

SUMMARY
Reactive oxygen species (ROS)-induced cysteine S-glutathionylation is an important posttranslational modification (PTM) that controls a wide range of intracellular protein activities. However, whether physiological ROS can modulate the function of extracellular components via S-glutathionylation is unknown. Using a screening approach, we identified ROS-mediated cysteine S-glutathionylation on several extracellular cytokines. Glutathionylation of the highly conserved Cys-188 in IL-1β positively regulates its bioactivity by preventing its ROS-induced irreversible oxidation, including sulfinic acid and sulfonic acid formation. We show this mechanism protects IL-1β from deactivation by ROS in an in vivo system of irradiation-induced bone marrow (BM) injury. Glutaredoxin 1 (Grx1), an enzyme that catalyzes deglutathionylation, was present and active in the extracellular space in serum and the BM, physiologically regulating IL-1β glutathionylation and bioactivity. Collectively, we identify cysteine S-glutathionylation as a cytokine regulatory mechanism that could be a therapeutic target in the treatment of various infectious and inflammatory diseases.
In Brief
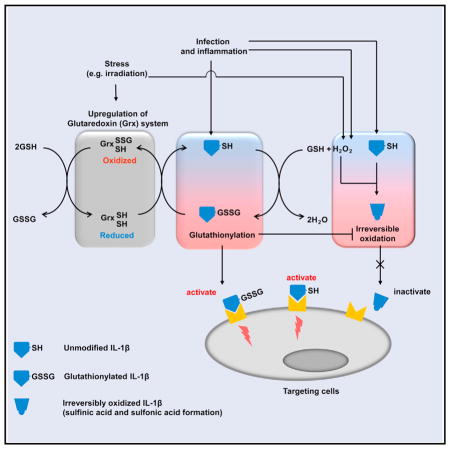
Zhang et al. reveal that cysteine S-glutathionylation of the highly conserved Cys-188 residue of IL-1β positively regulates its bioactivity by preventing its irreversible ROS-elicited deactivation. ROS-induced cysteine glutathionylation and its modulation by Glutaredoxin 1 (Grx1) are key physiological regulatory mechanisms controlling IL-1β activity, providing a potential therapeutic target in the treatment of infectious and inflammatory diseases.
INTRODUCTION
There is constant turnover of reactive oxygen species (ROS) in biological systems. ROS level is drastically elevated during infection and inflammation. Endogenous sources of ROS in mammals include NADPH oxidases (NOXs), the mitochondrial respiratory chain, the flavoenzyme ERO1 in the ER, xanthine oxidase, lipoxygenases, cyclooxygenases, cytochrome P450s, demethylase, and nitric oxide synthases (NOSs). Although classically the function of infection-induced ROS is to aid in the capability of phagocytes to kill pathogens, more recent studies have shed new light on unconventional roles for ROS in cellular signal transduction and functionality (Finkel, 2011; Nathan and Cunningham-Bussel, 2013). Redox regulation of cell function often involves the conversion of reactive thiols on specific cysteine residues from reduced to oxidized forms (Janssen-Heininger et al., 2008). The major types of thiol modifications that have been shown to play an important redox-dependent role include glutathionylation, sulfenic acid formation, nitrosylation, and disulfide bond formation (Hurd et al., 2005; Janssen-Heininger et al., 2008).
In several cell types, ROS production has been shown to drive the glutathionylation of free thiol groups (-SH) on cysteine residues of proteins to form protein-glutathione mixed disufide adducts (Pr-SSG) (Ghezzi, 2013; Grek et al., 2013; Murphy, 2012; Pastore and Piemonte, 2012). Early studies of a normal liver found that at least 1% of the total cellular glutathione is always bound to protein (Brigelius et al., 1983). Under oxidative conditions, this has been known to increase to as high as 20%–50% of total glutathione (Gilbert, 1984). Similar to other posttranslational modifications (PTMs) such as phosphorylation and acetylation, cysteine S-glutathionylation can affect the function of intracellular structural and signaling proteins and transcription factors, including actin, protein tyrosine kinases and phosphatases, Ras, integrins, and transcription factors such as NF-κB, and hence modulate cellular function (Alegre-Cebollada et al., 2014; Chen et al., 2010; Clavreul et al., 2006; Dalle-Donne et al., 2009; Klatt and Lamas, 2000; Shelton and Mieyal, 2008). In a recent study, we showed that chemotactic signal-elicited ROS production promotes actin glutathionylation and the subsequent depolymerization, establishing ROS-induced actin glutathionylation and its modulation by glutaredoxin as key physiological regulatory mechanisms controlling actin dynamics in neutrophils (Sakai et al., 2012).
ROS are increasingly viewed as important regulators of intra-cellular signaling. However, whether physiological ROS can modulate the function of extracellular components remains elusive. In current study, in an initial assessment of extracellular redox events, we detected significant amounts of glutathione (GSH) in the bone marrow (BM) extracellular space. Moreover, serum and BM fluid was more oxidized than intracellular compartments. Thus, we speculated that cysteine S-glutathionylation may also be an important mechanism of ROS-mediated regulation of extracellular protein function. Indeed, glutathionylation has been implicated in the regulation of membrane anchored molecules such as ICAM-1 and VLA-4, as well as secreted and circulating proteins such as Hmgb1 and PON-1, although the physiological significance of such modifications remains elusive (Shelton and Mieyal, 2008).
Here, using a screening approach to select the prototypical proinflammatory cytokine IL-1β (Dinarello, 2011; Garlanda et al., 2013) as the exemplar, we revealed that glutathionylation causally affected IL-1β bioactivity via the highly conserved Cys-188 residue by preventing its irreversible ROS-elicited deactivation. Glutaredoxin 1 (Grx1), an enzyme that regulates deglutathionylation (Aesif et al., 2011; Chung et al., 2010; Kuipers et al., 2012), was present and active in the extracellular space in serum and BM, including in patients receiving chemotherapy, with Grx1 physiologically regulating IL-1β glutathionylation. Thus, we establish ROS-induced cysteine glutathionylation and its modulation by Grx1 as key physiological regulatory mechanisms controlling IL-1β bioactivity, providing a potential therapeutic target in the treatment of various infectious and inflammatory diseases.
RESULTS
Identification of Extracellular ROS-Mediated Cysteine S-Glutathionylation in Cytokines
In an initial assessment of extracellular redox events, we found that a significant amount of GSH existed extracellularly in the bone marrow (BM). The level of glutathione, including both reduced (GSH) and oxidized (GSSG) forms, in the BM was almost comparable to those inside the cells (>200 μM). Moreover, serum and BM fluid was more oxidized compared to intra-cellular compartments, with the ratio of GSH to GSSG much smaller than those observed inside the cells (Figures S1A and S1B). Thus, we considered the possibility that glutathionylation might also be a mechanism for ROS-mediated regulation of extracellular proteins such as cytokines.
Similar to protein phosphorylation, S-glutathionylation is site specific, occurring only at certain cysteine residues in certain proteins. The determinants of site and protein specificity are, however, unclear. Since cytokines are major components of the extracellular microenvironment and putative targets for PTMs, we examined whether various human cytokines are glutathionylated in vitro. The exact catalytic mechanism driving glutathionylation is still debated; one commonly proposed mechanism is the formation of Pr-SSG or S-glutathionylated proteins by thiol-disulfide exchange between reactive thiolate anions on specific protein cysteine residues and oxidized glutathione (GSSG) (Cooper et al., 2011). Therefore, to specifically detect protein cysteine S-glutathionylation, we synthesized biotin-labeled oxidized GSH (BioGSSG, Figure S1C), and glutathionylation was probed with streptavidin-horseradish peroxidase (HRP) (Sakai et al., 2012). Although GSH-induced glutathionylation was observed only in the presence of H2O2, BioGSSG-induced glutathionylation occurred even in the absence of H2O2, supporting the notion that GSSG is an S-glutathionylation reaction intermediate (Figure S1D). Thirty cytokines/chemokines were screened, 14 of which could be glutathionylated (Figure S2; Table S1). However, in seven of these positive hits, dithiothreitol (DTT) failed to reduce glutathionylation, instead significantly augmenting it. All these cytokines contain one or more disulfide bonds; thus, it is likely that DTT reduced disulfide bonds, and the newly generated free cysteines were then glutathionylated. Thus, glutathionylation of these cytokines under such conditions may not be physiologically relevant. Glutathionylation of the other seven cytokines (IL-1β, IL-6, IL-9, IL-16, IL-17, IL-18, and IL-23) occurred in the absence of DTT, and DTT treatment caused a drastic decrease in glutathionylation signal to background levels, consistent with the reduction of GSH mixed disulfides. We surmised that glutathionylation is likely to occur on pre-existing free cysteines and may play a role in regulating the structure and function of cytokines.
The Most Conserved Cysteine in Mature IL-1β Can Be S-Glutathionylated
Interleukin-1β (IL-1β) is an important and archetypal proinflammatory cytokine; we chose to study its glutathionylation further (Dinarello, 2011). During infection and inflammation, IL-1β is expressed as an inactive precursor (pro-IL-1β, about 31 kDa), which is then cleaved to mature active form (mature IL-1β, about 17 kDa) by proteases such as caspase 1. Since only the cleaved mature IL-1β can be secreted to the extracellular space to regulate immune responses, we focused on glutathionylation of mature IL-1β in current study.
At a physiologically relevant GSH concentration (250 μM), both human (Figure 1A) and mouse mature IL-1β (Figure S3A) were highly sensitive to H2O2-induced S-glutathionylation, even at 0.05 μM of H2O2. Cysteine S-glutathionylation could also be detected when physiological GSSG (without biotin label) was used and the modification was probed with a GSH-specific antibody (Figure 1B). To verify glutathionylation and determine the exact site of glutathionylation, the modified IL-1β protein was subjected to mass spectrometry (MS) (Figure 1C). Human IL-1β contains two cysteine residues, Cys-125 and Cys-188. H2O2-induced sulfinic acid and sulfonic acid modifications were detected on both. However, S-glutathionylation was detected only on Cys-188 in GSSG-treated, but not H2O2-treated, samples (Figure 1D). Similarly, GSSG-induced Cys-188 S-glutathionylation was observed in mouse IL-1β (Figure S3B).
Figure 1. The Most Conserved Cysteine in Mature IL-1β Can Be S-Glutathionylated.

(A) Immunoblots of biotinylated glutathione (BioGSH)-modified human IL-1β with streptavidin-HRP. Total protein loading was evaluated with colloidal blue dye and IL-1β-specific antibodies.
(B) Immunoblots of S-glutathionylated human IL-1β using GSH-specific antibodies with total protein loading evaluated by IL-1β-specific antibodies.
(C) Graphical representation of the mass spectrometry (MS) analytical strategy for human IL-1β S-glutathionylation.
(D) Oxidation-induced cysteine modifications in human IL-1β detected by MS.
(E) Amino acid sequence alignments of full-length IL-1β. Gray highlight, IL-1β propiece; green highlight, conserved cysteines in mature IL-1β; yellow highlight, non-conserved cysteines in mature IL-1β; underline, cysteine-containing peptides detected in MS analysis.
(F) Amino acid sequences of WT and mutant forms of IL-1β.
(G) Immunoblots of S-glutathionylated WT and mutant forms of IL-1β using streptavidin-HRP. IL-1β was treated with oxidized biotinylated GSH BioGSSG (250 μM) in the presence or absence of DTT (5 mM). Total protein loading was evaluated by colloidal blue staining.
Cys-188 is the most evolutionarily conserved cysteine residue in IL-1β, suggesting that it plays a key role in the regulation of IL-1β activity and function (Figure 1E). Cys-188 is localized to the protein surface and should, therefore, be easily accessible for modification (Figure S3C). To further explore the role of Cys-188 in IL-1β glutathionylation, two IL-1β mutants were generated (Figure 1F). Mutation of Cys-188 to Ser (IL-1β 188C/S) or Ala (IL-1β 188C/A) abolished IL-1β glutathionylation (Figure 1G), confirming that the GSSG-induced modification is Cys-188 specific.
Cysteine S-Glutathionylation Protects IL-1β from H2O2-Induced Deactivation
To evaluate how S-glutathionylation might modulate IL-1β function, the bioactivities of modified and unmodified IL-1β were assessed by monitoring IκB degradation as an intracellular surrogate of IL-1β activity (Figures 2A and 2B), by using a D10 cell proliferation assay (Figures S4A–S4C), and by monitoring IL-1β-induced IL-6 production by mouse embryonic fibroblast (MEF) cells (Figures S5A–S5F). IL-1β glutathionylation is relatively stable in culture medium with cells (Figure S5A). Interestingly, S-glutathionylation alone did not affect IL-1β bioactivity per se, with glutathionylated IL-1β possessing the same bioactivity as unmodified IL-1β (Figures 2A, 2B, S4B, and S5B). We therefore sought other mechanisms by which glutathionylation may regulate IL-1β function. It is well known that ROS can deactivate various proinflammatory chemokines/chemoattractants such as C5a, fMLP (Clark and Klebanoff, 1979), LTB4 (Segal et al., 2002), and IL-8 (Lekstrom-Himes et al., 2005). Similarly, H2O2 significantly inhibited IL-1β bioactivity, reducing the EC50 by 5-fold (Figures 2C, 2D, and S5C). To rule out the possibility that H2O2 may inhibit IL-1β via H2O2-induced degradation of IL-1β protein, we compared IL-1β protein levels in H2O2 treated and untreated samples and found no differences (Figure S4D). Instead, H2O2-elicited suppression of IL-1β bioactivity was attributable to Cys-188, since IL-1β 188C/S and IL-1β 188C/A were fully active even in the presence of H2O2 (Figures 2E–2H and S5D). Thus, H2O2-induced deactivation of IL-1β is most likely mediated by a PTM on Cys-188. Since S-glutathionylation did not affect IL-1β bioactivity and H2O2 treatment also induced irreversible oxidation of Cys-188, including sulfinic acid and sulfonic acid formation (Figure 1D), we concluded that H2O2-elicited suppression of IL-1β bioactivity was mainly due to irreversible oxidation of Cys-188.
Figure 2. H2O2, but Not Cysteine S-Glutathionylation, Induces Cys-188-Dependent IL-1β Deactivation.

(A) The effect of glutathionylation on IL-β were pretreated with or without GSSG (250 μM) for 1 hr at 37°C before adding to HeLa cell cultures. Figures are representative of at least three independent experiments.
(B) Densitometry results expressed as ratios to the value of untreated cells (PBS alone). Results are the means (±SD) of three independent experiments. The IL-1β concentration inducing half the maximal response (EC50) is indicated.
(C) The effect of H2O2-induced oxidation on IL-1β bioactivity assessed by the IκB degradation assay. Indicated amounts of IL-1β were pretreated with H2O2 (100 μM) for one h at 37°C before adding to HeLa cell cultures.
(D) Densitometry results (means ±SD of three independent experiments).
(E) Bioactivity of mutant forms of IL-1β assessed by the IκB degradation assay. Representative immunoblots are shown.
(F) Densitometry results (means ±SD of three independent experiments).
(G) The effect of H2O2-induced oxidation on the bioactivity of mutant IL-1β assessed by the IκB degradation assay. Indicated amounts of mutant IL-1β were pretreated with or without H2O2 (100 μM) for one hr at 37°C before adding to HeLa cell cultures. Representative immunoblots for IκBα are shown.
(H) Densitometry results (means ±SD of three independent experiments). EC50 for each mutant IL-1β are indicated.
Although IL-1β Cys-188 is highly conserved, it was not required for IL-1β bioactivity per se as mutant forms of IL-1β (IL-1β 188C/S and IL-1β 188C/A) were as bioactive as wild-type (Figures 2E, 2F, S4E, and S5D). Cys-188 is, therefore, likely to act as a regulator of IL-1β function. Cysteine S-glutathionylation is reversible and may, therefore, alter cell signaling in response to redox and preserve reversible protein functionality by protecting it from irreversible modifications in response to overoxidation (van Bergen et al., 2014). To test this hypothesis, we next sought to examine whether Cys-188 S-glutathionylation can negatively regulate H2O2-induced irreversible oxidation of IL-1β. Quantitative MS revealed that, while sulfinic and sulfonic acid (Finkel, 2011; Holmström and Finkel, 2014; Jeong et al., 2012) were the major Cys-188 modifications on H2O2-treated IL-1β, pretreatment with GSSG converted the major modification to S-glutathionylation and significantly reduced sulfinic acid and sulfonic acid modifications (Figure 3A). In addition, GSSG pretreatment blocked H2O2-induced suppression of IL-1β bioactivity, further verifying that H2O2-induced IL-1β deactivation is mediated by cysteine sulfinic acid and sulfonic acid modifications and that GSSG negatively regulates this inhibitory mechanism (Figures 3B, 3C, and S5E). Collectively, these results demonstrate that cysteine S-glutathionylation is an efficient mechanism to modulate IL-1β bioactivity in the presence of ROS.
Figure 3. Cysteine S-Glutathionylation Protects IL-1β from H2O2-Induced Deactivation.

(A) The effect of glutathionylation on H2O2-induced irreversible oxidation of IL-1β assessed by MS. IL-1β was treated with or without GSSG (250 μM) and then H2O2 (100 μM) for another 30 min at 37°C. The cysteine modification was detected by MS, and the relative amount of each cysteine modification was assessed using Skyline software. Representative plots of precursor, precursor (M+1), and precursor (M+2) intensities for each indicated modification are shown. The molecular weights (MW) of peptides containing the indicated modified cysteine are shown.
(B) The effect of glutathionylation on H2O2-induced deactivation of IL-1β assessed by the IκB degradation assay. Indicated amounts of IL-1β were pretreated overnight with GSSG (250 μM) and then H2O2 (100 μM) for another 30 min at 37°C before adding to HeLa cell cultures. Representative immunoblots for IκBα are shown.
(C) Densitometry results. Shown are the means (±SD) of three independent experiments.
Cysteine S-Glutathionylation of IL-1β Occurs In Vivo and Is Required for Maintaining IL-1β Bioactivity In Vivo
The apparent role of cysteine S-glutathionylation in regulating IL-1β bioactivity ex vivo prompted us to determine whether glutathionylation can protect IL-1β from ROS-induced irreversible deactivation in vivo. ROS regulate various proteins and pathways; thus, it is unlikely that all ROS-elicited outcomes are solely mediated by IL-1β. To specifically explore the effect of ROS-elicited glutathionylation on IL-1β in vivo, we utilized a system in which intraperitoneally (i.p.) injected recombinant IL-1β significantly improves BM recovery and survival in irradiated mice (Tiberghien et al., 1993) (Figures 4A and S6). In this setup, the outcome of the experiments will solely depend on the bioactivity of the recombinant IL-1β; thus, we would be able to directly test the effect of cysteine S-glutathionylation on IL-1β bioactivity in vivo.
Figure 4. IL-1β Is S-Glutathionylated In Vivo.

(A) Survival of mice treated with 6Gy whole-body γ-irradiation. The indicated amounts of recombinant mouse IL-1β were administered i.p. 20 hr before irradiation. Survival was analyzed using Kaplan-Meier survival curves and the log-rank test. *p < 0.005 versus control.
(B) H2O2 concentrations in the bone marrow (BM) of irradiated mice. Data are means ± SD of n = 3 mice. *p < 0.005 versus time 0.
(C) IL-1β-elicited ROS elevation in the BM. Data shown are means ± SD of n = 3 mice. *p < 0.005 versus time 0.
(D) S-glutathionylation of i.p.-injected recombinant mouse IL-1β detected by western blotting. His-tagged recombinant mouse IL-1β (1 μg/mouse) was i.p.-injected into WT mice. Whole-body γ-irradiation was conducted 24 hr after injection. Recombinant IL-1β was pulled down from the serum or BM lavage using Ni-NTA agarose beads at the times indicated after irradiation. Precipitated IL-1β was assessed by western blotting using anti-mouse IL-1β antibody. S-glutathionylation of recombinant IL-1β was detected using an anti-GSH antibody. Representative immunoblots are shown.
(E) Cysteine modification of i.p.-injected recombinant mouse IL-1β analyzed by MS. Recombinant IL-1β (1 μg) was injected 24 hr before irradiation and was pulled down from serum using Ni-NTA agarose beads 3 days after irradiation. Mice not injected with recombinant IL-1β were used as controls. Representative graphs of total intensity of precursor, precursor (M+1), and precursor (M+2) for each indicated modification are shown.
Protein cysteine S-glutathionylation is initiated by ROS. Accordingly, we first examined whether IL-1β treatment and/or irradiation can induce ROS production in the BM. ROS levels in the BM were measured using Amplex Red, a colorless substrate that reacts with H2O2 with 1:1 stoichiometry to produce highly fluorescent resorufin (Kwak et al., 2015; Zhu et al., 2017). Both irradiation (Figure 4B) and IL-1β (Figure 4C) significantly increased H2O2 in the BM extracellular space. Consistent with the elevated ROS production and the high GSH level in the BM, the injected recombinant IL-1β was robustly and consistently glutathionylated in peripheral blood and the BM (Figures 4D, S7A, and S7B).
Recombinant IL-1β was relatively stable in recipient mice: the protein and its glutathionylation were still readily detected 3 days after injection (Figure 4D). Noticeably, since the recombinant IL-1β was injected i.p., it reached the maximal level 24 hr after the injection. Quantitative MS confirmed in vivo glutathionylation of recombinant IL-1β. S-glutathionylation of Cys-188 was identified to be the major modification in recombinant IL-1β. A smaller but significant level of irreversible Cys-188 oxidation, including sulfinic and sulfonic acid, was also detected (Figure 4E).
Our ex vivo data revealed that Cys-188 S-glutathionylation can block irreversible oxidation of IL-1β and thus prevent oxidation-mediated IL-1β deactivation. Since IL-1β was highly glutathionylated in vivo, we speculated that Cys-188 glutathionylation might be a physiological mechanism to maintain IL-1β bioactivity in vivo. We next explored whether cysteine S-glutathionylation can protect IL-1β from H2O2-induced deactivation in vivo. We first examined whether irreversible Cys-188 oxidation could indeed negatively regulate IL-1β bioactivity in vivo. Mice were i.p. injected with wild-type (WT) or mutant (188C/S) IL-1β. Compared to PBS or WT IL-1β controls, mice injected with IL-1β 188C/S displayed a significant increase in survival at both 0.5-μg/mouse and 0.2-μg/mouse doses (Figure 5A). Thus, Cys-188 is critical for the negative regulation of IL-1β bioactivity in vivo. It is noteworthy that 0.2 μg/mouse of IL-1β 188C/S displayed even higher in vivo bioactivity than 0.5 μg/mouse WT IL-1β, indicating that this Cys-188-mediated PTM mechanism should be an efficient and effective way for controlling IL-1β bioactivity under physiopathological conditions.
Figure 5. S-Glutathionylation of Cys188 Is Required for Maintaining IL-1β Bioactivity In Vivo.

(A) Kaplan-Meier survival curves indicate that mutant IL-1β (188C/S) has higher bioactivity than WT IL-1β. The bioactivities of i.p.-injected WT and mutant IL-IL-1β (188C/S) have higher bioactivity than WT IL-1β. The bioactivities of i.p.-injected WT and mutant IL-1β were assessed at two doses: 0.5 μg/mouse and 0.2 μg/mouse. *p ≤ 0.005 versus WT IL-1β, log-rank test.
(B) The effect of Grx1 on cysteine modification of IL-1β analyzed by MS. His-tagged recombinant mouse IL-1β (1 μg) and recombinant Grx1 (1 μg) were injected i.p. into WT mice. Whole-body γ-irradiation was conducted 24 hr after injection. Recombinant IL-1β was pulled down from serum using Ni-NTA agarose beads 3 days after injection, separated by SDS-PAGE, and then processed for MS. Representative graphs for each indicated modification are shown.
(C) Kaplan-Meier survival curves showing that co-injection of Grx1 reduces WT IL-1β, but not mutant IL-1β (188C/S), bioactivity. *p ≤ 0.005 versus IL-1β (WT or mutant form) alone, log-rank test. n.s., not significant.
Protein glutathionylation is dynamic and reversible, and deglutathionylation is tightly regulated by glutaredoxin (Grx) (Lillig et al., 2008; Subramanian and Luo, 2009). Co-injection of enzymatically active recombinant Grx1 (Figure S7C) consistently reduced IL-1β glutathionylation in recipients. Consequently, the degree of irreversible Cys-188 oxidation, including sulfinic and sulfonic acid, was significantly augmented (Figure 5B). Subsequent studies of irradiated mice demonstrated that co-injection of Grx1 inhibited the protective effect of WT but not mutant (188C/S) IL-1β (Figure 5C). Mutant (188C/S) IL-1β displayed significantly higher bioactivity even in the presence of Grx1, confirming that the effect elicited by Grx1 was mediated by S-glutathionylation at Cys-188. Taken together, these in vivo responses parallel the ex vivo assays and support the idea that irreversible Cys-188 oxidation deactivates IL-1β and reversible Cys-188 glutathionylation protects IL-1β from oxidative deactivation.
Extracellular Grx Is a Physiological Modulator of Protein Cysteine S-Glutathionylation
Grx is a thiol disulfide oxido-reductase (thioltransferase) and a key regulator of intracellular protein glutathionylation (Lillig et al., 2008; Meyer et al., 2009; Subramanian and Luo, 2009). Glutathionylation can be reversed by Grx. Mammalian cells express two dithiol Grx isoforms, Grx1 and Grx2. Grx1 is cytosolic, whereas Grx2 resides in mitochondria. Grx has been detected in human plasma and sputum, so it is conceivable that Grx-mediated deglutathionylation of extracellular proteins also occurs (Lundberg et al., 2004; Nakamura et al., 1998; Peltoniemi et al., 2006). Consistent with this, Grx was also significantly active in the serum (Figure 6A) and BM extracellular space (Figure 6B). Upon irradiation, Grx activity drastically increased to reach maximum levels on day 7 in the serum and day 11 in the BM and declined thereafter.
Figure 6. Grx1 Is a Physiological Modulator of Protein Cysteine S-Glutathionylation.

(A) Grx activity measured in the serum of irradiated mice. Serum samples were collected at the indicated times after 4 Gy whole-body irradiation. One unit was defined as consumption of 1 μmol of NADPH per min. Data are means ± SD of n = 3 mice.
(B) Grx activity measured in the BM extracellular space of irradiated mice (number of units per milligram BM protein). Data are means ± SD of n = 3 mice.
(C) Grx staining in BM biopsies of acute myeloid leukemia (AML) patients. Images are representative of BM before (left) and 1 month after (right) chemotherapy induction.
(D) The percentage of patients showing increased Grx1 expression after induction chemotherapy (n = 27).
(E) Glutathionylation staining of WT and Grx1 KO mouse BM in the presence or absence of GSH. Representative fluorescent images are shown. Addition of excess reduced GSH to the primary GSH antibody mix abrogated glutathionylation.
(F) Quantitative analysis of protein glutathionylation in WT and Grx1 KO mouse BM. Mean fluorescence intensities (±SD) are shown. *p ≤ how versus WT.
(G) Glutathionylation staining of the BM of irradiated WT and Grx1 KO mice (4 Gy whole-body irradiation). Representative fluorescent images are shown.
(H) Quantitative analysis of protein glutathionylation in irradiated WT and Grx1 KO mouse BM. Mean fluorescence intensities (±SD) are shown. *p ≤ 0.005 versus WT.
Grx upregulation may be a general adaptive response to stress, so we also examined its expression in the clinical context. Chemotherapy is commonly used to treat various malignant diseases and induces significant BM damage. We wondered whether chemotherapy-induced BM stress can lead to Grx1 up-regulation in the BM. In this study, all 27 patients were diagnosed with acute myeloid leukemia (AML). Standard chemotherapy induction was usually initiated shortly after diagnosis. Expression of Grx1 was assessed by immunostaining using a specific human Grx1 antibody. The percentage of BM cells expressing Grx1 was counted. Upregulation of Grx1 was detected in over 70% of AML patients after chemotherapy induction (Figures 6C and 6D), with a large proportion of Grx1 localizing to the BM extracellular matrix. For those patients without Grx increases, Grx1 was already highly expressed even prior to chemotherapy, which may be due to unidentified pre-existing stress responses or a BM stress directly elicited by leukemia (Table S2).
Significant amounts of extracellular Grx activate being present in the serum and BM. In addition, other key components for deglutathionylation machinery such as GSH reductase (Kim et al., 2016; Kum-Tatt et al., 1975) and NADPH (Billington et al., 2006; Hibbs et al., 2016) exist extracellularly. Thus, we surmised that Grx may physiologically regulate protein glutathionylation in vivo. We tested this directly in a Grx1 knockout (KO) mouse with completely abolished Grx1 protein expression (Ho et al., 2007). Consistent with its deglutathionylation activity, immunofluorescence revealed that loss of Grx1 significantly enhanced protein glutathionylation in the BM (Figures 6E and 6F). Addition of free GSH to the primary antibody mix completely inhibited fluorescence, confirming that the glutathionylation staining was specific. Consistent with the elevated generation of ROS in the BM of irradiated mice, γ-irradiation increased overall protein glutathionylation with maximum levels reached at day 2 after irradiation. Grx1 disruption further augmented glutathionylation levels at each time point examined (Figures 6G and 6H). Thus, Grx, particularly Grx1, is a physiological regulator of protein glutathionylation in vivo. Noticeably, although Grx1 level was significantly upregulated upon irritation, Grx1 disruption could also elevate overall protein glutathionylation in unchallenged (day 0) mice, suggesting that Grx1 is a key regulator for modulating protein glutathionylation under both stress and resting conditions.
Grx1 Is a Physiological Modulator of IL-1β Cysteine S-Glutathionylation and Regulates IL-1β Bioactivity In Vivo
We have shown that cysteine S-glutathionylation protects IL-1β from H2O2-induced deactivation and thus regulates IL-1β bioactivity. Since Grx1 is a physiological modulator of protein cysteine S-glutathionylation, we hypothesize that Grx1 may also physiologically regulate glutathionylation and thus the activity of IL-1β in vivo. To test this, we first investigated whether loss of Grx1 function can increase IL-1β glutathionylation in vivo. His-tagged recombinant mouse IL-1β was injected i.p. 24 hr before γ-irradiation. Recombinant IL-1β was pulled down from the serum using Ni-NTA agarose beads at the times indicated. Precipitated IL-1β was assessed by western blotting using anti-mouse IL-1β antibody. S-glutathionylation of recombinant IL-1β was detected using an anti-GSH antibody. Similar to what was observed in Figure 4D, the levels of recombinant IL-1β in the serum reached the peak 24 hr after the injection. The same amount of recombinant IL-1β was recovered from WT and Grx1 KO mice; however, glutathionylation of recombinant IL-1β was more profound in Grx1 KO than WT mice at every time point after irradiation (Figures 7A–7C). In WT mice, IL-1β glutathionylation peaked 48 hr post-irradiation and then marginally reduced by 72 hr. In contrast, IL-1β glutathionylation kept increasing and remained high even 72 hr after irradiation in Grx1 KO mice. Thus Grx1 appears to act as a physiological modulator of IL-1β glutathionylation, with its deglutathionylation activity maintaining homeostasis under stress conditions (Figures 7A–7C).
Figure 7. Grx1 Is a Physiological Regulator of IL-1β Cysteine S-Glutathionylation and IL-1β Bioactivity.

(A) S-glutathionylation of i.p.-injected recombinant IL-1β in WT and Grx1 KO mice detected by western blotting. His-tagged recombinant mouse IL-β in WT and Grx1 KO mice was detected by western blotting. His-tagged recombinant mouse IL-1β (1 μg/mouse) was injected i.p. 24 hr before γ-irradiation. Recombinant IL-1β was pulled down from the serum using Ni-NTA agarose beads at the times indicated.
(B) Quantitative analysis of protein glutathionylation. Densitometry of the blots performed using ImageJ software. Results are the means ±SD of three independent experiments. *p ≤ 0.005 versus WT.
(C) Quantitative analysis of total IL-1β protein level.
(D) Kaplan-Meier survival curves showing that disruption of Grx1 augments IL-1β bioactivity in mice. Recombinant mouse IL-1β (0.5 μg/mouse) was administered i.p. 20 hr before irradiation. *p ≤ 0.005 versus WT mice, log-rank test.
(E) S-glutathionylation of endogenously produced IL-1β. Mice were challenged with LPS (10 mg/kg, i.p. injection). The serum was collected 6 hr post-injection. Endogenously produced IL-1β in the serum (1 mL, pooled from three mice) was immunoprecipitated using Hamster anti-IL-1β mAb (clone B122).
(F) Disruption of Grx1 augmented S-glutathionylation of endogenously produced IL-1β. The experiment was conducted as described above. Samples were treated with or without DTT (10 mM) before loading onto the SDS-PAGE.
Finally, we examined whether Grx1 can regulate the bioactivity of IL-1β in vivo. Mice were challenged with a lethal dose of whole-body gamma irradiation. Without IL-1β pretreatment, both WT and Grx1 KO mice died in 2 weeks. In terms of survival rate, there was no significant difference between the two groups. When injected with the same amount of IL-1β (0.5 μg/mouse), Grx1 KO mice survived significantly longer than WT mice after lethal irradiation, indicating that IL-1β was more bioactive in the Grx1 KO mice. This further supports the finding that S-glutathionylation protects IL-1β from oxidative deactivation (Figure 7D). Collectively, our results demonstrated that Grx1, an enzyme that catalyzes deglutathionylation, was present and active in the extracellular space in serum and BM, physiologically regulated IL-1β glutathionylation, and maintained IL-1β bioactivity in vivo.
S-Glutathionylation of Endogenously Produced IL-1β
Recombinant IL-1β could be glutathionylated in vivo in live animals. We next explored whether endogenously produced IL-1β can be modified in a similar way. One challenge in detecting endogenous IL-1β is the extremely low level of IL-1β production, even in mice stimulated with LPS. To overcome this obstacle, we immunoprecipitated endogenous IL-1β from the serum using an anti-IL-1β antibody (Figure 7E). Like the injected recombinant IL-1β, the endogenously produced IL-1β could also be readily glutathionylated in the serum of LPS-challenged mice. Consistent with the regulatory role of Grx1, the level of IL-1 β glutathionylation was increased significantly in Grx1 KO mice serum. DTT treatment caused a drastic decrease in glutathionylation signal to background levels, consistent with the reduction of GSH mixed disulfides (Figure 7F). Taken together, these results further demonstrated that cysteine S-glutathionylation is a physiologically relevant regulatory mechanism that controls IL-1β bioactivity in vivo.
DISCUSSION
Historically viewed as toxic byproducts, ROS such as hydrogen peroxide are now increasingly viewed as important second messengers that can regulate intracellular signal transduction under a variety of physiological and pathophysiological conditions. However, whether physiological ROS can also modulate the function of extracellular components remains elusive. Large-scale proteomic studies have identified numerous redox-sensitive proteins including some extracellular proteins. For instance, two recent elegant studies identified a set of glutathionylated proteins released during inflammation (Checconi et al., 2015; Mullen et al., 2015). Nevertheless, likely due to the extremely low levels of production, none of the cytokines (including IL-1β) were identified as glutathionylation targets in the previous studies. In current study, we revealed that physiological ROS-mediated cysteine S-glutathionylation could effectively and efficiently control in vivo bioactivity of an extracellular cytokine IL-1β, which was achieved by preventing ROS-induced irreversible oxidation and deactivation of IL-1β. This is the first demonstration of regulation of extracellular cytokines by physiological ROS-mediated S-glutathionylation.
Similar to protein phosphorylation, S-glutathionylation is site specific, occurring only at certain cysteine residues in certain proteins (Ghezzi, 2013; Grek et al., 2013). A common mechanism proposed for protein glutathionylation is thiol-disulfide exchange between a reactive thiolate anion on a specific protein cysteine residue and oxidized glutathione (GSSG), leading to formation of Pr-SSG or S-glutathionylated proteins. An alternative mechanism involves the initial oxidative modification of a reduced protein thiol to an activated protein, which may then react with GSH to the mixed disulfide (Dalle-Donne et al., 2009). Both reactions imply that an increase of ROS level or a decrease in GSH/GSSG ratio during oxidative stress or respiratory burst promotes protein glutathionylation. Nevertheless, not all protein thiols are efficient redox sensors. Most protein thiols have pKa values >8.0 at neutral pH and exist in protanated forms (-SH), which are more resistant to oxidation. However, specific thiols in redox-sensitive proteins are more prone to attack by oxidants, since they are more accessible to the oxidant and have a decreased pKa value as a result of specific amino acid charge distributions in their vicinity, allowing them to exist as an oxidation prone thiolate anion (-S–) even at neutral pH within the cytosol or in the extracellular space. Consistently, among the 30 cytokines/chemokines screened in current study, only seven could be efficiently glutathionylated. Another possible mechanism for protein glutathionylation is generation of protein mixed disulfides via thiol-hydrogen peroxide (H2O2) oxidation products, such as sulphenic acid (Pr-SOH) intermediates. Although, Pr-SOH modifications are reversible, they can be readily oxidized to generate sufinic (Pr-SO2H) and sulfonic acids (Pr-SO3H), which are known to be irreversible modifications causing permanent loss of protein activity. Alternate routes of protein glutathionylation have also been proposed, including partially oxidized thiyl radical (generated by reaction of Pr-SH and hydroxyl radical), mechanisms promoted by NO resulting in S-nitrosoglutathione production, and direct interaction between GSH and the cysteine residue triggered by oxidants such as diamide (Gallogly and Mieyal, 2007). Although the exact catalytic mechanism driving protein glutathionylation has not been identified and multiple mechanisms may account for production of protein mixed-disufides in vivo, the irreversible modifications such as Pr-SO2H and Pr-SO3H can be prevented by immediate glutathionylation of the free cysteine or sulfenic acid intermediates.
Here, we show that Cys-188-mediated posttranslational glutathionylation mechanism was an efficient and effective way to control IL-1β bioactivity under physiopathological conditions. Intriguingly, Cys-188 glutathionylation did not affect IL-1β bioactivity directly. Instead, it protected IL-1β from ROS-induced irreversible deactivation. GSH is a hydrophilic, low-molecular-weight tri-peptide that is produced directly from three amino acids, glycine, cysteine, and glutamate (γ-Glu-Cys-Gly). Currently, it is not known why irreversible modifications such as Pr-SO2H and Pr-SO3H formation could lead to IL-1β deactivation, while S-glutathionylation failed to do so. It is possible that, due to the reversibility, glutathionylated IL-1β was converted to free IL-1β before binding to its receptors on target cells.
Levels of protein glutathionylation can be controlled by enzymatic regulators of protein deglutathionylation. The family of enzymes that can perform this function consist of Grx/Glutaredoxin Reductase system that can deglutathionate glutathionylated proteins and thiol disulfide bonds (Holmgren, 1989), Thioredoxins (Trx)/Thioredoxin Reductase system that can reduce thiol disulfides and sulfenic acids (Haendeler, 2006), and Sulfiredoxins (Srx) that also mediate deglutathionylation of glutathionylated proteins (Findlay et al., 2006). All these enzymes are known to mediate reduction of protein thiols, by concomitantly oxidizing their own cysteines. They serve as intracellular antioxidants that can reverse the oxidation of protein thiols, so that protein function can be recovered upon release of oxidative stress or termination of redox processes. The reversible feature of protein-GSH mixed disulfide adducts and other modifications, such as disulfide and sulfenic acids, makes these modifications robust redox regulators of intracellular signaling. In current study, we demonstrated that Grx, particularly Grx1, was a physiological regulator of IL-1β deglutathionylation in vivo. Whether Trx and Srx can also modulate the glutathionylation and thus the bioactivity of IL-1β in vivo needs to be further investigated.
IL-1β is a prototypic proinflammatory cytokine and key regulator of host inflammatory and immune responses to infection. It mediates the expression of a vast array of genes involved in secondary inflammation. IL-1-responsive genes coordinate all aspects of local inflammation and also attract and activate cells of the adaptive immune system at sites of infection. IL-1β on the one hand activates monocytes, macrophages, and neutrophils, and on the other hand induces Th1 and Th17 adaptive cellular responses (Dinarello, 2011). Mechanisms of IL-1β regulation have traditionally focused on pattern recognition receptor-induced gene transcription and inflammasome-mediated cleavage of pro-IL-1β. However, the complete IL-1β cytokine regulatory repertoire is still largely unknown. Here, we reveal that cysteine S-glutathionylation plays a previously unidentified role in modulating cytokine activity. It causally affects IL-1β bioactivity via the highly conserved Cys-188 residue by preventing its irreversible oxidation. This is the first demonstration of regulation of IL-1β by physiological ROS-mediated S-glutathionylation. Infection, inflammation, and tissue damage are associated with elevated ROS generation and cytokine release. Cysteine S-glutathionylation is likely to regulate the bioactivity of endogenously generated IL-1β during infection and inflammation.
Cytokines are major players in host defense against invading bacteria and other pathogens. Conversely, excessive cytokine production and/or activation can be detrimental to the system, resulting in unwanted and exaggerated tissue inflammation. Hence, the bioactivity of cytokines needs to be well controlled during infection and information. Here, we show that, besides IL-1β, several other cytokines also appear to be glutathionylated (Table S1), suggesting that protein cysteine S-glutathionylation may be a more general physiological regulatory mechanism controlling cytokine bioactivity, providing a potential therapeutic target for the treatment of various infectious and inflammatory diseases.
EXPERIMENTAL PROCEDURES
Mice
Grx1−/− mice were kindly provided by Dr. Y.S. Ho and were backcrossed for more than 16 generations onto a C57BL/6 background (Ho et al., 2007). In all experiments, 8- to 12-month-old male mice were utilized. Age-matched C57BL/6 mice (Jackson Laboratories) were used as WT controls. All experiments involving equal treatments in WT and mutant samples and animals were conducted by experimenters blind to conditions. All animal manipulations were conducted in accordance with the Animal Welfare Guidelines of the Children’s Hospital Boston. All procedures were approved and monitored by the Children’s Hospital Animal Care and Use Committee.
Biotin-GSSG-Biotin Synthesis
Biotin-GSSG-biotin was synthesized as described in Figure S1C. All reagents including (+)-biotin and GSH were obtained from Sigma-Aldrich, except for the solvents (dimethylformamide [DMF]), which were from Thermo Fisher Scientific.
S-Glutathionylation of Human Cytokines and Chemokines
Proteins were prepared by 1:16 dilution with distilled water (32 μL for every 2 μg of protein). 10 μL of protein solution was divided between three test tubes: DMSO (1 μL) was added to the first tube, and BioGSSG (biotinylated oxidized GSH, 1 μL) was added to the second and third tubes prior to incubation at 37°C for 30 min. 1 μL of DTT (DTT, 60 mM stock) was then added to the third test tube, and 1 μL of distilled water was added to the first and second tubes. Samples were incubated at room temperature (RT) for another 30 min before being mixed with 12 μL 2 × LDS loading buffer (without reducing agents, Bio-Rad; catalog number 161-0737) and boiled for 7 min. Proteins were resolved on non-reducing 4%–12% gradient gels and probed by western blotting using an α-GSH antibody (Virogen; 1:1,000). Total protein loading was evaluated by colloidal blue staining (Life Technologies).
MS Analysis
Samples were digested with trypsin at 37°C for 2 hr in buffer containing 2 M urea and 50 mM ammonium bicarbonate, acidified with glacial acetic acid to a final concentration of 2%, and desalted by ZipTip (Millipore). Tryptic peptides were analyzed by highly sensitive nanospray liquid chromatography-tandem mass spectrometry (LC-MS/MS) using an LTQ-Orbitrap mass spectrometer (Thermo Fisher Scientific).
Statistical Analysis
For most experiments, the two-tailed, unpaired, Student’s t test was used to compare groups (Microsoft Excel or Prism software). Data were presented as means (±SD). A p value ≤0.05 was considered statistically significant. Statistical power analysis was used to justify the sample size. We assumed that data were normally distributed, since most outcome values were symmetrically distributed around the mean value within each group. The variance was similar between groups as determined by the F test. No samples or animals subjected to successful procedures and/or treatments were excluded from the analysis. No randomization was used for animal studies, since it was not applicable in this case. For survival analysis, Kaplan-Meier survival curves were generated using survival data and groups were compared by log-rank analysis using Prism software (GraphPad Software).
Supplementary Material
Highlights.
The most conserved cysteine in mature IL-1β, Cys-188, can be S-glutathionylated
Cysteine S-glutathionylation protects IL-1β from H2O2-induced deactivation
S-glutathionylation of Cys188 is required for maintaining IL-1β bioactivity in vivo
Grx1 is a physiological negative regulator of IL-1β cysteine S-glutathionylation
Acknowledgments
The authors thank Li Chai, Leslie Silberstein, Joris Messens, John Lucky, and John Manis for helpful discussions. Y.X. is supported by grants from the National Basic Research Program of China (2015CB964903), the CAMS Innovation Fund for Medical Sciences (2016-12M-1-003), and the Chinese National Natural Science Foundation (31471116). P.L. is supported by a grant from the Chinese National Natural Science Foundation (81600083) and the PUMC Youth Fund (3332015184). H.R.L. is supported by NIH grants R01AI103142, R01HL092020, and P01 HL095489 and a grant from the FAMRI (CIA130004).
Footnotes
Supplemental Information includes Supplemental Experimental Procedures, eight figures, and two tables and can be found with this article online at http://dx.doi.org/10.1016/j.celrep.2017.05.070.
AUTHOR CONTRIBUTIONS
X.Z., P.L., C.Z., D.C., J.L, A.V., F.Z., H.Y., K.Y.L., Z.Z., W.Z., H.Y., H.Z., L.Z., J.S., and Y.C. designed and carried out experiments, analyzed data, interpreted results, and prepared the manuscript. B.B., B.X., Y.X., T.C., L.E.S., and K.Y.L. helped with designing experiments, analyzing data, and evaluating manuscript. Y.-S.H. provided Grx1−/− mice. H.R.L. designed experiments, analyzed data, and wrote the paper. All authors gave their final approval to the manuscript.
References
- Aesif SW, Anathy V, Kuipers I, Guala AS, Reiss JN, Ho YS, Janssen-Heininger YM. Ablation of glutaredoxin-1 attenuates lipopolysaccharide-induced lung inflammation and alveolar macrophage activation. Am J Respir Cell Mol Biol. 2011;44:491–499. doi: 10.1165/rcmb.2009-0136OC. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alegre-Cebollada J, Kosuri P, Giganti D, Eckels E, Rivas-Pardo JA, Hamdani N, Warren CM, Solaro RJ, Linke WA, Fernández JM. S-glutathionylation of cryptic cysteines enhances titin elasticity by blocking protein folding. Cell. 2014;156:1235–1246. doi: 10.1016/j.cell.2014.01.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Billington RA, Bruzzone S, De Flora A, Genazzani AA, Koch-Nolte F, Ziegler M, Zocchi E. Emerging functions of extracellular pyridine nucleotides. Mol Med. 2006;12:324–327. doi: 10.2119/2006-00075.Billington. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brigelius R, Muckel C, Akerboom TP, Sies H. Identification and quantitation of glutathione in hepatic protein mixed disulfides and its relationship to glutathione disulfide. Biochem Pharmacol. 1983;32:2529–2534. doi: 10.1016/0006-2952(83)90014-x. [DOI] [PubMed] [Google Scholar]
- Checconi P, Salzano S, Bowler L, Mullen L, Mengozzi M, Hanschmann EM, Lillig CH, Sgarbanti R, Panella S, Nencioni L, et al. Redox proteomics of the inflammatory secretome identifies a common set of redoxins and other glutathionylated proteins released in inflammation, influenza virus infection and oxidative stress. PLoS ONE. 2015;10:e0127086. doi: 10.1371/journal.pone.0127086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CA, Wang TY, Varadharaj S, Reyes LA, Hemann C, Talukder MA, Chen YR, Druhan LJ, Zweier JL. S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature. 2010;468:1115–1118. doi: 10.1038/nature09599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung S, Sundar IK, Yao H, Ho YS, Rahman I. Glutaredoxin 1 regulates cigarette smoke-mediated lung inflammation through differential modulation of IkappaB kinases in mice: Impact on histone acetylation. Am J Physiol Lung Cell Mol Physiol. 2010;299:L192–L203. doi: 10.1152/ajplung.00426.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark RA, Klebanoff SJ. Chemotactic factor inactivation by the myeloperoxidase-hydrogen peroxide-halide system. J Clin Invest. 1979;64:913–920. doi: 10.1172/JCI109557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clavreul N, Adachi T, Pimental DR, Ido Y, Schöneich C, Cohen RA. S-glutathiolation by peroxynitrite of p21ras at cysteine-118 mediates its direct activation and downstream signaling in endothelial cells. FASEB J. 2006;20:518–520. doi: 10.1096/fj.05-4875fje. [DOI] [PubMed] [Google Scholar]
- Cooper AJ, Pinto JT, Callery PS. Reversible and irreversible protein glutathionylation: Biological and clinical aspects. Expert Opin Drug Metab Toxicol. 2011;7:891–910. doi: 10.1517/17425255.2011.577738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalle-Donne I, Rossi R, Colombo G, Giustarini D, Milzani A. Protein S-glutathionylation: A regulatory device from bacteria to humans. Trends Biochem Sci. 2009;34:85–96. doi: 10.1016/j.tibs.2008.11.002. [DOI] [PubMed] [Google Scholar]
- Dinarello CA. Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood. 2011;117:3720–3732. doi: 10.1182/blood-2010-07-273417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findlay VJ, Townsend DM, Morris TE, Fraser JP, He L, Tew KD. A novel role for human sulfiredoxin in the reversal of glutathionylation. Cancer Res. 2006;66:6800–6806. doi: 10.1158/0008-5472.CAN-06-0484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkel T. Signal transduction by reactive oxygen species. J Cell Biol. 2011;194:7–15. doi: 10.1083/jcb.201102095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallogly MM, Mieyal JJ. Mechanisms of reversible protein glutathionylation in redox signaling and oxidative stress. Curr Opin Pharmacol. 2007;7:381–391. doi: 10.1016/j.coph.2007.06.003. [DOI] [PubMed] [Google Scholar]
- Garlanda C, Dinarello CA, Mantovani A. The interleukin-1 family: Back to the future. Immunity. 2013;39:1003–1018. doi: 10.1016/j.immuni.2013.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezzi P. Protein glutathionylation in health and disease. Biochim Biophys Acta. 2013;1830:3165–3172. doi: 10.1016/j.bbagen.2013.02.009. [DOI] [PubMed] [Google Scholar]
- Gilbert HF. Redox control of enzyme activities by thiol/disulfide exchange. Methods Enzymol. 1984;107:330–351. doi: 10.1016/0076-6879(84)07022-1. [DOI] [PubMed] [Google Scholar]
- Grek CL, Zhang J, Manevich Y, Townsend DM, Tew KD. Causes and consequences of cysteine S-glutathionylation. J Biol Chem. 2013;288:26497–26504. doi: 10.1074/jbc.R113.461368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haendeler J. Thioredoxin-1 and posttranslational modifications. Antioxid Redox Signal. 2006;8:1723–1728. doi: 10.1089/ars.2006.8.1723. [DOI] [PubMed] [Google Scholar]
- Hibbs JB, Jr, Vavrin Z, Cox JE. Complex coordinated extra-cellular metabolism: Acid phosphatases activate diluted human leukocyte proteins to generate energy flow as NADPH from purine nucleotide ribose. Redox Biol. 2016;8:271–284. doi: 10.1016/j.redox.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho YS, Xiong Y, Ho DS, Gao J, Chua BH, Pai H, Mieyal JJ. Targeted disruption of the glutaredoxin 1 gene does not sensitize adult mice to tissue injury induced by ischemia/reperfusion and hyperoxia. Free Radic Biol Med. 2007;43:1299–1312. doi: 10.1016/j.freeradbiomed.2007.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren A. Thioredoxin and glutaredoxin systems. J Biol Chem. 1989;264:13963–13966. [PubMed] [Google Scholar]
- Holmström KM, Finkel T. Cellular mechanisms and physiological consequences of redox-dependent signalling. Nat Rev Mol Cell Biol. 2014;15:411–421. doi: 10.1038/nrm3801. [DOI] [PubMed] [Google Scholar]
- Hurd TR, Costa NJ, Dahm CC, Beer SM, Brown SE, Filipovska A, Murphy MP. Glutathionylation of mitochondrial proteins. Antioxid Redox Signal. 2005;7:999–1010. doi: 10.1089/ars.2005.7.999. [DOI] [PubMed] [Google Scholar]
- Janssen-Heininger YM, Mossman BT, Heintz NH, Forman HJ, Kalyanaraman B, Finkel T, Stamler JS, Rhee SG, van der Vliet A. Redox-based regulation of signal transduction: Principles, pitfalls, and promises. Free Radic Biol Med. 2008;45:1–17. doi: 10.1016/j.freeradbiomed.2008.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeong W, Bae SH, Toledano MB, Rhee SG. Role of sulfiredoxin as a regulator of peroxiredoxin function and regulation of its expression. Free Radic Biol Med. 2012;53:447–456. doi: 10.1016/j.freeradbiomed.2012.05.020. [DOI] [PubMed] [Google Scholar]
- Kim JS, Kwon WY, Suh GJ, Kim KS, Jung YS, Kim SH, Lee SE. Plasma glutathione reductase activity and prognosis of septic shock. J Surg Res. 2016;200:298–307. doi: 10.1016/j.jss.2015.07.044. [DOI] [PubMed] [Google Scholar]
- Klatt P, Lamas S. Regulation of protein function by S-glutathiolation in response to oxidative and nitrosative stress. Eur J Biochem. 2000;267:4928–4944. doi: 10.1046/j.1432-1327.2000.01601.x. [DOI] [PubMed] [Google Scholar]
- Kuipers I, Bracke KR, Brusselle GG, Aesif SW, Krijgsman R, Arts IC, Wouters EF, Reynaert NL. Altered cigarette smoke-induced lung inflammation due to ablation of Grx1. PLoS ONE. 2012;7:e38984. doi: 10.1371/journal.pone.0038984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kum-Tatt L, Tan IK, Seet AM. A new colorimetric method for the determination of NADH/NADPH dependent glutathione reductase in erythrocytes and in plasma. Clin Chim Acta. 1975;58:101–108. doi: 10.1016/s0009-8981(75)80002-7. [DOI] [PubMed] [Google Scholar]
- Kwak HJ, Liu P, Bajrami B, Xu Y, Park SY, Nombela-Arrieta C, Mondal S, Sun Y, Zhu H, Chai L, et al. Myeloid cell-derived reactive oxygen species externally regulate the proliferation of myeloid progenitors in emergency granulopoiesis. Immunity. 2015;42:159–171. doi: 10.1016/j.immuni.2014.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lekstrom-Himes JA, Kuhns DB, Alvord WG, Gallin JI. Inhibition of human neutrophil IL-8 production by hydrogen peroxide and dysregulation in chronic granulomatous disease. J Immunol. 2005;174:411–417. doi: 10.4049/jimmunol.174.1.411. [DOI] [PubMed] [Google Scholar]
- Lillig CH, Berndt C, Holmgren A. Glutaredoxin systems. Biochim Biophys Acta. 2008;1780:1304–1317. doi: 10.1016/j.bbagen.2008.06.003. [DOI] [PubMed] [Google Scholar]
- Lundberg M, Fernandes AP, Kumar S, Holmgren A. Cellular and plasma levels of human glutaredoxin 1 and 2 detected by sensitive ELISA systems. Biochem Biophys Res Commun. 2004;319:801–809. doi: 10.1016/j.bbrc.2004.04.199. [DOI] [PubMed] [Google Scholar]
- Meyer Y, Buchanan BB, Vignols F, Reichheld JP. Thioredoxins and glutaredoxins: Unifying elements in redox biology. Annu Rev Genet. 2009;43:335–367. doi: 10.1146/annurev-genet-102108-134201. [DOI] [PubMed] [Google Scholar]
- Mullen L, Seavill M, Hammouz R, Bottazzi B, Chan P, Vaudry D, Ghezzi P. Development of ‘Redox Arrays’ for identifying novel glutathionylated proteins in the secretome. Sci Rep. 2015;5:14630. doi: 10.1038/srep14630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP. Mitochondrial thiols in antioxidant protection and redox signaling: Distinct roles for glutathionylation and other thiol modifications. Antioxid Redox Signal. 2012;16:476–495. doi: 10.1089/ars.2011.4289. [DOI] [PubMed] [Google Scholar]
- Nakamura H, Vaage J, Valen G, Padilla CA, Björnstedt M, Holmgren A. Measurements of plasma glutaredoxin and thioredoxin in healthy volunteers and during open-heart surgery. Free Radic Biol Med. 1998;24:1176–1186. doi: 10.1016/s0891-5849(97)00429-2. [DOI] [PubMed] [Google Scholar]
- Nathan C, Cunningham-Bussel A. Beyond oxidative stress: An immunologist’s guide to reactive oxygen species. Nat Rev Immunol. 2013;13:349–361. doi: 10.1038/nri3423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pastore A, Piemonte F. S-Glutathionylation signaling in cell biology: Progress and prospects. Eur J Pharm Sci. 2012;46:279–292. doi: 10.1016/j.ejps.2012.03.010. [DOI] [PubMed] [Google Scholar]
- Peltoniemi MJ, Rytilä PH, Harju TH, Soini YM, Salmenkivi KM, Ruddock LW, Kinnula VL. Modulation of glutaredoxin in the lung and sputum of cigarette smokers and chronic obstructive pulmonary disease. Respir Res. 2006;7:133. doi: 10.1186/1465-9921-7-133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakai J, Li J, Subramanian KK, Mondal S, Bajrami B, Hattori H, Jia Y, Dickinson BC, Zhong J, Ye K, et al. Reactive oxygen species-induced actin glutathionylation controls actin dynamics in neutrophils. Immunity. 2012;37:1037–1049. doi: 10.1016/j.immuni.2012.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segal BH, Kuhns DB, Ding L, Gallin JI, Holland SM. Thioglycollate peritonitis in mice lacking C5, 5-lipoxygenase, or p47(phox): Complement, leukotrienes, and reactive oxidants in acute inflammation. J Leukoc Biol. 2002;71:410–416. [PubMed] [Google Scholar]
- Shelton MD, Mieyal JJ. Regulation by reversible S-glutathionylation: Molecular targets implicated in inflammatory diseases. Mol Cells. 2008;25:332–346. [PMC free article] [PubMed] [Google Scholar]
- Subramanian KK, Luo HR. T.M.P. Classification and R.H.S.K. Pathology, editor. Granulocytes. Nova Science Publishers; 2009. Non-classical roles of NADPH-oxidase dependent Reactive Oxygen Species in Phagocytes. [Google Scholar]
- Tiberghien P, Laithier V, Mabed M, Racadot E, Reynolds CW, Angonin R, Loumi R, Pavy JJ, Cahn JY, Noir A, et al. Interleukin-1 administration before lethal irradiation and allogeneic bone marrow transplantation: Early transient increase of peripheral granulocytes and successful engraftment with accelerated leukocyte, erythrocyte, and platelet recovery. Blood. 1993;81:1933–1939. [PubMed] [Google Scholar]
- van Bergen LA, Roos G, De Proft F. From thiol to sulfonic acid: Modeling the oxidation pathway of protein thiols by hydrogen peroxide. J Phys Chem A. 2014;118:6078–6084. doi: 10.1021/jp5018339. [DOI] [PubMed] [Google Scholar]
- Zhu H, Kwak HJ, Liu P, Bajrami B, Xu Y, Park SY, Nombela-Arrieta C, Mondal S, Kambara H, Yu H, et al. Reactive oxygen species-producing myeloid cells act as a bone marrow niche for sterile inflammation-induced reactive granulopoiesis. J Immunol. 2017;198:2854–2864. doi: 10.4049/jimmunol.1602006. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.