of adjuvant 5-FU in CIMP+ patients. Finally, Jo et al. (51) investigated 150 patients with locally advanced rectal cancer, and showed poor DFS in CIMP+ patients, even if CIMP-high was found in only 10% of patients.
CIMP+ advanced CRC has been correlated with poor prognosis (52, 53, 54), although Ogino et al. (55) showed that in 649 stage I-IV colon cancer patients, CIMP-high status appears to be an independent predictor of a low colon cancer-specific mortality.
More recently, a comprehensive meta-analysis (56) has demonstrated that CIMP is significantly associated with shorter DFS (pooled HR estimate 1.45; 1.07–1.97) and OS (pooled HR estimate 1.43; 1.18–1.73) among CRC patients irrespective of MSI status.
The predictive value of CIMP remains controversial, although different studies showed improved OS or DFS in CIMP+ CRC patients after 5-FU treatment compared to non-CIMP patients (43, 50, 57).
Lately, a new Consensus Molecular Subtypes (CMS) has been developed, integrating six previously independent molecular classifications (58): CIMP-H status is enriched in CMS1 among other characteristics (such as hypermutational load, MSI, BRAF mut, female sex, right-sided). Importantly, CMS1 has been related to a worse OS and progression free survival (PFS) in comparison to the other CMS subtypes (OS 15.0 months in CMS1 vs 40.3 in CMS2, p <.0001; PFS 7.1 months vs 13.4 in CMS2, p <.0001 in CALGB 80405 clinical trial), showing a benefit from bevacizumab vs cetuximab treatment (59).
Despite CIMP status being one of the most promising prognostic and predictive factors in CRC, controversial results, along with the lack of a global consensus regarding CIMP status definition make it difficult to utilize CIMP status in clinical practice.
6. Other Epigenetic alterations in CRC
6.1 MicroRNA (miRNA)
In the last decade, our knowledge about miRNAs and their role in the context of carcinogenesis has increased. miRNAs are single-stranded short RNA molecules of 20–25 nucleotides in length that regulate post-transcriptional silencing by either binding to the target mRNA and causing its degradation or inhibiting its translation into protein (60). Each mRNA can be targeted by more than one miRNA, and each miRNA can target hundreds of different transcripts. The first description and demonstration of the role of miRNA in cancer were in 2002 when Calin and coworkers investigated that miR-15 and miR-16 were located in a region frequently deleted in chronic lymphocytic leukemia (61).
miRNAs are regulated at different levels and their expression is altered in human cancers by different mechanisms, one of which is epigenetic alteration: miRNA are modulated both by hypermethylation and hypomethylation.
The first evidence that methylation status affects noncoding RNAs, was provided by Saito and colleagues (62): they showed that miR-127 expression was about 49 times greater in cells treated with both a DNA demethylating agent (5-aza-20-deoxycytidine, 5-AZA) and a histone deacetylases (HDAC) inhibitor (4-phenylbutyric acid), and that reduced the expression of the oncogene BCL6, one of its direct target genes.
Hundreds of miRNAs have been shown to be related to cancer development and progression and their deregulation can lead to alteration of the expression of important oncogenes and tumor-suppressor genes.
For instance, the most important pathways involved in CRC pathogenesis are regulated by miRNAs. These include the WNT pathway, which is regulated by miR-145, miR135b; RAS/MAPK pathway regulated by miR143, and let-7 and PI3K/AKT pathways regulated by miR-1 and miR21 (reviewed in 63).
Recently, several studies have shown that miRNAs are frequently deregulated in CRC via aberrant DNA methylation. In addition, aberrant DNA methylation of miRNA genes shows promising clinical applications, such as biomarkers for early screening, prognosis, and therapeutic targets in CRC (64).
mi-R124a has been the first miRNA silenced by DNA hypermethylation to be described (65) in CRC. After that, many other studies demonstrated this way to silence miRNA that eventually leads to dysregulation of the expression of crucial genes involved in CRC carcinogenesis. For instance, miR-342 silencing by DNA hypermethylation resulted in a dramatic reduction of the expression of DNMT1: this, in turn, reactivated ADAM23, Hint1, RASSF1A and RECK genes via promoter demethylation (66). In addition to this, methylation of the miR-137 promoter is an early event in CRC carcinogenesis, and since one of its target is Lysine specific demethylase 1 (LSD1), a histone demethylase, this provides novel evidence for the cross-talk between miRNAs and other components of the epigenetic machinery (67).
A recent and comprehensive review incorporated 103 studies from 36 articles with a total of 3124 CRC patients and 2579 healthy individuals. It revealed that miRNAs can exert relatively high screening and diagnostic accuracy for CRC with an overall sensitivity of 0.769, specificity of 0.806, and AUC of 0.857 (68). Undoubtedly miRNA deregulation is involved in CRC carcinogenesis, but to what extent is still unclear. This promising and fascinating field of research needs further studies in order to use miRNAs in clinical practice.
6.2 Histone modification
Another important epigenetic modification is chromatin alteration, mainly due to the acetylation level of histone proteins. While hypoacethylation histone is found in transcriptionally inactive heterochromatin, histone acetylation is a hallmark of euchromatin (active regions). The first evidence of histone modification in CRC was discovered by Fraga and colleagues (69) and since then, other studies suggested that histone alterations can lead to a dysregulation of oncogenic pathways. This has shed lights on the complex interplay between chromatin alteration and carcinogenesis. For example, histone covalent modifications can be affected by oncogenic RAS pathways to regulate the expression of target genes like Cyclin D1 or E-cadherin (70).
Methylation of histone tails has been largely documented in CRC primary tumors and cell lines, including loss of trimethylation of H3K20, and di- and tri-methylation of H3K4 (H3K4me2/me3), H3K9 (H3K9me2/me3) and H3K27 (H3K27me2/me3) (71). In addition to this, it has been discovered that H3K27me2 (72) and H3K4me2 (73) correlated with poorer survival rates in mCRC.
As mentioned above, LSD1 is a histone demethyltransferase protein that catalyzes the demethylation of monomethylated and dimethylated histone H3 lysine4 (H3K4) and H3 lysine 9 (H3K9) through a redox process, and it showed a significantly higher expression, in colon cancer specimens classified as high TNM stage lesions and with distant metastasis. Moreover, it has been related to a worse colon cancer prognosis (74).
However, protein modifications are less stable biomarkers than DNA methylation, and the diagnostic potential of histone tail modifications in CRC is limited when compared with DNA methylation biomarkers that can be easily detected in circulating blood DNA (75). Nonetheless, the development of histone deacetylases inhibitors (HDACi) led to the FDA approval of vorinostat, romidepsin and belinostat for hematological malignancies, and some of them are under investigation in combination with other compounds in CRC and other solid tumors (clinicaltrials.gov).
7. Clinical implications for CRC patients and future directions
7.1 Biomarkers for early CRC detection
Technological advancements have considerably augmented our capacity to detect a wide number of epigenetic alterations that can be eventually used as clinical biomarkers for early detection, prognostic and predictive purposes in patients with CRC. Promoter DNA hypermethylation of specific CRC driver genes may be identified in tools, urine and blood as diagnostic biomarkers and some of them are already available for clinical use. Some examples of these genes are MLH1, CDKN2A, MGMT, among others that have been identified in human stools sample in several studies (76); VIM, WIF1, ALX4 and NDRG4 have been detected in urine (77) and MLH1, APC, MGMT, RASSF2A and TMEFF2 among others in blood samples. Investigating the methylation status of circulating DNA, some of them (SEPT9, ALX4, SDC2, RUNX3, TMEFF2, NEUROG1) present high sensitivity and specificity for CRC detection during initial stages (78). Epigenetic and genetic alterations can be detected in blood sample, as circulation cell-free DNA (cfDNA) and circulating tumor cell (CTC). The incredible progress made in “liquid biopsies” has provided us with new cost- effective and sensitive biomarkers. However, the clinical value of all these factors is not defined, and larger studies are warranted to elucidate the role and the potential application in the patient management, in terms of early diagnosis, but also prognosis and treatment resistance.
Currently, the most investigated and well-established blood-based diagnostic biomarker is the detection of aberrant hypermethylation at the promoter region of SEPT9 gene V2 transcript. The first description by Grützmann et al. (79) showed a sensitivity of 72% and specificity of 90% in detection in plasma of CRC patients. Since then, numerous studies have investigated the performance of SEPT9 assay, where the sensitivity varies between 54.1% and 95.6%, with specificity between 81.5% and 99.1% (reviewed in 80) (Tab. 1). Recently in 2016, FDA approved Epi proColon® for CRC screening test, and it was already available in Europe and some other countries. In order to improve both sensitivity and specificity of early CRC detection, many researchers tried to combine SEPT9 with others methylated genes. For instance, Kostin et al (81) showed that the panel of integrated genes (HTLF, ALX4 and SEPT9) had a diagnostic sensitivity of 74-88% and a specificity of 90-96%.
Tab. 1.
Currently available epigenetics biomarkers for CRC.
| Biomarker | Test | Source and purpose | Sensitivity % |
Specificity % |
ref |
|---|---|---|---|---|---|
| SEPT9 | proColon® | blood-based diagnostic test | 54-96 | 82-99 | 80 |
| VIM | ColoSure™ | stool-based diagnostic test | 72-77 | 83-94 | 85 |
| KRAS, NDRG4, MBP3 | Cologuard™ | stool-based diagnostic test | 92 | 73 | 86 |
More recently, Tham et al. (82) have demonstrated that high serum methylation levels of TAC1 at 6-month follow-up and SEPT9 at 1-year follow-up were independent predictors for tumor recurrence and unfavorable cancer-specific survival (CSS) (P <.05 in all tests), in 150 patients with stage I-III CRC. Combined test with multiple biomarkers including SEPT9 methylation may be a future option to improve CRC screening performance and compliance, although more data are needed before it can be considered part clinical practice.
Since blood-based DNA methylation assays are non-invasive and cost-effective in comparison to colonoscopy, they may be used as a substitute for screening and diagnostic purposes in the near future, leading to greater screening compliance in the general population. Regarding the fecal-based tools, an expanding list of hypermethylated genes in stool has been validated during the past decades (reviewed in 83). Among them, methylated SFRP2 has been identified as a diagnostic biomarker with a sensitivity of 77-90% and a specificity of 90%. More importantly, CRC patients with SFRP2 hypermethylation in tumor, stool and serum samples had a significantly shorter OS than those negative for SFRP2 methylation (p=0.0216, 0.0219, and 0.0255, respectively) (84).
Two stool-based CRC screening tests are currently available: ColoSure™ (Lab Corp, Burlington, NC) and Cologuard™, which recently received FDA approval (Tab. 1).
ColoSure™ is a single-marker test that detects methylation of the vimentin gene (VIM) (85). Several studies have shown methylated VIM as a good diagnostic biomarker in stool samples both for adenomas detection and cancer detection, even if sensitivity and specificity among the studies varied considerably. In addition, the clinical validity of this assay is still unclear, and its use within the general (average-risk) population is not recommended.
Cologuard™ is a stool-based DNA diagnostic test that includes quantitative molecular assays for KRAS mutations, aberrant NDRG4 and BMP3 methylation, β-actin and a hemoglobin immunoassay (86). In this study, almost 10,000 patients were enrolled and this assay was compared with fecal immunochemical test (FIT). The sensitivity for detecting CRC was 92.3% (93.3% for stage I-III) with DNA testing and 73.8% with FIT (73.3% for stage I-III) (p=0.002). Nonetheless, the specificity was 86.6% with DNA testing and 94.9% with FIT. People who prefer non-invasive testing may consider this test as a valuable option, although colonoscopy remains the gold standard for CRC screening.
7.2 Epigenetics modifications with prognostic and/or predictive values
Hundreds of genes have already been shown to be hypermethylated in CRC, whereas it is thought that only a small number of these genes can have a distinct role in CRC carcinogenesis.
In addition, DNA methylation occurs early in CRC and some of these genes commonly undergo an age-dependent methylation, such as HPP1, p16, APC, AXIN2, SFRP1, SFRP2, SFRP4, N33 and DKK1. Other common hypermethylated genes in CRC include MLH1, MGMT, RASSF1, SLC5A8, RUNX3, CDH-1 and 13, CDKN2A, CXCL12, VIM, SEP9, among others (Tab. 2).
Tab. 2.
Examples of commonly aberrantly methylated genes in CRC
| Gene | Protein | Effect of aberrant methylation |
|---|---|---|
| APC | Adenomatous polyposis coli | increased of WNT signaling |
| ADAM23 | Disintegrin and metalloproteinase domain- containing protein 23 | alteration in cell-cell and cell-matrix interactions |
| CDKN2A | p14/ARF | decreased p53 activation |
| CDKN2A | p16/INK4a | increased cell proliferation |
| CDH1 | E-cadherin | loss of cell adhesion |
| CDH13 | cadherin-13 | increased PI3K/Akt/mTOrR signaling |
| CXCL12 | C-X-C Motif Chemokine Ligand 12 | increased tumor cell metastases |
| DCC | deleted in colorectal cancer | inhibition of apoptosis |
| DAPK | death associated protein kinase | interferon gamma signaling and TNF alpha signaling |
| HLTF | Helicase-like transcriptor factor | alteration in chromatin structure |
| HACE1 | HECT domain and ankyrin repeat-containing ubiquitin ligase | loss of tumor suppressor gene |
| IRF8 | Interferon Regulatory Factor 8 | interferon signaling |
| MLHl† | MutL Homolog 1 | microsatellite instability |
| MGMT | O-6-Methylguanine-DNA Methyltransferase | Increased G>A mutation frequency |
| MINT clones | Methylated in tumor locus | NA |
| RASSFla | Ras association domain family 1 (isoform A) | Increased RAS/RAF/MAPK signaling |
| RUNX3*† | Runt-related transcription factor 3 | Decreased TGF-beta signaling |
| SOCS1*† | Suppressor Of Cytokine Signaling 1 | increased of JAK/STAT3 signaling |
| SFRP1-2 | Secreted Frizzled Related Protein 1 | Increased WNT signaling |
| SEPT9 | Septin 9 | Impaired cytokinesis |
| SERF1 | Small EDRK-Rich Factor 1A | Increased WNT signaling |
| VIM | Vimentin | NA |
| NEUROG1*† | Neurogenin 1 | Increased WNT signaling |
| CRABPl† | Cellular Retinoic Acid Binding Protein 1 | Increased WNT signaling |
| IGF2*† | Insulin Like Growth Factor 2 | Increased WNT signaling |
| CACNA1G*† | Calcium Voltage-Gated Channel Subunit Alpha1 G | Increased WNT signaling |
| SLC5A8 | Solute Carrier Family 5 Member 8 | NA |
Ogino’s panel to define CIMP status
Weisenberger’s panel to define CIMP status
Khambata-Ford et al. (87) have discovered that patients with overexpression of the EGFR ligands epiregulin and amphiregulin are more likely to have disease control on cetuximab treatment and significantly longer PFS. These results have been confirmed by Jacobs et al. (88) who have shown a significant association between AREG/EREG expression and Cetuximab response and outcome. More recently, Lee et al. (89) have highlighted that EREG and AREG expression had a strong inverse correlation with methylation and was inversely associated with right-sided tumor, BRAF mutation and CIMP-high status. They also noticed that treatment with hypomethylating agents (azacitidine or decitabine) increased expression of EREG. In addition, CIMP-high status was associated with inferior PFS, also in BRAF/NRAS wild-type patients. Discovering that promoter DNA methylation is the main regulatory mechanism of AREG/EREG expression may explain the association between CIMP-status, right-sided tumor location and anti-EGFR response in mCRC. In addition, Scartozzi et al. (90) previously reported that EGFR promoter DNA methylation occurs in 58% of primary colon tumors and that it is strongly correlated with shorter OS and PFS (PFS 2.4 vs 7.4 months, p<0.0001; OS 6.1 vs 17.8 months, p<0.0001). Only circa 50% of patients with RAS-WT mCRC benefit from anti-EGFR treatment, and DNA methylation may partially account for this resistance. However, the exact reason for EGFR-resistance is still unclear. Altogether, these evidences suggest that combination of demethylation agents with anti-EGFR compounds may have a synergistic therapeutic effect.
RASSF1A is a tumor suppressor gene involved in RAS signaling pathway and Hippo pathway. In addition, it has been found to regulate the EGFR ligand amphiregulin by hippo pathway activation. The most common contributor to loss or reduction of RASSF1A function is promoter DNA methylation. RASSF1A promoter methylation occurs predominantly in KRAS wild-type CRCs. In addition, DNA methylation of the RASSF1A, associated with p14ARF and APC1A genes, defines a poor prognostic subset of CRC patients independent of both tumor stage and differentiation (91). The central role of RASSF1A in both RAS and Hippo pathways makes it an intriguing candidate for novel drug development, especially for KRAS WT CRC patients. Furthermore, Hippo pathway activation through YAP1 oncogene has been related to Cetuximab resistance (92). Since loss of RASSF1A is necessary for YAP1 activation, this reveals another potential cause for anti-EGFR resistance in patients with KRAS WT CRC, strengthening the rationale for combining demethylating agents with anti- EGFR compounds. Future analyses are warranted to evaluate if RASSF1A can be used as a biomarker for predicting efficacy of cetuximab in KRAS WT patients.
Of special interest is the DNA repair gene O6-methylguanine-DNA methyltransferase (MGMT) that removes alkylating groups from O6-guanine and that is frequently methylated in CRC. The DNA repair protein encoded by the MGMT gene is involved in defending cells against alkylating agents and has been shown to be silenced by promoter hypermethylation in more than 40% of mCRC and in normal adjacent mucosa (93). Alterations in the MGMT gene impair the ability of the MGMT protein to remove alkyl groups, potentially enhancing the susceptibility to the cytotoxic effects of alkylating agents, such as temozolomide and dacarbazine. For this reason, different phase II clinical trials (reviewed in 94) showed promising results, evaluating the efficacy of alkylating agents in mCRC, using MGMT deficiency as a predictive biomarker.
As highlighted above, numerous other epigenetics alterations (e.g. CIMP status, LINE1 hypomethylation, histone modification, miRNA) showed promising results, but unfortunately, none of them has had direct impact in clinical practice so far. Further validating studies are warranted in order to be able to use these biomarkers in clinical practice.
7.3 Epigenetics and immuno-oncology
In the last few years, immuno-oncology has revolutionized the treatment strategies of many types of cancer (e.g. melanoma, NSCLC, head and neck, kidney, Hodgkin lymphoma among others). Immune-checkpoint inhibitors (ICI) have shown striking and impressive results, although only a minority of patients are sensitive to these drugs. In fact, the causes of the non-responsiveness remain unknown. A recent work by Ghoneim et al. (95) showed the crucial role of de novo DNA methylation in maintaining T cell exhaustion status that contributes to ICI non-responsiveness. Previous studies demonstrated that the use of DNA demethylation agents can enhance CTLA-4 blockade-mediated T cell responses (96). In addition, it has been reported that treatment of epithelial cancer cell lines (including CRC cells) with demethylating agents, namely 5-azacitidine, leads to a significant enrichment of immunomodulatory pathways (interferon signaling, antigen processing and presentation, and cytokines/chemokines) (97). Lately, Brocks et al. (98) demonstrated the genome-wide transcriptional and epigenomic consequences of DNA methyltransferases inhibitors (DNMTi) and histone deacetylase inhibitors (HDACi): cryptic transcription of thousands of treatment-induced non-annotated transcriptional start sites (TINATs) may contribute to cancer immunogenicity due to novel translated potential antigenic proteins.
Altogether, accumulating evidence shows a strong immunomodulatory role of DNA demethylating agents in cancer cells, strengthening the rationale to combine these compounds with immunotherapy (such as ICI, allogenic cancer cell vaccine, immunomodulatory compounds, etc) in cancer patients. In addition, these results shed light on the crucial interplay between epigenetic modifications, immune cells and cancer cells, revealing new potential strategies to overcome both ICI non-responsiveness and acquired resistance.
Indeed, early phase clinical trials combining ICI and demethylating agents are ongoing in different type of cancers, including CRC (tab 3).
Tab. 3.
Main ongoing clinical trials investigating DNA methyltransferases inhibitors (DNMTi) and histone deacetylase inhibitors (HDACi) in colorectal cancer.
| N | Drug | Phase | Design | Status | Trial Identifiers |
|---|---|---|---|---|---|
| 1 | Azacitidine and Entinostat | II | Open Label; Azacitidine and Entinostat in Patients with mCRC | Completed | NCT01105377 |
| 2 | Decitabine | I/Proof of Principle Study | Open Label; Pre-operative Decitabine in Colon Cancer to assess whether short-course preoperative treatment with decitabine can increase Wnt target gene expression as measured in resected tumors compared to pretreatment biopsies | Recruiting | NCT01882660 |
| 3 | Cyclophosphamide (CY), Allogenic Colon Cancer Cell Vaccine (GVAX), and SGI-110 | I/Pilot Study | Randomized; SGI-110 in Combination with GVAX and CY in mCRC as Maintenance Therapy | Recruiting | NCT01966289 |
| 4 | Azacitidine and CAPOX | I/II | Open label; to assess MTD of Azacitidine and CAPOX and its efficacy in mCRC Enriched for Hypermethylation of CpG Promoter Islands | Completed | NCT01193517 |
| 5 | Pembrolizumab and Azacitidine | II | Open Label; to assess the activity, safety, and tolerability of Pembrolizumab + Azacitidine in chemo-refractory mCRC | On-going, but not recruiting | NCT02260440 |
| 6 | Epacadostat in Combination with Pembrolizumab and Azacitidine | I/II | Open label, single-arm; to assess safety, tolerability and anti-tumor efficacy of Epacadostat in combination with Pembrolizumab plus Azacitidine in patients with chemo-refractory MSS mCRC. | Not open yet | NCT03182894 |
| 7 | Azacitidine and Durvalumab | II/Basket Study | Open label; to assess the antitumor activity of azacitidine with durvalumab in MSS-CRC and others solid tumors | Recruiting | NCT02811497 |
| 8 | Decitabine and Panitumumab | I | Open label; to assess safety, feasibility and activity of Decitabine and Panitumumab in 2nd or 3rd line for KRAS WT mCRC | Completed | NCT00879385 |
| 9 | Decitabine by HAI | I/II | Open label; to establish the recommended dose and activity of Decitabine by HAI in liver-predominant mCRC | Recruiting | NCT02316028 |
| 10 | SGI-110 Combined With Irinotecan vs Regorafenib or TAS-102 | I/II | Randomized open label; to assess safety and activity of SGI-110 Combined with Irinotecan versus Regorafenib or TAS-102 in refractory mCRC | Recruiting | NCT01896856 |
mCRC: metastatic colorectal cancer; CY: cyclophosphamide; GVAX: Allogenic Colon Cancer Cell Vaccine; CAPOX: capecitabine and oxaliplatin; MTD:
Maximum Tolerated Dose; MSS: microsatellite stable; WT: Wild Type; HAI: Hepatic Arterial Infusion.
8. Therapeutic challenges
Epigenetic alterations are involved in every phase of CRC development and progression, from pre-neoplastic lesion to metastatic disease. Mounting evidence and wider knowledge about DNA methylation, histone modifications and miRNA regulation suggested that CRC patient may benefit from epigenetic therapies.
Among DNA methylation inhibitors the most investigated agents are the DNMTi: 5- azacytidine (azacitidine or 5-azaCR or Vidaza) and its deoxy derivative 5-aza-2- deoxycytidine (5-azaCdR or decitabine). Both incorporate into DNA and form irreversible covalent bonds with DNA-methyltransferases at cytosine sites targeted for methylation. Vidaza and Decitabine were approved by FDA in 2004 and 2006, respectively, for the treatment of myelodysplastic syndrome (MDS). Later, another compound enlarged this family: Guadecitabine (SGI-110) (formerly S110).
HDACi target the catalytic sites of histone deacetylase, leading to the accumulation of acetylated histones, which induce transcriptional and related molecular effects culminating in cycle arrest and apoptosis. Vorinostat has been the first of the new HDACi to be approved by the FDA for the treatment of cutaneous T-cell lymphoma patients. Numerous evidences have shown that HDACs play a crucial role in the regulation of colon cancer, therefore it is believed that CRC patients may benefit from this therapeutic strategy. Indeed, LaBonte et al. (99) have demonstrated the potential activity of dual EGFR/HER2 inhibitor (lapatinib) in combination with the HDACi Panobinostat in colon cancer cells, warranting further clinical investigation to evaluate the efficacy of this combination for CRC treatment.
In 2013, Garrido-Laguna et al. (100) investigated safety and efficacy of decitabine in combination with panitumumab (monoclonal antibody - mAb anti-EGFR) in 20 wild-type KRAS mCRC patients, showing 10% of partial response (PR) and 50% stable disease (SD). Unfortunately, two subsequent and more recent studies have not found any clinical activity. The first study investigated the combination of 5-Aza-CR and CAPOX (capecitabine + oxaliplatin) in CIMP-high CRC patients (101), and the second study evaluated the combination of 5-Aza-CR and entinostat, a histone deacetylase inhibitor (102).
Vitamin C has gained widespread attention in the last few years due to its impact in CRC. Indeed, vitamin C selectively kills KRAS and BRAF mutated CRC cells through inhibition of glyceraldehyde 3-phosphate dehydrogenase (GAPDH), which leads the cell towards metabolic stress and eventual apoptosis (103). In addition, Vitamin C and 5-azacitidine showed synergistic inhibition of cancer-cell proliferation and increased apoptosis. This synergistic effect is likely the result of both passive DNA demethylation by 5-azacitidine and active TET-dependent DNA methylation at long terminal repeat (LTR) regions of endogenous retroviruses (ERV) (104) by vitamin C. Since vitamin C is orally available, cost- effective and has a very low toxicity profile, it may represent a therapeutic option in combination with DNMTi, especially for patients with CRC KRAS and BRAF mutations.
There are many phase I/II clinical trials in mCRC patients (Tab. 3) investigating the efficacy of methylation inhibitors (5-Aza-CR or 5-Aza-CdR or SGI-110) in combination with either immunotherapy (such as durvalumab, pembrolizumab), or standard chemotherapy, or anti- EGFR (such as panitumumab), or Allogenic Colon Cancer Cell Vaccine-GVAX and Cyclophosphamide. These trials are currently active or recently completed, suggesting that epigenetic therapies may become an exciting new option for CRC patients in the near future.
9. Conclusion
Our understanding of epigenetic alterations in CRC is rapidly growing and has the potential for great implications in clinical practice. Methylated genes and miRNA have been shown to be reliable biomarkers for diagnostic, prognostic and predictive purposes, and may be part of the future tools to improve: a) patients’ compliance to screening programs (non-invasive test vs colonoscopy); b) accuracy of patients’ prognosis and c) prediction of therapeutic response. Nevertheless, only a few of them are currently available for clinical use, and much effort and further studies are warranted in order to validate biomarkers with higher sensitivity and specificity.
The heterogeneity and complexity of epigenetic alterations in CRC remain a considerable issue, which may explain why the “bench-to-beside” process is so arduous, and why none of the discoveries have dramatically changed our ability to take care of CRC patients so far. Although CIMP status is a promising prognostic and predictive marker in both early and advanced disease, further investigations are needed to standardize CIMP status definition. Stratifying patients as CIMP-H/CIMP-L may be important to predict which patient will benefit the most from epigenetic therapies. Other DNA-methylation signatures, such as RASSF1A, may be better prognostic and predictive biomarkers than CIMP status. The tight correlation between CIMP-H, BRAF mutation and MSI-H status highlights the importance of epigenetic alterations in clinical practice. In addition, this subset of patients may benefit from specific combination treatments, whose efficacy needs to be validated in clinical trials.
Finally, Vitamin C, DNA methylation inhibitors and histone deacetylase inhibitors have already shown promising results as new therapeutic options for patients with CRCs, especially in association with standard chemotherapy, immunotherapy and targeted therapy. In fact, these new compounds are in early phase clinical trials and may become part of our clinical practice in the near future.
Acknowledgments
The project described was supported in part by award number P30CA014089 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. Martin D. Berger received a grant from the Swiss Cancer League (BIL KLS-3334-02-2014) and the Werner and Hedy Berger-Janser Foundation for cancer research. Ryuma Tokunaga received a grant from the Uehara Memorial Foundation (201630045).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
The authors declare no conflict of interest.
References
- 1.Ferlay J, Soerjomataram I, Ervik M, et al. GLOBOCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase No. 11 [Internet] Lyon, France: International Agency for Research on Cancer; 2013. [Google Scholar]
- 2.Arnold M, Sierra MS, Laversanne M, et al. Global patterns and trends in colorectal cancer incidence and mortality. doi: 10.1136/gutjnl-2015-310912. Gut Published Online First: 27 Jan 2016. [DOI] [PubMed] [Google Scholar]
- 3.Fearon ER, Vogelstein BA. Genetic Model for Colorectal Tumorigenesis. Cell. 1990;61:759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 4.Toyota M, Ahuja N, Ohe-Toyota M, et al. CpG island methylator phenotype in colorectal cancer. (Medical Sciences).Proc Natl Acad Sci USA. 1999;96:8681–8686. doi: 10.1073/pnas.96.15.8681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grady WM, Carethers JM. Genomic and Epigenetic Instability in Colorectal Cancer Pathogenesis. Gastroenterology. 2008;135(4):1079–1099. doi: 10.1053/j.gastro.2008.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bettington M, Walker N, Clouston A, et al. The serrated pathway to colorectal carcinoma: current concepts and challenges. Histopathology. 2013;62:367–386. doi: 10.1111/his.12055. [DOI] [PubMed] [Google Scholar]
- 7.Boland CR, Goel A. Microsatellite Instability in Colorectal Cancer. Gastroenterology. 2010;138(6):2073–2087. doi: 10.1053/j.gastro.2009.12.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lao VV, Grady WM. Epigenetics and Colorectal Cancer. Nat Rev Gastroenterol Hepatol. 2011;8(12):686–700. doi: 10.1038/nrgastro.2011.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mojarad EN, Kuppen PJK, Aghdaei HA, et al. The CpG island methylator phenotype (CIMP) in colorectal cancer. Gastroenterol Hepatol Bed Bench. 2013;6(3):120–128. [PMC free article] [PubMed] [Google Scholar]
- 10.Sharma S, Kelly TK, Jones PA. Epigenetics in cancer. Carcinogenesis. 2010;31:27–36. doi: 10.1093/carcin/bgp220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Du J, Johnson LM, Jacobsen SE, et al. DNA methylation pathways and their crosstalk with histone methylation. Nat Rev Mol Cell Biol. 2015;16(9):519–32. doi: 10.1038/nrm4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dawson MA, Kouzarides T. Cancer Epigenetics: from Mechanism to Therapy. Cell. 2012;150(1):12–27. doi: 10.1016/j.cell.2012.06.013. [DOI] [PubMed] [Google Scholar]
- 13.Jones PA. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet. 2012;13:484–492. doi: 10.1038/nrg3230. [DOI] [PubMed] [Google Scholar]
- 14.Hellman A, Chess A. Gene body-specific methylation on the active X chromosome. Science. 2007;315:1141–1143. doi: 10.1126/science.1136352. [DOI] [PubMed] [Google Scholar]
- 15.Jin B, Robertson KD. DNA Methyltransferases (DNMTs), DNA Damage Repair, and Cancer. Adv Exp Med Biol. 2013;754:3–29. doi: 10.1007/978-1-4419-9967-2_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin RK, Wang YC. Dysregulated transcriptional and post-translational control of DNA methyltransferases in cancer. Cell Biosci. 2014;4(1):46. doi: 10.1186/2045-3701-4-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai H-C, Baylin SB. Cancer epigenetics: linking basic biology to clinical medicine. Nat Publ Gr. 2011;2124(21):502–517. doi: 10.1038/cr.2011.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakai E, Nakajima A, Kaneda A. Accumulation of aberrant DNA methylation during colorectal cancer development. World J Gastroenterol. 2014;20(4):978–987. doi: 10.3748/wjg.v20.i4.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Feinberg AP, Vogelstein B. Hypomethylation distinguishes genes of some human cancers from their normal counterparts. Nature. 1983;301:89–92. doi: 10.1038/301089a0. [DOI] [PubMed] [Google Scholar]
- 20.Iyer LM, Tahiliani M, Rao A, et al. Prediction of novel families of enzymes involved in oxidative and other complex modifications of bases in nucleic acids. Cell Cycle. 2009;8:1698–1710. doi: 10.4161/cc.8.11.8580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pastor WA, Aravind L, Rao A. TETonic shift-biological roles of TET proteins in DNA demethylation and transcription. Nat Rev Mol Cell Biol. 2013;14(6):341–56. doi: 10.1038/nrm3589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cortellino S, Xu J, Sannai M, et al. Thymine DNA Glycosylase Is Essential for Active DNA Demethylation by Linked Deamination-Base Excision Repair. Cell. 2011;146:67–79. doi: 10.1016/j.cell.2011.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.He YF, Li BZ, Li Z, et al. Tet-Mediated Formation of 5-Carboxylcytosine and Its Excision by TDG in Mammalian DNA. Science. 2011;333(6047):1303–1307. doi: 10.1126/science.1210944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dang L, White DW, Gross S, et al. Cancer-associated IDH1 mutations produce 2-hydroxyglutarate. Nature. 2010;465(7300):966. doi: 10.1038/nature09132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xu W, Yang H, Liu Y, et al. Oncometabolite 2-hydroxyglutarate is a competitive inhibitor of alpha- ketoglutaratae-dependent dioxygenases. Cancer cell. 2011;19(1):17–30. doi: 10.1016/j.ccr.2010.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cairns RA, Mak TW. Oncogenic Isocitrate Dehydrogenase Mutations: Mechanisms, Models, and Clinical Opportunities. Cancer Discov. 2013;3(7):730–41. doi: 10.1158/2159-8290.CD-13-0083. [DOI] [PubMed] [Google Scholar]
- 27.Gaidzik VI, Paschka P, Spath D, et al. TET2 Mutations in Acute Myeloid Leukemia (AML): Results from a Comprehensive Genetic and Clinical Analysis of the AML Study Group. J Clin Oncol. 2012;30:1350–1357. doi: 10.1200/JCO.2011.39.2886. [DOI] [PubMed] [Google Scholar]
- 28.Neri F, Dettori D, Incarnato D, et al. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway. Oncogene. 2015;34:4168–4176. doi: 10.1038/onc.2014.356. [DOI] [PubMed] [Google Scholar]
- 29.Ogino S, Kawasaki T, Nosho K, et al. LINE-1 hypomethylation is inversely associated with microsatellite instability and CpG island methylator phenotype (CIMP) in colorectal cancer. Int J Cancer. 2008;122(12):2767–2773. doi: 10.1002/ijc.23470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ogino S, Nosho K, Kirkner GJ, et al. A cohort study of tumoral LINE-1 hypomethylation and prognosis in colon cancer. J Natl Cancer Inst. 2008;100:1734–1738. doi: 10.1093/jnci/djn359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kawakami K, Matsunoki A, Kaneko M, et al. Long interspersed nuclear element-1 hypomethylation is a potential biomarker for the prediction of response to oral fluoropyrimidines in microsatellite stable and CpG island methylator phenotype-negative colorectal cancer. Cancer Sci. 2011;102:166–174. doi: 10.1111/j.1349-7006.2010.01776.x. [DOI] [PubMed] [Google Scholar]
- 32.Ang PW, Loh M, Liem N, et al. Comprehensive profiling of DNA methylation in colorectal cancer reveals subgroups with distinct clinicopathological and molecular features. BMC Cancer. 2010;10:227. doi: 10.1186/1471-2407-10-227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Weisenberger DJ, Levine AJ, Long TI, et al. Association of the Colorectal CpG Island Methylator Phenotype with Molecular Features, Risk Factors, and Family History. Cancer Epidemiol Biomarkers Prev. 2015;24(3):515–519. doi: 10.1158/1055-9965.EPI-14-1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ogino S, Kawasaki T, Kirkner GJ, et al. CpG Island Methylator Phenotype-Low (CIMP-Low) in Colorectal Cancer: Possible Associations with Male Sex and KRAS Mutations. J Mol Diagn. 2006;8:582–588. doi: 10.2353/jmoldx.2006.060082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen L, Toyota M, Kondo Y, et al. Integrated genetic and epigenetic analysis identifies three different subclasses of colon cancer. Proc Natl Acad Sci USA. 2007;104:18654–18659. doi: 10.1073/pnas.0704652104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Yagi K, Akagi K, Hayashi H, et al. Three DNA methylation epigenotypes in human colorectal cancer. Clin Cancer Res. 2010;16:21–33. doi: 10.1158/1078-0432.CCR-09-2006. [DOI] [PubMed] [Google Scholar]
- 37.Hinoue T, Weisenberger DJ, Lange CP, et al. Genome-scale analysis of aberrant DNA methylation in colorectal cancer. Genome Res. 2012;22:271–282. doi: 10.1101/gr.117523.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ogino S, Kawasaki T, Kirkner GJ, et al. Evaluation of Markers for CpG Island Methylator Phenotype (CIMP) in Colorectal Cancer by a Large Population-Based Sample. Journal of Molecular Diagnostics. 2007;9(3):305–314. doi: 10.2353/jmoldx.2007.060170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Weisenberger DJ, Siegmund KD, Campan M, et al. CpG island methylator phenotype underlies sporadic microsatellite instability and is tightly associated with BRAF mutation in colorectal cancer. Nature Genetics. 2006;38(7):787–793. doi: 10.1038/ng1834. [DOI] [PubMed] [Google Scholar]
- 40.Berg M, Hagland HR, Soreide K. Comparison of CpG Island Methylator Phenotype (CIMP) Frequency in Colon Cancer Using Different Probe- and Gene-Specific Scoring Alternatives on Recommended Multi-Gene Panels. PLOS ONE. 2014;9(1):e86657. doi: 10.1371/journal.pone.0086657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Puccini A, Berger MD, Zhang W, et al. What we know about stage II and III colon cancer: it’s still not enough. Target Oncol. 2017 May 15; doi: 10.1007/s11523-017-0494-5. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shiovitz S, Bertagnolli MM, Renfro LA, et al. CpG island methylator phenotype is associated with response to adjuvant irinotecan-based therapy for stage III colon cancer. Gastroenterology. 2014;147:637–645. doi: 10.1053/j.gastro.2014.05.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Van Rijnsoever M, Elsaleh H, Joseph D, et al. CpG island methylator phenotype is an independent predictor of survival benefit from 5-fluorouracil in stage III colorectal cancer. Clin Cancer Res. 2003;9:2898–2903. [PubMed] [Google Scholar]
- 44.Ahn JB, Chung WB, Maeda O, et al. DNA methylation predicts recurrence from resected stage III proximal colon cancer. Cancer. 2011;117(9):1847–1854. doi: 10.1002/cncr.25737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Koo DH, Hong YS, Kim K, et al. CpG island methylator phenotype and KRAS mutation status as prognostic markers in patients with resected colorectal cancer. J Clin Oncol. 2011;29(Suppl) abstr 3595. [Google Scholar]
- 46.Zanutto S, Pizzamiglio S, Lampis A, et al. Methylation status in patients with early stage colon cancer: a new prognostic marker? Int J Cancer. 2012;130:488–489. doi: 10.1002/ijc.26011. [DOI] [PubMed] [Google Scholar]
- 47.Wang Y, Long Y, Xu Y, et al. Prognostic and predictive value of CpG Island Methylator phenotype in Patients with locally advanced non metastatic sporadic colorectal cancer. Gastroenterol Res Pract. 2014;436985 doi: 10.1155/2014/436985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jover R, Nguyen TP, Perez-Carbonell L, et al. 5-Fluorouracil adjuvant chemotherapy does not increase survival in patients with CpG island methylator phenotype colorectal cancer. Gastroenterology. 2011;140:1174–1181. doi: 10.1053/j.gastro.2010.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee S, Cho NY, Choi M, et al. Clinicopathological features of CpG island methylator phenotype-positive colorectal cancer and its adverse prognosis in relation to KRAS/BRAF mutation. Pathol Int. 2008;58:104–113. doi: 10.1111/j.1440-1827.2007.02197.x. [DOI] [PubMed] [Google Scholar]
- 50.Donada M, Bonin S, Barbazza R, et al. Management of stage II colon cancer—the use of molecular biomarkers for adjuvant therapy decision. BMC Gastroenterol. 2013;13:36. doi: 10.1186/1471-230X-13-36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jo P, Jung K, Grade M, et al. CpG island methylator phenotype infers a poor disease-free survival in locally advanced rectal cancer. Surgery. 2012;151:564–570. doi: 10.1016/j.surg.2011.08.013. [DOI] [PubMed] [Google Scholar]
- 52.Shen L, Catalano PJ, Benson AB, 3rd, et al. Association between DNA methylation and shortened survival in patients with advanced colorectal cancer treated with 5-fluorouracil based chemotherapy. Clin Cancer Res. 2007;13:6093–6098. doi: 10.1158/1078-0432.CCR-07-1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Simons CC, Hughes LA, Smits KM, et al. A novel classification of colorectal tumors based on microsatellite instability, the CpG island methylator phenotype and chromosomal instability: implications for prognosis. Ann Oncol. 2013;24:2048–2056. doi: 10.1093/annonc/mdt076. [DOI] [PubMed] [Google Scholar]
- 54.Kim JH, Shin SH, Kwon HJ, et al. Prognostic implications of CpG island hypermethylator phenotype in colorectal cancers. Virchows Arch. 2009;455:485–494. doi: 10.1007/s00428-009-0857-0. [DOI] [PubMed] [Google Scholar]
- 55.Ogino S, Nosho K, Kirkner GJ, et al. CpG island methylator phenotype, microsatellite instability, BRAF mutation and clinical outcome in colon cancer. Gut. 2009;58:90–96. doi: 10.1136/gut.2008.155473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Juo YY, Johnston FM, Zhang DY, et al. Prognostic value of CpG island methylator phenotype among colorectal cancer patients: a systematic review and meta-analysis. Ann Oncol. 2014;25:2314–2327. doi: 10.1093/annonc/mdu149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Min BH, Bae JM, Lee EJ, et al. The CpG island methylator phenotype may confer a survival benefit in patients with stage II or III colorectal carcinomas receiving fluoropyrimidine-based adjuvant chemotherapy. BMC cancer. 2011;11:344. doi: 10.1186/1471-2407-11-344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guinney J, Dienstmann R, Wang X. The consensus molecular subtypes of colorectal cancer. Nat Med. 2015 Nov;21(11):1350–6. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lenz H-J, Ou F-S, Venook AP, et al. Impact of consensus molecular subtyping (CMS) on overall survival (OS) and progression free survival (PFS) in patients (pts) with metastatic colorectal cancer (mCRC): Analysis of CALGB/SWOG 8040 5 (Alliance) J Clin Oncol. 35:2017. (suppl; abstr 3511) [Google Scholar]
- 60.Kita Y, Vincent K, Natsugoe S, et al. Epigenetically regulated microRNAs and their prospect in cancer diagnosis. Expert Review of Molecular Diagnostics. 2014;14(6):673–683. doi: 10.1586/14737159.2014.925399. [DOI] [PubMed] [Google Scholar]
- 61.Calin GA, Dumitru CD, Shimizu M, et al. Frequent deletions and down-regulation of micro- RNA genes miR15 and miR16 at 13q14 in chronic lymphocytic leukemia. Proc Natl Acad Sci USA. 2002;99(24):15524–9. doi: 10.1073/pnas.242606799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Saito Y, Liang G, Egger G, et al. Specific activation of microRNA-127 with downregulation of the protooncogene BCL6 by chromatin-modifying drugs in human cancer cells. Cancer Cell. 2006;9(6):435–43. doi: 10.1016/j.ccr.2006.04.020. [DOI] [PubMed] [Google Scholar]
- 63.Okugawa Y, Grady WM, Goel A. Epigenetic Alterations in Colorectal Cancer: Emerging Biomarkers. Gastroenterology. 2015;149(5):1204–1225. doi: 10.1053/j.gastro.2015.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kaur S, Johanna E, Lotsari-Salomaa JE, et al. MicroRNA Methylation in Colorectal Cancer. Advances in Experimental Medicine and Biology. 2016;937:109–122. doi: 10.1007/978-3-319-42059-2_6. [DOI] [PubMed] [Google Scholar]
- 65.Lujambio A, Ropero S, Ballestar E, et al. Genetic unmasking of an epigenetically silenced microRNA in human cancer cells. Cancer Res. 2007;67(4):1424–9. doi: 10.1158/0008-5472.CAN-06-4218. [DOI] [PubMed] [Google Scholar]
- 66.Wang H, Wu J, Meng X, et al. MicroRNA-342 inhibits colorectal cancer cell proliferation and invasion by directly targeting DNA methyltransferase. Carcinogenesis. 2011;32(7):1033–42. doi: 10.1093/carcin/bgr081. [DOI] [PubMed] [Google Scholar]
- 67.Balaguer F, Link A, Lozano JJ, et al. Epigenetic Silencing of miR-137 Is an Early Event in Colorectal Carcinogenesis. Cancer Res. 2010;70(16):6609–18. doi: 10.1158/0008-5472.CAN-10-0622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yan L, Zhao W, Yu H, et al. A Comprehensive Meta-Analysis of MicroRNAs for Predicting Colorectal Cancer. Medicine (Baltimore) 2016;95(9):e2738. doi: 10.1097/MD.0000000000002738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fraga MF, Ballestar E, Villar-Garea A, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nature Genetics. 2005;37:391–400. doi: 10.1038/ng1531. [DOI] [PubMed] [Google Scholar]
- 70.Pelàez IM, Kalogeropoulou M, Ferraro A, et al. Oncogenic RAS alters the global and gene-specific histone modification pattern during epithelial-mesenchymal transition in colorectal carcinoma cells. Int J Biochem Cell Biol. 2010 Jun;42(6):911–20. doi: 10.1016/j.biocel.2010.01.024. [DOI] [PubMed] [Google Scholar]
- 71.Gargalionis AN, Piperi C, Adamopoulos C, Papavassiliou AG. Histone modifications as a pathogenic mechanism of colorectal tumorigenesis. Int J Biochem Cell Biol. 2012;44(8):1276–89. doi: 10.1016/j.biocel.2012.05.002. [DOI] [PubMed] [Google Scholar]
- 72.Tamagawa H, Oshima T, Shiozawa M, et al. The global histone modification pattern correlates with overall survival in metachronous liver metastasis of colorectal cancer. Oncol Rep. 2012 Mar;27(3):637–42. doi: 10.3892/or.2011.1547. [DOI] [PubMed] [Google Scholar]
- 73.Tamagawa H, Oshima T, Numata M, et al. Global histone modification of H3K27 correlates with the outcomes in patients with metachronous liver metastasis of colorectal cancer. Eur J Surg Oncol. 2013;39(6):655–61. doi: 10.1016/j.ejso.2013.02.023. [DOI] [PubMed] [Google Scholar]
- 74.Jie D, Zhongmin Z, Guoqing L, et al. Positive expression of LSD1 and negative expression of E-cadherin correlate with metastasis and poor prognosis of colon cancer. Dig Dis Sci. 2013;58(6):1581–9. doi: 10.1007/s10620-012-2552-2. [DOI] [PubMed] [Google Scholar]
- 75.Coppedè F. The role of epigenetics in colorectal cancer. Expert Review of Gastroenterology & Hepatology. 2014;8(8):935–948. doi: 10.1586/17474124.2014.924397. [DOI] [PubMed] [Google Scholar]
- 76.Petko Z, Ghiassi M, Shuber A, et al. Aberrantly methylated CDK2NA, MGMT, and MLH1 in colon polyps and in fecal DNA from patients with colorectal polyps. Clin Cancer Res. 2005 Feb 1;11(3):1203–9. [PubMed] [Google Scholar]
- 77.Altobelli E, Angeletti PM, Latella G. Role of Urinary Biomarkers in the Diagnosis of Adenoma and Colorectal Cancer: A Systematic Review and Meta-Analysis. Journal of Cancer. 2016;7(14):1984–2004. doi: 10.7150/jca.16244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Galanopoulos M, Tsoukalas N, Papanikolaou IS, et al. Abnormal DNA methylation as a cell-free circulating DNA biomarker for colorectal cancer detection: A review of literature. World J Gastrointest Oncol. 2017 Apr 15;9(4):142–152. doi: 10.4251/wjgo.v9.i4.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Grützmann R, Molnar B, Pilarsky C, et al. Sensitive detection of colorectal cancer in peripheral blood by septin 9 DNA methylation assay. PLoS One. 2008;3:e3759. doi: 10.1371/journal.pone.0003759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Song L, Yu H, Jia J, Li Y. A systematic review of the performance of the SEPT9 gene methylation assay in colorectal cancer screening, monitoring, diagnosis and prognosis. Cancer Biomarkers. 2017;18:425–432. doi: 10.3233/CBM-160321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Kostin PA, Zakharzhevskaia NB, Generozov EV, et al. Hypermethylation of the CDH1, SEPT9, HLTF and ALX4 genes and their diagnostic significance in colorectal cancer. Vopr Onkol. 2010;56:162–168. [PubMed] [Google Scholar]
- 82.Tham C, Chew M, Soong R, et al. Postoperative Serum Methylation Levels of TAC1 and SEPT9 Are Independent Predictors of Recurrence and Survival of Patients with Colorectal Cancer. Cancer. 2014;120:3131–41. doi: 10.1002/cncr.28802. [DOI] [PubMed] [Google Scholar]
- 83.Xue M, Lai SC, Xu ZP, Wang LJ. Noninvasive DNA methylation biomarkers in colorectal cancer: A systematic review. Journal of Digestive Diseases. 2016;16:699–712. doi: 10.1111/1751-2980.12299. [DOI] [PubMed] [Google Scholar]
- 84.Tang D, Liu J, Wang DR, Yu HF, Li YK, Zhang JQ. Diagnostic and prognostic value of the methylation status of secreted frizzled-related protein 2 in colorectal cancer. Clin Invest Med. 2011;34:E88–95. doi: 10.25011/cim.v34i1.15105. [DOI] [PubMed] [Google Scholar]
- 85.Ned RM, Melillo S, Marrone M. Fecal DNA testing for Colorectal Cancer Screening: the ColoSure™ test. PLoS Curr. 2011 Mar 22;3:RRN1220. doi: 10.1371/currents.RRN1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Imperiale TF, Ransohoff DF, Itzkowitz SH, et al. Multitarget Stool DNA Testing for Colorectal-Cancer Screening. N Engl J Med. 2014;370:1287–97. doi: 10.1056/NEJMoa1311194. [DOI] [PubMed] [Google Scholar]
- 87.Khambata-Ford S, Garrett CR, Meropol NJ, et al. Expression of Epiregulin and Amphiregulina and K-ras Mutation status predict disease control in metastatic colorectal cancer patients treated with Cetuximab. J Clin Oncol. 2007;25:3230–3237. doi: 10.1200/JCO.2006.10.5437. [DOI] [PubMed] [Google Scholar]
- 88.Jacobs B, De Roock W, Piessevaux H, et al. Amphiregulin and Epiregulin mRNA expression in primary tumors predict outcome in metastatic colorectal cancer treated with Cetuximab. J Clin Oncol. 2009;27:5068–5074. doi: 10.1200/JCO.2008.21.3744. [DOI] [PubMed] [Google Scholar]
- 89.Lee MS, McGuffey EJ, Morris JS, et al. Association of CpG island methylator phenotype and EREG/AREG methylation and expression in colorectal cancer. Br J Cancer. 2016;114:1352–1361. doi: 10.1038/bjc.2016.87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Scartozzi M, Bearzi I, Mandoles A, et al. Epidermal growth factor receptor (EGFR) gene promoter methylation and cetuximab treatment in colorectal cancer patients. Br J Cancer. 2011;104:1786–1790. doi: 10.1038/bjc.2011.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Nilsson TK, Löf-Öhlin ZM, Sun XF. DNA methylation of the p14ARF, RASSF1A and APC1A genes as an independent prognostic factor in colorectal cancer patients. Int J Cancer. 2013;42:127–133. doi: 10.3892/ijo.2012.1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lee KW, Lee SS, Kim SB, et al. Significant Association of Oncogene YAP1 with Poor Prognosis and Cetuximab Resistance in Colorectal Cancer Patients. Clin Cancer Res. 2015;21(2):357–64. doi: 10.1158/1078-0432.CCR-14-1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Shen L, Kondo Y, Rosner GL, et al. MGMT Promoter Methylation and Field Defect in Sporadic Colorectal Cancer. J Natl Cancer Inst. 2005;97:1330–1338. doi: 10.1093/jnci/dji275. [DOI] [PubMed] [Google Scholar]
- 94.Cremolini C, Pietrantonio F. How the lab is changing our view of colorectal cancer. Tumori. 2016;102(6):541–547. doi: 10.5301/tj.5000551. [DOI] [PubMed] [Google Scholar]
- 95.Ghoneim HE, Fan Y, Moustaki A, et al. De Novo Epigenetic Programs Inhibit PD-1 Blockade-Mediated T Cell Rejuvenation. Cell. 2017;170(1):1–16. doi: 10.1016/j.cell.2017.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chiappinelli KB, Strissel PL, Desrichard A, Li, et al. Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell. 2016;164:1073. doi: 10.1016/j.cell.2015.10.020. [DOI] [PubMed] [Google Scholar]
- 97.Li H, Chiappinelli KB, Guzzetta AA, et al. Immune regulation by low doses of the DNA methyltransferase inhibitor 5-azacitidine in common human epithelial cancer. Oncotarget. 2014;5:587–598. doi: 10.18632/oncotarget.1782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Brocks D, Schmidt CR, Daskalakis M, et al. DNMT and HDAC inhibitors induce cryptic transcription start sites encoded in long terminal repeats. Nat Genet. 2017;49(7):1052–1060. doi: 10.1038/ng.3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.LaBonte MJ, Wilson PM, Fazzone W, et al. The dual EGFR/HER2 inhibitor lapatinib synergistically enhances the antitumor activity of the histone deacetylase inhibitor panobinostat in colorectal cancer models. Cancer Res. 2011;71(10):3635–3648. doi: 10.1158/0008-5472.CAN-10-2430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Garrido-Laguna I, McGregor KA, Wade M, et al. A phase I/II study of decitabine in combination with panitumumab in patients with wild-type (wt) KRAS metastatic colorectal cancer. Invest New Drugs. 2013;31:1257–1264. doi: 10.1007/s10637-013-9947-6. [DOI] [PubMed] [Google Scholar]
- 101.Overman MJ, Morris V, Moinova H. Phase I/II study of azacitidine and capecitabine/oxaliplatin (CAPOX) in refractory CIMP-high metastatic colorectal cancer: evaluation of circulating methylated vimentin. Oncotarget. 2016;7:67495–67506. doi: 10.18632/oncotarget.11317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Azad NS, El-Khoueiry A, Yin J, et al. Combination epigenetic therapy in metastatic colorectal cancer (mCRC) with subcutaneous 5-azacitidine and entinostat; a phase 2 consortium/stand Up 2 cancer study. Oncotarget. 2017 Feb 5; doi: 10.18632/oncotarget.15108. [Epub ahead of print] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Yun J, Mullarky E, Lu C, et al. Vitamin C selectively kills KRAS and BRAF mutant colorectal cancer cells by targeting GAPDH. Science. 2015;350(6266):1391–1396. doi: 10.1126/science.aaa5004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Liu M, Ohtani H1, Zhou W, et al. Vitamin C increases viral mimicry induced by 5-aza-2′-deoxycytidine. Proc Natl Acad Sci U S A. 2016;113(37):10238–44. doi: 10.1073/pnas.1612262113. [DOI] [PMC free article] [PubMed] [Google Scholar]