

Abstract
Introduction:
Joubert syndrome (JS) is a rare autosomal recessive inherited disease belonging to ciliopathy with the causative mutation of genes. Except for X-linked inheritance, the high recurrence rate of a family is about 25%. After birth, it may cause a series of neurological symptoms, even with retina, kidney, liver, and other organ abnormalities, which is defined as Joubert syndrome and related disorders (JSRD). Molecular genetics research contributes to disease prediction and genetic counseling. Prenatal diagnosis is rare. Magnetic resonance imaging (MRI) is usually the first-choice diagnostic modality with typical brain images characterized by the molar tooth sign. We describe a case of JS prenatally and Dandy-Walker malformation for the differential diagnosis based on ultrasonograms. We also review the etiology, imaging features, clinical symptoms, and diagnosis of JSRD.
Case presentation:
A 22-year-old woman was pregnant at 27 1/7 weeks’ gestation with fetal cerebellar vermis hypoplasia. Fetal ultrasonography and MRI confirmed a diagnosis of JS at our center. The couple finally opted to terminate the fetus, which had a normal appearance and growth parameters. The couple also had an AHI1 gene mutation on chromosome 6.
Conclusions:
Currently, a diagnosis of JS is commonly made after birth. Fewer cases of prenatal diagnosis by ultrasonography have been made, and they are more liable to be misdirected because of some nonspecial features that also manifest in Dandy-Walker malformation, cranio-cerebello-cardiac syndrome, and so on.
Keywords: cilia, genetic mutation, JSRD, MTS, prenatal diagnosis
1. Introduction
Joubert syndrome (JS), which is a rare congenital nervous system developmental disorder, was first discovered by Marie Joubert in 1969.[1] Pure JS can manifest with intermittent dyspnea or pause, developmental delay, ataxia, muscle tone loss, oculomotor apraxia, and other abnormalities of the nervous system, but not retinal, kidney, liver, or other organ disorders.[2] The most common characteristic brain image of JS is the molar tooth sign (MTS) on the axial plane, which can reflect thickened superior cerebellar peduncles (SCPs), cerebellar vermis (CV) hypoplasia, and a deepened interpeduncular fossa.[3] In recent years, it has been reported that JS is part of a spectrum of diseases characterized by the MTS with overlapping features and standing for distinct syndromes, such as cerebello-oculo-renal syndrome, combination of cerebellar vermis hypoplasia, oligophrenia of intelligence, ataxia, coloboma of ocular and hepatic fibrosis syndrome (COACH), Varadi-Papp syndrome (also known as oral-facial-digital syndrome type VI), Senior-Loken syndrome (in few patients), Dekaban-Arima syndrome, and Malta syndrome.[3–5] Finally, the term Joubert syndrome and related disorders (JSRD) has been defined as all disorders showing the MTS on brain imaging studies. According to the different organs involved, Francesco et al[2] made an easy nosology to divide JSRD into 6 clinical subtypes, including pure JS, JS with ocular defect, JS with renal defect, JS with oculorenal defects, JS with hepatic defect, and JS with orofaciodigital defects. Each subtype corresponds with many different genotypes, and 1 special gene mutation can cause different subtypes.[6] Here, we describe a woman with a singleton fetus, the evaluation of which resulted in a diagnosis of JS.
2. Case report
A woman, 22 years old, gravida 1, para 0, was referred to our center for a prenatal ultrasonographic consultation and fetal head magnetic resonance imaging (MRI) examination at 27 1/7 weeks’ gestation after fetal CV hypoplasia had been diagnosed at an outside facility based on a prenatal morphology ultrasonogram. The final 2 test results prompted the possibility of fetal JS. Fetal blood sampling revealed about 320 to 400 bands, and G with horizontal analysis showed no obvious abnormalities. The couple finally opted to terminate the pregnancy of a female fetus with a normal appearance and growth parameters.
The husband and wife were healthy and married with no consanguinity. The family members of both sides did not have developmental delay, mental retardation, or other nervous system abnormalities, and no other of their family members had a fetus with JS or JSRD.
After following up with the family, we detected that the couple had AHI1 gene mutation by Illumina HiSeq sequencing and Sanger sequencing. In the wife, the site of deletion was chromosome 6:135778533–135784544, involving the AHI1 gene 5–6 exon and belonging to a heterozygous deletion; the husband had a heterozygous mutation of AHI1 gene 11 exon c.1799_1802delAACA on chromosome 6. These 2 mutations are theoretically pathogenic and can lead to a change in the amino acid coding of cilia.
For some reason, the parents refused to perform genetic testing and autopsy on the fetus. Finally, combined with the fetal ultrasonogram, the MRI findings and genetic test results of the couple confirmed a diagnosis of JS. If the wife becomes pregnant again, prenatal counseling and screening of JS are recommended.
2.1. Imaging findings
2.1.1. Ultrasonography
With the last menstrual period as baseline, fetal ultrasonographic measurements were within the normal reference range. A biparietal diameter of 7.1 cm (appropriate for 28 4/7 weeks’ gestation), head circumference of 25.7 cm (28 weeks), abdominal circumference of 25.4 cm (29 4/7 weeks), and femur length of 5.0 cm (26 6/7 weeks) were detected by 2-dimensional ultrasonography. The fetal heart rate and frequency of movement were normal. The placenta, amniotic fluid, and other fetal appendages were well-developed. All aforementioned numbers were measured with the GE Voluson E10 ultrasound machine (GE Healthcare). Except for the fetal brain abnormalities, there were no other structural abnormalities, such as polycystic kidney disease (involving renal parenchymal echo enhancement and multiple renal cysts), polydactyly, and so on.
2.1.2. Fetal head ultrasonography
There was an inferior CV missing with the fourth ventricle (4V) communicating with the posterior fossa (Fig. 1A, C). The MTS was shown on the axial plane, which was made up of the brain stem (BS) and bilaterally thickened, elongated SCPs (Fig. 1A). The shape of the 4V changed greatly (Fig. 1A–D) and was enlarged in the midsagittal section without a fastigium (Fig. 1C).
Figure 1.
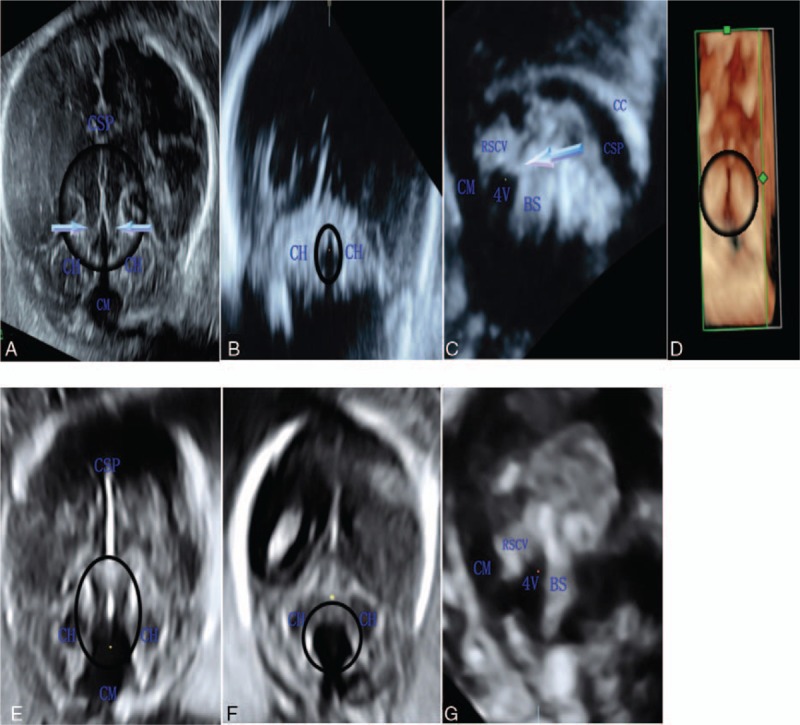
Prenatal head ultrasonograms of Joubert syndrome (A–D) and the differential diagnosis of Dandy-Walker malformation (E–G). (A) In the axial plane, the molar tooth sign (circled) is shown with superior cerebellar peduncles (SCPs) bilaterally (arrow), a narrow fourth ventricle (4V) with a sharp front and anteroposterior diameter greater than the maximum transverse diameter (the circled anechoic dark area), and communication between the 4V and cisterna magna (CM). (B) In the coronal plane, the 4V (circled) is abnormal compared with F. (C) In the midsagittal plane, the remnant of the superior cerebellar vermis (RSCV) is measured with an area of 0.71 cm2 and circumference of 4.01 cm, which was less than normal, the thickened SCP can be seen perpendicular to the brain stem (BS) (arrow), and 4V is dilated and changing in shape without a normal fastigium. (D) In the axial plane, bilaterally thickened, elongated, and horizontally-oriented SCPs and a narrow 4V are found with 3-dimensional reconstruction (circled). (E) In the axial plane, the 4V had cystic enlargement without thickened SCPs, and both cerebellar hemisphere (CHs) are separated (inside of the circle). (F) In the coronal plane, the 4V has cystic enlargement (circled anechoic dark area). (G) The 4V in the midsagittal plane compared to that in C. CC = corpus callosum.
2.1.3. MRI
The MTS was observed at the pontomesencephalic level (Fig. 2A). There was a dilated cisterna magna (CM) measuring 1.7 cm (Fig. 2A, B). In the midsagittal section, a remnant of the superior cerebellar vermis (RSCV) existed, and the 4V was enlarged and communicating with the CM (Fig. 2B).
Figure 2.
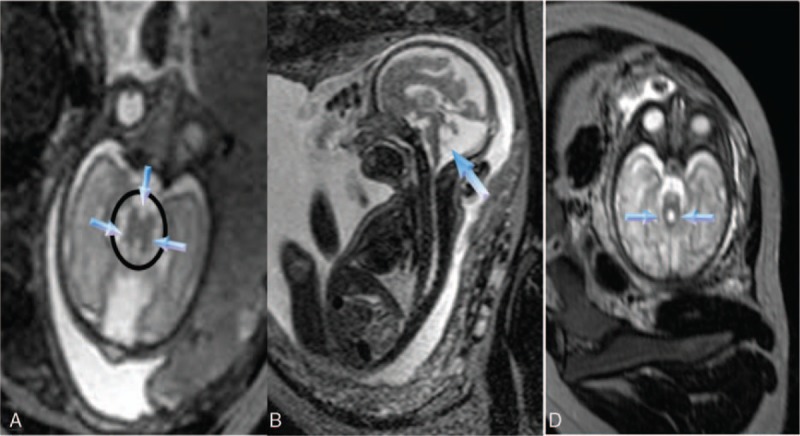
(A) The molar tooth sign (circled) with a deeper interpeduncular fossa (down arrow); bilaterally thickened, elongated, and parallel superior cerebellar peduncles (SCPs) (other arrows), and a dilated cisterna magna are shown on axial T2-weighted imaging. (B) The enlarged fourth ventricle (arrow) remains on midsagittal T2-weighted imaging. (C) Two big curved SCPs (arrows) in another case. SCP = superior cerebellar peduncle.
2.2. Informed consent
The patient has agreed to publish this case and informed consent has been obtained.
3. Discussion
Joubert syndrome and related disorders, a rare syndrome, is usually autosomal recessive inheritance, except for the rare mutation of the OFD1 gene with X mode of inheritance and male predominance.[7] In most cases, apart from X-linked inheritance, recurrence in the same family has a high risk of about 25%.[4] JSRD has genetic heterogeneity and belongs to the primary cilia disease. In recent years, the research on primary cilia has been a hot topic because many inherited human diseases are related to cilia abnormality.[8] In the human body, cilia, which are little, hair-like organelles on cell surfaces, appear everywhere, and they have an important role in the early stages of embryonic development and homeostasis.[9] Its molecular structure is complex with hundreds of different proteins. Genetic studies in mice support that cilia are necessary for diverse signaling pathways such as the Hedgehog signal transduction and critical for organizing the body plan and organ formation.[8,10] Cilia can be divided into motile cilia and immotile cilia, so the cilia-related human diseases can also be mainly divided into motile ciliopathies (eg, Kartagener syndrome[11]) and immotile ciliopathies (eg, JSRD, nephronophthisis, Bardet-Biedl syndrome, and Meckel syndrome).[12–14] Until now, all ciliopathies have been caused by genetic mutations.[14] Defections of ciliary proteins or intraflagellar transport affecting signaling pathways are the main causes of disorders by genetic mutations. Of note, as the motile cilia diseases produce many of the same symptoms, immotile ciliopathies also have pleiotropy and overlapping phenotypes, including polycystic kidney disease, retinal dysplasia or blindness, polydactyly, and liver disease (usually congenital liver fibrosis).[14,15] These ciliary dysfunctions can be a single tissue defect, such as polycystic kidney disease, retinal dysplasia, or syndromic multiple organ disorders.[14,16]
Joubert syndrome is a classic syndrome that is a unique brain neurological disorder featuring the MTS as the hallmark of the disease, and it is associated with primary ciliary protein defects, usually accompanied by other organ abnormalities. Primary cilia that most cells have for receiving and transmitting signals are like tiny legs, and they belong to immotile cilia. These substances play an important role in the growth and function of certain types of cells, involving neuronal cells in the mammalian brain, skeleton, retina photoreceptors, kidney tubules and collecting ducts, palate, lip, and liver.[13,14] So far, more than 30 causative genes have been found for the various subtypes of JSRD.[17] Additionally, as Bachmann-Gagescu et al[18] reported, TMEM67, C5orf42, CC2D2A, CEP290, and AHI1 genes were the most frequent ones. Enza et al[19] also believed that the AHI1 gene was a common mutant gene that caused JSRD. AHI1 is abundant in the brain and kidney and weakly expressed in the liver.[20] According to Valente et al,[13] the most common subtype of JSRD associated with the AHI1 gene mutation is JS with a retinal defect. Several mutant genes of JSRD could also exist in other syndromes, such as Meckel syndrome (mutations of RPGRIP1L and CEP290 genes[21]), and share some features with JSRD.[12] This phenomenon suggests that they have similar or identical pathogenic ways. Genetic analysis of JSRD and the study of mutant gene functions are of great value in predicting disease development and genetic counseling.[3,5] However, this molecular detection is still significantly challenging. Moreover, advanced sequencing techniques such as targeted next-generation sequencing technology and molecular inversion probe technology have been found to improve the detection rate.[17]
Joubert syndrome and related disorders is a special deformity of midbrain-hindbrain structures, with distinct brain findings of the MTS, CV hypoplasia (usually inferior vermis hypoplasia), and the changing shape of the 4V. However, the degree of vermis hypoplasia and the form of the MTS are variable with an accession from mild to obvious. Additionally, abnormal BS structures existed in 30% of patients, involving the pons and mesencephalon.[22] First, the MTS in axial planes, initially recognized in JS, results from the combination of bilateral thickened, elongated SCPs, an abnormally deeper IF with narrow brainstem isthmus (a junction of the midbrain and pons),[12,13] and CV hypoplasia. In axial planes at the midbrain and pons level, the direction of both superior peduncles may differ, involving a parallel, V-like, curved, and A-like structure.[22] In median parasagittal planes, thick SCPs can also be seen perpendicular to the BS, even in the fetal period.[2,12,13] Second, the vermis hypoplasia or absence with normal cerebellar hemispheres (CHs) exists in most patients. Furthermore, the enlarged posterior fossa sometimes communicates with the 4V.[4,22] Quarello et al's[12] study showed that a large communication was seen between CHs in 6 of 7 patients, whereas thin communication was seen in 1 patient. Finally, morphological changes of the 4V appeared in patients with JSRD. In the normal fetal period, the 4V can be clearly visible after 18 weeks of pregnancy, which is a liquid area shaped like a trapezoid (Fig. 3A) in axial planes, with the maximum transverse diameter usually greater than the anteroposterior diameter. The 4V has 4 walls (the front wall of the brain stem, posterior wall of the cerebellum, and 2 lateral walls of the superior and inferior cerebellum peduncles, which link the BS to the cerebellar). In JSRD, the 4V is abnormal because of the MTS in the axial planes.[12] The shape of the 4V with the first 3 aforementioned SCPs in the axial planes usually has an anteroposterior diameter greater than the maximum transverse diameter, with a sharp front unlike a trapezoid, whereas the last SCP has an enlarged 4V like a triangle or even a “batwing shape.”[10,22,23] In the midsagittal plane, the 4V was dilated and paramorphia, unlike a triangle with a fastigium. In addition to the MTS and vermis hypoplasia, JSRD can also be combined with other central nervous system deformities, involving mesencephalon hypoplasia, white matter cysts, white matter in high signal lesions, corpus callosum dysgenesis, hydrocephalus, polymicrogyria, encephalocele, and occipital cerebral edema.[13,22]
Figure 3.
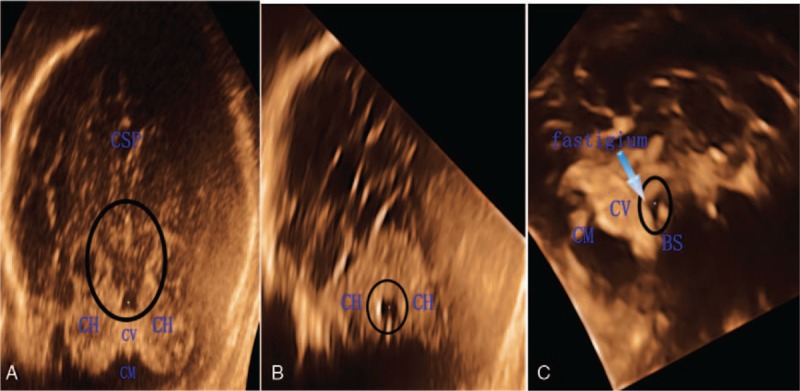
Normal fourth ventricle (4V) (A–C: circled) in the fetal period after 18 weeks of pregnancy. (A) In the axial plane, the anechoic dark area without molar tooth sign (MTS) is like a trapezoid. (B) Normal 4V in the coronal plane. (C) In the midsagittal plane, the anechoic dark area is like a triangle with a fastigium (arrow). BS = brain stem, CH = cerebellar hemisphere, CM = cisterna magna, CSP = cavity of septum pellucidum, CV = cerebellar vermis.
The clinical symptoms of JSRD vary from mild to severe. Increasing evidence suggests that congenital ataxia with broad-based gait, abnormal eye movement, decreased muscle tone, neonatal intermittent dyspnea or pause, mental retardation, and various growth retardations are common nervous manifestations.[24] The BS, which is an important part of respiratory function, can cause central dyspnea or pause, threatening an individual's life, and it can be affected by other complex neurogenic and metabolic control mechanisms.[3,25] It is also well-known that JSRD is associated with hypoplasia or the absence of vermis, which has an important effect on balance control, the regulation of muscle tension, motor ability, and rapid eye movement.[26] Defects of additional extra-nervous systems involve polycystic kidney disease, retinal degeneration, skeletal defects (polydactyly is most frequently postaxial[27]), and liver disorder (usually congenital hepatic fibrosis of abnormal development of the bile duct and portal vein during the embryonic period).[2,27] It has been reported that the retina is the most frequently involved organ in JSRD, followed by kidney defects (25%), polydactyly of the hands or toes (8%–16%), and hepatic fibrosis such as COACH syndrome (in the minority).[2,13] Pure JS occurs in a minority of patients, whereas multiorgan involvement is common.[18] Dempsey et al[28] found that a breathing problem (35%, 14/40 patients) was the most common cause of death in patients younger than 6 years, and kidney failure (37.5%, 15/40) was a more common cause of death in older people. However, these symptoms do not necessarily appear. The prognoses of patients with JSRD vary from each other and are too difficult to predict, such as mental impairment (mild or severe), vision (normal to blind), normal walking and speaking, or neonatal death (the most serious).[4,16,27] Elhassanien and Alghaiaty[24] reported that the initial symptom of JSRD was hyperpnoea from the first 10 days to 5 months, and it was especially worse in patients with a poor emotional status; additionally, retinal dysplasia might gradually worsen and even cause blindness. Therefore, breathing and feeding problems in newborns and infants are particularly concerning; however, respiratory distress will spontaneously improve with age and recover completely.[2,4,16]
Nowadays, a diagnosis of JSRD is made more often after birth, with the MTS detected by MRI, and the clinical symptoms of hypomyotonia and developmental delay or mental retardation.[24,29] The MTS is the pathognomonic hallmark of JSRD. When the MTS and CV hypoplasia are found, further assessment of other organ abnormalities is required.[2,5] Physicians can also suggest patients to undergo genetic testing for JSRD, which will be helpful for genetic analysis and prenatal counseling.
A diagnosis of JSRD made by prenatal sonography is rare and difficult to make. Ultrasonographic physicians usually lack knowledge of the syndrome and can merely observe nonspecific features, such as the widened posterior fossa and CV dysplasia. However, these signs can also be found in other malformations, such as the Dandy-Walker malformation, which has a very low risk of recurrence in the same family (Fig. 1E–G), pontocerebellar hypoplasia, and cranio-cerebello-cardiac syndrome.[26] In these disorders, dysplasia or the absence of CV is visually the most striking ultrasonographic finding. Further, ultrasonographic physicians should improve the chance of detecting the MTS. Sagittal planes can be observed well with 3-dimensional multiplanar ultrasonography. In a normal pregnancy, CV is still in the developmental stage before 18 weeks. Ultrasonographic observation after 18 weeks of pregnancy will be helpful to make a correct diagnosis.[4]
4. Conclusions
Joubert syndrome and related disorders is a congenital anomaly with a high risk of family recurrence, and MTS detected by prenatal sonography enables physicians to differentiate JSRD from other midbrain-hindbrain malformations.[12] Prenatal genetic analyses of the fetus and couple are essential to make a definitive diagnosis.
Acknowledgments
We gratefully acknowledge the help received from Dr Xin Liu and Dr Chunyan He, as they provided valuable assistance in preparing this case report.
Footnotes
Abbreviations: 4V = fourth ventricle, BS = brain stem, CC = corpus callosum, CH = cerebellar hemisphere, CM = cisterna magna, COACH = cerebellar vermis hypoplasia, oligophrenia of intelligence, ataxia, coloboma of ocular and hepatic fibrosis syndrome, CSP = cavity of septum pellucidum, CV = cerebellar vermis, IF = interpeduncular fossa, JS = Joubert syndrome, JSRD = Joubert syndrome and related disorders, MRI = magnetic resonance imaging, MTS = molar tooth sign, RSCV = remnant of the superior cerebellar vermis, SCP = superior cerebellar peduncle.
The authors disclose no conflicts of interest.
References
- [1].Joubert M, Eisenring JJ, Robb JP, et al. Familial agenesis of the cerebellar vermis. A syndrome of episodic hyperpnea, abnormal eye movements, ataxia, and retardation. Neurology 1969;19:813–25. [DOI] [PubMed] [Google Scholar]
- [2].Brancati F, Dallapiccola B, Valente EM. Joubert Syndrome and related disorders. Orphanet J Rare Dis 2010;5:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Louie CM, Gleeson JG. Genetic basis of Joubert syndrome and related disorders of cerebellar development. Hum Mol Genet 2005;2(14 Spec No):R235–42. [DOI] [PubMed] [Google Scholar]
- [4].Buke B, Canverenler E, İpek G, et al. Akkaya diagnosis of Joubert syndrome via ultrasonography. J Med Ultrason 2017;44:197–202. [DOI] [PubMed] [Google Scholar]
- [5].Kroes HY, Monroe GR, van der Zwaag B, et al. Joubert syndrome: genotyping a Northern European patient cohort. Eur J Hum Genet 2016;24:214–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Parisi MA. Clinical and molecular features of Joubert syndrome and related disorders. Am J Med Genet C Semin Med Genet 2009;151C:326–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Field M, Scheffer IE, Gill D, et al. Expanding the molecular basis and phenotypic spectrum of X-linked Joubert syndrome associated with OFD1 mutations. Eur J Hum Genet 2012;20:806–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Goetz SC, Anderson KV. The primary cilium: a signalling centre during vertebrate development. Nat Rev Genet 2010;11:331–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Watson CM, Crinnion LA, Berry IR, et al. Enhanced diagnostic yield in Meckel-Gruber and Joubert syndrome through exome sequencing supplemented with split-read mapping. BMC Med Genet 2016;17:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Huppke P, Wegener E, Böhrer-Rabel H, et al. Tectonic gene mutations in patients with Joubert syndrome. Eur J Hum Genet 2015;23:616–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Praveen K, Davis EE, Katsanis N. Unique among ciliopathies: primary ciliary dyskinesia, a motile cilia disorder. F1000Prime Rep 2015;7:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Quarello E, Molho M, Garel C, et al. Prenatal abnormal features of the fourth ventricle in Joubert syndrome and related disorders. Ultrasound Obstet Gynecol 2014;43:227–32. [DOI] [PubMed] [Google Scholar]
- [13].Valente EM, Brancati F, Dallapiccola B. Genotypes and phenotypes of Joubert syndrome and related disorders. Eur J Med Genet 2008;51:1–23. [DOI] [PubMed] [Google Scholar]
- [14].Brown JM, Witman GB. Cilia and diseases. Bioscience 2014;64:1126–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mok CA, Héon E, Zhen M. Ciliary dysfunction and obesity. Clin Genet 2010;77:18–27. [DOI] [PubMed] [Google Scholar]
- [16].Sattar S, Gleeson JG. The ciliopathies in neuronal development: a clinical approach to investigation of Joubert syndrome and Joubert syndrome-related disorders. Dev Med Child Neurol 2011;53:793–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Vilboux T, Doherty DA, Glass IA, et al. Molecular genetic findings and clinical correlations in 100 patients with Joubert syndrome and related disorders prospectively evaluated at a single center. Genet Med 2017;19:875–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Bachmann-Gagescu R, Dempsey JC, Phelps IG, et al. Joubert syndrome: a model for untangling recessive disorders with extreme genetic heterogeneity. J Med Genet 2015;52:514–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Valente EM, Brancati F, Silhavy JL, et al. AHI1 gene mutations cause specific forms of Joubert syndrome-related disorders. Ann Neuro 2006;59:527–34. [DOI] [PubMed] [Google Scholar]
- [20].Ferland RJ, Eyaid W, Collura RV, et al. Abnormal cerebellar development and axonal decussation due to mutations in AHI1 in Joubert syndrome. Nat Genet 2004;36:1008–13. [DOI] [PubMed] [Google Scholar]
- [21].Delous M, Baala L, Salomon R, et al. The ciliary gene RPGRIP1L is mutated in cerebello-oculo-renal syndrome (Joubert syndrome type B) and Meckel syndrome. Nat Genet 2007;39:875–81. [DOI] [PubMed] [Google Scholar]
- [22].Poretti A, Huisman T, Scheer I, et al. Joubert syndrome and related disorders: spectrum of neuroimaging findings in 75 patients. Am J Neuroradiol 2011;32:1459–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Baldawa S. An unusual association of classical Joubert syndrome with retrocerebellar arachnoid cyst. Childs Nerv Syst 2016;32:1181–2. [DOI] [PubMed] [Google Scholar]
- [24].Elhassanien AF, Alghaiaty HAA. Joubert syndrome: clinical and radiological characteristics of nine patients. Ann Indian Acad Neurol 2013;16:239–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Wolfe L, Lakadamyali H, Mutlu GM. Joubert syndrome associated with severe central sleep apnea. J Clin Sleep Med 2010;6:384–8. [PMC free article] [PubMed] [Google Scholar]
- [26].Millen KJ, Gleeson JG. Cerebellar development and disease. Curr Opin Neurobiol 2008;18:12–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Doherty D. Joubert syndrome: insights into brain development, cilium biology, and complex disease. Semin Pediatr Neurol 2009;16:143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Dempsey JC, Phelps IG, Bachmann-Gagescu R, et al. Mortality in Joubert syndrome. Am J Med Genet 2017;173:1237–42. [DOI] [PubMed] [Google Scholar]
- [29].Szymanska K, Hartill VL, Johnson CA. Unraveling the genetics of Joubert and Meckel-Gruber syndromes. J Pediatr Genet 2014;3:65–78. [DOI] [PMC free article] [PubMed] [Google Scholar]