

Supplemental Digital Content is available in the text
Keywords: African Americans, autoantibodies, systemic sclerosis
Abstract
Racial differences exist in the severity of systemic sclerosis (SSc). To enhance our knowledge about SSc in African Americans, we established a comprehensive clinical database from the largest multicenter cohort of African American SSc patients assembled to date (the Genome Research in African American Scleroderma Patients (GRASP) cohort).
African American SSc patients were enrolled retrospectively and prospectively over a 30-year period (1987–2016), from 18 academic centers throughout the United States. The cross-sectional prevalence of sociodemographic, clinical, and serological features was evaluated. Factors associated with clinically significant manifestations of SSc were assessed using multivariate logistic regression analyses.
The study population included a total of 1009 African American SSc patients, comprised of 84% women. In total, 945 (94%) patients met the 2013 American College of Rheumatology/European League Against Rheumatism (ACR/EULAR) classification criteria for SSc, with the remaining 64 (6%) meeting the 1980 ACR or CREST (calcinosis, Raynaud's phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia) criteria. While 43% were actively employed, 33% required disability support. The majority (57%) had the more severe diffuse subtype and a young age at symptom onset (39.1 ± 13.7 years), in marked contrast to that reported in cohorts of predominantly European ancestry. Also, 1 in 10 patients had a severe Medsger cardiac score of 4. Pulmonary fibrosis evident on computed tomography (CT) chest was present in 43% of patients and was significantly associated with anti-topoisomerase I positivity. 38% of patients with CT evidence of pulmonary fibrosis had a severe restrictive ventilator defect, forced vital capacity (FVC) ≤50% predicted. A significant association was noted between longer disease duration and higher odds of pulmonary hypertension, telangiectasia, and calcinosis. The prevalence of potentially fatal scleroderma renal crisis was 7%, 3.5 times higher than the 2% prevalence reported in the European League Against Rheumatism Scleroderma Trials and Research (EUSTAR) cohort.
Our study emphasizes the unique and severe disease burden of SSc in African Americans compared to those of European ancestry.
1. Introduction
There is evidence that racial differences exist in the susceptibility to and severity of systemic sclerosis (scleroderma; SSc). African Americans have a higher age-specific incidence and prevalence of SSc compared to European Americans.[1,2] Moreover, the most current national report of SSc-associated mortality in the United States (US) noted death rates that peaked a decade earlier in the African American population, with age-adjusted mortality significantly higher in African Americans compared to European Americans.[3]
The leading cause of mortality in SSc is attributed to pulmonary complications, which occur in 70% to 90% of patients.[4] The 10-year survival for SSc patients with interstitial lung disease (ILD) is only 60%.[5] African ancestry is an independent predictor of lung involvement in SSc.[6] Furthermore, the incidence of SSc-associated severe ILD[7] and pulmonary hypertension[8,9] is reported to be higher in African Americans than other ethnic groups.
Socioeconomic factors and impaired access to health care have not fully accounted for the predilection of African Americans to poor health outcomes.[10] Attempts to elucidate the factors influencing increased disease severity have been hindered by the relatively small size of available African American SSc cohorts.[11,12]
Accordingly, a multicenter SSc cohort database, the Genome Research in African American Scleroderma Patients (GRASP) clinical database, was established to enhance our understanding of the phenotype of SSc in African Americans and identify factors contributing to the severity of their disease. The GRASP cohort consists of more than 1000 extensively phenotyped African American SSc patients enrolled from academic centers throughout the US. It is currently the largest multicenter cohort of African American SSc patients. Consequently, the comprehensive clinical database and significant size of the GRASP cohort provides adequate statistical power to perform informative multivariate analyses.
In this paper, we describe the clinical and serological characteristics of the GRASP cohort and report the results of multivariate analyses, to identify factors associated with clinically significant and severe manifestations of SSc in African Americans. Additionally, we compare the findings in the GRASP cohort to that reported in a multicenter cohort of predominantly European ancestry.
2. Methods
2.1. Study population
The GRASP clinical database was established in May 2013 and includes socio-demographic and clinical characteristics of a US cohort of exclusively African American SSc patients, enrolled retrospectively and prospectively over a 30-year period (1987–2016). African American race was ascertained by patient self-identification. All patients met the 1980 American College of Rheumatology (ACR) or 2013 ACR/EULAR (European League Against Rheumatism) classification criteria for systemic sclerosis; or had at least 3 of 5 features of the CREST (calcinosis, Raynaud's phenomenon, esophageal dysmotility, sclerodactyly, telangiectasia) syndrome.[13,14]
2.2. Study protocol
Patients were enrolled from a total of 18 academic centers throughout the US. The study was conducted in accordance with the Declaration of Helsinki[15] and participating centers secured local ethics committee approval prior to participant enrollment. GRASP investigators documented clinical, serological and socio-demographic data, including age, sex, dates of SSc diagnosis and symptom onset, smoking status, immunosuppressive medication use, history of malignancy and autoantibody status. The presence of an overlapping autoimmune disease (rheumatoid arthritis, systemic lupus erythematosus, inflammatory myopathy, Sjögren's syndrome) was ascertained based on established classification criteria.[16–20]
Data from diagnostic studies including pulmonary function tests (PFTs), echocardiograms, chest radiographs, and high-resolution computed tomography (CT) scans of the chest and right heart cardiac catheterizations were obtained. All data were subsequently assembled in a clinical database, maintained at the Johns Hopkins University coordinating site.
Disease onset was defined as the occurrence of the first ever symptom attributed to SSc (Raynaud's or non-Raynaud's). Disease duration was defined as the time from disease onset to the date of sample collection for genetic analysis. SSc subtype was classified as diffuse (dcSSc) or limited (lcSSc) based on the extent of cutaneous involvement, as described by LeRoy et al.[21] Patients were classified as having diffuse SSc if there was clinical evidence of cutaneous fibrosis extending proximal to the elbows or knees, at any time during the disease course.
The pattern of skin involvement was further classified into 4 subsets (Type 0,1,2,3) as previously defined by Cottrell et al.[22] The degree of cutaneous fibrosis was quantitatively assessed using the physician assigned modified Rodnan Skin Score (mRSS).[23] The maximum mRSS and worst ever organ specific severity scores were recorded for each patient. Organ-specific severity scores were assigned in accordance with the revised Medsger Severity Score for SSc.[24]
Target organ involvement was deemed to be present if the respective Medsger Severity Score was ≥ 1. Severe organ involvement was defined as a Medsger Severity Score of 3 (severe) or 4 (end stage).[24] Accordingly, severe peripheral vascular involvement was defined as the presence of digital tip ulcerations or digital gangrene. An mRSS ≥30 was indicative of severe cutaneous involvement. Severe gastrointestinal disease included malabsorption syndrome, episodes of pseudo-obstruction or the requirement of total parenteral nutrition. For renal disease, severe involvement was defined by a serum creatinine level ≥3.0 mg/dL, or the requirement for dialysis or renal transplant. Skeletal muscle involvement was deemed to be severe if proximal muscle weakness with less than grade 3/5 power was evident on physical examination, or the patient required ambulation aids. Severe cardiac disease was defined as a left ventricular ejection fraction <40%, clinical signs of heart failure, an arrhythmia requiring treatment, or heart transplant. Severe pulmonary involvement was characterized by the presence of at least one of the following: forced vital capacity (FVC) < 50% of predicted, diffusing capacity of the lung for carbon monoxide (DLCO) < 50% of predicted, moderate to severe pulmonary hypertension, requirement for oxygen due to SSc-associated pulmonary disease or lung transplant.
2.3. Statistical analysis
The cross-sectional prevalence of clinical and serological features in the GRASP cohort was determined using the data obtained at the time of study enrollment. Clinical and socio-demographic characteristics were compared between groups based on sex, serological profile, and SSc subtype respectively, using t-test, chi-square test, Fisher's exact test, and one-way analysis of variance (ANOVA) as appropriate. Factors associated with clinical manifestations of SSc and severe organ involvement were identified using multivariable logistic regression analyses including covariates: sex (male versus female), SSc subtype (dcSSc versus lcSSc), SSc-associated autoantibody status (anti-centromere, anti-topoisomerase I or anti-RNA polymerase III positivity), age at first symptom onset and disease duration in years. These variables were fixed in all analyses because of their clinical relevance.
The date of onset of the first symptom attributed to SSc (Raynaud's or non-Raynaud's) was used to calculate disease duration. Sensitivity analyses were performed to ensure the choice of disease onset (Raynaud's onset versus first non-Raynaud's symptom onset) did not impact the magnitude or significance of observed associations with relevant clinical outcomes. The covariate of smoking status (ever versus never smoked cigarettes) was included in multivariable logistic regression analysis to determine factors associated with vascular and cardiopulmonary involvement. Assumptions in the statistical analyses were verified using normal probability and leverage plots. Statistical significance was defined as a 2-sided P value ≤ .05. The dataset was analyzed using Stata Statistical Software version 14.2 (College Station, TX).
3. Results
3.1. Patient characteristics and sociodemographic features
As of November 2016, a total of 1009 African American SSc patients were enrolled in the GRASP cohort from the 18 participating US academic centers (Supplementary Table 1). Comprehensive clinical and serological data were provided for most patients (Supplementary Table 2). The sociodemographic features of the GRASP cohort are summarized in Table 1.
Table 1.
Socio-demographic characteristics of the Genome Research in African American Scleroderma Patients cohort.

There was a female predominance, 843 (84%) women. The majority of patients were insured. More than 50% completed a college or post-graduate education. While 43% were actively employed, 33% required disability support. At the time of study enrollment, 35% of patients had a history of or currently smoked cigarettes.
3.2. Disease characteristics
In total 94% patients met the 2013 ACR/EULAR classification criteria for SSc, with the remaining 6% meeting the 1980 ACR or CREST criteria (Table 2 ). The majority of patients (57%) were classified as dcSSc.
Table 2.
Clinical and serological characteristics of the Genome Research in African American Scleroderma Patients cohort by sex.
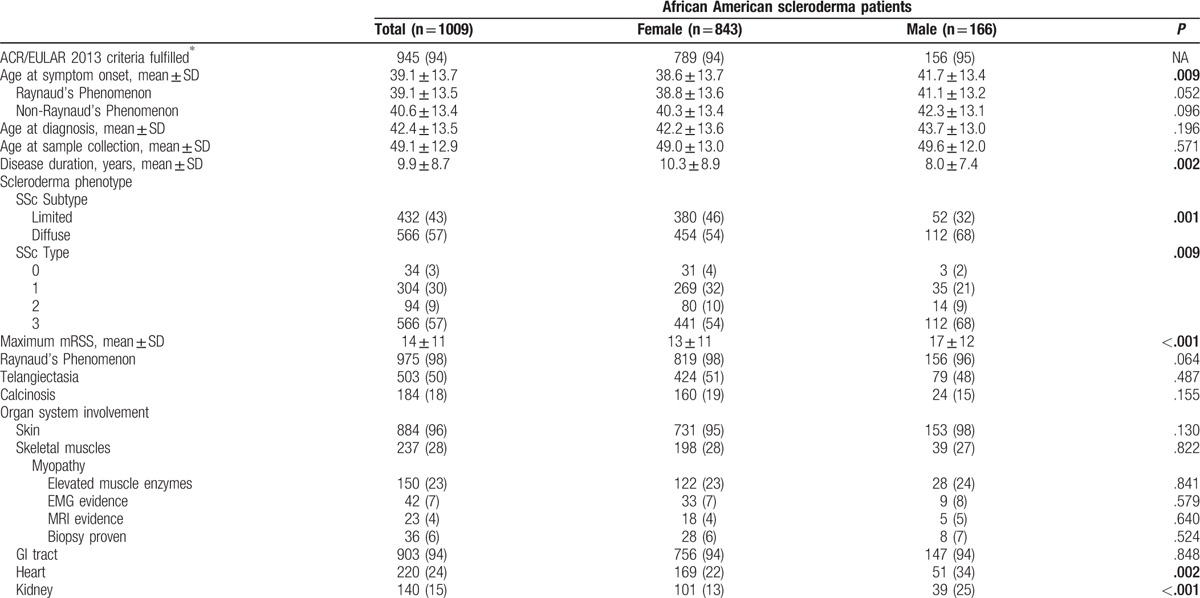
The mean age at SSc diagnosis was 42.4 ± 13.5 years, with an average time to diagnosis of 3.4 ± 6.0 years from the onset of the first symptom attributed to SSc. The mean age at onset of the first symptom attributed to SSc was 39.1 ± 13.7 years (Table 2 ).
An assessment of the general health status was scored by the Medsger general severity scale, which uses weight loss and hematologic measures to define disease burden.[24] 10% exhibited a severe grade 4 disease burden, with weight loss ≥44 pounds or anemia with hematocrit <25% (Fig. 1).
Figure 1.
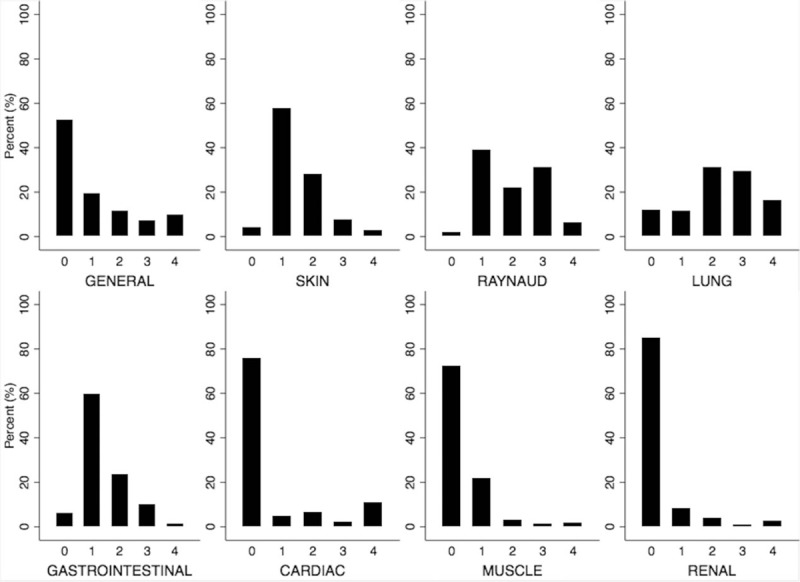
Distribution of respective severity scores for organ involvement in the GRASP cohort∗. ∗Respective severity scores assigned in accordance with the revised Medsger Severity Score for systemic sclerosis.[24] GRASP = Genome Research in African American Scleroderma Patients cohort.
3.3. Organ involvement
3.3.1. Cutaneous
A high prevalence of diffuse disease (57%) was noted, and a predilection for the diffuse subtype was observed in both men and women (Table 2 ). The mean maximum mRSS for patients with dcSSc was 20 ± 10, and 5 ± 5 in patients with lcSSc (Table 3). Anti-topoisomerase I (adjusted odds ratio [OR] 1.67, 95% confidence interval [95% CI] 1.08–2.58) and anti-RNA polymerase III (adjusted OR 2.54, 95% CI 1.36–4.75) positivity were significantly associated with the diffuse subtype, whereas anti-centromere positivity was found to be protective (adjusted OR 0.14, 95% CI 0.05–0.38) (Table 4).
Table 2 (Continued).
Clinical and serological characteristics of the Genome Research in African American Scleroderma Patients cohort by sex.
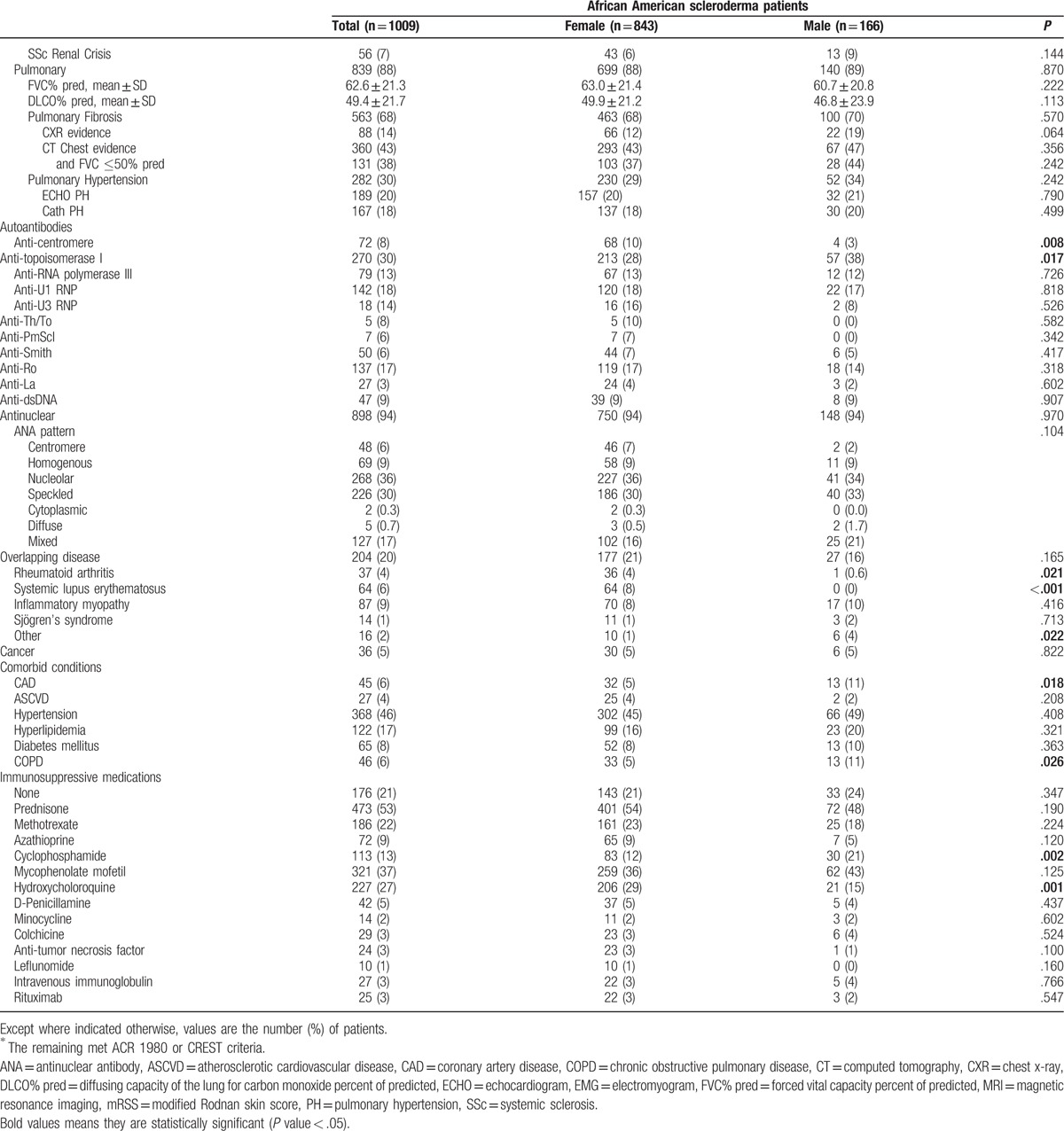
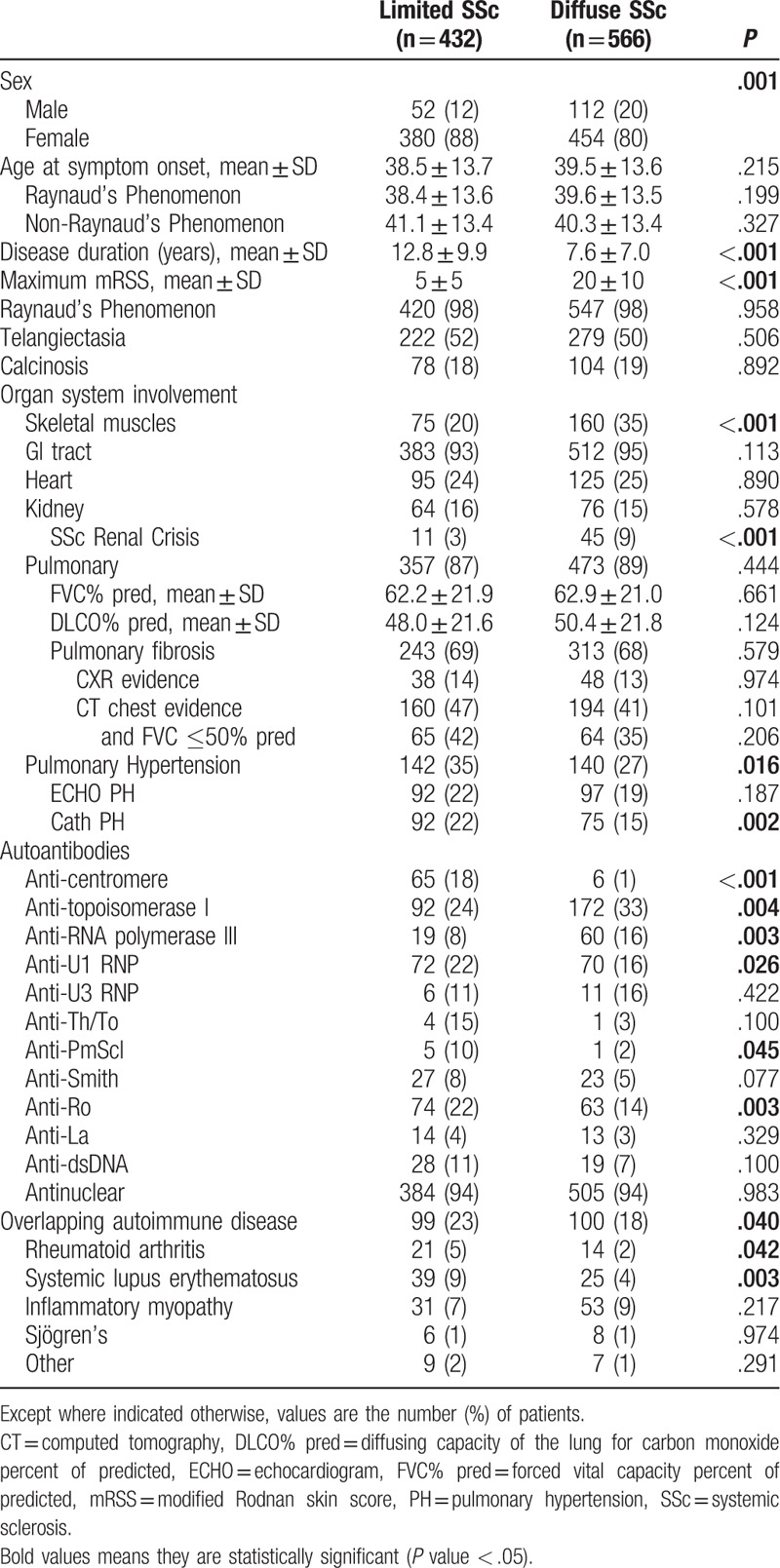
Table 3.
Clinical and serological characteristics of the Genome Research in African American Scleroderma Patients cohort by Scleroderma subtype.
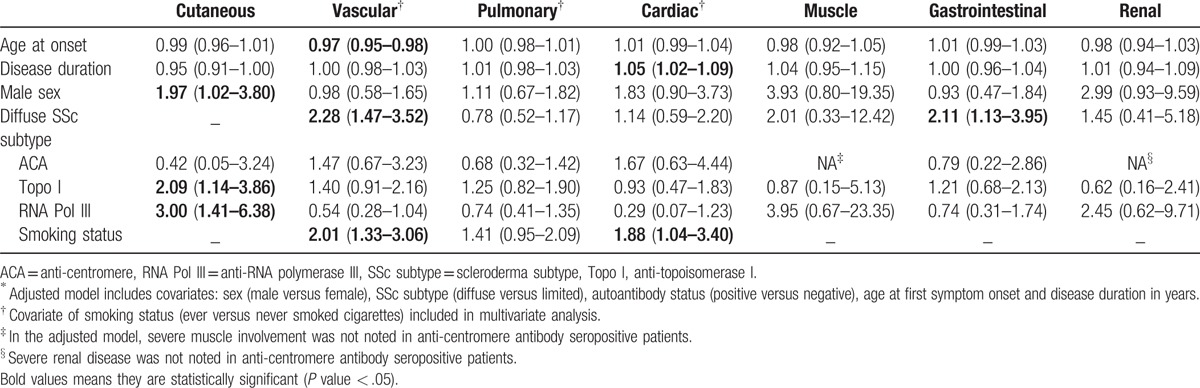
In bivariate analyses, higher mRSS values were associated with male sex (difference in mRSS 4, P < .001), anti-topoisomerase I positivity (difference in mRSS 4, P < .001) and anti-RNA polymerase III positivity (difference in mRSS 7, P < .001). Conversely, anti-centromere antibody positivity was associated with lower mRSS values (difference in mRSS 8, P < .001). In multivariate analyses, severe cutaneous involvement (mRSS ≥ 30) was significantly associated with male sex, anti-topoisomerase I, and anti-RNA polymerase III positivity (Table 5).
Table 4.
Factors associated with Clinical Manifestations of Systemic Sclerosis, adjusted odds ratio (95% confidence interval)∗.
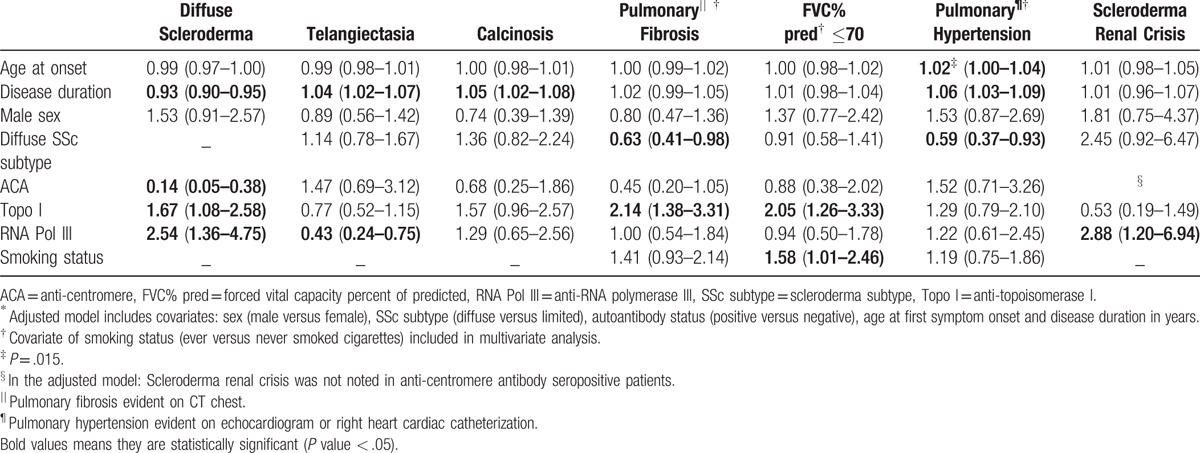
Table 5.
Factors associated with Severe Organ Involvement, adjusted odds ratio (95% confidence interval)∗.
50% of the GRASP cohort had telangiectasia (Table 2 ). In the adjusted models, the relative odds of having telangiectasia was 4% higher for each year of disease duration and anti-RNA polymerase III positivity was associated with over 50% lower odds of exhibiting telangiectasia (Table 4).
18% of patients had a history of calcinosis. Longer disease duration was the only factor significantly associated with calcinosis (Table 4).
3.3.2. Raynaud's phenomenon
The mean age at onset of Raynaud's phenomenon (RP) was 39.1 ± 13.5 years (Table 2 ). 98% of patients reported a history of RP. 31% of patients experienced digital tip ulcerations and 6% reported complications of digital gangrene. Diffuse disease and cigarette smoking were significantly associated with severe vascular complications of digital ulcers and gangrene (Table 5). Older age at symptom onset was associated with significantly lower odds of experiencing these severe vascular complications (Table 5).
3.3.3. Pulmonary
More than half of the cohort (66%) had a restrictive ventilatory defect with FVC ≤70% of predicted. The overall mean FVC percent predicted was 63%, and mean DLCO percent predicted was 49% (Table 2 ). By report 68% of patients had pulmonary fibrosis. 43% of patients had evidence of pulmonary fibrosis on CT chest and 38% of these patients had a severe restrictive ventilatory defect (FVC ≤50% predicted) (Table 2 ).
Anti-topoisomerase I positivity was associated with significantly higher odds of pulmonary fibrosis on CT chest, and 2-fold higher odds of a restrictive ventilatory defect with FVC≤70% predicted (Table 4). The presence of a restrictive ventilatory defect was significantly associated with cigarette smoking (Table 4). Notably, patients with dcSSc compared to lcSSc had approximately equal odds of demonstrating a restrictive ventilatory defect (Table 4).
30% of patients had evidence of pulmonary hypertension on echocardiogram or right heart cardiac catheterization. Moreover, these patients had severe disease with a mean DLCO 42% of predicted. In multivariate analyses, older age at symptom onset was significantly associated with pulmonary hypertension (Table 4). Furthermore, a 6% increase in odds of pulmonary hypertension was noted with each 1 year increase in disease duration. The diffuse subtype was protective and associated with 41% lower odds of pulmonary hypertension (Table 4).
3.3.4. Cardiac
Echocardiographic or electrocardiogram evidence of cardiac disease was noted in 24% of patients. 11% of patients had a Medsger cardiac severity score of 4, indicative of either clinical signs of heart failure, a left ventricular ejection fraction of <30%, an arrhythmia requiring treatment or a heart transplant (Fig. 1).
In multivariate analyses, a significant association was noted between cardiac involvement and older age at symptom onset (adjusted OR 1.03 per 1 year increase in age, 95% CI 1.01–1.05), longer disease duration (adjusted OR 1.05 per year, 95% CI 1.02–1.08), and male sex (adjusted OR 2.00, 95% CI 1.14–3.48). Moreover, the significance of the observed associations was maintained, even after adjustment for atherosclerotic cardiovascular disease and cardiovascular risk factors including diabetes mellitus, hypertension, hyperlipidemia, and cigarette smoking.
The odds of severe cardiac involvement increased by 5% per annum of disease duration (Table 5). The magnitude and significance of this association was maintained in multivariate analysis adjusting for atherosclerotic cardiovascular disease and cardiovascular risk factors (adjusted OR 1.07 per 1 year increase in age, 95% CI 1.03–1.12). Cigarette smoking was associated with 88% higher odds of severe cardiac involvement (Table 5).
3.3.5. Gastrointestinal tract
94% of patients had a history of gastrointestinal tract involvement of varying severity. In the adjusted model, anti-RNA polymerase III positivity was associated with 63% lower odds of gastrointestinal involvement including gastrointestinal reflux, abnormal small bowel series, small intestinal bacterial overgrowth, malabsorption syndrome, episodes of pseudo-obstruction or the requirement of total parenteral nutrition (adjusted OR 0.37, 95% CI 0.14–0.96).
Notably, 11% of patients experienced severe gastrointestinal complications such as malabsorption syndrome, pseudo-obstruction, or required total parenteral nutrition. The diffuse subtype was associated with over 2-fold higher odds of severe gastrointestinal complications (Table 5).
3.3.6. Renal
Normal renal function was noted in 85% of patients. The prevalence of scleroderma renal crisis (SRC) in the GRASP cohort was 7% (Table 2 ). In bivariate analyses, SRC was significantly associated with the diffuse subtype (OR 3.30, 95% CI 1.68–6.47). Additionally, anti-RNA polymerase III positivity was associated with more than 4-fold increased odds of SRC (OR 4.03, 95% CI 1.85–8.77). The significant association with anti-RNA polymerase III positivity, but not diffuse subtype, was maintained in the adjusted model (Table 4).
3.3.7. Muscle
237 (28%) patients had an abnormal muscle severity score indicating weakness. Patients with dcSSc had a higher prevalence of skeletal muscle involvement (Table 3). The diffuse subtype was associated with a 2-fold higher odds of skeletal muscle involvement compared to limited disease (adjusted OR 2.00, 95% CI 1.20–3.33).
3.4. Serological profile
Clinical characteristics of the GRASP cohort by the serological profile are summarized in Supplementary Table 3. 30% of patients were seropositive for anti-topoisomerase I while only 8% were anticentromere positive (Table 2 ). A low prevalence of anti-RNA polymerase III positivity (13%) was observed; however, it is important to note that anti-RNA polymerase III data were missing in 40% of patients, likely because this assay was not commercially available until 2007.
A predilection for anti-topoisomerase I positivity was noted in men, while a significantly higher prevalence of anti-centromere positivity was observed in women (Table 2 ). A variety of antinuclear antibody (ANA) patterns were observed, with the nucleolar pattern being the most common, noted in 36% of patients (Table 2 ).
3.5. Overlapping autoimmune diseases
In total, 1 in 5 patients were diagnosed with an overlapping autoimmune disease of which systemic lupus erythematosus (SLE) and inflammatory myopathies were the most common (Table 2 ). In the multivariate model which also included anti-U1RNP positivity, seropositivity for anti-topoisomerase I was associated with significantly lower odds of an overlapping autoimmune disease (adjusted OR 0.53, 95% CI 0.29–0.96). Anti-U1RNP positivity was associated with an approximately 4-fold higher odds of exhibiting an overlapping autoimmune disease (adjusted OR 3.99, 95% CI 2.33–6.84).
3.6. Immunosuppressive therapy
A history of exposure to immunosuppressive therapy was obtained. The immunosuppressive agents administered are summarized in Table 2 . The most commonly prescribed immunosuppressive agents were prednisone (53%) and mycophenolate mofetil (37%), while cyclophosphamide was used in 13% of patients (Table 2 ).
4. Discussion
Our study highlights sociodemographic, clinical and serological features of the largest multicenter cohort of African American patients with SSc. It provides insight into the factors associated with clinically significant and severe manifestations of SSc in African Americans and emphasizes the unique clinical features of SSc in African Americans that differ from that reported in cohorts of European ancestry.
The European League Against Rheumatism Scleroderma Trials and Research (EUSTAR) cohort is the largest multinational SSc cohort, comprised of over 7000 patients of predominantly European ancestry.[25] Genetic studies have been conducted using this cohort to identify SSc disease susceptibility loci.[26–28] However, patients in the EUSTAR cohort differ in fundamental clinical characteristics from those enrolled in GRASP. The inferences made from genetic studies in the EUSTAR and other cohorts of predominantly European ancestry[29–31] may not be applicable to African American patients represented in the GRASP cohort.
The mean age at onset of RP and the first non-RP symptom was 39.1 and 40.6 years, respectively, in the GRASP cohort. In contrast in the EUSTAR cohort, the mean age of onset of RP and non-RP symptoms occurred 3 and 4 years later, respectively.[25] This is consistent with prior studies, in which the average age at onset of SSc-associated symptoms and subsequent diagnosis was reported to be significantly younger in African Americans than European Americans.[2,10,32–35]
Diffuse cutaneous SSc was present in 57% of the GRASP cohort, in contrast to 37% in the EUSTAR cohort.[25] This is of particular importance, as patients with dcSSc have more extensive cutaneous fibrosis affecting the trunk and proximal limbs, and are noted to exhibit a higher frequency of cardiac, pulmonary, and renal involvement especially within the first 3 years of disease onset.[36]
Pulmonary complications are a prominent source of morbidity and mortality in SSc[5,37–39] and African ancestry is reported to be an independent predictor of early pulmonary involvement[6] and severe pulmonary fibrosis.[35] Compared to the EUSTAR cohort, participants in the GRASP cohort had a lower mean FVC % predicted[25] (63% vs 92%). Additionally, 16% of patients in the GRASP cohort required supplemental oxygen therapy compared to 3% in the EUSTAR cohort.
Prior to the advent of initiation of angiotensin converting enzyme (ACE) inhibitors for management of SRC, the 5-year cumulative survival of patients with this potentially fatal complication was <10%.[40] The prevalence of SRC in the GRASP cohort was 3.5 times that observed in the EUSTAR cohort (7% vs 2%).[25] This difference may potentially be attributed to the higher frequency of dcSSc in the GRASP cohort or the higher seroprevalence of anti-RNA polymerase III in the GRASP (13% vs 2%) compared to the EUSTAR cohort.[25]
The frequency of anti-topoisomerase I positivity in African American SSc cohorts is estimated to range from 16% to 39%.[2,8,10,32,33,35,41,42] 30% of patients in the GRASP cohort were anti-topoisomerase I positive. Interestingly, a slightly higher prevalence of anti-topoisomerase I positivity (37%) was observed in the EUSTAR cohort.[25] Additionally, while the prevalence of anti-RNA polymerase III positivity in the GRASP cohort (13%) was consistent with prior estimates from other African American SSc cohorts[32,35,41] it was 6 times higher than that observed in the EUSTAR cohort (13% vs 2%).[25] This suggests that differences exist in the serological profiles among SSc patients of African and European ancestry, thereby limiting the generalizability of the EUSTAR reports.
Anti-centromere antibody positivity was lower (8% vs 32%) and anti-U1 RNP positivity was higher (18% vs 8%) in the GRASP compared to EUSTAR cohort.[25] Unfortunately, serological data on anti-U3 RNP and other nucleolar autoantibodies were not uniformly available for patients in the GRASP cohort, limiting our ability to make reliable inferences about the prevalence of these autoantibodies. Anti-U3 RNP antibodies are reported to be highly specific for SSc and more prevalent in African Americans,[43,44] exhibiting associations with diffuse disease, skeletal muscle involvement, and primary pulmonary arterial hypertension[43,45] as well as aggressive gastrointestinal disease.[35] Of note anti-U3 RNP antibodies typically demonstrate a nucleolar pattern on indirect immunofluorescence. This pattern was observed in 36% of the GRASP cohort.
African American patients with SSc are reported to have overall lower sociodemographic status and significantly fewer years of education than European Americans.[8,10] It is noted that 97% of the GRASP cohort obtained a high school education with 59% completing college or post-graduate education. Furthermore, participants in the GRASP cohort were almost universally insured, with 97% having Medicare, private insurance or medical assistance. In light of this, the high disease burden in the GRASP cohort is unlikely to be substantially attributed to socioeconomic factors and impaired access to healthcare.
There are limitations to our study, primarily related to missing data stemming from the retrospective collection of information. In particular, while DNA samples have been uniformly provided, for some patients the corresponding clinical and serological information are incomplete. Nevertheless, the data gleaned from the GRASP cohort is comprehensive, providing the most complete phenotypic characterization of SSc in African American patients to date.
Evidence of distinct clinical and serological differences between SSc patients of African and European ancestry underscores the critical importance of further research in African Americans, who otherwise may not benefit from precision medicine through new clinical and technological advancements in the treatment of their disease.[46] Our study highlights sociodemographic, clinical and serological features of this multicenter cohort of African American patients and emphasizes the severe disease burden of SSc in African Americans. Furthermore, GRASP provides a unique cohort to facilitate future investigations probing the role of genetic factors in SSc disease susceptibility and severity in African Americans.
Supplementary Material
Acknowledgments
The authors would like to thank Gwendolyn Leatherman and Adrianne Woods for excellent database management support. They would also like to acknowledge the Martha McCrory endowment.
Footnotes
Abbreviations: ACE = angiotensin converting enzyme, ACR/EULAR = American College of Rheumatology/European League Against Rheumatism, ANA = antinuclear antibody, ANOVA = analysis of variance, CI = confidence interval, CREST = calcinosis, CT = computed tomography, dcSSc = diffuse cutaneous systemic sclerosis, DLCO = diffusing capacity of the lung for carbon monoxide, esophageal dysmotility, EUSTAR = European League Against Rheumatism Scleroderma Trials and Research, EUSTAR = European League Against Rheumatism Scleroderma Trials and Research, FVC = forced vital capacity, GRASP = Genome Research in African American Scleroderma Patients cohort, ILD = interstitial lung disease, lcSSc = limited cutaneous systemic sclerosis, mRSS = modified Rodnan Skin Score, Musculoskeletal and Skin Diseases, NIAMS = National Institute of Arthritis, NIH = National Institutes of Health, OR = odds ratio, PFTs = pulmonary function tests, Raynaud's phenomenon, RP = Raynaud's phenomenon, sclerodactyly, SLE = systemic lupus erythematosus, SRC = scleroderma renal crisis, SSc = systemic sclerosis, telangiectasia, US = United States.
PG and FB equally contributed to this study and are co-senior authors.
Authorship: NDM, AAS, FW, FB: analysis and interpretation of data and drafting of the article. All authors: conception and study design; critical review and revision of the article to ensure important intellectual content, and final approval of the version to be published.
Ethics approval: Approval from respective local ethics committees was obtained by each participating center.
Funding: The GRASP consortium was supported by research funding from the Scleroderma Research Foundation and the Intramural Research Programs of the National Human Genome Research Institute and the National Institute of Arthritis and Musculoskeletal and Skin Diseases.
Dr Nadia Morgan was supported by the National Institute of Arthritis, Musculoskeletal and Skin Diseases (NIAMS) of the National Institutes of Health (NIH) under Award Number T32AR048522 and the Rheumatology Research Foundation Scientist Development Award.
Dr Ami Shah was supported by NIAMS of the NIH under Award Number K23AR061439.
Dr Maureen Mayes was supported by grants from NIAMS of the NIH Centers of Research Translation P50-AR054144, NIH grant N01-AR-02251 and R01-AR-055258; and the Department of Defense Congressionally Directed Medical Research Program W81XWH-07–1–011 and WX81XWH-13–1-0452.
Dr Paul Ramos was supported by grants from the NIH K01 AR067280, R03 AR065801, P60 AR062755, and the South Carolina Clinical and Translational Research Institute, with an academic home at the Medical University of South Carolina, through NIH Grants numbers UL1 RR029882 and UL1 TR000062.
Dr Richard Silver was supported by grants from the NIH P60 AR062755 and the South Carolina Clinical and Translational Research Institute, with an academic home at the Medical University of South Carolina, through NIH Grants numbers UL1 RR029882 and UL1 TR000062.
Dr Dinesh Khanna was supported by grants from NIAMS of the NIH K24 AR063120, has received investigator-initiated grants and acts as a consultant to Actelion, BMS, Bayer, Corbus, Cytori, ChemoMab, Eicos, GSK, Genentech/Roche, Sanofi-Aventis, and UCB.
Dr Chris Derk was supported by research funding from Gilead, Actelion and Cytori.
Dr Francesco Boin was supported by research funding from Nina Ireland Program for Lung Health.
The authors have no conflicts of interest to disclose.
Supplemental Digital Content is available for this article.
References
- [1].Steen VD, Oddis CV, Conte CG, et al. Incidence of systemic sclerosis in Allegheny County, Pennsylvania. A twenty-year study of hospital-diagnosed cases, 1963–1982. Arthritis Rheum^^ 1997;40:441–5. [DOI] [PubMed] [Google Scholar]
- [2].Mayes MD, Lacey JV, Jr, Beebe-Dimmer J, et al. Prevalence, incidence, survival, and disease characteristics of systemic sclerosis in a large US population. Arthritis Rheum 2003;48:2246–55. [DOI] [PubMed] [Google Scholar]
- [3].Mendoza F, Derk CT. Systemic sclerosis mortality in the United States: 1999-2002 implications for patient care. J Clin Rheumatol 2007;13:187–92. [DOI] [PubMed] [Google Scholar]
- [4].Akter T, Silver RM, Bogatkevich GS. Recent advances in understanding the pathogenesis of scleroderma-interstitial lung disease. Curr Rheumatol Rep 2014;16:411. [DOI] [PubMed] [Google Scholar]
- [5].Steen VD, Medsger TA. Changes in causes of death in systemic sclerosis, 1972–2002. Ann Rheum Dis 2007;66:940–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].McNearney T, Reveille JD, Fischbach M, et al. Pulmonary involvement in systemic sclerosis: associations with genetic, serologic, sociodemographic, and behavioral factors. Arthritis Rheum 2007;57:318–26. [DOI] [PubMed] [Google Scholar]
- [7].Silver RM, Bogatkevich G, Tourkina E, et al. Racial differences between blacks and whites with systemic sclerosis. Curr Opin Rheumatol 2012;24:642–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Reveille JD, Fischbach M, McNearney T, et al. Systemic sclerosis in 3 US ethnic groups: a comparison of clinical, sociodemographic, serologic, and immunogenetic determinants. Semin Arthritis Rheum 2001;30:332–46. [DOI] [PubMed] [Google Scholar]
- [9].Blanco I, Mathai S, Shafiq M, et al. Severity of systemic sclerosis-associated pulmonary arterial hypertension in African Americans. Medicine 2014;93:177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nietert PJ, Mitchell HC, Bolster MB, et al. Racial variation in clinical and immunological manifestations of systemic sclerosis. J Rheumatol 2006;33:263–8. [PubMed] [Google Scholar]
- [11].Arnett FC, Gourh P, Shete S, et al. Major histocompatibility complex (MHC) class II alleles, haplotypes and epitopes which confer susceptibility or protection in systemic sclerosis: analyses in 1300 Caucasian, African–American and Hispanic cases and 1000 controls. Ann Rheum Dis 2010;69:822–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Hudson LL, Silver RM, Pandey JP. Ethnic differences in cytotoxic T lymphocyte associated antigen 4 genotype associations with systemic sclerosis. J Rheumatol 2004;31:85–7. [PubMed] [Google Scholar]
- [13].Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum 1980;23:581–90. [DOI] [PubMed] [Google Scholar]
- [14].van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 2013;72:1747–55. [DOI] [PubMed] [Google Scholar]
- [15].World Medical Association Declaration of Helsinki: Ethical principles for medical research involving human subjects. JAMA 2013;310:2191–4. [DOI] [PubMed] [Google Scholar]
- [16].Petri M, Orbai AM, Alarcon GS, et al. Derivation and validation of the Systemic Lupus International Collaborating Clinics classification criteria for systemic lupus erythematosus. Arthritis Rheum 2012;64:2677–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Aletaha D, Neogi T, Silman AJ, et al. 2010 rheumatoid arthritis classification criteria: an American College of Rheumatology/European League Against Rheumatism collaborative initiative. Ann Rheum Dis 2010;69:1580–8. [DOI] [PubMed] [Google Scholar]
- [18].Bohan A, Peter JB. Polymyositis dermatomyositis (first of two parts). N Engl J Med 1975;292:344–7. [DOI] [PubMed] [Google Scholar]
- [19].Targoff IN, Miller FW, Medsger TA, Jr, et al. Classification criteria for the idiopathic inflammatory myopathies. Curr Opin Rheumatol 1997;9:527–35. [DOI] [PubMed] [Google Scholar]
- [20].Shiboski SC, Shiboski CH, Criswell L, et al. American College of Rheumatology classification criteria for Sjogren's syndrome: a data-driven, expert consensus approach in the Sjogren's International Collaborative Clinical Alliance cohort. Arthritis Care Res (Hoboken) 2012;64:475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].LeRoy EC, Black C, Fleischmajer R, et al. Scleroderma (systemic sclerosis): classification, subsets and pathogenesis. J Rheumatol 1988;15:202–5. [PubMed] [Google Scholar]
- [22].Cottrell TR, Wise RA, Wigley FM, et al. The degree of skin involvement identifies distinct lung disease outcomes and survival in systemic sclerosis. Ann Rheum Dis 2014;73:1060–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Clements P, Lachenbruch P, Siebold J, et al. Inter and intraobserver variability of total skin thickness score (modified Rodnan TSS) in systemic sclerosis. J Rheumatol 1995;22:1281–5. [PubMed] [Google Scholar]
- [24].Medsger TA, Jr, Bombardieri S, Czirjak L, et al. Assessment of disease severity and prognosis. Clin Exp Rheumatol 2003;21(3 suppl 29):S42–46. [PubMed] [Google Scholar]
- [25].Meier FM, Frommer KW, Dinser R, et al. Update on the profile of the EUSTAR cohort: an analysis of the EULAR Scleroderma Trials and Research group database. Ann Rheum Dis 2012;71:1355–60. [DOI] [PubMed] [Google Scholar]
- [26].Allanore Y, Borderie D, Airo P, et al. Lack of association between three vascular endothelial growth factor gene polymorphisms and systemic sclerosis: results from a multicenter EUSTAR study of European Caucasian patients. Ann Rheum Dis 2007;66:257–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Alizadeh BZ, Broen J, Rueda B, et al. Functional variants of Fc gamma receptor (FCGR2A) and FCGR3A are not associated with susceptibility to systemic sclerosis in a large European study (EUSTAR). J Rheumatol 2010;37:1673–9. [DOI] [PubMed] [Google Scholar]
- [28].Koumakis E, Wipff J, Dieude P, et al. TGFbeta receptor gene variants in systemic sclerosis-related pulmonary arterial hypertension: results from a multicentre EUSTAR study of European Caucasian patients. Ann Rheum Dis 2012;71:1900–3. [DOI] [PubMed] [Google Scholar]
- [29].Mayes MD, Bossini-Castillo L, Gorlova O, et al. Immunochip analysis identifies multiple susceptibility loci for systemic sclerosis. Am J Human Genet 2014;94:47–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Lopez-Isac E, Campillo-Davo D, Bossini-Castillo L, et al. Influence of TYK2 in systemic sclerosis susceptibility: a new locus in the IL-12 pathway. Ann Rheum Dis 2016;75:1521–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Coustet B, Bouaziz M, Dieude P, et al. Independent replication and meta analysis of association studies establish TNFSF4 as a susceptibility gene preferentially associated with the subset of anticentromere-positive patients with systemic sclerosis. J Rheumatol 2012;39:997–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Gelber A, Manno R, Shah A, et al. Race and association with disease manifestations and mortality in scleroderma: a 20-year experience at the Johns Hopkins Scleroderma Center and review of the literature. Medicine 2013;92:191–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Laing TJ, Gillespie BW, Toth MB, et al. Racial differences in scleroderma among women in Michigan. Arthritis Rheum 1997;40:734–42. [DOI] [PubMed] [Google Scholar]
- [34].Nashid M, Khanna P, Furst D, et al. Gender and ethnicity differences in patients with diffuse systemic sclerosis—analysis from three large randomized clinical trials. Rheumatology 2011;50:335–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Steen VD, Domsic R, Lucas M, et al. A clinical and serologic comparison of African American and Caucasian patients with systemic sclerosis. Arthritis Rheum 2012;64:2986–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Denton CP. Systemic sclerosis: from pathogenesis to targeted therapy. Clin Exp Rheumatol 2015;33(4 suppl 92):S3–7. [PubMed] [Google Scholar]
- [37].Pakas I, Ioannidis JP, Malagari K, et al. Cyclophosphamide with low or high dose prednisolone for systemic sclerosis lung disease. J Rheumatol 2002;29:298–304. [PubMed] [Google Scholar]
- [38].Ioannidis JP, Vlachoyiannopoulos PG, Haidich AB, et al. Mortality in systemic sclerosis: an international meta-analysis of individual patient data. Am J Med 2005;118:2–10. [DOI] [PubMed] [Google Scholar]
- [39].Diot E, Giraudeau B, Diot P, et al. Is anti-topoisomerase I a serum marker of pulmonary involvement in systemic sclerosis? Chest 1999;116:715–20. [DOI] [PubMed] [Google Scholar]
- [40].Steen VD, Medsger TA., Jr Long-term outcomes of scleroderma renal crisis. Ann Intern Med 2000;133:600–3. [DOI] [PubMed] [Google Scholar]
- [41].Krzyszczak M, Li Y, Ross S, et al. Gender and ethnicity differences in the prevalence of scleroderma-related autoantibodies. Clin Rheumatol 2011;30:1333–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Steen VD. Autoantibodies in systemic sclerosis. Semin Arthritis Rheum 2005;35:35–42. [DOI] [PubMed] [Google Scholar]
- [43].Okano Y, Steen VD, Medsger TA. Autoantibody to U3 Nucleolar Ribonucleoprotein (Fibrillarin) in patients with systemic sclerosis. Arthritis Rheum 1992;35:95–100. [DOI] [PubMed] [Google Scholar]
- [44].Masataka K, Yutaka O, Junichi K, et al. Racial differences in the distribution of systemic sclerosis-related serum antinuclear antibodies. Arthritis Rheum 1994;37:902–6. [DOI] [PubMed] [Google Scholar]
- [45].Aggarwal R, Lucas M, Fertig N, et al. Anti-U3 RNP autoantibodies in systemic sclerosis. Arthritis Rheum 2009;60:1112–8. [DOI] [PubMed] [Google Scholar]
- [46].Ramos PS, Silver RM, Feghali-Bostwick CA. Genetics of systemic sclerosis: recent advances. Curr Opin Rheumatol 2015;27:521–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.